

Background: Benzylsuccinate synthase (BSS) catalyzes the formation of a C—C bond between toluene and fumarate by a radical mechanism.
Results: BSS binds substrates in a buried active site and uses conformational changes to gate access.
Conclusion: BSS and related enzymes precisely position substrates for chemistry.
Significance: These structures reveal the molecular basis for BSS radical chemistry and substrate specificity.
Keywords: bacterial metabolism, biodegradation, enzyme catalysis, enzyme mechanism, substrate specificity, x-ray crystallography, glycyl radical enzyme, radical chemistry
Abstract
Various bacteria perform anaerobic degradation of small hydrocarbons as a source of energy and cellular carbon. To activate non-reactive hydrocarbons such as toluene, enzymes conjugate these molecules to fumarate in a radical-catalyzed, C—C bond-forming reaction. We have determined x-ray crystal structures of the glycyl radical enzyme that catalyzes the addition of toluene to fumarate, benzylsuccinate synthase (BSS), in two oligomeric states with fumarate alone or with both substrates. We find that fumarate is secured at the bottom of a long active site cavity with toluene bound directly above it. The two substrates adopt orientations that appear ideal for radical-mediated C—C bond formation; the methyl group of toluene is positioned between fumarate and a cysteine that forms a thiyl radical during catalysis, which is in turn adjacent to the glycine that serves as a radical storage residue. Toluene is held in place by fumarate on one face and tight packing by hydrophobic residues on the other face and sides. These hydrophobic residues appear to become ordered, thus encapsulating toluene, only in the presence of BSSβ, a small protein subunit that forms a tight complex with BSSα, the catalytic subunit. Enzymes related to BSS are able to metabolize a wide range of hydrocarbons through attachment to fumarate. Using our structures as a guide, we have constructed homology models of several of these “X-succinate synthases” and determined conservation patterns that will be useful in understanding the basis for catalysis and specificity in this family of enzymes.
Introduction
Microbial degradation is one route for the mitigation of hydrocarbon pollution (1–3). Bacterial species have devised numerous means of degrading inert hydrocarbon molecules (4). In the presence of oxygen, a wide range of bacteria and some fungi initiate metabolism of aromatic hydrocarbons and alkanes through direct oxidation by mono- or dioxygenases followed by β oxidation of the resulting hydroxylated fragments. In the absence of oxygen, hydrocarbon functionalization is more challenging, and different strategies are required. Experiments with the denitrifying bacterium Thauera aromatica have revealed the details of a microbial toluene degradation pathway. The first step in this pathway is reaction of toluene with fumarate to give R-benzylsuccinate, which is catalyzed by the enzyme benzylsuccinate synthase (BSS)2 (Fig. 1A) (5). The addition of fumarate serves to activate the benzylic carbon of toluene, allowing it to be metabolized through β oxidation to benzoyl-CoA and then reductively dearomatized by benzoyl-CoA reductase before further oxidation (4, 6).
FIGURE 1.
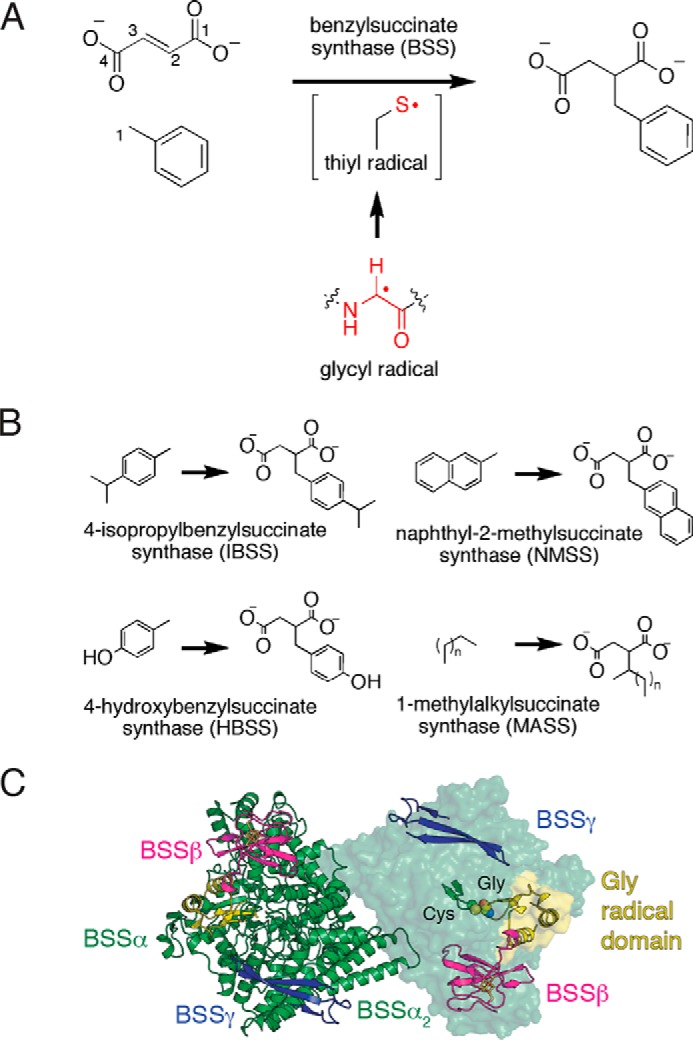
Components of the BSS enzyme complex. A, BSS catalyzes a C—C bond forming reactions between fumarate and toluene. The enzyme contains a stable glycyl radical that is thought to generate a transient active site thiyl radical. B, characterized XSSs utilize aryl or alkyl hydrocarbons (X) to generate the corresponding X-succinate conjugates with fumarate (not shown) as a co-substrate. C, the BSS (αβγ)2 heterohexamer contains a dimeric BSSα complex, with BSSβ and BSSγ binding at independent sites within a single BSSα protomer. The Cys loop and Gly loop (right, ribbons) are buried within BSSα The Gly loop resides on the glycyl radical domain, which inserts into the barrel on one face. BSSβ rests on top of the glycyl radical domain. Panel C is adapted from Funk et al. (23).
A group of enzymes closely related to BSS, collectively dubbed aryl-succinate synthase (aryl-SS) enzymes, are known to function on toluene-like aromatic hydrocarbons, including p-cymene (7, 8), p-cresol (9, 10), and 2-methylnaphthalene (Fig. 1B) (11, 12). Additionally, alkyl-SSs, which function on alkyl chains including n-hexane, have also been discovered (13, 14), although there is less known about the substrate specificity and mechanism of these enzymes. Together these enzymes form a large family of X-succinate synthases (XSSs) responsible for functionalization of a wide variety of hydrocarbon substrates, and the range of substrates is still growing.
The C—C bond-forming reaction performed by BSS and its relatives requires an oxygen-sensitive radical cofactor. BSS is a member of the glycyl radical enzyme (GRE) family and contains a backbone glycyl radical in its activated form. Other members of this family include the central metabolic enzymes pyruvate formate lyase and anaerobic ribonucleotide reductase and several radical eliminases (15, 16). All of these radical enzymes are believed to generate a substrate-based radical that undergoes rearrangement in the active site to generate a product-based radical, which is reduced by the enzyme to regenerate the glycyl radical. In all GREs thus far studied, an enzyme-based thiyl radical is proposed to be an intermediary between the glycyl- and substrate-based radical species (17, 18). The cysteine proposed to form the thiyl radical initiates radical chemistry on the substrate, whereas the glycine is the radical storage site.
Radical chemistry in GREs occurs deep within the core of a 10-stranded β/α barrel where reactive radical species are protected. At the barrel core, a loop containing the essential glycine (the Gly loop) is juxtaposed with the loop containing the catalytic cysteine residue (the Cys loop) allowing for hydrogen atom transfer between the two. Residues surrounding these loops compose the active site. BSS exists as an (αβγ)2 heterohexamer (Fig. 1C). BSSα contains the 10-stranded barrel and is the catalytic subunit, whereas BSSβ and BSSγ are both small, [4Fe-4S]-cluster-containing proteins of unknown function (19, 20). Despite the fact that the roles of BSSβ and BSSγ are unclear, both proteins are required for viable growth of T. aromatica strain T1 on toluene (21, 22). We previously solved structures of the BSSαβγ and BSSαγ complexes, revealing that removal of BSSβ from the complex with BSSαγ permits a large conformational change within BSSα (23). The core barrel opens, exposing the active site, and a C-terminal domain that harbors the Gly loop (the glycyl radical domain) shifts a few ångströms out of the active site but does not completely vacate the barrel. Movement of the glycyl radical domain away from the barrel core is expected to be required for the initial installation of the glycyl radical by the cognate S-adenosylmethionine (AdoMet) radical-activating enzyme (24). Thus, in the absence of BSSβ, the enzyme adopts an open state that appears amenable to activation, whereas in the presence of BSSβ the enzyme adopts a closed state that appears ready for catalysis, with the glycyl radical domain positioned for radical transfer. We identified a channel that substrates could take to enter the active site; however, BSSβ appeared to block the channel entrance, suggesting either that substrates cannot bind in the presence of BSSβ or that BSSβ plays a role in gating active site accessibility.
Here we have turned again to crystallography, this time to explore substrate binding in BSS. Given that neither we nor other groups (25) have been able to purify the requisite activating enzyme for BSS, thus impeding mutagenesis and other in vitro biochemical studies, crystallography has proven to be an excellent tool to probe what would otherwise be a largely intractable enzyme system. In particular, our current study investigates how BSS binds to fumarate and toluene in both αβγ and αγ complexes. We find that both substrates can bind to the BSSαβγ complex, thus demonstrating that the presence of BSSβ does not prevent substrate access into the active site. Fumarate binding to BSSαγ partially shifts the barrel from the open state observed in the substrate-free BSSαγ structure toward the closed state seen in BSSαβγ. However, ordering of BSSα into the fully closed, catalytic state does not occur in this structure in the absence of BSSβ; only structures with BSSαβγ depict the fully closed, catalytically competent state. In this closed state, both substrates are bound at the bottom of the proposed channel in an orientation in which toluene is ideally positioned to undergo hydrogen atom abstraction by the putative, transient thiyl radical followed by C—C bond formation between toluene and fumarate. Finally, we have constructed homology models of other members of the XSS family based on the structure of BSS to predict the determinants of specificity in these enzymes. This analysis will aid in characterization of the diverse communities of microbes known to cooperate in the degradation of hydrocarbons.
Experimental Procedures
Protein Production and Crystallization
BSSαβγ and BSSαγ were purified (20) and crystallized (23) as previously reported. Initial crystals were discovered using screens dispensed by a TPP Labtech Mosquito liquid-handling robot housed in a room-temperature, N2-filled, MBraun anaerobic chamber with O2 < 0.1 ppm. All subsequent crystallizations were performed in this chamber using the sitting drop vapor diffusion method and reagents from Hampton Research. In particular, BSSαβγ at 8 mg ml−1 in buffer containing 50 mm Tris, pH 7.6, 15% (v/v) glycerol, and 200 mm NaCl was added at a 2:1 ratio to well solution containing 25% (w/v) PEG 3350, 100 mm Tris, pH 8.5, 60 mm KCl, and 5 mm fumarate. 1–2 μl of toluene was added to the bottom of the well and allowed to diffuse slowly into the protein drop. Diffraction-quality crystals grew over the course of 3 weeks. Crystals were soaked in a cryoprotection solution containing 50 mm Tris, pH 8.5, 25% (w/v) PEG 3350, 10% (v/v) glycerol, 50 mm fumarate for 30 s before cryocooling. Crystals indexed in space group P21212 (a = 141.5, b = 115.4, c = 121.7 Å) with two molecules per asymmetric unit. This crystal form is different from that observed before for substrate-free BSSαβγ (space group I222; a = 113.4, b = 120.4, c = 136.0 Å), but the packing interactions are virtually identical, with small changes accounting for the lower crystallographic symmetry. BSSαγ at 15 mg ml−1 in buffer containing 20 mm HEPES, pH 7.6, 100 mm NaCl, and 5 mm fumarate was mixed in a 1:1 ratio with precipitant solution containing 20% (w/v) PEG 400, 50 mm Bis-Tris, pH 6.5, 25 mm Tris pH 8.0 and incubated over a well of the same solution at room temperature. Crystals formed in several days and were pale yellow. Crystals were soaked in a solution containing 30% (w/v) PEG 400, 50 mm Bis-Tris 6.5, 25 mm Tris 8.0, and 50 mm sodium fumarate for 5–10 min before cryocooling. Crystals indexed in space group P43212 (a = b = 154.9, c = 82.1 Å) with one molecule per asymmetric unit. Crystals were cryocooled in liquid nitrogen in a Coy anaerobic chamber filled with 95% Ar, 5% H2.
Data Collection and Processing
All data were collected at the National Synchrotron Light Source beamline X26C on a Quantum-4 ASDC CCD detector at a wavelength of 1.000 Å (Table 1). Data were indexed and scaled in HKL2000 (26). The diffraction data for fumarate-bound BSSαγ were strongly anisotropic along the c axis as determined by the programs Truncate and phenix.xtriage. Due to this anisotropic diffraction, many of the high resolution reflections measured along the c axis displayed weak intensities, affecting both the signal-to-noise ratio in the high resolution bin and the overall completeness of data with intensities above zero (Table 1). To compensate for this issue with the data, all map calculations were performed with anisotropy corrections in Phenix, which improved map quality. Inclusion of the weak high resolution data to 2.0 Å resolution yielded maps with finer detail than when these data were truncated. The usefulness of the high resolution data is also indicated by the CC1/2 value of ∼0.7, which is obtained for all data to 2.0 Å resolution. The fumarate- and toluene-bound BSSαβγ diffraction data were also anisotropic but to a less severe degree.
TABLE 1.
X-ray diffraction data processing statistics
Values in parentheses represent the highest resolution shell.
| Data collection | Fumarate- and toluene-bound BSSαβγ | Fumarate-bound BSSαγ |
|---|---|---|
| Resolution (Å) | 40.0-3.30 (3.36-3.30) | 35.0-2.00 (2.03-2.00) |
| Completeness,a all data | 98.9 (95.4) | 97.0 (80.4) |
| Completeness, I ≥ 0 | 91.7 (75.6) | 86.9 (76.7)a |
| 〈I/σI〉 | 9.6 (1.3) | 11.8 (1.4) |
| Rmerge | 0.10 (>1) | 0.09 (0.90) |
| Unique reflections | 30311 | 65770 |
| Redundancy | 3.8 (3.5) | 5.6 (2.8) |
| CC1/2 | (0.669) | (0.700) |
a The data sets were anisotropic along the c-axis, resulting in a large number of weak reflections (see “Experimental Procedures”).
The structure of fumarate-bound BSSαγ was solved initially at 2.3 Å resolution by molecular replacement using the Phenix implementation of Phaser (27) with the previously solved structure of BSSαγ as a model (23). Manual rebuilding of the model was performed in Coot (28) followed by refinement with phenix.refine (29) at 2.0 Å resolution. Translation/libration/screw refinement of B factors improved Rfree substantially. The final model (Table 2) contains residues 6–708 and 716–865 (of 865 native residues) in BSSα and 9–47 (of 60) in BSSγ. Residues 709–715, which compose part of β8 and α8′, are disordered. The 14-residue C-terminal linker and His6 tag are not observed in BSSα. The model contains 97.1% of residues in the most favorable region of the Ramachandran plot, 2.7% in additionally allowed regions, and 0.2% (4 residues) in outlier regions. As in the previously reported structure (23), the [4Fe-4S] cluster and three of the four cysteine residues coordinating it from BSSγ are not observed in the electron density. Parameter files for fumaric acid (Protein Data Bank ligand ID FUM) were obtained from the CCP4 database.
TABLE 2.
Model and refinement statistics
Values in parentheses represent the highest resolution shell. r.m.s.d., root mean squared deviation.
| Refinement | Fumarate- and toluene-bound BSS | Fumarate-bound BSS |
|---|---|---|
| Resolution (Å) | 38.3–3.30 | 34.6–2.00 |
| Rwork | 0.212 | 0.195 |
| Rfree | 0.237a | 0.223b |
| Protein atoms | 15350 | 7063 |
| Toluene atoms | 14 | NA |
| Fumarate atoms | 16 | 8 |
| Other ligand atoms | 16c | 71d |
| Water molecules | 0 | 415 |
| r.m.s.d. | ||
| Bond length (Å) | 0.003 | 0.003 |
| Bond angle (°) | 0.913 | 0.705 |
| Coordinate error (Å) | 0.44 | 0.25 |
| Rotamer outliers (%) | 0.37 | 0.13 |
| Average B factors (Å2) | ||
| BSSα | 111.1 | 53.7 |
| BSSβ | 128.6 | NA |
| BSSγ | 130.0 | 53.9 |
| Water atoms | NA | 42.8 |
| Toluene | 95.2 | NA |
| Fumarate | 94.2 | 41.6 |
| Other ligands | 130.3c | 61.0d |
a 6% of reflections (1809) were set aside for cross-validation.
b 5% of reflections (3271) were set aside for cross validation.
c [4Fe-4S] clusters are present within the two BSSβ subunits.
d Six polyethylene glycol molecules and a molecule of Tris buffer are present at the protein surface.
The structure of toluene- and fumarate-bound BSSαβγ was solved by molecular replacement at 3.30 Å resolution in the Phenix implementation of Phaser using the published 2.00 Å resolution, chloride-bound BSSαβγ structure (23) as an initial model. Due to the low resolution of this dataset, refinement was tightly restrained with the high resolution model as a reference structure in phenix.refine. The final model (Table 2) contains residues 9–865 (of 865 native residues) in BSSα, 13–81 (of 81) in BSSβ, and 11–47 (of 60) BSSγ. The model contains 97.4% of residues in the most favorable region of the Ramachandran plot, 2.2% in additionally allowed regions, and 0.4% (8 residues) in outlier regions. The 14-residue C-terminal linker and His6 tag are not observed in BSSα. Composite omit maps calculated by phenix.autobuild were used to verify positions and identity of ligands and active site residues in both structures. A [4Fe-4S] cluster is present in BSSβ, but no density is present in BSSγ for a cluster. Structure figures were generated in PyMOL v1.4.1 (Schrödinger, LLC). Parameter files for toluene (methylbenzene, Protein Data Bank Ligand ID MBN) were obtained from the CCP4 database. The atomic coordinates and structure factors for the fumarate-bound structure of BSSαγ and fumarate- and toluene-bound structure of BSSαβγ have been deposited in the Protein DataBank under accession numbers 5BWD and 5BWE.
Homology Modeling of XSS Family Members
Models of the catalytic subunit of 4-isopropylbenzyl-SS (IBSS; GenBankTM ID AIS23708.1), 4-hydroxybenzyl-SS (HBSS; CCK78655.1), naphthyl-2-methyl-SS (NMSS; ADB04297.1), and 1-methylalkyl)succinate synthase (MASS; CAO03074.1) were constructed with Modeler v9.1 (30) using the substrate-bound structure of BSSαβγ as a starting model. The models have Discrete Optimized Protein Energy scores ranging from −96,600 to −10,700. Ligand models for docking were prepared in phenix.eLBOW. Docking of aryl-SS substrates was performed manually based on the position of toluene in the active site of BSSαβγ.
Results
Substrates Can Access the Active Site in the BSSαβγ Complex
To investigate how substrates bind, we incubated BSSαβγ with fumarate and toluene before crystallization. Crystals produced by this method diffract to 3.3 Å resolution (Table 1). Omit electron density peaks in the central active site can be modeled as molecules of fumarate and toluene (Fig. 2A). Substrate-bound BSSαβγ adopts the same closed state observed previously in substrate-free BSSαβγ (23) with virtually no structural changes observable within the active site or on the exterior of the protein (BSSα Cα root mean square deviation: 0.057 Å). As expected, the substrates are bound at the bottom of the proposed access channel. This channel is formed by two helices outside the core barrel, α3′ and α8′, and a loop between α1′ and β1. Together these elements contribute residues Val-185, Glu-189, Ala-308, Tyr-381, Ile-384, and Leu-711 (Fig. 3), forming a contraction at the midway point that appears to restrict movement of substrates and solvent in or out of the active site. In addition to residues from BSSα, a hairpin loop from BSSβ is inserted within the putative access channel and appears to completely block access to the active site. Although these structural data show that substrates can bind to BSS in the presence of BSSβ, it remains unclear what conformational changes are required for passage of substrates into the active site within the full BSSαβγ complex.
FIGURE 2.
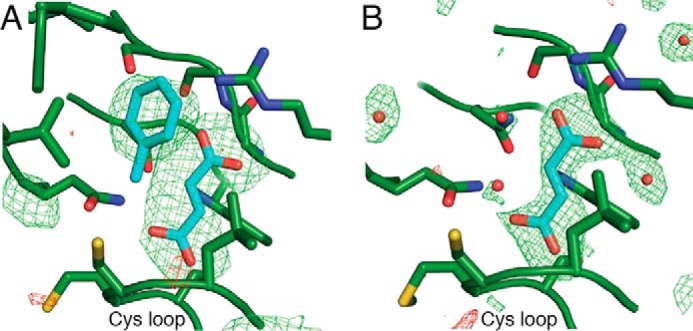
Electron density peaks from bound substrates are present in BSS structures. A, fumarate and toluene co-crystallization in BSSαβγ. B, fumarate co-crystallization in BSSαγ. In both panels, mFo − DFc omit density is shown at 3σ (green) and −3σ (red) from the fully refined structures after exclusion of the substrates and water molecules.
FIGURE 3.
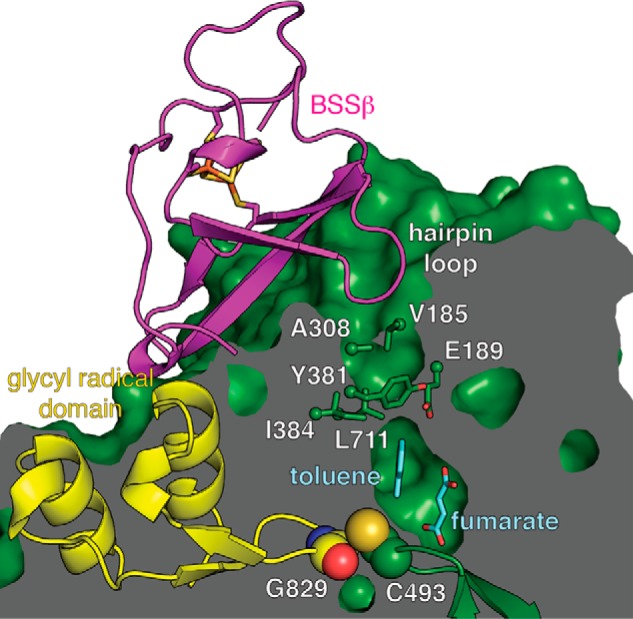
A putative substrate access channel in BSS. The van der Waals surface of BSS is shown as a cutaway with substrates (cyan) bound in the central active site cavity above the Cys loop. As noted before (23), BSSβ binds both adjacent to the glycyl radical domain (yellow) and within a surface cavity that leads to the proposed channel. In particular, a hairpin loop of BSSβ inserts into this cavity, filling it completely. The channel also has a bottleneck at the midway point (labeled residues are shown as sticks). With BSSβ bound, the glycyl radical domain appears locked in the active site for catalysis. This structure should represent the catalytically active form were the glycyl radical present.
Fumarate Binding Stabilizes a Partially Closed State of BSSαγ
We co-crystallized BSSαγ with 5 mm fumarate to determine if the binding of fumarate alone can convert the open enzyme state, as previously observed in the structure of substrate-free BSSαγ (23), to the closed state observed in both substrate-free and substrates-bound BSSαβγ structures (Fig. 4A). The fumarate-bound BSSαγ structure was determined to 2.0 Å resolution (Table 1) and showed unambiguous density for fumarate in the active site cavity (Fig. 2B). We found that the overall structure of fumarate-bound BSSαγ is an intermediate between the closed and open states observed for BSSαβγ and substrate-free BSSαγ, respectively (Fig. 4, A and B). Instead of generating a fully closed enzyme form, the binding of fumarate to BSSαγ stabilizes β strands β7, β8, and β9, which allows the C-terminal helix bundle covering the top half of the BSS active site to adopt a more closed conformation (Fig. 4A). The molecular basis for the 3–4 Å shift in this region of the protein is revealed by inspection of the active site cavity. In particular, Trp-613 adopts a different rotamer conformation when fumarate is present, and this rotamer shift of Trp-613 allows for β9 to become ordered (Fig. 5, A and B). Fumarate also forms hydrogen bonds with residues on β7 and β8, additionally stabilizing both β8 and β9. The repositioning and ordering of these β strands allows helices of the C-terminal helical domain to shift toward the core of the structure (Fig. 5, C and D). Although connections between β strands do not typically require stabilization by substrate binding, in BSS β7, β8, and β9 are short and thus only form a small number of interstrand hydrogen bonds. Moreover, conserved proline residues block some of the hydrogen bonds that would normally be present in a more traditional β sheet or barrel. Given the weakness of the interactions in this region of the protein without substrate, fumarate binding appears to exert a large, stabilizing effect on the BSS core barrel, which could prepare the active site to bind toluene. Our previous work showed that BSSβ binding stabilizes the barrel in a completely closed conformation that appears more suited to catalysis (23). Thus, both fumarate and BSSβ appear to contribute to stabilizing the barrel in the closed conformation, but BSSβ appears to cause a more complete closure.
FIGURE 4.
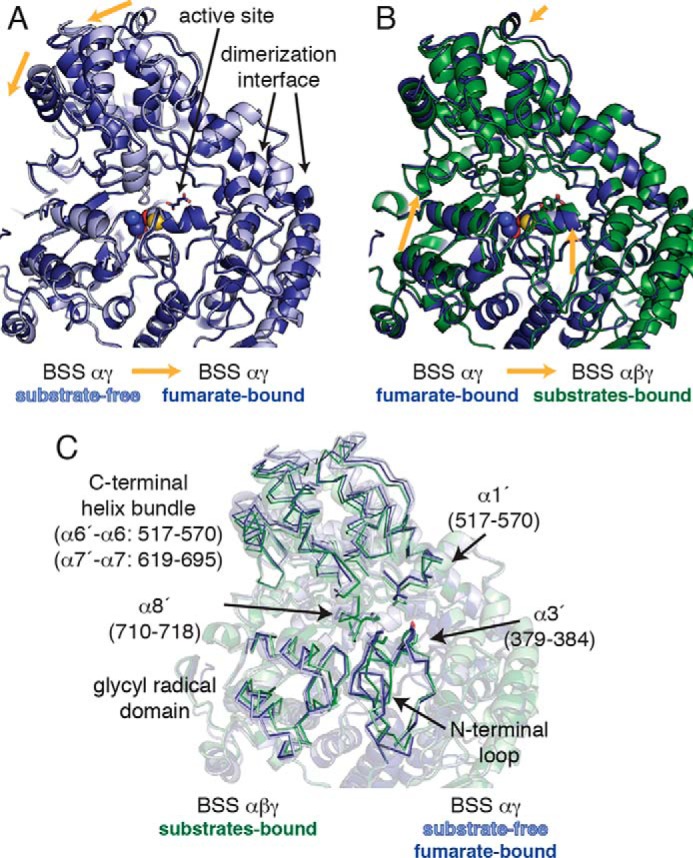
The structure of BSSα is influenced by the presence of substrates and by the binding of BSSβ. A, the C-terminal helical bundle of fumarate-bound BSSαγ is shifted toward the center of the protein relative to the substrate-free BSSαγ structure (PDB ID 4PKC; Ref. 23), leading to partial closure of the enzyme. However, fumarate binding only very slightly shifts the conformation of the glycyl radical domain within this complex. B, the C-terminal helical bundle of fumarate-bound BSSαγ is similar to that observed in BSSαβγ structures. However, the glycyl radical domain must shift inward to form the fully closed conformation needed for catalysis. Only BSSα is shown in A and B; there were no changes in the structure of BSSγ. The structures were aligned based on the dimerization interface, which does not change between BSSαγ and BSSαβγ structures. C, transparent overlay of all structures shown in A and B with regions of interest highlighted for clarity (ribbon).
FIGURE 5.
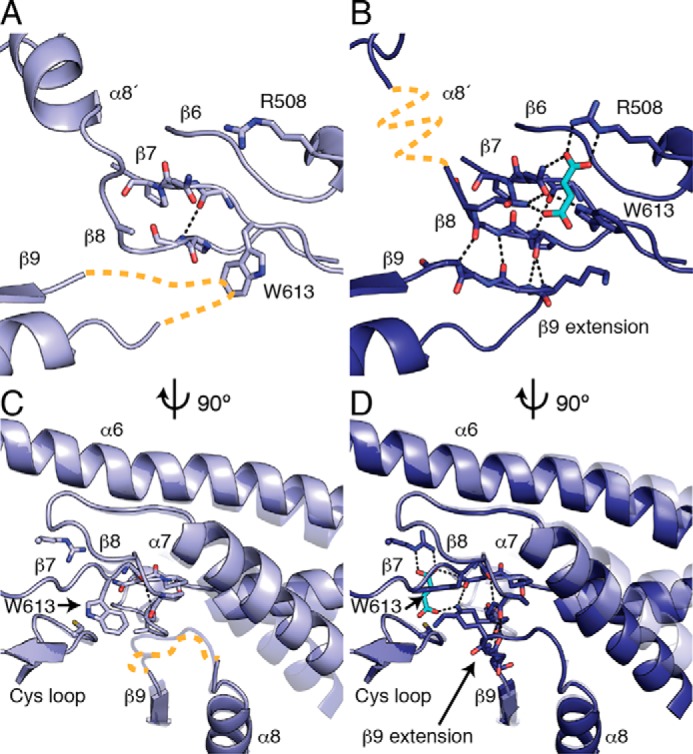
Fumarate binding stabilizes a partially closed conformation in BSSαγ. A, in the absence of fumarate, contacts between β7 and β8 are disrupted, and β9 is completely disordered (orange dashed line). Conserved proline residues in β7 and β8 contribute to the especially weak interstrand contacts in this region. B, a rotamer flip of Trp-613 enables fumarate binding and enables a chain of conformational changes within the barrel. A network of hydrogen bonds is created between Trp-613, fumarate, and the residues on β7 and β8. Fumarate binding presumably stabilizes β8 in a more traditional β sheet conformation, thus allowing an extension of β9 to contribute interstrand hydrogen bonds to β8. α8′ is disordered in fumarate-bound BSSαγ (orange dashed line) despite the restoration of the closed barrel (see Fig. 6). Panels C and D are side views of panels A and B. C, the core helices α7, α8, and α9 are shown with a transparent copy of the helices from the other structure for comparison. The rotamer flip of Trp-613 is correlated with movement and disorder in β9 and the β9 extension (orange dashed line), as the indole side chain would clash with backbone residues in this conformation. The movement in α7 is likewise correlated with the changes in β9 extension, as these residues come in close contact with each other. D, fumarate binding stabilizes the positions of Trp-613 and β9 in the closed state.
Despite this large, fumarate-induced shift in the barrel, the active site in fumarate-bound BSSαγ remains open to solvent, mainly due to the unfolding of helix α8′ (residues 710–718) (Fig. 6A) as well as a shift in the position of helices α3′ (residues 379–384) and α1′ (residues 176–184) (Fig. 4C) away from the glycyl radical domain. Helix α8′ residues are also poorly ordered in the substrate-free BSSαγ, although it was possible to model the residues based on the structure of BSSαβγ in this case (Fig. 6B). Interestingly, toluene soaks of fumarate-bound BSSαγ crystals did not lead to a toluene-bound structure, suggesting that this open form of the enzyme cannot stably bind the second substrate.
FIGURE 6.
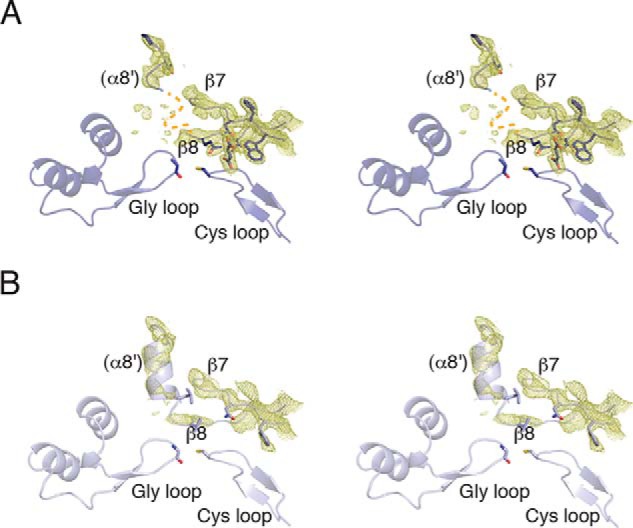
Helix α8′ is disordered in BSSαγ structures. A, Walleye stereoimage showing 2mFo − DFc electron density contoured at 1σ around the non-core helix α8′ and the core strands β7 and β8. There is density present, suggesting that helix α8′ is present but structurally heterogeneous. The missing residues have been marked (orange dashed line) B, in substrate-free BSSαγ, α8′ is better ordered despite deterioration in the density for β7 and β8.
Unlike the C-terminal helix bundle, binding of fumarate does not change the position of glycyl radical domain and associated N-terminal loop relative to substrate-free BSSαγ. These regions remain shifted out of the active site by ∼2 Å relative to their position in BSSαβγ (Fig. 4B). There are no contacts between fumarate and the glycyl radical domain, and it appears that the partial closure of the barrel between β8 and β9 is independent of the inward shift of the glycyl radical domain.
BSS Lines Up Fumarate and Toluene for Radical Chemistry
BSS binds fumarate through both of its carboxylates. The C4 carboxylate of fumarate forms hydrogen bonds to the backbone amides of the Cys loop (NH of residues Leu-492, Met-494, and Ser-495), replacing a chloride ion observed in the substrate-free structure (23), and Arg-508 anchors the C1 carboxylate of fumarate in a bidentate salt bridge (Fig. 7A). Fumarate is held nearly planar, with contacts to Trp-613 in back and Leu-492 in front ensuring the re face of the C2 carbon of fumarate faces out toward toluene and Cys-493 (which becomes the putative, transient thiyl radical). A hydrogen-bonding network that is formed by residues Asn-615 and Gln-707 (on β7 and β8, respectively) and fumarate anchors both carboxylates of the substrate, securing fumarate into position at the back of the active site. Trp-613 joins this hydrogen-bonding network with an indole nitrogen contact to Asn-615 but also forms an edge-to-face interaction with the double bond of fumarate. The orientation of fumarate is unambiguous in the 2.0 Å resolution structure of BSSαγ, with C2 facing out into the active site cavity. The enzyme actively discriminates against dicarboxylates other than fumarate; modeling of succinate in the active site reveals steric clashes with Leu-492 and Trp-613 (Fig. 8, A and B). Accordingly, even after soaking crystals of BSSαγ in 100 mm succinate, no density was observed in the active site.
FIGURE 7.
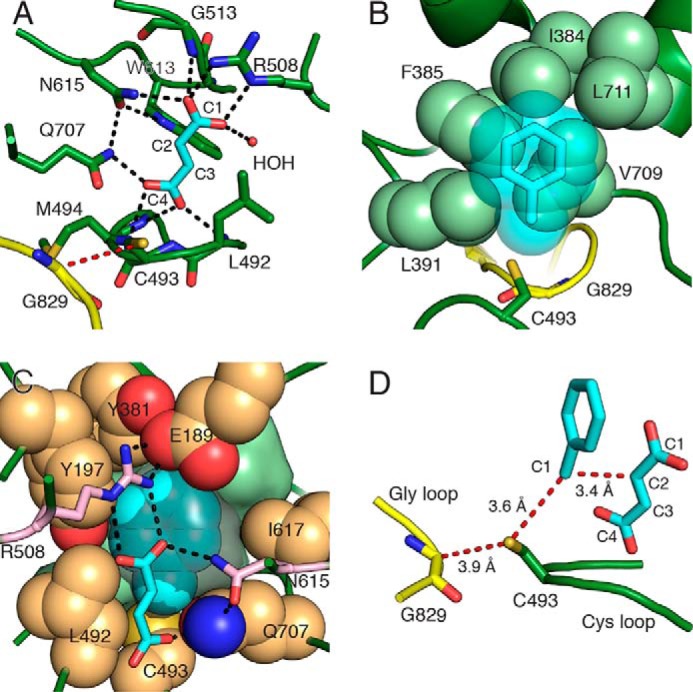
Substrates bind BSS adjacent to the active site Cys loop. A, BSSαγ with fumarate (cyan) bound above the Cys loop. The glycyl radical domain (yellow) is shown with the radical transfer path (red dashes) between the Gly-829 and Cys-493. Hydrogen bonds (black dashes) are shown for polar distances less than 3.2 Å. A single water molecule (HOH) participates in fumarate binding. B, van der Waals sphere representation of the hydrophobic wall (light green spheres) involved in binding toluene (cyan transparent spheres and sticks). The orientation is 180° along the y axis from that in panel A. C, the toluene binding site is shown with residues contributing to the sides of the binding pocket (tan spheres). The hydrophobic wall is present behind toluene (light green surface). Fumarate (cyan sticks) is shown in the foreground with residues that provide hydrogen bonds and contribute to the toluene pocket (pink sticks). The cavity contours around toluene are shown (gray surface) to illustrate the tight packing. D, the toluene/fumarate co-complex positions substrates such that the expected hydrogen atom transfer distances and C—C bond-forming distance (red dashes) are minimized. The orientation is 90° along y axis from that in panel A.
FIGURE 8.
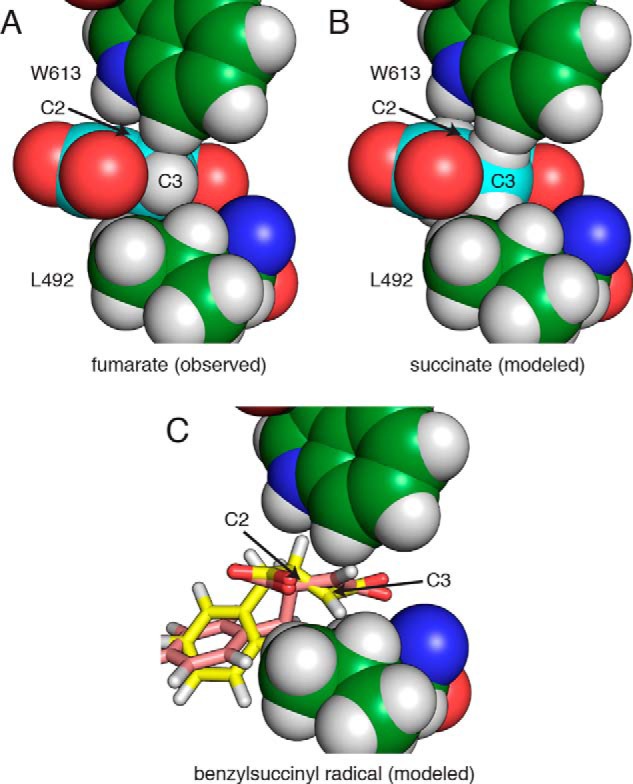
The BSS active site discriminates against non-planar dicarboxylates. A, Trp-613 and Leu-492 in the BSSαγ structure pack against C3 of fumarate with an edge-to-face interaction and van der Waals contacts, respectively. B, modeled succinate clashes with both residues through hydrogen atoms. C, two different conformations of the R-benzylsuccinyl radical modeled into the fumarate-bound active site clash with either Trp-613 (yellow) or Leu-492 (pink).
Toluene contains no polar functional groups to facilitate its positioning during catalysis. The binding site is thus created primarily by aliphatic and aromatic residues. Residues from two regions, α3′ (Ile-384, Phe-385, Leu-391) and the loop between β8 and α8′ (Val-709, Leu-711), create what we have called a “hydrophobic wall” (23) that forms one side of the active site (Fig. 7B), opposite fumarate. A ring of hydrophobic (Tyr-197, Tyr-381, Leu-492, Ile-617) and polar (Glu-189, Gln-707) residues forms the sides of the toluene binding pocket, matching the contours of toluene perfectly (Fig. 7C). The final side of the binding pocket is created by fumarate itself, with the methyl group of toluene stacked against C2 of fumarate and the aromatic ring of toluene stacked against the C1 carboxylate of fumarate. This orientation of toluene is consistent with the production of (R)-benzylsuccinate, as previously observed (31, 32), as it places C1 of toluene directly adjacent to C2 of fumarate (Fig. 7D). The observed distances between Cα of Gly-829 (the putative glycyl radical), Sγ of Cys-493 (the putative transient thiyl radical), and the C1 of toluene are 3.4–3.9 Å, consistent with observed donor-acceptor distances in a number of other radical systems (33). The two substrates are thus observed prearranged to permit C—C bond formation with little to no movement.
Two Active Site Regions Contribute to Specificity in X-succinate Synthases
Most residues that contribute to fumarate binding are conserved within putative XSSs but not in other GRE families (Fig. 9). Within the aryl-SS enzymes IBSS, HBSS, and NMSS, the Cys loop and residues within strand β6, including Arg-508, are virtually identical to BSS, suggesting that they may bind fumarate and their aryl hydrocarbon substrates in similar orientations as observed in BSS. 1-Methylalkyl-SS has some substitutions in these regions that may have catalytic relevance (discussed below). However, among both aryl- and alkyl-SS enzymes there is much less sequence conservation within residues that are predicted to determine hydrocarbon specificity (Fig. 10).
FIGURE 9.

Alignment of Cys loop and adjacent residues in GREs. XSS enzymes (top five sequences) are distinguished by the presence of a CM[A/S]P motif followed by an absolutely conserved arginine (Arg-508) that contacts fumarate directly (see Fig. 7A). Conserved residues in the active site of BSS are in bold. The alignment was performed in Clustal Omega (43). HPD, 4-hydroxyphenylacetate decarboxylase from Clostridium difficile; PFL2, pyruvate formate lyase-like protein from Archeoglobus fulgidus; GDH, glycerol dehydratase from Clostridium butyricum; CutC, choline trimethylamine lyase from Desulfovibrio alaskensis; PFL, pyruvate formate lyase from Escherichia coli.
FIGURE 10.

Sequence alignment of characterized XSS enzymes. The alignment has been sectioned into regions important for substrate binding, as described in Figs. 5, 7, and 11. Structural features of BSS are highlighted within the sequences: fumarate binding residues (yellow), residues that appear to contribute to aryl versus alkyl specificity (cyan), and other residues that contribute to hydrocarbon specificity (green). Residues conserved between BSS and related enzymes are also highlighted. The active site glycine and cysteine residues (red) are indicated. The alignment was performed in Clustal Omega (43).
To assess how the residues of the hydrophobic wall could contribute to substrate specificity, we constructed homology models of four characterized XSS family members based on the structure of BSSαβγ (Fig. 11A) and manually docked their known substrates into the models. IBSS is closest in sequence conservation to BSS (72% identity) and has only 2 substitutions (L711V, I384A) of 13 active site residues (Fig. 11B). Both residues are found at the far end of the active site, away from the site of hydrogen atom abstraction and directly adjacent to the isopropyl group of the modeled substrate, p-cymene (4-isopropyltoluene). The predicted result of these two substitutions is an enlarged binding site tailored perfectly for the desired substrate. Similarly, HBSS (54% identity) contains four substitutions that cluster at the end of the active site near the phenolic oxygen of the modeled substrate, p-cresol (Fig. 11C). The net effect of these substitutions is expected to be a slightly larger binding site for p-cresol in HBSS than for the smaller toluene in BSS. A unique E189Q substitution in the HBSS subfamily would appear to place a glutamine adjacent to the phenolic oxygen of the substrate. These differences between BSS and HBSS are not dramatic, and it is, therefore, not surprising that BSS also accepts p-cresol as a substrate, although with reduced proficiency relative to toluene (34).
FIGURE 11.
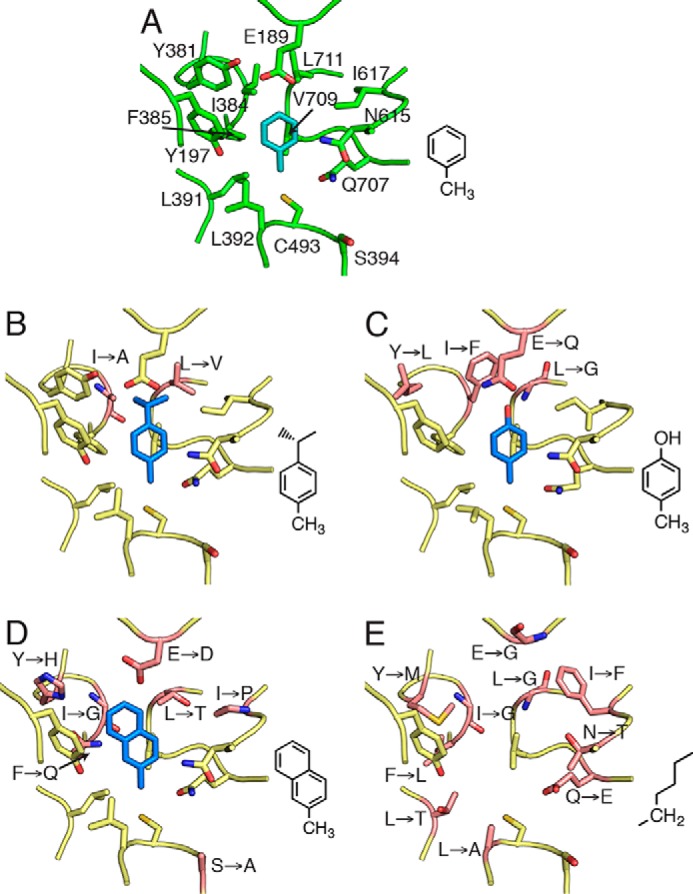
Homology models of XSS family members displaying the hydrocarbon binding site. A, Crystal structure of BSSαβγ bound to toluene (cyan). For clarity, bound fumarate is not shown. B, model of IBSS with p-cymene. C, model of HBSS with p-cresol. D, model of NMSS with 2-methylnapthalene. E, model of MASS; due to the substantial changes in the residues lining the active site, modeling of a known substrate, n-hexane, is not appropriate. For each model, substrates (blue, diagramed at the right) were manually modeled according to changes in the active site due to amino acid substitutions (pink). Residues altered from BSS are labeled.
NMSS (49% identity) has six substitutions (E189D, I384G, F385Q, L711T, I617P), all of which decrease the size of the amino acid side chains in the active site, thus allowing the second aryl ring of the substrate, 2-methylnaphthalene, to fit in the cavity (Fig. 11D). In contrast to the top face of the active site, which varies substantially between different enzymes, aryl-SSs have a core set of conserved residues at the bottom of the active site, which position the methyl group of toluene between the Sγ of Cys-493 and C2 of fumarate in BSS. This pattern of conservation suggests that these enzymes maintain the same substrate geometry around the methylbenzene moiety of the substrate to perform the required chemistry. Thus, hydrocarbon specificity appears governed by substitution of residues at the top of the active site, whereas residues at the bottom of the active site involved in fumarate positioning are conserved.
A distinct subset of XSS enzymes has been shown to generate various 1-methylalkylsuccinate products instead of arylsuccinates. We created a homology model of MASS (35% identity to BSS); however, the sequence is likely too diverged from BSS to model with confidence in the precise side chain placement or orientation of the substrate (Fig. 11E). This model nonetheless reveals key differences from the other aryl-SS enzymes. Only two residues that make up the toluene binding pocket in BSS are conserved in MASS (Val-709 and Tyr-197) and three residues crucial to the binding of toluene in BSS (Leu-711, Ile-384, Glu-189) are replaced by glycines, suggesting the active site may be completely remodeled to bind to linear alkyl chains. Although MASS retains two key residues for binding fumarate in BSS, Arg-508 and Trp-613, it is notable that there are substitutions in other residues (G513A, Q707E, N615T) that contribute to binding of fumarate (Fig. 7A). The disruption of these important contacts, and especially substitution at glycine, may indicate that large structural changes in the architecture of the MASS active site reposition fumarate within the active site cavity to allow for its interaction with a non-aryl substrate.
Discussion
Although GREs perform a diverse set of chemical transformations, they utilize a common architecture for radical storage and to enable handling of radical intermediates. These enzymes are faced with the challenge of alternatively needing to adopt open and closed barrel states. The open state is essential to expose the Gly loop to install the radical within unactivated enzymes. The enzyme must then return the barrel to the closed state to protect the newly formed glycyl radical and the subsequent radical intermediates thought to be generated during catalysis. In addition, XSS enzymes have the special task of securing hydrocarbons like toluene or n-hexane that have no functional groups that can serve as anchor points. These molecules must be held in the active site with precision so that hydrogen atom transfer can take place while also allowing for an access channel to open for substrate binding and product release. Thus, closed and open states of the putative substrate-access channel are also required once the enzyme has been activated.
XSS enzymes are unusual among GREs in their ability to bind two small subunits, BSSβ and BSSγ, both of which appear to directly impact the stability of the protein (20, 23). BSSγ is most important for the solubility of BSSα, whereas BSSβ is proposed to regulate the conformational dynamics of BSSα (23). In particular, we previously showed that the absence of BSSβ leads to an unfolding of the active site barrel such that the glycyl radical domain can exit and access to the active site for substrates is not hindered, whereas in the presence of BSSβ the active site is sealed. To determine if substrate binding to the BSSβ-free complex leads to a conformational change that seals the active site, we determined a fumarate-bound structure of BSSαγ. Although we found that fumarate binding did lead to a more compact structure, the resulting partially closed barrel was not ready for catalysis as regions of the protein important for toluene binding, including the hydrophobic wall, were still disordered in this structure. Furthermore, we were unable to capture a structure with toluene bound to BSSαγ, suggesting that the closing of the channel and ordering of the hydrophobic toluene binding pocket, as observed in the BSSαβγ structure, may be necessary to retain toluene in the active site. Additionally, the binding of fumarate alone to the open BSSαγ complex did not change the position of the glycyl radical domain or shorten the distance between the active site Gly and Cys loops, suggesting that extra factors besides fumarate binding are necessary to generate a form of the enzyme capable of radical transfer. One of these factors may be activation itself, which is expected to change the backbone conformation of the Gly loop and thus may allow for deeper packing of the glycyl radical domain within the active site. Alternatively, the binding of BSSβ may be the trigger that is needed for both the correct positioning of the glycyl radical domain and for the formation of the toluene binding pocket, readying the enzyme for catalysis.
Before this work, it was not clear if the BSSαβγ complex would be able to bind substrates as the channel appears to be blocked in the presence of BSSβ. Here we find that substrates do bind to BSSαβγ and appear to be positioned perfectly for catalysis. In retrospect, it makes sense that toluene could only be stably bound in an active site that is largely sealed. With no functional groups to hold onto, the enzyme must create a binding pocket around the toluene to position it for catalysis, which is what we observe. Although it seems that some rearrangement or partial dissociation of BSSβ must occur to allow for substrate access, these structures suggest that BSSβ will be bound during enzyme turnover.
Due to the experimental difficulty of generating the catalytically essential glycyl radical in vitro, the order of substrate binding in BSS has not yet been determined kinetically; however, there are numerous reasons to believe that fumarate will bind before toluene. First, fumarate lies deep within the active site of BSS, whereas toluene binds above it, adjacent to residues thought to form the channel (Fig. 3). Second, our structures show that in BSSαγ fumarate can bind in the absence of toluene and that fumarate binding does not completely order the active site in this complex. Third, fumarate forms one side of the toluene binding pocket, indicating that tight binding of toluene requires the presence of fumarate (Fig. 7C). Finally, deuterium labeling studies have shown that hydrogen atom abstraction first occurs from C1 of toluene (31, 32), so obligate binding in this order would prevent off-pathway production of the free benzyl radical, which could damage the enzyme.
Taken together, our data allow us to propose a structure-based mechanism (Fig. 12) that builds on proposals from previous biochemical and computational experiments (35, 36). We show fumarate binding first (Fig. 12, i) and toluene binding second (ii) for the reasons discussed above. Once toluene enters the active site, Leu-492, Gln-707, and residues of the hydrophobic wall position the C1 of toluene perfectly between Sγ of Cys-493 and C2 of fumarate. Hydrogen atom abstraction from C1 of toluene can, therefore, result in immediate formation of the C—C bond between toluene and fumarate, as the singly occupied p-orbital of the benzyl radical is already adjacent to the double bond of fumarate (iii–iv). The resulting benzylsuccinyl-C3-radical intermediate would need to change orientation in the active site to regenerate the thiyl radical at Cys-493 (v–vi). This change in orientation may be facilitated by the adjacent Leu-492 and Trp-613, both of which pack tightly against fumarate (Fig. 8A). Modeling the proposed benzylsuccinyl radical intermediate suggests that the loss of planarity at C2 after C—C bond formation and movement of the C2 hydrogen toward Trp-613 may force the C4 carboxylate to shift, thus facilitating hydrogen atom abstraction from Cys-493 by the C3 radical (Fig. 8C). Experimental support for such a carboxylate shift comes from the observation that if maleate (the cis isomer of fumarate) is used as a substrate, the addition of toluene across the double bond occurs in an anti-, rather than syn-, orientation (31, 32). This result implies that the benzylsuccinyl radical can undergo rotation about the newly formed single bond before the hydrogen atom is abstracted from Cys-493. The exit of benzylsuccinate (vi–vii) may be facilitated by Leu-492 and Trp-613, which are arranged in the fumarate-bound structure such that they should form unfavorably close interactions with the sp3-hybridized C2 and C3 of the product, benzylsuccinate.
FIGURE 12.

Structure-based mechanistic proposal for BSS. i, conserved residues facilitate fumarate binding above the Cys loop. ii, toluene binds after fumarate; binding and specificity are effected by the hydrophobic wall and surrounding residues. Binding of both substrates facilitates hydrogen atom transfer from Cys-493 to the Gly-829 glycyl radical cofactor. iii, the transient thiyl radical abstracts a hydrogen atom from toluene to generate a benzyl radical. iv, C—C bond formation is facilitated by preorganizing substrates directly adjacent to each other. v, the putative benzylsuccinyl radical intermediate clashes with Trp-613 causing a rearrangement that places C3 close to Cys-493. vi, hydrogen atom re-abstraction from Cys-493 forms benzylsuccinate in an overall syn addition. vii, hydrogen atom re-abstraction from Gly-829 regenerates the radical storage cofactor. Clashes with Leu-492 and Trp-613 help eject benzylsuccinate. viii, once the radical has returned to the Gly loop, the product is free to exit the active site. Although the steps are shown as sequential, there is no direct evidence for accumulation of radical intermediates, and steps ii–iv and vi–viii could be concerted.
Our structures of BSS with substrates bound provide a fresh perspective on sequence motifs of putative XSS enzymes in genomic databases. Specifically, a Cys loop sequence motif of LCM[A/S]P appears to be diagnostic of aryl-SSs, whereas an [A/S]CMSP motif would seem to specify an alkyl-SSs (Fig. 9). In both of these classes of XSS enzymes, the small side chains of alanine or serine two residues after the active site cysteine create space for fumarate to bind to the backbone amides of the Cys loop. Instead of alanine or serine, a glutamate is commonly found in this position in GREs that perform dehydration reactions and is thought to participate in catalysis as a general base (37). The conserved methionine and proline residues are likely important for the overall structure of the Cys loop as similar residues are observed in non-XSS GREs (Fig. 9).
We suspect that the difference in the amino acid identity one residue upstream of the cysteine in the Cys loop (leucine for aryl-SSs and alanine for alkyl-SSs) has to do with positioning of the substrate. In aryl-SS enzymes, the larger leucine positions the aryl substrate further from cysteine, allowing for C1 hydrogen atom abstraction. The initial hydrogen atom abstraction from alkanes by MASS is predicted to be substantially more difficult than that proposed for aromatic hydrocarbons (38) due to the lack of resonance delocalization in the product-based radical. The growth rate of alkane-degrading species is notably much slower than related aromatic-hydrocarbon-degrading species (39), perhaps in part due to the difficulty of the initial activation reaction, in which a saturated hydrocarbon C-H bond is broken rather than the moderately activated aryl C-H bond. The elevated barrier for this hydrogen atom abstraction may be somewhat mitigated by performing radical abstraction at the subterminal methylene carbon (C2) rather than the terminal methyl (C1). For the reaction to occur with this regiochemistry, the methyl group of the alkyl chain must be positioned differently such that C2 is adjacent to the active site cysteine. In alkyl-SS enzymes, the substitution of alanine for leucine directly adjacent to the presumed binding site of the substrate could make room for the terminal methyl group to shift away from the catalytic cysteine, thus allowing for hydrogen atom abstraction at C2. There have also been proposals of terminal hydrogen atom abstraction in short chain alkanes (40) and even the proposal that methane could be activated by an XSS (41). However, the ability of an alkyl-SS to perform such a dramatically uphill C-H bond cleavage (42) has not been substantiated in vitro to date, and no candidate XSS has been proposed for this reaction.
Within BSS, residues in the Cys loop are important for fumarate binding, the stereochemistry of the reaction, and the reaction mechanism, which is likely to be conserved in related aryl-SSs. However, residues at the upper end of the active site appear to vary considerably and thus govern substrate specificity. BSS, HBSS, and IBSS have toluene or toluene-like substrates and are similar enough in sequence that homology modeling allows us to propose residue substitutions that could account for their different substrate specificities. The much lower sequence identity of NMSS with BSS makes rationalizing its substrate preference less certain, but a conservative interpretation is that residue substitutions at the top of the active site and along the hydrophobic wall serve to enlarge the active site to accommodate the larger substrate of NMSS. The XSS family member MASS shows the greatest divergence in sequence, which is consistent with its role as an alkyl-SS as these substrates require a change not only in the hydrocarbon binding pocket but also in the region of the active site that governs specificity of hydrogen atom abstraction. The very low sequence identity to BSS within the active site strands and loops of MASS makes drawing specific inferences about the chemistry occurring within alkyl-SS enzymes untenable. Although our homology models are useful for proposing general trends in active site architecture, detailed biochemical and structural investigation is needed for more members of the XSS family to fully understand how this class of GREs recognizes substrates and performs radical chemistry.
The structures of BSS with bound substrates presented here greatly advance our knowledge of a key reaction in the anaerobic degradation of hydrocarbons. We have identified residues that may determine specificity in XSS enzymes and thus provide a genetic handle for determining the hydrocarbon degrading potential of microbial communities. Although hydrocarbon degradation is a beneficial process in natural environments, in industrial settings oil fouling by microbes can be detrimental to oil quality as well as to physical infrastructure. Inhibitors of anaerobic hydrocarbon degradation inspired by these structures could, therefore, be useful additives. These structures also provide a starting place for future efforts to characterize the BSS reaction computationally, especially to investigate the series of rearrangements occurring after C—C bond formation that return the radical to its storage site within the protein backbone.
Author Contributions
M. A. F. and C. L. D. designed the research, analyzed the data, and wrote the paper. M. A. F. performed the research. E. N. G. M. contributed new reagents. All authors reviewed and edited the manuscript and approved of the final version.
Acknowledgments
Data were collected at Beamline X26A, National Synchrotron Light Source, Brookhaven National Laboratory. Beamline X26A is supported by the Department of Energy Geosciences (DE-FG02-92ER14244 to the University of Chicago Center for Advanced Radiation Sources). The National Synchrotron Light Source is supported by the Department of Energy Office of Basic Energy Sciences under Contract DE-AC02-98CH10886.
This work was supported, in whole or in part, by National Institutes of Health Grant GM093088 (to E. N. G. M.). This work was also supported in part by a National Science Foundation Graduate Research Fellowship under Grant 0645960 (to M. A. F.). The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (codes 5BWD and 5BWE) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- BSS
- benzylsuccinate synthase
- aryl-SS
- aryl-succinate synthase
- XSS
- X-succinate synthase
- GRE
- glycyl radical enzyme
- HBSS
- 4-hydroxybenzyl-SS
- NMSS
- naphthyl-2-methyl-SS
- MASS
- (1-methylalkyl)succinate synthase
- IBSS
- 4-isopropylbenzyl-SS.
References
- 1. Leahy J. G., Colwell R. R. (1990) Microbial degradation of hydrocarbons in the environment. Microbiol. Rev. 54, 305–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ocean Studies and Marine Board (2003) in Oil in the Sea III: Inputs, Fates, and Effects, pp. 89–118, National Academies Press, Washington, D. C. [PubMed] [Google Scholar]
- 3. Atlas R., Bartha R. (1992) Hydrocarbon biodegradation and oil spill bioremediation. In Adv. Microb. Ecol. (Marshall K. C. ed.) pp. 287–338, Springer-Verlag New York Inc., New York [Google Scholar]
- 4. Fuchs G. (2008) Anaerobic metabolism of aromatic compounds. Ann. N.Y. Acad. Sci. 1125, 82–99 [DOI] [PubMed] [Google Scholar]
- 5. Biegert T., Fuchs G., Heider J. (1996) Evidence that anaerobic oxidation of toluene in the denitrifying bacterium Thauera aromatica is initiated by formation of benzylsuccinate from toluene and fumarate. Eur. J. Biochem. 238, 661–668 [DOI] [PubMed] [Google Scholar]
- 6. Leuthner B., Heider J. (2000) Anaerobic toluene catabolism of Thauera aromatica: the bbs operon codes for enzymes of beta oxidation of the intermediate benzylsuccinate. J. Bacteriol. 182, 272–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Strijkstra A., Trautwein K., Jarling R., Wöhlbrand L., Dörries M., Reinhardt R., Drozdowska M., Golding B. T., Wilkes H., Rabus R. (2014) Anaerobic activation of p-cymene in denitrifying betaproteobacteria: methyl group hydroxylation versus addition to fumarate. Appl. Environ. Microbiol. 80, 7592–7603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Harms G., Rabus R., Widdel F. (1999) Anaerobic oxidation of the aromatic plant hydrocarbon p-cymene by newly isolated denitrifying bacteria. Arch. Microbiol. 172, 303–312 [DOI] [PubMed] [Google Scholar]
- 9. Müller J. A., Galushko A. S., Kappler A., Schink B. (2001) Initiation of anaerobic degradation of p-cresol by formation of 4-hydroxybenzylsuccinate in Desulfobacterium cetonicum. J. Bacteriol. 183, 752–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wöhlbrand L., Jacob J. H., Kube M., Mussmann M., Jarling R., Beck A., Amann R., Wilkes H., Reinhardt R., Rabus R. (2013) Complete genome, catabolic sub-proteomes, and key-metabolites of Desulfobacula toluolica Tol2, a marine, aromatic compound-degrading, sulfate-reducing bacterium. Environ. Microbiol. 15, 1334–1355 [DOI] [PubMed] [Google Scholar]
- 11. Annweiler E., Materna A., Safinowski M., Kappler A., Richnow H. H., Michaelis W., Meckenstock R. U. (2000) Anaerobic degradation of 2-methylnaphthalene by a sulfate-reducing enrichment culture. Appl. Environ. Microbiol. 66, 5329–5333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Selesi D., Jehmlich N., von Bergen M., Schmidt F., Rattei T., Tischler P., Lueders T., Meckenstock R. U. (2010) Combined genomic and proteomic approaches identify gene clusters involved in anaerobic 2-methylnaphthalene degradation in the sulfate-reducing enrichment culture N47. J. Bacteriol. 192, 295–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Callaghan A. V., Wawrik B., Ní Chadhain S. M., Young L. Y., Zylstra G. J. (2008) Anaerobic alkane-degrading strain AK-01 contains two alkylsuccinate synthase genes. Biochem. Biophys. Res. Commun. 366, 142–148 [DOI] [PubMed] [Google Scholar]
- 14. Grundmann O., Behrends A., Rabus R., Amann J., Halder T., Heider J., Widdel F. (2008) Genes encoding the candidate enzyme for anaerobic activation of n-alkanes in the denitrifying bacterium, strain HxN1. Environ. Microbiol. 10, 376–385 [DOI] [PubMed] [Google Scholar]
- 15. Craciun S., Balskus E. P. (2012) Microbial conversion of choline to trimethylamine requires a glycyl radical enzyme. Proc. Natl. Acad. Sci. U.S.A. 109, 21307–21312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. O'Brien J. R., Raynaud C., Croux C., Girbal L., Soucaille P., Lanzilotta W. N. (2004) Insight into the mechanism of the B12-independent glycerol dehydratase from Clostridium butyricum: preliminary biochemical and structural characterization. Biochemistry 43, 4635–4645 [DOI] [PubMed] [Google Scholar]
- 17. Becker A., Kabsch W. (2002) X-ray structure of pyruvate formate lyase in complex with pyruvate and CoA: How the enzyme uses the Cys-418 thiyl radical for pyruvate cleavage. J. Biol. Chem. 277, 40036–40042 [DOI] [PubMed] [Google Scholar]
- 18. Becker A., Fritz-Wolf K., Kabsch W., Knappe J., Schultz S., Volker Wagner A. F. (1999) Structure and mechanism of the glycyl radical enzyme pyruvate formate lyase. Nat. Struct. Biol. 6, 969–975 [DOI] [PubMed] [Google Scholar]
- 19. Leuthner B., Leutwein C., Schulz H., Hörth P., Haehnel W., Schiltz E., Schägger H., Heider J. (1998) Biochemical and genetic characterization of benzylsuccinate synthase from Thauera aromatica: a new glycyl radical enzyme catalysing the first step in anaerobic toluene metabolism. Mol. Microbiol. 28, 615–628 [DOI] [PubMed] [Google Scholar]
- 20. Li L., Patterson D. P., Fox C. C., Lin B., Coschigano P. W., Marsh E. N. (2009) Subunit structure of benzylsuccinate synthase. Biochemistry 48, 1284–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bhandare R., Calabro M., Coschigano P. W. (2006) Site-directed mutagenesis of the Thauera aromatica strain T1 tutE tutFDGH gene cluster. Biochem. Biophys. Res. Commun. 346, 992–998 [DOI] [PubMed] [Google Scholar]
- 22. Coschigano P. W. (2002) Construction and characterization of insertion/deletion mutations of the tutF, tutD, and tutG genes of Thauera aromatica strain T1. FEMS Microbiol. Lett. 217, 37–42 [DOI] [PubMed] [Google Scholar]
- 23. Funk M. A., Judd E. T., Marsh E. N., Elliott S. J., Drennan C. L. (2014) Structures of benzylsuccinate synthase elucidate roles of accessory subunits in glycyl radical enzyme activation and activity. Proc. Natl. Acad. Sci. U.S.A. 111, 10161–10166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vey J. L., Yang J., Li M., Broderick W. E., Broderick J. B., Drennan C. L. (2008) Structural basis for glycyl radical formation by pyruvate formate lyase activating enzyme. Proc. Natl. Acad. Sci. U.S.A. 105, 16137–16141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hilberg M. (2012) Anaerober Toluol-Stoffwechsel in Thauera aromatica: Biochemische und spektroskopische Untersuchungen zur Reaktion der (R)-Benzylsuccinat Synthase. Ph.D. thesis, Philipps-Universität Marburg [Google Scholar]
- 26. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 27. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Eswar N., Webb B., Marti-Renom M. A., Madhusudhan M. S., Eramian D., Shen M. Y., Pieper U., Sali A. (2006) Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinformatics, Chapter 5, Unit 5.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li L., Marsh E. N. (2006) Mechanism of benzylsuccinate synthase probed by substrate and isotope exchange. J. Am. Chem. Soc. 128, 16056–16057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Qiao C., Marsh E. N. (2005) Mechanism of benzylsuccinate synthase: stereochemistry of toluene addition to fumarate and maleate. J. Am. Chem. Soc. 127, 8608–8609 [DOI] [PubMed] [Google Scholar]
- 33. Vey J. L., Drennan C. L. (2011) Structural insights into radical generation by the radical SAM superfamily. Chem. Rev. 111, 2487–2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Verfürth K., Pierik A. J., Leutwein C., Zorn S., Heider J. (2004) Substrate specificities and electron paramagnetic resonance properties of benzylsuccinate synthases in anaerobic toluene and m-xylene metabolism. Arch. Microbiol. 181, 155–162 [DOI] [PubMed] [Google Scholar]
- 35. Li L., Marsh E. N. (2006) Deuterium isotope effects in the unusual addition of toluene to fumarate catalyzed by benzylsuccinate synthase. Biochemistry 45, 13932–13938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Himo F. (2005) C—C bond formation and cleavage in radical enzymes, a theoretical perspective. Biochim. Biophys. Acta 1707, 24–33 [DOI] [PubMed] [Google Scholar]
- 37. Lehtiö L., Goldman A. (2004) The pyruvate formate lyase family: sequences, structures and activation. Protein Eng. Des. Sel. 17, 545–552 [DOI] [PubMed] [Google Scholar]
- 38. Bharadwaj V. S., Vyas S., Villano S. M., Maupin C. M., Dean A. M. (2015) Unravelling the impact of hydrocarbon structure on the fumarate addition mechanism: a gas-phase ab initio study. Phys. Chem. Chem. Phys. 17, 4054–4066 [DOI] [PubMed] [Google Scholar]
- 39. Spormann A. M., Widdel F. (2000) Metabolism of alkylbenzenes, alkanes, and other hydrocarbons in anaerobic bacteria. Biodegradation 11, 85–105 [DOI] [PubMed] [Google Scholar]
- 40. Kniemeyer O., Musat F., Sievert S. M., Knittel K., Wilkes H., Blumenberg M., Michaelis W., Classen A., Bolm C., Joye S. B., Widdel F. (2007) Anaerobic oxidation of short-chain hydrocarbons by marine sulphate-reducing bacteria. Nature 449, 898–901 [DOI] [PubMed] [Google Scholar]
- 41. Duncan K. E., Gieg L. M., Parisi V. A., Tanner R. S., Tringe S. G., Bristow J., Suflita J. M. (2009) Biocorrosive thermophilic microbial communities in Alaskan North Slope oil facilities. Environ. Sci. Technol. 43, 7977–7984 [DOI] [PubMed] [Google Scholar]
- 42. Beasley K. K., Nanny M. A. (2012) Potential energy surface for anaerobic oxidation of methane via fumarate addition. Environ. Sci. Technol. 46, 8244–8252 [DOI] [PubMed] [Google Scholar]
- 43. Sievers F., Higgins D. G. (2014) Clustal Omega, accurate alignment of very large numbers of sequences. Methods Mol. Biol. 1079, 105–116 [DOI] [PubMed] [Google Scholar]