

Background: The current therapeutic strategy cannot suppress multiple cytokines in human disease.
Results: We developed a new strategy to selectively block γc-family cytokines.
Conclusion: Our novel strategy was proven effective both in vitro and in vivo.
Significance: This approach will fill in a gap in the existing therapeutic strategy.
Keywords: cell proliferation, cytokine action, drug design, interleukin, peptide interaction
Abstract
The common γ molecule (γc) is a shared signaling receptor subunit used by six γc-cytokines. These cytokines play crucial roles in the differentiation of the mature immune system and are involved in many human diseases. Moreover, recent studies suggest that multiple γc-cytokines are pathogenically involved in a single disease, thus making the shared γc-molecule a logical target for therapeutic intervention. However, the current therapeutic strategies seem to lack options to treat such cases, partly because of the lack of appropriate neutralizing antibodies recognizing the γc and, more importantly, because of the inherent and practical limitations in the use of monoclonal antibodies. By targeting the binding interface of the γc and cytokines, we successfully designed peptides that not only inhibit multiple γc-cytokines but with a selectable target spectrum. Notably, the lead peptide inhibited three γc-cytokines without affecting the other three or non-γc-cytokines. Biological and mutational analyses of our peptide provide new insights to our current understanding on the structural aspect of the binding of γc-cytokines the γc-molecule. Furthermore, we provide evidence that our peptide, when conjugated to polyethylene glycol to gain stability in vivo, efficiently blocks the action of one of the target cytokines in animal models. Collectively, our technology can be expanded to target various combinations of γc-cytokines and thereby will provide a novel strategy to the current anti-cytokine therapies against immune, inflammatory, and malignant diseases.
Introduction
The four-helical bundle structure is a common feature shared by many cytokines (1). Another shared feature is the sharing of the receptor and signaling components that account for “pleiotropy” and “redundancy,” the two important characteristics (2) of cytokines, and provide the reason to classify multiple cytokinases into a family. Examples include the IL-6 family that uses the gp130 molecule; the γc-family (IL-2, -4, -7, -9, -15, and -21) that uses γc, and the IL-3 family that uses the common β-molecule. The γc-cytokines diversely control normal immune responses (3) as exemplified by defective immune functions of knock-out mice or humans lacking individual γc-cytokines, receptors, or signaling components (4–7). Moreover, each γc-cytokine is connected with various immune and inflammatory disorders in humans. IL-2 has been implicated in inflammatory bowel diseases (IBD)3 (8–11). Likewise, IL-4 has been implicated in asthma (12); IL-7 has been implicated in multiple sclerosis, ulcerative colitis, and sarcoidosis (13), and IL-9 has been implicated in allergic inflammation (14) and in asthma (15). Overexpression of IL-15 in mice (16, 17) and in humans (18) is associated with T/NK leukemia. IL-15 has also been strongly connected with celiac disease (19–26). IL-21 (27) is a recent addition and is involved in the differentiation of B and follicular helper T cells (28), which has been implicated in IBD/celiac disease, psoriasis, uveitis (29), atopic dermatitis, systemic lupus erythematosus, multiple sclerosis, and type I diabetes (30, 31).
In these cases, involvement of more than two cytokines from a family (Table 1) has been implicated. Surprisingly, efficient treatment of these cases may prove challenging to the current anti-cytokine strategy that relies on monoclonal antibodies (one specific target) or signal inhibitors (many targets but poor specificity) (32). Because of the sigmoid nature of the cellular response to cytokines, a neutralization of only one factor in a combination of redundant factors would have negligible inhibitory effect when each factor is causing a near-saturation effect. Thus, we decided to design inhibitors that can target more than two γc-cytokines for clinical use. To this end, we took advantage of the recent advancement in the structural information on the binding of cytokines and receptors and chose to target the interface of the cytokine and γc interaction. We combined the natural sequences of γc-cytokines and synthesized composite peptides to be tested by biological assays. This design strategy not only allowed us to design inhibitors to target multiple γc-cytokines, but provided us the capacity to choose target cytokines by design. Here, we demonstrate that our lead peptide (BNZ132-1) selectively inhibits IL-2, IL-9, and IL-15 of the γc-cytokines but no other cytokines (a proof-of-concept). Additionally, we provide evidence for translational merit of our concept using ex vivo cells from a human disease. Our concept may be expandable to eventually creating a library of compounds to target any human diseases that pathogenically involve multiple γc-cytokines.
TABLE 1.
List of human diseases involving multiple γc-cytokines and human diseases associated with multiple γc-cytokines
The table was compiled through literature survey. Representative references for each disease listed is as follows: HAM-TSP (43, 58); celiac disease/IBD (19–21, 26); uveitis (29); asthma (14, 15, 55, 56); multiple sclerosis (61–63); and rheumatoid arthritis (64–67).
| Disease | Cytokines |
|---|---|
| HAM-TSP | IL-2, IL-15, (IL-9?) |
| Celiac disease/IBD | (IL-2), IL-15, IL-21 |
| Uveitis | IL-2, IL-21 |
| Asthma/chronic obstructive pulmonary disease | IL-4, IL-9 IL-5, IL-13 |
| Multiple sclerosis | IL-2, IL-9, IL-15, IL-21 |
| Rheumatoid arthritis | IL-7, IL-15, IL-21, IL-6 |
Experimental Procedures
Cell Culture and Proliferation Assay
Cells were maintained in 10% fetal bovine serum (FBS, Gemini Bio, West Sacramento, CA), RPMI 1640 medium (Invitrogen) supplemented with 2 mm glutamine, penicillin, and streptomycin in a humidified 5% CO2 incubator at 37 °C. For cytokine proliferation assay, cells were plated at 0.2 million/ml (for cell lines and 2 million/ml for PBMCs) in 0.2 ml of media containing appropriate cytokines in 96-well plates in triplicate for 20 h. One μCi of [3H]thymidine (GE Healthcare) or 20 μl of WST-1 (Clontech) reagent was added for 6 h before measurements. PBMCs were isolated from blood by the Ficoll-Paque density gradient (GE Healthcare), stimulated by 5 μg/ml PHA-P (Sigma), and expanded by 1 nm IL-2 (PeproTech, Rocky Hill, NJ) for 48 h before setting up a proliferation assay. On some occasions, T cells were negatively enriched from the expanded PBMCs using magnetic-activated cell sorting (Miltenyi, Cambridge, MA). PT-18 and derivative clones (33) were maintained with 1 nm human IL-3 (supplemented as a conditioned medium of NIH3T3 fibroblasts transfected with the human IL-3 gene prepared in-house).
Materials Derived from Patients
Samples were obtained from HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP) patients upon written and informed consent. The study was reviewed and approved by the institutional review board of the NINDS, National Institutes of Health.
Peptide Synthesis and FITC Conjugation
Peptides were synthesized and FITC-conjugated by Bachem (Torrance CA). The control peptide is a 19-amino acid scrambled sequence (QITISILSQINRVFHEKFI) of BNZ132-1. The F/P ratio (0.5) was determined based on A495 and A280.
Alanine Substitution
A library of alanine-substituted mutant peptides of BNZ132-1 were synthesized by Mimotopes (Melbourne, Australia) (34). The working stocks were prepared in PBS (pH 6.5). The peptides were added to the CTLL-2 cells at 1 or 10 μm (final concentrations) with 100 pm IL-2 or IL-15. The proliferation was measured 24 h thereafter. The assays were performed in triplicate and repeated three times.
Neutralizing Antibodies
Neutralizing antibodies against cytokines were purchased from the R & D Systems (Minneapolis, MN) as follows: human IL-2; MAB202, mouse IL-4; MAB404, human IL-7; MAB207, mouse IL-9; MAB409, human IL-15; MAB247, mouse IL-21; AF594 (polyclonal Ab), mouse IL-3; MAB403, mouse GM-CSF; MAB415, mouse stem cell factor; MAB455, and human FLT3-L; MAB308. Antibodies used for flow experiments (details in the supplemental material) were purchased from BioLegend (San Diego) and eBioscience (San Diego).
Immunoblotting
Cellular proteins were extracted using the RIPA buffer (0.5% Nonidet P-40, 10 mm Tris-Cl (pH 8.0), 1 mm EDTA, 0.5 mm EGTA, 0.1% sodium deoxycholate, 150 mm NaCl/proteinase inhibitor mixture (Roche Applied Science)). Samples were resolved on Tris-glycine gels (8–16% gradient gel, Invitrogen) under reducing conditions, transferred to a PVDF membrane (Millipore, Billerica, MA), and blocked overnight in the Superblock solution (Pierce). Primary Ab was diluted at 1:2000 in 0.05% Tween 20/PBS (T-PBS) for 1 h of incubation at 37 °C with the membrane. After five washes (5 min each) in T-PBS, the blot was incubated with horseradish peroxidase-conjugated secondary Abs against rabbit/mouse IgG (Cell Signaling Technologies, Danvers, MA, used at 1:3000) for 1 h at room temperature and visualized using chemiluminescence (ECL Plus, PerkinElmer Life Sciences) according to the manufacturer's instructions. Sample loadings were verified by reprobing blots using anti-vinculin Ab (clone; hVin-1, Sigma).
Flow Cytometry
About 2 × 105 cells were washed in 1% FBS/PBS (flow cytometry media) and resuspended in a 10-μl Ab mixture that contains appropriately (determined by prior titration experiments) diluted individual antibodies in flow cytometry media. Incubation was performed for 45 min on ice, and the pellets were then washed twice in flow cytometry media. Photomultiplier tube of the FACSAria II (BD Biosciences) was calibrated using isotype control antibodies with matching fluorochromes. Antibodies were purchased from BioLegend (San Diego). Data were analyzed using the FlowJo software (FlowJo, Ashland, OR).
Protein Alignment
For the structural comparison of cytokines, the T-coffee algorithm was used (35).
PEG Conjugation of BNZ132-1
BNZ132-1 was synthesized with a specific linker (CGSGG), conjugated with methoxy-PEG (20, 40, and 60 kDa)-maleimide molecules, and purified by a reverse phase HPLC. Conjugation was performed on the N and C termini of the peptide. Upon testing their inhibitory action using CTLL-2 cells,4 we decided to focus on the PEG40-conjugated BNZ132-1 (BNZ132-1–40, PEG40-linker, BNZ132-1).
In vivo dose of BNZ132-1-40 was determined based on the estimated IC50 values (∼0.5 nm), to be at or around 0.8 mg/kg. Separate pharmacokinetic studies indicated that the PEG40-BNZ132-1 (BNZ132-1–40) shows an 84.4-h half-life (data not shown) in mice, supporting the theory that two injections per week would maintain effective biological concentrations necessary for effective inhibition of target cytokines.
Mouse in Vivo Experiments
All mouse experiments have been conducted following a protocol approved by the IACUC of University of Maryland School of Medicine, and the mouse housing and injection part of the experiments were conducted at Bioqual Inc. (Rockville, MD).
Age-matched (12 weeks old) female congenic Thy 1.1-C57B6 mice (The Jackson Laboratory) were used for the preclinical animal studies. T-reg expansion by IL-2 was induced in these mice by injecting 150 μg of recombinant human IL-2 (PeproTech) intraperitoneally on days 1 and 4. To observe the therapeutic rather than prevention effects of our compounds, intervening compounds such as neutralizing anti-IL-2 antibody (R & D Systems, MAB 202, 100 μg per injection) or BNZ132-1–40 (1 mg/kg, ∼25 μg per mouse) were injected intraperitoneally to expect slow release. Venous blood was collected on day 7 to purify PBMCs by the Ficoll-Paque density separation. Cells were stained by a mixture of antibodies (FITC-CD8; clone 53-6.7, PE-CD44; clone Im7, PerCP/Cy5.5-CD4; clone RM4-5, APC-CD25; clone 3C7, Brilliant Violet 421-CD3; clone 145-2C11) to identify major T subsets including the T-regs.
For the IL-15 leukemia study, an established cell line producing its own IL-15 (from IL-15 transgenic mice on the Thy 1.2 C57B6 background (17)) has been maintained in vitro in 10% FBS/RPMI 1640 medium without any exogenous cytokine in culture. One million cells were injected in 150 μl of sterile PBS intravenously (i.v.) via the tail vein on day 0. Intervening compounds (anti-IL-15 Ab and BNZ132-1–40, low at 1 mg/kg and high at 10 mg/kg) were injected on days 1, 4, 8, and 11. Tofacitinib was continuously administered using a subcutaneous osmotic pump (ALZET, Cupertino, CA) as described before (36). Body weight of the mice was monitored every week to detect signs of general distress. With mice showing over 20% of body weight loss, blood was collected for flow analysis to detect the expansion of Thy 1.1+ leukemic cells. Antibodies used for flow cytometry were Brilliant Violet 421-Thy 1.1 (CD90.1), clone OX-7, and Brilliant Violet 510-Thy 1.2 (CD90.2), clone 53–2.1.
Results
Targeting the Interface of the γc and Cytokine Interaction
Based on data mining, we compiled a table of human diseases in which more than two members of the γc-cytokines have been implicated (Table 1). We aimed at designing antagonists against γc-cytokines that target multiple but select members of this family. As a start, we chose IL-2 and IL-15. The D-helices (last of the four α-helices) of γc-cytokines primarily interact with the γc (37), and the critical residues of IL-2 or IL-15 that interact with each receptor subunit (i.e. α, β, or γc) have been identified (38). Consistently, the D-helices, among all four helices, show the highest conservation across mammalian species (Table 2). The alignment of D-helices from human γc-cytokines demonstrated a mildly conserved motif within this region (the γc-box, Fig. 1A). We also noticed an additional short motif (the IL-2/15-box, Fig. 1B) involving IL-2 and IL-15 (IL-21 may be included, albeit weakly). These two form a subfamily in the γc-family due to the sharing of another receptor subunit IL-2/IL-15Rβ between them (39, 40). Our findings may indicate that these D-helices of two cytokines spatially overlap more closely than with D-helices from other γc-cytokines in their binding to the γc.
TABLE 2.
Conservation of D-helices among γc-cytokines across mammalian species
Left, homology/similarity of IL-2 at the amino acid level were compared among mammalian species for each of the four helices (A–D). Right, amino acid sequences of 6 human γc-cytokines were compared (similarity level, && denotes no significant homology was observed) at each of the four helices (A–D). The similarity/homology scores were determined by the T-coffee algorithm (35).
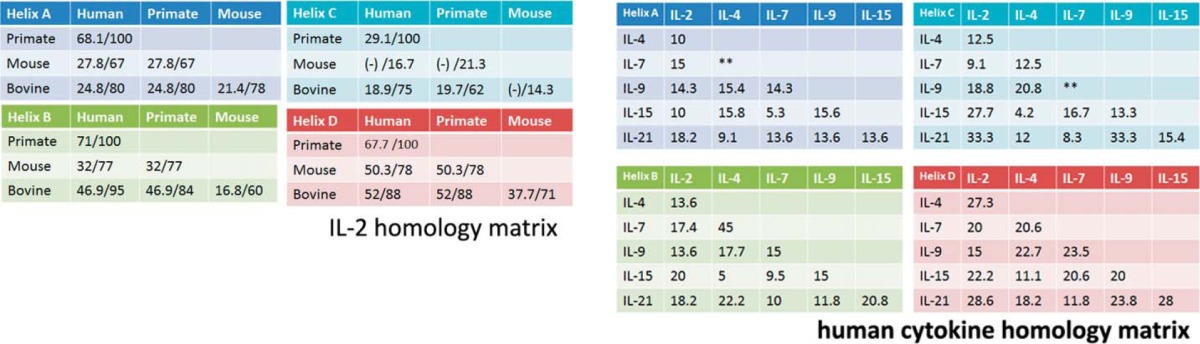
FIGURE 1.
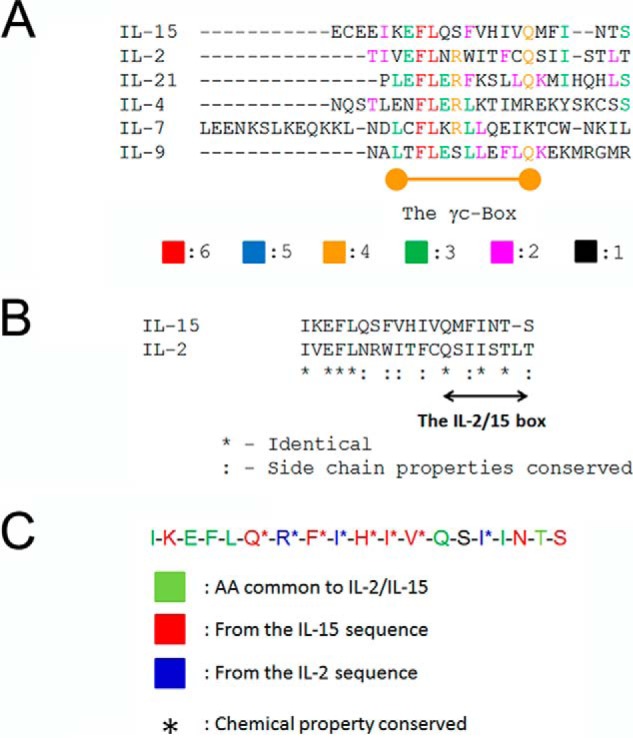
Conserved primary structures of the D-helices from γc-cytokines. A, alignment of the six human γc-cytokines. Amino acid sequences of the D-helices from human γc-cytokines (Table 2) have been aligned using the T-coffee algorithm (35). A region with moderate conservation (identical amino acids or conservative substitutions) was named as the γc-Box. Color codes represent how many human γc-cytokines share the indicated amino acids. B, alignment of human IL-2 and IL-15. Between IL-2 and IL-15, another conserved region was identified and named the “IL-2/15 box.” * means identical between IL-2 and -15, and : conservative substitutions. C, design of the BNZ132-1 peptide. Based on a logic described for supplemental Fig. 1, 120 candidate peptides (supplemental T a b l e S 2) were designed and run through a series of computer simulations (supplemental Fig. S1). The resultant 39 peptides were examined by biological assays (examples shown in Fig. 3) to choose a final peptide shown in C. Color codes are as follows: green, common to IL-2/15; red, from the IL-15 sequence; blue, from the IL-2 sequence.
Design of BNZ132-1, a Specific Inhibitor for the IL-2/IL-15 Subfamily
Although our ultimate goal is to prepare a library of compounds that could cover all potential combinations of γc-cytokines (see supplemental Table 1 for detail), we first attempted to design a common inhibitor for IL-2 and IL-15 to test our concept, because these two cytokines are structurally most related among γc-cytokines (39, 40). The three-dimensional structures of the D-helix of IL-2 and IL-15 are nearly super-imposable, but fine differences exist in their binding to γc (38). We hypothesized that by leveraging differences in their primary structures, we could design an inhibitory peptide that equally blocks IL-2 and IL-15. Our design logic was as follows: 1) such peptide(s) should form a helical structure; 2) it should contain key amino acids responsible for the binding of target cytokines to γc; and 3) peptides with composite sequence derived nearly equally from IL-2 and IL-15 (minimum bias to either cytokine) should be tested. Of the 120 peptides (supplemental Table 2) that were designed and screened by a computer docking simulation (supplemental Fig. 1), 39 showed appropriate docking. They were all synthesized in small scale and tested for inhibition on IL-2 and IL-15. BNZ132-1 (Fig. 1C) was finally chosen as the most potent inhibitor of IL-2 and IL-15 actions.
Direct Binding of BNZ132-1 to the γc (CD132) Molecule
To prove that BNZ132-1 directly binds to the γc molecule (CD132), we conducted a binding study. Human γc-molecule was expressed on murine PT-18 cells by gene transfection (PT-18huγ, Fig. 2, F and H). BNZ132-1 and a scrambled control peptide (see under “Experimental Procedures”) were purchased as fluorescein isothiocyanate (FITC)-conjugated form and used in binding studies. Notably, PT-18 parental cells express marginal levels of mouse γc (Fig. 2, E and F versus H) (33) that are almost undetectable by a commercial antibody (TUGm2). Similarly, FITC-BNZ132-1 failed to show binding (Fig. 2A) due to the low F/P ratio (0.5) of FITC peptides, leaving only the transfected human γc as the detectable target on PT-18huγ for FITC peptides. Under these conditions, FITC-BNZ132-1 only showed binding to PT-18huγ cells (Fig. 2, B and C) and not with PT-18 parental cells (Fig. 2A). FITC control peptides failed to show detectable binding to any cells used (Fig. 2, A and B). To test the specificity of the binding, FITC-BNZ132-1 was incubated with 4-fold excess unlabeled BNZ132-1 before binding to target cells, and the binding was nearly completely inhibited (Fig. 2D) in a dose-dependent manner. These results collectively confirmed our original hypothesis that BNZ132-1 directly binds to the γc subunit.
FIGURE 2.

Binding of the FITC-BNZ132-1 peptide to the γc-molecule. A, no binding of FITC-BNZ132-1 to the parental PT-18 cell line that lacks high expression of mouse γc. PT-18 cells express marginal levels of the mouse γc-molecule (33), which is insufficient to be detected by the FITC-BNZ132-1 binding due to the low F/P ratio of this conjugate. No binding was observed by the FITC-control peptide. B, binding of FITC-BNZ132-1 but not that of FITC-control peptide to PT-18huγ. PT-18huγ is a subclone of PT-18 that expresses extremely high levels of human γc (CD132) due to gene transfection. FITC-BNZ132-1, but not FITC-control peptide, shows binding to these cells. C, dose-dependent binding of FITC-BNZ132-1 to PT-18huγ. To test the specificity of the FITC-BNZ132-1 binding, various concentrations of FITC-BNZ132-1 were incubated with target cells, showing the dose-dependent nature of the binding. D, inhibition of FITC-BNZ132-1 binding by the unlabeled BNZ132-1 peptide. To further test the specificity of the binding of BNZ132-1, a competition with cold (unlabeled) ligand was performed. The results show dose-dependent inhibition of the binding of FITC-BNZ132-1 by the unlabeled peptide. E and F, only marginal staining was observed in these cells by anti-mouse CD132 (γ-c) antibody. G and H, anti-human CD132 antibody did not stain mouse PT-18 cells but showed a strong staining with the same cells transfected with the human CD132 (γ-c) gene.
Inhibitory Profile of BNZ132-1 to Cytokines
Fig. 3A shows the efficient inhibition of IL-2 and IL-15 by BNZ132-1, which is an affirmation of the screening process described above. To our knowledge, this is the first example of a single peptide equally blocking two cytokines from this family. We next tested whether BNZ132-1 inhibits other γc-cytokines using murine PT-18 cells (33, 41). Fig. 3B demonstrates that BNZ132-1 does not inhibit IL-4 in PT-18 (p = 0.32), but it partially inhibits IL-9 (p = 0.001), albeit not as robust as its inhibition on IL-2 or IL-15. A recent report mapped IL-9Rα structurally close to IL-2/IL-15Rβ (42) among the private chains for γc-cytokines, and this may account for the unexpected inhibition of IL-9 by BNZ132-1. Finally, we tested whether the two remaining γc-cytokines (IL-7 and -21) could be inhibited by BNZ132-1. We established a PT-18 subclone that responds to human IL-21 by transfecting human IL-21Rα. Although hIL-21Rα+PT-18 cells did not robustly proliferate to IL-21 (Fig. 3B), IL-21 prevented their apoptotic death following the withdrawal of IL-3 from the culture (Fig. 3C, p = 0.001), and this process was not inhibited by BNZ132-1 (p = 0.59, not significant). Next, we tested whether our peptide blocks the IL-7 function. Curiously, the human-mouse chimeric IL-7 receptor (hIL-7Rα+m γc) generated on PT-18 by gene transfection failed to confer response to human IL-7.5 Thus, we used ex vivo human peripheral T cells to address the question. As shown in Fig. 3D, human T cells demonstrated moderate proliferative response to 10 nm IL-7 (versus no cytokine; p = 0.002), which was not significantly inhibited by BNZ132-1 (p = 0.30). Collectively, the inhibitory capacity of BNZ132-1 seems restricted to IL-2, -15, and -9 but excludes IL-4, -7, or -21.
FIGURE 3.

Inhibitory spectrum of BNZ132-1 over γc- and non-γc-cytokines. A, inhibition of IL-2 and IL-15 by BNZ132-1; CTLL-2 proliferation assay. CTLL-2 has been incubated with 1 nm human IL-2 or human IL-15 (PeproTech) in the presence of indicated concentrations (micromolars) of BNZ132-1. Neutralizing antibodies against each cytokine (R & D Systems, 5 μg/ml) were included as controls. After 20 h, the cellular proliferation was determined by the WST-1 reagent. A 19-mer control peptide was used as a negative control at 5 μm (abbreviated as Ctrl pep throughout the figures). B, inhibition of IL-9 but not of IL-4 by BNZ132-1; PT-18 proliferation assay. PT-18β (subclone of PT-18 transfected with human IL-2/IL-15Rβ (CD122) (33)) or PT-18h21Rα (subclone transfected with human IL-21Rα) cells were incubated with 1 nm mouse IL-4, mouse IL-9, or mouse IL-21 in the presence or absence of the indicated doses (micromolars) of BNZ132-1. Neutralizing antibodies to each cytokine (5 μg/ml, except anti-mouse IL-21 Ab at 15 μg/ml) and a control peptide (5 μm) were included as negative controls. WST-1 assay was conducted as described above. C, no inhibition of IL-21 by BNZ132-1; PT-18 h21Rα survival assay. PT-18h21Rα cells do not proliferate robustly to human IL-21 (B). Both parental and hIL-21Rα-positive PT-18 cells were washed and incubated without IL-3 for 12 h before the assay that would cause apoptotic death within 24 h due to death by neglect. 1 nm human IL-21 (PeproTech) was added in the presence or absence of 5 μm BNZ132-1 or control peptide for 24 h, and cellular apoptosis was determined by the staining of phycoerythrin-annexin V (BD Biosciences). Only hIL-21Rα transfected, but not the parental cells, showed survival by human IL-21 (p = 0.001), which was not blocked by BNZ132-1 (p = 0.48, nonsignificant). D, no inhibition of IL-7 by BNZ132-1; human PBMC proliferation assay. The ex vivo T cells were prepared from human PBMCs as described in the text. IL-2 was withdrawn from the culture 12 h prior to the assay. Cells were incubated with 10 nm human IL-7 (Gemini) for 24 h in the presence or absence of the indicated amounts (micromolars) of BNZ132-1. Human IL-2 or IL-15 (1 nm each) was used as positive controls. As negative controls, neutralizing antibodies (5 μg/ml) against each cytokine and a control peptide (5 μm) were tested. WST-1 proliferation assay was conducted as described above. E, no inhibition of non-γc-cytokines by BNZ132-1; PT-18 proliferation assay. PT-18β cells were depleted of IL-3 for 12 h prior to the assay. Indicated cytokines (IL-15/Flt3 ligand from human, others from mouse sequences, each at 1 nm) were added to the fasted PT-18β for 20 h with or without 15 μm BNZ132-1 and then pulsed with [3H]thymidine (GE Healthcare). Neutralizing antibodies (5 μg/ml, except anti-mSCF Ab at 20 μg/ml) to each cytokine and a control peptide were included as controls. The y axis represents counts/min values measured by a β-counter. F–H, dose-dependent inhibition of human IL-2, IL-15, and IL-9 in NK92 cells by BNZ132-1. NK92 cells were cultured with human IL-2. Cells were withdrawn from IL-2 overnight, and 2 × 105 cells (in 200 μl volume) were incubated with the indicated concentrations of cytokines and BNZ132-1 for 24 h and then 20 μl of WST-1 reagent Clontech) was added for 6 h to measure A450. I, dose-dependent co-inhibition of human IL-2 and IL-15 in human peripheral T cells by BNZ132-1. Human PBMCs were prepared as described above and enriched by a magnetic-activated cell sorting (Miltenyi) negative selection. Purified T cells were cultured without IL-2 overnight and then 4 × 105 cells (in 200 μl of culture) were incubated with cytokines and BNZ132-1 at the indicated concentrations for 24 h and then with WST-1 reagent (Clontech) for 12 h before A450 measurements. Experiments were conducted in triplicate, and results represent one of at least three independent occasions (applies to all experiments in Fig. 3).
Off-target Effects of BNZ132-1 on Non-γc-cytokines
Next, we tested several non-γc-cytokines to examine the potential off-target effects of our peptide. We again used PT-18, which natively responds to an array of non-γc-cytokines (33). As shown in Fig. 3E, BNZ132-1, even at an excess dose of 15 μm, showed no inhibition on these non-γc-cytokines.
Dose Inhibition Profile of BNZ132-1
Next, we estimated the affinity of BNZ132-1 binding to γc by analyzing the dose inhibition response. As the Cheng-Prusoff equation (43, 44) dictates, IC50 is closely associated to the binding affinity of the antagonist to the cellular receptor. Fig. 3, F–I, demonstrates that BNZ132-1 inhibits IL-2 and -15 by 50% at or around 50–500 nm in human cells (NK92 cell line and ex vivo human T cells, see Fig. 4 D, although doses for efficient biological inhibition varied between cell types, including murine cells that are not shown here), which gives ∼25–250 nm binding affinity of BNZ132-1 to target cells. The inhibition of IL-9 was less efficient as a micromolar concentration of peptide was needed for efficient blocking. These results, however, show some discrepancy from those obtained from direct binding assays (see under “Discussion”).
FIGURE 4.

Effective inhibition of a combined function of γc-cytokines by BNZ132-1. A, dose response of human ex vivo T cells to recombinant human IL-2 or IL-15. Human ex vivo peripheral T cells were prepared as described under “Experimental Procedures.” Four hundred thousand cells (in 200 μl culture) were incubated with the cytokines, antibodies, and BNZ132-1 at the indicated concentrations for 24 h, and cellular proliferation was determined using the WST-1 reagent (Clontech). The y axis represents A450 values. The graph indicates the EC50 concentrations of each cytokine (∼50 pm). B, dose inhibition response of anti-IL-2 and anti-IL-15 antibodies used in this study. Approximately 1 μg/ml inhibition of each antibody was sufficient to suppress the saturating effect of each target cytokine on ex vivo human T cells, with no cross-reactivity to the other cytokine at a higher 5 μg/ml (right panel). C, antibody titration and dose inhibition relationship of single and combined Abs on IL-2, IL-15, or a combination of both. Each of anti-IL-2 or anti-IL-15 antibodies had marginal effects (25–30%) in inhibiting the equimolar mixture of IL-2 and IL-15 (100 pm each, to achieve saturating response of T cells as a combination) at high concentrations (5 and 10 μg/ml). When each cytokine was combined at close to the ED50 concentrations, the inhibition by the single Ab was discernibly better (40–45%). The numbers on the x axis for the mixture of antibodies indicate those for each antibody (i.e. 1.25 = 1.25 μg/ml anti-IL-2 + 1.25 μg/ml anti-IL-15). The mixture of Abs reached close to 100% inhibition irrespective of the concentration of mixed cytokines. D, combinatorial effects of IL-2 and IL-15 to human ex vivo T cells were effectively inhibited by BNZ132-1. Shown is the dose inhibition curve of combined IL-2 + IL-15 effects on ex vivo human T cells by BNZ132-1. Notably, the inhibition by BNZ132-1 is similar to those by anti-IL-2 + anti-IL-15 antibodies shown in C. E, translational potential of BNZ132-1 for HAM-TSP. PBMCs of HAM-TSP patients were tested in a spontaneous proliferation assay as described previously (60). The cellular proliferation was measured by the thymidine incorporation assay. The y axis represents counts/min values. Doses on the x axis for BNZ132-1 are 3 and 0.3 μm. Antibodies against IL-2 or IL-15 (1 and 10 μg/ml of each, R & D Systems) are shown. The p values are as follows: between no treatment and BNZ132-1(0.3), 0.018; between no treatment and BNZ132-1 (3), 0.0018; between BNZ132-1 (3) and anti-IL-2 + IL-15 (10), 0.29. The assay was performed in triplicate, and the figure represents a typical result out of five different HAM-TSP patients.
Efficient Inhibition by BNZ132-1 of the Combined Cytokines Sharing Redundant Function
So far, we have shown that BNZ132-1 inhibits not only one or all γc-cytokines but three of these cytokines (IL-2, -9, and -15). We next asked whether BNZ132-1 can effectively block the combined effect of two γc-cytokines (i.e. IL-2 and IL-15). To this end, we conducted a series of experiments using human ex vivo T cells to determine basic conditions such as the saturating cytokine concentration and antibody concentrations necessary to achieve full inhibition of target cytokines (Fig. 4, A–C). Notably, even a 50:50 mixture of IL-2 and IL-15 cannot be inhibited by 50% using either anti-IL-2 or anti-IL-15 antibody alone if the combined cytokines make a near-saturating response (Fig. 4B, 100 pm + 100 pm). Naturally, each antibody could more efficiently block the mixture if the single and combined effects of cytokines are on the linear phase of the response (50 pm + 50 pm). This can be explained by two factors. 1) The gradient of the dose-response curve at the semi-linear phase is not equal to 1 (Fig. 4A, 25 pm, 10%; 50 pm, 45%; 100 pm, 75%). 2) At near saturation dose, doubling the cytokine concentration (e.g. from 100 to 100 + 100 combination) could only increase the biological response from 75 to 95% of saturation; therefore, an Ab to either factor would block about 20% of the response. These demonstrations suggest that a single Ab treatment may fail to show discernible effects in vivo against a mixture of factors even if the target cytokine represented nearly 50% of the biological activity in the mixture. BNZ132-1 per se blocked the T-cell proliferation as efficiently as the combined antibodies at two doses of the stimulation, demonstrating the unique and efficient therapeutic potential of our compound for multicytokine diseases.
Translational Merit for BNZ132-1 in a Human Disease
We and others previously reported that freshly isolated T lymphocytes from HAM-TSP patients spontaneously proliferate ex vivo without external activations (45, 46) and that this proliferation is mediated by the formation of an autocrine loop involving at least IL-2 and IL-15 as a consequence of transcriptional trans-activation by Tax of HTLV-1 (46, 47). Above, we have shown that one Ab against each cytokine would have limited inhibitory capacity to the overall cellular responses to a combination of functionally redundant cytokines. A similar result was observed in HAM-TSP cases (46). As shown in Fig. 4E, BNZ132-1 indeed inhibited HAM-TSP T cells as effectively as the combined anti-IL-2 and -IL-15 antibodies, which underscores two major arguments of our study. 1) There are human diseases that pathogenically involve more than two members of a cytokine family. 2) BNZ132-1 (and similar peptides from our concept) would be a superior inhibitor to a single Ab for such circumstances.
Inhibition of the Signaling Events by BNZ132-1 Is Specific for and Limited to Ones Generated by the Target Cytokine
Cellular proliferation often represents a combination of distinct signaling pathways; therefore, inhibiting one branch of the signaling could stop the proliferation while keeping other branches activated and leaving nonproliferative activation machineries of the cell in operation. Thus, we tested whether BNZ132-1 comprehensively silenced multiple signaling cascades triggered by its target cytokines. Fig. 5 shows the inhibition by BNZ132-1 of the tyrosine phosphorylation of key molecules representing major branches of the γc-cytokine signaling, i.e. JAK-STAT, PI3K-Akt, and MAPK axes. A complete inhibition by BNZ132-1 of these signaling cascades by human IL-15 (Fig. 5A in PT-18β (33)), mouse IL-9 (Fig. 5B in PT-18), and human IL-2 (Fig. 5C in human peripheral T cells) has been observed. Cellular proliferation shown in Fig. 3 paralleled the pattern of inhibition. IL-4, a nontarget cytokine for BNZ132-1, utilizes STAT6 (Fig. 5B), although a marginal phosphorylation of STAT5 is known to happen (Fig. 5B), but BNZ132-1 did not inhibit this action of IL-4, although similar phosphorylation events caused by IL-9 in the same cell were completely blocked, strongly illustrating its specificity (no off-target effects even on a γc-cytokine outside of the target spectrum). This observation further implies that BNZ132-1 may have less severe side effects than small signal inhibitors (see under “Discussion”).
FIGURE 5.
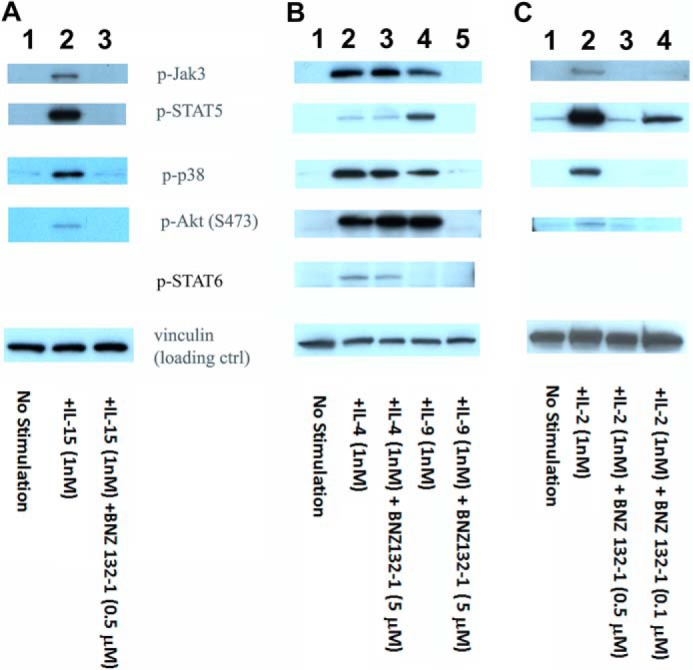
Comprehensive inhibition by BNZ132-1 of signaling events caused by target γc-cytokines. A, comprehensive inhibition of IL-15 signaling in PT-18β by BNZ132-1. Cells had been withdrawn from IL-3 for 12 h and then stimulated with 1 nm human IL-15 with or without 0.5 μm BNZ132-1 for 15 min before extracting cellular proteins. Phosphorylation of targets was detected using commercial antibodies (Cell Signaling), followed by ECL. Lane 1, no stimulation; lane 2, IL-15(1 nm); and lane 3, IL-15 (1 nm) + BNZ132-1 (0.5 μm). B, signal transduction of IL-4 in PT-18 was not inhibited, whereas the IL-9 signal was inhibited by BNZ132-1. Cells were fasted from IL-3 for 12 h then stimulated with 1 nm mouse IL-4 or mouse IL-9 in the presence or absence of an excess dose of BNZ132-1 (5 μm) for 15 min before extracting cellular proteins. Phosphorylation of the indicated proteins was detected using phosphospecific antibodies (Cell Signaling), followed by ECL. IL-4 is the only γc-cytokine that induces the phosphorylation of STAT6. As shown before, only IL-4, but not IL-9, induced the tyrosine phosphorylation of STAT6. Conversely, IL-4 showed only marginal phosphorylation of STAT5, whereas IL-9 induced a strong STAT5 phosphorylation. Notably, only IL-9 action was inhibited by BNZ132-1. Lane 1, no stimulation; lane 2, IL-4; lane 3, IL-4 + BNZ132-1; lane 4, IL-9; and lane 5, IL-9 + BNZ132-1. C, IL-2 signal in human PBMC was blocked by BNZ132-1. Human ex vivo T cells were prepared as described in the text. After 24 h of IL-2 depletion, cells were stimulated by 1 nm IL-2 with or without BNZ132-1 (0.5 or 0.1 μm) for 20 min before lysis. Tyrosine phosphorylation of targets was detected by antibodies (Cell Signaling), followed by ECL. Lane 1, no stimulation; lane 2, IL-2 stimulation; lane 3, IL-2 + BNZ132-1 (0.5 μm); and lane 4, IL-2 + BNZ132-1 (0.1 μm). In all blots, anti-vinculin Ab (Sigma) was used to validate equal protein loadings.
Dissection of the Function-Structure Aspects of the BNZ132-1 Peptide
To investigate the structure-function relationship of BNZ132-1, we next conducted the alanine scanning mutation analysis (48) on BNZ132-1. As shown in Fig. 6, A and B, substitution of each residue resulted in various perturbations of the cytokine inhibition by BNZ132-1. We used an excess dose of mutants (10 μm) to ascertain the saturated effect of them. A summary of our observations is as follows. 1) No substitution augmented the inhibitory capacity of the peptide beyond that of the original peptide. 2) IL-2 competition by BNZ132-1 is more susceptible to Ala single mutation than the IL-15 competition. 3) The N terminus seems more critical for the inhibitory function as substitutions of the first seven amino acids led to a profound loss of inhibition for both IL-2 and IL-15, although these substitutions more specifically affect the inhibition on IL-15. 4) The C terminus seems slightly dispensable for the inhibitory function on IL-15. Curiously, IL-2 inhibition seems to require this region. 5) Gln-13 appears to have special importance, which is consistent with a structural demonstration that Gln-13 is a conserved residue because it is crucial for the binding of many γc-cytokines to γc (38). In general, replacement of structurally important amino acids (i.e. those involved in the physical contact with the γc) with Ala shows detrimental effects on the inhibitory activity of BNZ132-1, suggesting that the binding of BNZ132-1 to the γc is mediated by a similar mechanism as has been demonstrated with IL-2 or IL-15 in a previous publication (38). Additional mutational and structure analyses are underway to decipher the unique nature of BNZ132-1 more in depth as a novel inhibitor of multiple γc-cytokines.
FIGURE 6.
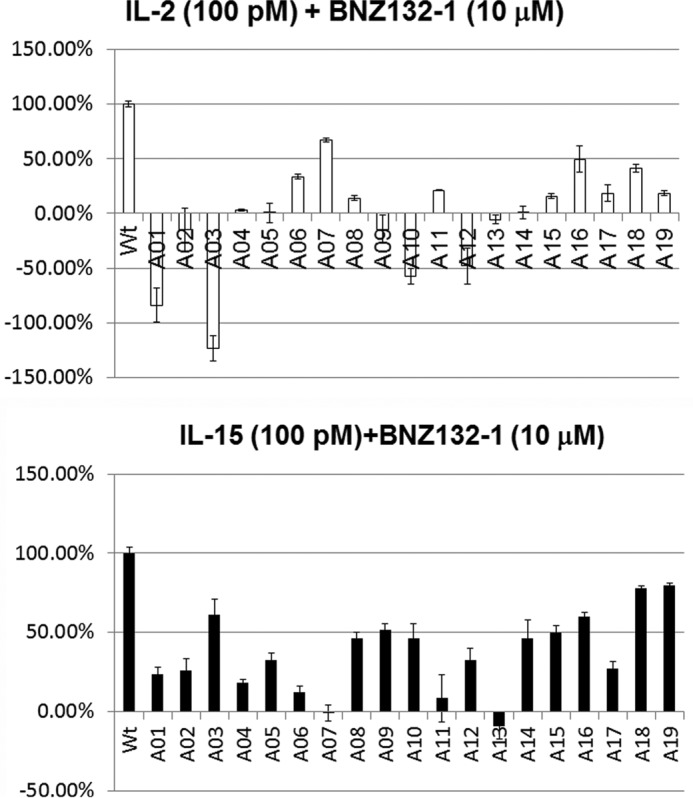
Mutational analysis to evaluate the functional importance of each of the 19 amino acids of BNZ132-1, alanine scanning. Alanine-substituted peptides were synthesized in small scale and were tested using the CTLL-2 cell assay if they inhibit the proliferative activity of IL-2 and IL-15, as described in the text. A05 (x axis), for example, denotes that the amino acid at the 5th position (leucine in IL-2 and IL-15) was replaced by alanine. The y axis of the graph represents the inhibitory activity of the mutated peptide relative to the original BNZ132-1. One hundred % indicates that the mutant is as potent as BNZ132-1 and 0% indicates that the peptide has lost the inhibitory function. In some cases, the peptides became agonistic (stimulating proliferation rather than inhibiting it) after the mutation, which are represented by negative numbers in the graph. Results represent three independent experiments.
Potent in Vivo Inhibitory Action of BNZ132-1 on the Action of IL-2, One of the Target Cytokines of This Peptide
To test the potential of BNZ132-1 for clinical application, we conducted a few animal studies. The continued presence of human IL-2 in mice, by transgenesis or cytokine administration, is known to cause a temporal expansion of CD4+CD25+ T-regs as reported by us and others (49, 50). We thus challenged C57B6 mice with recombinant human IL-2 and tested whether the subsequent expansion of T-regs would be blocked by BNZ132-1. To elongate the in vivo half-life of BNZ132-1, we conjugated the peptide to PEG (40-kDa average molecular mass) at the N terminus of the peptide (see “PEG Conjugation of BNZ132-1” under “Experimental Procedures” for detail). As shown in Fig. 7A, the PEG conjugation hardly changed the inhibitory nature of BNZ132-1. Human recombinant IL-2 was administered (intraperitoneally) into C57BL6 mice on days 1 and 4 (150 μg in 150 μl of sterile PBS per injection) into three groups of mice (n = 5). One additional group (n = 3) was injected with PBS alone as a negative control. Out of the three IL-2-challenged groups, one group received no intervention (received only PBS injection); another group received intervention by BNZ132-1–40 (5 mg/kg), and the last group received an intervention by neutralizing anti-IL-2 antibody (R & D Systems) as shown in Fig. 7B. Blood was collected on day 7 and was analyzed by polychromatic flow cytometry to trace the changes of T-regs in the CD4 T cell compartment. The experimental design is illustrated in Fig. 7B. In Fig. 7C, a representative flow cytometry pattern indicates the temporal expansion of T-regs by IL-2 and the concurrent inhibition of the expansion by BNZ132-1–40. Notably, BNZ132-1–40 was as efficient as anti-IL-2 antibody by this assessment. Fig. 7D shows the statistical comparison of the results obtained from individual group.
FIGURE 7.

Effective in vivo inhibition of IL-2-mediated T-reg expansion in mice by BNZ132-1–40. A, intact biological characteristics of BNZ132-1 following PEG40 conjugation. BNZ132-1 peptide was conjugated to PEG40 at the N terminus of the peptide as described under “Experimental Procedures” to prolong the half-life in vivo. To test that the conjugation did not affect the inhibitory action of the peptide on target cytokines, a proliferation assay was conducted using the IL-2 and CTLL-2 cell line. The y axis represents the A450 reading (cellular proliferative response) after the addition of WST-1 reagent. At 100–1000 nm, the peptide and the PEG40-conjugated form (BNZ132-1–40) showed a similar inhibitory profile on the action of IL-2. Similar results were obtained with IL-15 and IL-9 actions (A. Basheer, unpublished observations). Control peptide (a 19-mer scrambled peptide shown above) showed no inhibition even at a higher dose of 5 μm. B, design of the cytokine injection study. Recombinant human IL-2 (PeproTech) was injected (intraperitoneally) to induce T-regs in mice. Intervention was performed by injecting relevant compounds (neutralizing anti-IL-2 antibody or BNZ132-1–40, intraperitoneally) on days 2 and 5. Venous blood was collected on day 7 to purify PBMCs for subsequent flow cytometry analyses. C, representative flow cytometry data. Following cytokine injections and interventions by anti-cytokine compounds, mouse PBMCs were analyzed by polychromatic flow cytometry. T cells were gated by anti-CD3 staining, and the percentages of CD25+CD4+ (T-regs), CD25−CD4+ (conventional CD4), and CD4−CD25−(CD8 T cells) were measured as shown. D, statistical evaluation of the T-reg data. Obtained percentages of T-regs in the CD4 compartment have been processed by a Medcalc software (MedCalc bvba, Ostend, Belgium), and resultant average and 95% confidence ranges are grouped by treatment and are shown.
BNZ132-1 Potently Prevented Mice from IL-15-mediated Experimental Leukemia
To further assess the therapeutic effect of BNZ132-1 in vivo, we have used a unique IL-15-mediated leukemic model for human large granular lymphocyte leukemia that we have established (17, 51). The leukemic cells were established from IL-15 transgenic mice (51), producing human IL-15 and sustaining their in vitro and in vivo growth (17). The cells quickly manifest secondary leukemia upon injection into syngeneic C57B6 normal mice (17). We have already demonstrated that interventions to IL-15 action (i.e. by neutralizing anti-IL-15 (17) or by tofacitinib, a JAK3 inhibitor (36)) prevents the IL-15-dependent expansion of leukemic cells in host mice. As shown in Fig. 8A, BNZ132-1–40 completely protected mice (n = 5) from leukemic death caused by the injection of IL-15 leukemic cells (one million cells injected i.v. on day 0). In contrast, untreated mice died within 30 days (n = 3). Neutralizing anti-IL-15 antibody, the positive control for the experiment, protected mice as well as we reported previously (17). Tofacitinib, which never perfectly protected host mice from leukemic death in our hands (36), only showed life-elongating effects. The experiment was designed in such a way that we could distinguish injected cells and host cells by the congenic marker Thy 1 (see “Mouse in Vivo Experiments” under “Experimental Procedures” for detail). This allowed us to track whether the injected leukemic cells (Thy 1.2+) expanded in the host (Thy 1.1+) environment. Fig. 8B clearly shows the lack of even traces of Thy 1.2+ cells in BNZ132-1–40 treated mice, whereas untreated mice or tofacitinib-treated mice near death showed massive to moderate expansion of Thy 1.2+ leukemic cells in their blood. Collectively, these two preclinical animal studies strongly suggest that the BNZ132-1 peptide, when appropriately modified to ensure in vivo stability, would efficiently block the pathogenic action of its target cytokines (i.e. IL-2 and IL-15).
FIGURE 8.
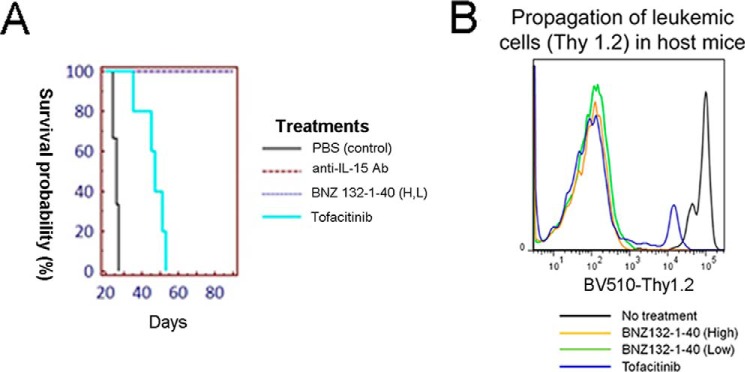
Protection of mice from IL-15-mediated CD8 T leukemia. A, survival of mice treated with BNZ132-1–40. B, lack of the expansion of Thy 1.2+ leukemic cells in surviving mice. We have previously reported the occurrence of fatal CD8 T cell leukemia in our IL-15 transgenic mice (17, 36, 51). We established a mouse leukemic cell line (K2) that produces human IL-15 and sustains perpetual growth into fatal secondary CD8 T cell leukemia when injected into syngeneic (C57B6) mice. One million K2 cells were injected (i.v.) into congenic Thy 1.1+ C57B6 mice. Without any treatment, mice died of leukemia from 20 to 30 days after injection (shown in the Kaplan-Meier survival plot in A). Blood collected from morbid mice show massive expansion of Thy 1.2+ K2 leukemic cells in Thy 1.1+ host mice (B). As we reported previously (17, 36), this leukemic death was completely prevented by repeated injections of neutralizing anti-IL-15 antibody (R & D system, MAB247, 150 μg per injection, intraperitoneally on days 1, 4, 8, and 11). Tofacitinib (Sigma), which was continuously administered using subcutaneous osmotic pump (ALZET), only prolonged the survival but did not prevent the death of mice as we described previously (36). BNZ132-1–40 was administered intraperitoneally on days 1, 4, 8, and 11 (low dose at 1 mg/kg and high dose at 10 mg/kg) and completely protected mice from leukemic death (A). No propagation of Thy 1.2+ K2 leukemic cells was observed in the PBMCs from BNZ132-1–40-treated mice (B).
Discussion
Recent studies demonstrate increasing examples of human diseases in which more than one cytokine is involved (Table 1). We would like to emphasize that the current anti-cytokine strategy relying on a specific monoclonal Ab lacks the capacity to cope with such situations as shown in Fig. 4A. To address this challenge, we set the following goals. 1) To design cytokine inhibitor peptides that target multiple related cytokines. 2) Such inhibitors should not block all γc-cytokines but should provide selectable target spectrum so we can tailor each of them to treat a particular disease. Using the γc-family as a model, we have constructed a rational algorithm to accomplish our goals as the lead peptide, BNZ132-1, showed potent inhibitory action on chosen members of the γc-family (i.e. IL-2, -9 and -15) but not on other γc- or non-γc-cytokines. Not only that, we designed two additional peptides with different target specificities (supplemental Table 1). We also demonstrated a novel translational potential of our peptide for a human disease (HAM-TSP), which is currently without an established treatment regimen (52). We are also pursuing the structural implications of our observations by extending the studies to the structural aspect of the binding between γc and various γc-cytokines. We hope that our approach could fill in the existing gap in the current anti-cytokine therapeutic strategy.
The functional redundancy among γc-cytokines is largely due to the sharing of the γc-subunit (53). Thus, we rationally chose to target the binding interface of cytokines and γc for inhibiting multiple γc-cytokines. Based on the structural studies on the γc-cytokine system (37, 38), we designed candidate peptides through a series of in silico and in vitro experiments (supplemental Fig. 1 and supplemental Table 2), and we synthesized “finalists,” including BNZ132-1. The selective target spectrum of BNZ132-1, as opposed to blocking all γc-cytokines, may appear counterintuitive, because it targets the shared binding interface. However, the binding of γc-cytokines and γc has been resolved for only select members (IL-2, -4, and -15) (38, 54). We hypothesized that binding interfaces of each γc-cytokine on γc overlap but are not identical, allowing BNZ132-1 only to compete with select targets. Computer-simulated docking pre-screening of candidate peptides showed that one-third of them displayed a competitive binding geometry with IL-2 or IL-15, but even they never fully displaced cytokines from their binding pockets on γc. A full-length γc-cytokine interacts with multiple receptor subunits that are assembled in a stepwise manner (53). It is plausible that the position of the D-helix in a full-length γc-cytokine is influenced by the private chain(s), although that of BNZ132-1 is not due to the lack of other helices (A–C). These may account for the selective target spectrum of BNZ132-1. Mutational analyses provided additional insights in support of our working hypothesis mentioned herein, because the mutation of some residues of BNZ132-1 differentially affected its inhibitory ability on IL-2 and on IL-15. It is interesting that mutations of a few residues that have been known to be critical for the binding of D-helices of IL-2 and IL-15 to the γc crippled the biological inhibitory effects of BNZ132-1, which seemed in disagreement with the in silico observations described above that the binding geometry of BNZ132-1 showed slight difference from that of natural cytokines. We believe that these residues are also critical to support the α-helical structure of the peptide. To address these issues, we are currently resolving the co-crystal structure of BNZ132-1 and γc to test the hypothesis.
The awareness that one Ab can be disappointingly ineffective in blocking the action of a combination of redundant cytokines (Fig. 4, A and B) alarmed us to propose a re-evaluation of previous clinical trials on asthma. Initially, linkage studies suggested the involvement of IL-4 and -9 in this disorder (12, 15). However, consequent clinical trials targeted each cytokine separately and produced unsatisfactory outcomes (55, 56). As IL-4 and IL-9 are functionally overlapping γc-cytokines, this may not be too surprising in light of our demonstration (Fig. 4). Perhaps targeting both cytokines could have generated a more successful outcome. However, clinical therapy involving more than two antibodies, although scientifically sound, will be extremely costly. We have a third peptide that targets IL-4 and IL-9 (supplemental Table S1), an interesting candidate for treating asthma. One important lesson from these observations would be that the lack of effective inhibition when only one Ab was employed does not exclude the target from the pathogenic entities in the disorder of interest. This prompts us to propose that strategies enabling simultaneous blocking of redundant cytokines should be seriously pursued.
Another clinical strategy to target multiple cytokines involves the use of signal inhibitors. An inhibitor to the JAK3 kinase has been developed (CP690,550, also known as tofacitinib) (42) and approved by the Food and Drug Administration for treating ulcerative colitis, rheumatoid arthritis, and graft versus host disease upon kidney transplant (57). However, the long-term use of tofacitinib may be problematic as it could cause immunodeficiency due to the inhibition of IL-7 (4, 7). Tofacitinib also inhibits other JAK kinases (JAK1 and -2) (58) and causes anemia (59) as JAK2 is essential for erythropoiesis. In contrast, BNZ132-1 has sharp target spectrum (Fig. 3) and acts only in the extracellular compartment (Fig. 5). Thus, our concept may provide a safe and versatile alternative to the current strategy.
The two preclinical experiments represented by Figs. 7 and 8 provided encouraging results that BNZ132-1 could be a strong candidate for clinical treatment of cytokine-mediated disorders. Despite the contrasting indications for the affinity of BNZ132-1 to the γc, the preclinical data suggested that the peptide effectively inhibits the target at submicromolar concentrations in vivo. Although the binding assay shown in Fig. 2 indicates a micromolar range of binding affinity, the biological inhibition shown in Figs. 3 and 4 suggests affinity ranging from 25 to 250 nm. We provide our working hypothesis that the binding of BNZ132-1 and γc-cytokines to the γc molecule may be stabilized by the presence of private receptor subunits. Nonetheless, preclinical studies may suggest that indeed BNZ132-1 seems effectively blocking target cytokines in the range of 100–500 nm in vivo. We have learned through parallel toxicology studies that BNZ132-1–40 shows negligible toxicity in vivo on hematopoietic cells even at a high dose of 10 mg/kg in mice and in cynomolgus monkeys.6
The γc-family is a mathematical group of six members. The unique combination of six members generates 63 subsets (6C6 + 6C5 + 6C4 + 6C3 + 6C2 + 6C1 = 63) (supplemental Table S1), and each one of these may represent a unique human disease. Eventually, it may be possible to complete a library of compounds (peptides, antibodies, and small chemicals) to cover all these subsets. Such a library would empower us to treat any human diseases that pathogenically involve unique combinations of γc-cytokines.
We limited our study to the γc-family cytokines, and one may wonder why we did not include factors from another family. Although this may be a future area to cover, we feel that intra-family redundancy is more relevant than inter-family cooperation of cytokines because factors from different families show less overlapping signaling pathways and thus contribute different cellular outcomes than to a single biological consequence.
In summary, we have developed a new technology that enables targeting more than two γc-cytokines with selectable target spectrum. We have hereby demonstrated the in vitro proof-of-concept. Additional in vivo preclinical studies would be a next major milestone leading to clinical applications. This technology clearly has advantages over the single monoclonal Ab therapy or ones that involve signal inhibitors such as tofacitinib.
The lead BNZ132-1 may be a good candidate to treat HAM-TSP, an orphan disease without existing treatment. We explored the therapeutic potential of BNZ132-1 on HAM-TSP with extensive analyses of the effective inhibition of ex vivo activation/proliferation of T cells from HAM-TSP patients by this peptide.7 In addition, we are testing whether BNZ132-1 could be applied to treating IBD. The BNZ132-1 peptide may have its own limitation. In some cases of refractory celiac disease, IL-15 and more recently IL-21 have been shown to be pathogenic factors (19–21, 23, 26). However, IL-2 may inhibit the progression of the disease by activating T-regs (53). Thus, BNZ132-1 is not a perfect fit for this disease, and what we need is a multi-inhibitor targeting IL-15 and -21 but not IL-2. We have developed BNZ132-2 (supplemental Table S1) to tailor this need. These examples would emphasize the conceptual justification for our long-term goal of constructing a library of multicytokine inhibitors with different target specificities as explained in supplemental Table S1. Testing these novel peptides in appropriate animal models would advance our understanding of the complex cytokine milieu that is associated with many human diseases. Finally, our research could provide safe and novel options for treating many more human necrotic conditions such as autoimmunity, inflammatory diseases, and cancer.
Author Contributions
T. N. determined the inhibitory properties of BNZ132-1, including the target spectrum, and the effects on intracellular signaling. A. B. determined the binding of BNZ132-1 and participated in the rational design and screening of BNZ132-1 and the alanine-scanning mutation analysis. T. N., A. B., F. C., R. v.B., R. M., S. J., N. A., and Y. T. designed the experiments, participated in the study discussion, and provided scientific inputs to the manuscript. T. N., A. B., F. C., R. v.B., R. M., and Y. T. conducted the experiments. T. N., A. B., N. A., and Y. T. wrote the manuscript.
Supplementary Material
Acknowledgments
We thank J. Hu-Li and W. Paul of the Laboratory of Immunology, NIAID, National Institutes of Health, for generously providing the CT.h4S cell line. We also thank Hanne Andersen and team members at Bioqual Inc. (Rockville, MD) for the excellent help in conducting mouse experiments. We are grateful for valuable discussions with W. Lu, E. Sundberg, and M. Patzgier of the Institute of Human Virology. The BNZ-γ peptides (including BNZ132-1,2,3) and the multicytokine targeting concept with selectable target spectrum are covered by a United States Patent 08455449 (68) and related intellectual properties belong to the BIONIZ Inc. (Irvine, CA).
This work was supported in part by research funding from BIONIZ (to T. N., F. C., and Y. T.) and in part by internal research funding from the Institute of Human Virology, University of Maryland School of Medicine. This work was authored, in whole or in part, by National Institutes of Health staff. A. B. is an employee of BIONIZ, Inc. N. A. is the founder of BIONIZ. BIONIZ holds the United States patent for the technology behind this work and the concept and peptides used in this paper. We also declare that these financial involvements of BIONIZ did not undermine the scientific objectivity and integrity of the presented work.

This article contains supplemental Fig. S1, Tables S1 and S2, and additional references.
Y. Tagaya and R. Massoud, unpublished observations.
Y. Tagaya, unpublished observations.
N. Azimi and Y. Tagaya, unpublished observations.
Massoud, R., Enose-Akahata, Y., Tagaya, Y., Azimi, N., and Jacobson, S. (2015) Proc. Natl. Acad. Sci. U.S.A., 10.1073/pnas.1412626112.
- IBD
- inflammatory bowel disease
- Ab
- antibody
- T-reg
- regulatory T cell
- h
- human
- PBMC
- peripheral blood mononuclear cell
- HAM/TSP
- HTLV-1-associated myelopathy/tropical spastic paraparesis.
References
- 1. Bazan J. F. (1992) Unraveling the structure of IL-2. Science 257, 410–413 [DOI] [PubMed] [Google Scholar]
- 2. Ozaki K., Leonard W. J. (2002) Cytokine and cytokine receptor pleiotropy and redundancy. J. Biol. Chem. 277, 29355–29358 [DOI] [PubMed] [Google Scholar]
- 3. Rochman Y., Spolski R., Leonard W. J. (2009) New insights into the regulation of T cells by γ(c) family cytokines. Nat. Rev. Immunol. 9, 480–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Macchi P., Villa A., Giliani S., Sacco M. G., Frattini A., Porta F., Ugazio A. G., Johnston J. A., Candotti F., O'Shea J. J. (1995) Mutations of Jak-3 gene in patients with autosomal severe combined immune deficiency (SCID). Nature 377, 65–68 [DOI] [PubMed] [Google Scholar]
- 5. Noguchi M., Yi H., Rosenblatt H. M., Filipovich A. H., Adelstein S., Modi W. S., McBride O. W., Leonard W. J. (1993) IL-2 receptor γ chain mutation results in X-linked severe combined immunodeficiency in humans. Cell 73, 147–157 [DOI] [PubMed] [Google Scholar]
- 6. Russell S. M., Tayebi N., Nakajima H., Riedy M. C., Roberts J. L., Aman M. J., Migone T. S., Noguchi M., Markert M. L., Buckley R. H., O'Shea J. J., Leonard W. J. (1995) Mutation of Jak3 in a patient with SCID: essential role of Jak3 in lymphoid development. Science 270, 797–800 [DOI] [PubMed] [Google Scholar]
- 7. Nosaka T., van Deursen J. M., Tripp R. A., Thierfelder W. E., Witthuhn B. A., McMickle A. P., Doherty P. C., Grosveld G. C., Ihle J. N. (1995) Defective lymphoid development in mice lacking Jak3. Science 270, 800–802 [DOI] [PubMed] [Google Scholar]
- 8. Malek T. R., Yu A., Vincek V., Scibelli P., Kong L. (2002) CD4 regulatory T cells prevent lethal autoimmunity in IL-2Rβ-deficient mice. Implications for the nonredundant function of IL-2. Immunity 17, 167–178 [DOI] [PubMed] [Google Scholar]
- 9. Glas J., Stallhofer J., Ripke S., Wetzke M., Pfennig S., Klein W., Epplen J. T., Griga T., Schiemann U., Lacher M., Koletzko S., Folwaczny M., Lohse P., Göke B., Ochsenkühn T., et al. (2009) Novel genetic risk markers for ulcerative colitis in the IL2/IL21 region are in epistasis with IL23R and suggest a common genetic background for ulcerative colitis and celiac disease. Am. J. Gastroenterol. 104, 1737–1744 [DOI] [PubMed] [Google Scholar]
- 10. Festen E. A., Goyette P., Scott R., Annese V., Zhernakova A., Lian J., Lefèbvre C., Brant S. R., Cho J. H., Silverberg M. S., Taylor K. D., de Jong D. J., Stokkers P. C., Mcgovern D., Palmieri O., et al. (2009) Genetic variants in the region harbouring IL2/IL21 associated with ulcerative colitis. Gut 58, 799–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Márquez A., Orozco G., Martínez A., Palomino-Morales R., Fernández-Arquero M., Mendoza J. L., Taxonera C., Díaz-Rubio M., Gómez-García M., Nieto A., López-Nevot M. A., de la Concha E. G., Martín J., Urcelay E. (2009) Novel association of the IL2-IL21 region with inflammatory bowel disease. Am. J. Gastroenterol. 104, 1968–1975 [DOI] [PubMed] [Google Scholar]
- 12. Steinke J. W., Borish L., Rosenwasser L. J. (2003) 5. Genetics of hypersensitivity. J. Allergy Clin. Immunol. 111, S495–S501 [DOI] [PubMed] [Google Scholar]
- 13. Lundström W., Fewkes N. M., Mackall C. L. (2012) IL-7 in human health and disease. Semin. Immunol. 24, 218–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shimbara A., Christodoulopoulos P., Soussi-Gounni A., Olivenstein R., Nakamura Y., Levitt R. C., Nicolaides N. C., Holroyd K. J., Tsicopoulos A., Lafitte J. J., Wallaert B., Hamid Q. A. (2000) IL-9 and its receptor in allergic and nonallergic lung disease: increased expression in asthma. J. Allergy Clin. Immunol. 105, 108–115 [DOI] [PubMed] [Google Scholar]
- 15. Nicolaides N. C., Holroyd K. J., Ewart S. L., Eleff S. M., Kiser M. B., Dragwa C. R., Sullivan C. D., Grasso L., Zhang L. Y., Messler C. J., Zhou T., Kleeberger S. R., Buetow K. H., Levitt R. C. (1997) IL9: a candidate gene for asthma. Proc. Natl. Acad. Sci. U.S.A. 94, 13175–13180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fehniger T. A., Suzuki K., Ponnappan A., VanDeusen J. B., Cooper M. A., Florea S. M., Freud A. G., Robinson M. L., Durbin J., Caligiuri M. A. (2001) Fatal leukemia in IL15 transgenic mice follows early expansions in natural killer and memory phenotype CD8+ T cells. J. Exp. Med. 193, 219–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sato N., Sabzevari H., Fu S., Ju W., Petrus M. N., Bamford R. N., Waldmann T. A., Tagaya Y. (2011) Development of an IL-15-autocrine CD8 T-cell leukemia in IL-15-transgenic mice requires the cis expression of IL-15Rα. Blood 117, 4032–4040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zambello R., Facco M., Trentin L., Sancetta R., Tassinari C., Perin A., Milani A., Pizzolo G., Rodeghiero F., Agostini C., Meazza R., Ferrini S., Semenzato G. (1997) IL-15 triggers the proliferation and cytotoxicity of granular lymphocytes in patients with lymphoproliferative disease of granular lymphocytes. Blood 89, 201–211 [PubMed] [Google Scholar]
- 19. Meresse B., Chen Z., Ciszewski C., Tretiakova M., Bhagat G., Krausz T. N., Raulet D. H., Lanier L. L., Groh V., Spies T., Ebert E. C., Green P. H., Jabri B. (2004) Coordinated induction by IL15 of a TCR-independent NKG2D signaling pathway converts CTL into lymphokine-activated killer cells in celiac disease. Immunity 21, 357–366 [DOI] [PubMed] [Google Scholar]
- 20. Tang F., Chen Z., Ciszewski C., Setty M., Solus J., Tretiakova M., Ebert E., Han J., Lin A., Guandalini S., Groh V., Spies T., Green P., Jabri B. (2009) Cytosolic PLA2 is required for CTL-mediated immunopathology of celiac disease via NKG2D and IL-15. J. Exp. Med. 206, 707–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Meresse B., Curran S. A., Ciszewski C., Orbelyan G., Setty M., Bhagat G., Lee L., Tretiakova M., Semrad C., Kistner E., Winchester R. J., Braud V., Lanier L. L., Geraghty D. E., Green P. H., et al. (2006) Reprogramming of CTLs into natural killer-like cells in celiac disease. J. Exp. Med. 203, 1343–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jabri B., de Serre N. P., Cellier C., Evans K., Gache C., Carvalho C., Mougenot J. F., Allez M., Jian R., Desreumaux P., Colombel J. F., Matuchansky C., Cugnenc H., Lopez-Botet M., Vivier E., et al. (2000) Selective expansion of intraepithelial lymphocytes expressing the HLA-E-specific natural killer receptor CD94 in celiac disease. Gastroenterology 118, 867–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bhagat G., Naiyer A. J., Shah J. G., Harper J., Jabri B., Wang T. C., Green P. H., Manavalan J. S. (2008) Small intestinal CD8+TCRγδ+NKG2A+ intraepithelial lymphocytes have attributes of regulatory cells in patients with celiac disease. J. Clin. Invest. 118, 281–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Devkota S., Wang Y., Musch M. W., Leone V., Fehlner-Peach H., Nadimpalli A., Antonopoulos D. A., Jabri B., Chang E. B. (2012) Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10−/− mice. Nature 487, 104–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yokoyama S., Perera P. Y., Waldmann T. A., Hiroi T., Perera L. P. (2013) Tofacitinib, a Janus kinase inhibitor demonstrates efficacy in an IL-15 transgenic mouse model that recapitulates pathologic manifestations of celiac disease. J. Clin. Immunol. 33, 586–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bodd M., Ráki M., Tollefsen S., Fallang L. E., Bergseng E., Lundin K. E., Sollid L. M. (2010) HLA-DQ2-restricted gluten-reactive T cells produce IL-21 but not IL-17 or IL-22. Mucosal Immunol. 3, 594–601 [DOI] [PubMed] [Google Scholar]
- 27. Leonard W. J., Spolski R. (2005) IL-21: a modulator of lymphoid proliferation, apoptosis and differentiation. Nat. Rev. Immunol. 5, 688–698 [DOI] [PubMed] [Google Scholar]
- 28. Chtanova T., Tangye S. G., Newton R., Frank N., Hodge M. R., Rolph M. S., Mackay C. R. (2004) T follicular helper cells express a distinctive transcriptional profile, reflecting their role as non-Th1/Th2 effector cells that provide help for B cells. J. Immunol. 173, 68–78 [DOI] [PubMed] [Google Scholar]
- 29. Amadi-Obi A., Yu C. R., Liu X., Mahdi R. M., Clarke G. L., Nussenblatt R. B., Gery I., Lee Y. S., Egwuagu C. E. (2007) TH17 cells contribute to uveitis and scleritis and are expanded by IL-2 and inhibited by IL-27/STAT1. Nat. Med. 13, 711–718 [DOI] [PubMed] [Google Scholar]
- 30. Costanzo A., Chimenti M. S., Botti E., Caruso R., Sarra M., Monteleone G. (2010) IL-21 in the pathogenesis and treatment of skin diseases. J. Dermatol. Sci. 60, 61–66 [DOI] [PubMed] [Google Scholar]
- 31. Sarra M., Pallone F., Monteleone G. (2013) IL-21 in chronic inflammatory diseases. BioFactors 39, 368–373 [DOI] [PubMed] [Google Scholar]
- 32. Changelian P. S., Flanagan M. E., Ball D. J., Kent C. R., Magnuson K. S., Martin W. H., Rizzuti B. J., Sawyer P. S., Perry B. D., Brissette W. H., McCurdy S. P., Kudlacz E. M., Conklyn M. J., Elliott E. A., Koslov E. R., et al. (2003) Prevention of organ allograft rejection by a specific Janus kinase 3 inhibitor. Science 302, 875–878 [DOI] [PubMed] [Google Scholar]
- 33. Tagaya Y., Burton J. D., Miyamoto Y., Waldmann T. A. (1996) Identification of a novel receptor/signal transduction pathway for IL-15/T in mast cells. EMBO J. 15, 4928–4939 [PMC free article] [PubMed] [Google Scholar]
- 34. Antal Z., Baker J. C., Smith C., Jarchum I., Babad J., Mukherjee G., Yang Y., Sidney J., Sette A., Santamaria P., DiLorenzo T. P. (2012) Beyond HLA-A*0201: new HLA-transgenic nonobese diabetic mouse models of type 1 diabetes identify the insulin C-peptide as a rich source of CD8+ T cell epitopes. J. Immunol. 188, 5766–5775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Notredame C., Higgins D. G., Heringa J. (2000) T-Coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302, 205–217 [DOI] [PubMed] [Google Scholar]
- 36. Ju W., Zhang M., Jiang J. K., Thomas C. J., Oh U., Bryant B. R., Chen J., Sato N., Tagaya Y., Morris J. C., Janik J. E., Jacobson S., Waldmann T. A. (2011) CP-690,550, a therapeutic agent, inhibits cytokine-mediated Jak3 activation and proliferation of T cells from patients with ATL and HAM/TSP. Blood 117, 1938–1946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang X., Rickert M., Garcia K. C. (2005) Structure of the quaternary complex of IL-2 with its α, β, and γc receptors. Science 310, 1159–1163 [DOI] [PubMed] [Google Scholar]
- 38. Ring A. M., Lin J. X., Feng D., Mitra S., Rickert M., Bowman G. R., Pande V. S., Li P., Moraga I., Spolski R., Ozkan E., Leonard W. J., Garcia K. C. (2012) Mechanistic and structural insight into the functional dichotomy between IL-2 and IL-15. Nat. Immunol. 13, 1187–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bamford R. N., Grant A. J., Burton J. D., Peters C., Kurys G., Goldman C. K., Brennan J., Roessler E., Waldmann T. A. (1994) The interleukin (IL) 2 receptor β chain is shared by IL-2 and a cytokine, provisionally designated IL-T, that stimulates T-cell proliferation and the induction of lymphokine-activated killer cells. Proc. Natl. Acad. Sci. U.S.A. 91, 4940–4944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Grabstein K. H., Eisenman J., Shanebeck K., Rauch C., Srinivasan S., Fung V., Beers C., Richardson J., Schoenborn M. A., Ahdieh M. (1994) Cloning of a T cell growth factor that interacts with the β chain of the IL-2 receptor. Science 264, 965–968 [DOI] [PubMed] [Google Scholar]
- 41. Vanderhoek J. Y., Tare N. S., Bailey J. M., Goldstein A. L., Pluznik D. H. (1982) New role for 15-hydroxyeicosatetraenoic acid. Activator of leukotriene biosynthesis in PT-18 mast/basophil cells. J. Biol. Chem. 257, 12191–12195 [PubMed] [Google Scholar]
- 42. Boulay J. L., O'Shea J. J., Paul W. E. (2003) Molecular phylogeny within type I cytokines and their cognate receptors. Immunity 19, 159–163 [DOI] [PubMed] [Google Scholar]
- 43. Cheng Y., Prusoff W. H. (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22, 3099–3108 [DOI] [PubMed] [Google Scholar]
- 44. Craig D. A. (1993) The Cheng-Prusoff relationship: something lost in the translation. Trends Pharmacol. Sci. 14, 89–91 [DOI] [PubMed] [Google Scholar]
- 45. Tendler C. L., Greenberg S. J., Blattner W. A., Manns A., Murphy E., Fleisher T., Hanchard B., Morgan O., Burton J. D., Nelson D. L. (1990) Transactivation of IL2 and its receptor induces immune activation in human T-cell lymphotropic virus type I-associated myelopathy: pathogenic implications and a rationale for immunotherapy. Proc. Natl. Acad. Sci. U.S.A. 87, 5218–5222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Azimi N., Jacobson S., Leist T., Waldmann T. A. (1999) Involvement of IL-15 in the pathogenesis of human T lymphotropic virus type I-associated myelopathy/tropical spastic paraparesis: implications for therapy with a monoclonal antibody directed to the IL-2/15R β receptor. J. Immunol. 163, 4064–4072 [PubMed] [Google Scholar]
- 47. Chen J., Petrus M., Bryant B. R., Phuc Nguyen V., Stamer M., Goldman C. K., Bamford R., Morris J. C., Janik J. E., Waldmann T. A. (2008) Induction of the IL-9 gene by HTLV-I Tax stimulates the spontaneous proliferation of primary adult T-cell leukemia cells by a paracrine mechanism. Blood 111, 5163–5172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Morrison K. L., Weiss G. A. (2001) Combinatorial alanine-scanning. Curr. Opin. Chem. Biol. 5, 302–307 [DOI] [PubMed] [Google Scholar]
- 49. Antony P. A., Paulos C. M., Ahmadzadeh M., Akpinarli A., Palmer D. C., Sato N., Kaiser A., Hinrichs C. S., Heinrichs C., Klebanoff C. A., Tagaya Y., Restifo N. P. (2006) IL-2-dependent mechanisms of tolerance and immunity in vivo. J. Immunol. 176, 5255–5266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Boyman O., Krieg C., Letourneau S., Webster K., Surh C. D., Sprent J. (2012) Selectively expanding subsets of T cells in mice by injection of interleukin-2/antibody complexes: implications for transplantation tolerance. Transplant. Proc. 44, 1032–1034 [DOI] [PubMed] [Google Scholar]
- 51. Marks-Konczalik J., Dubois S., Losi J. M., Sabzevari H., Yamada N., Feigenbaum L., Waldmann T. A., Tagaya Y. (2000) IL-2-induced activation-induced cell death is inhibited in IL-15 transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 97, 11445–11450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Matsuura E., Yamano Y., Jacobson S. (2010) Neuroimmunity of HTLV-I infection. J. Neuroimmune Pharmacol. 5, 310–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Liao W., Lin J. X., Leonard W. J. (2013) IL-2 at the crossroads of effector responses, tolerance, and immunotherapy. Immunity 38, 13–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. LaPorte S. L., Juo Z. S., Vaclavikova J., Colf L. A., Qi X., Heller N. M., Keegan A. D., Garcia K. C. (2008) Molecular and structural basis of cytokine receptor pleiotropy in the IL-4/13 system. Cell 132, 259–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Oh C. K., Raible D., Geba G. P., Molfino N. A. (2011) Biology of the IL-9 pathway and its therapeutic potential for the treatment of asthma. Inflamm. Allergy Drug Targets 10, 180–186 [DOI] [PubMed] [Google Scholar]
- 56. Steinke J. W. (2010) Current prospective of anti-IL-4, -IL-9, and -IL-13 therapies in allergic disease. Recent Pat. Inflamm. Allergy Drug Discov. 4, 222–230 [DOI] [PubMed] [Google Scholar]
- 57. West K. (2009) CP-690550, a JAK3 inhibitor as an immunosuppressant for the treatment of rheumatoid arthritis, transplant rejection, psoriasis and other immune-mediated disorders. Curr. Opin. Investig. Drugs 10, 491–504 [PubMed] [Google Scholar]
- 58. Jiang J. K., Ghoreschi K., Deflorian F., Chen Z., Perreira M., Pesu M., Smith J., Nguyen D. T., Liu E. H., Leister W., Costanzi S., O'Shea J. J., Thomas C. J. (2008) Examining the chirality, conformation and selective kinase inhibition of 3-((3R,4R)-4-methyl-3-(methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)piperidin-1-y l)-3-oxopropanenitrile (CP-690,550). J. Med. Chem. 51, 8012–8018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Garber K. (2011) Pfizer's JAK inhibitor sails through phase 3 in rheumatoid arthritis. Nat. Biotechnol. 29, 467–468 [DOI] [PubMed] [Google Scholar]
- 60. Azimi N., Nagai M., Jacobson S., Waldmann T. A. (2001) IL-15 plays a major role in the persistence of Tax-specific CD8 cells in HAM/TSP patients. Proc. Natl. Acad. Sci. U.S.A. 98, 14559–14564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Nowak E. C., Weaver C. T., Turner H., Begum-Haque S., Becher B., Schreiner B., Coyle A. J., Kasper L. H., Noelle R. J. (2009) IL-9 as a mediator of Th17-driven inflammatory disease. J. Exp. Med. 206, 1653–1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Li H., Nourbakhsh B., Ciric B., Zhang G. X., Rostami A. (2010) Neutralization of IL-9 ameliorates experimental autoimmune encephalomyelitis by decreasing the effector T cell population. J. Immunol. 185, 4095–4100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Saikali P., Antel J. P., Pittet C. L., Newcombe J., Arbour N. (2010) Contribution of astrocyte-derived IL-15 to CD8 T cell effector functions in multiple sclerosis. J. Immunol. 185, 5693–5703 [DOI] [PubMed] [Google Scholar]
- 64. McInnes I. B., Liew F. Y. (2005) Cytokine networks–towards new therapies for rheumatoid arthritis. Nat. Clin. Pract. Rheumatol. 1, 31–39 [DOI] [PubMed] [Google Scholar]
- 65. Hartgring S. A., Bijlsma J. W., Lafeber F. P., van Roon J. A. (2006) Interleukin-7 induced immunopathology in arthritis. Ann. Rheum. Dis. 65, Suppl. 3, 69–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kishimoto T. (2010) IL-6: from its discovery to clinical applications. Int. Immunol. 22, 347–352 [DOI] [PubMed] [Google Scholar]
- 67. Young D. A., Hegen M., Ma H. L., Whitters M. J., Albert L. M., Lowe L., Senices M., Wu P. W., Sibley B., Leathurby Y., Brown T. P., Nickerson-Nutter C., Keith J. C. Jr., Collins M. (2007) Blockade of the interleukin-21/interleukin-21 receptor pathway ameliorates disease in animal models of rheumatoid arthritis. Arthritis Rheum. 56, 1152–1163 [DOI] [PubMed] [Google Scholar]
- 68. Tagaya Y., Azimi N. ( June 4, 2013) U. S. Patent 08455449
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.