

Background: C1q binds to apoptotic cells, but the binding sites are unclear.
Results: C1q binds to the nucleolus in advanced apoptotic cells, and the C1q-associated C1r/C1s proteases degrade nucleolar proteins.
Conclusion: C1q recruits C1r/C1s to specific structures in dead cells, leading to cellular antigen degradation.
Significance: This helps to explain why C1r/C1s and C1q deficiency cause autoimmunity.
Keywords: apoptosis, autoimmunity, complement, dendritic cell, nucleolus, C1q, C1s
Abstract
In infection, complement C1q recognizes pathogen-congregated antibodies and elicits complement activation. Among endogenous ligands, C1q binds to DNA and apoptotic cells, but whether C1q binds to nuclear DNA in apoptotic cells remains to be investigated. With UV irradiation-induced apoptosis, C1q initially bound to peripheral cellular regions in early apoptotic cells. By 6 h, binding concentrated in the nuclei to the nucleolus but not the chromatins. When nucleoli were isolated from non-apoptotic cells, C1q also bound to these structures. In vivo, C1q exists as the C1 complex (C1qC1r2C1s2), and C1q binding to ligands activates the C1r/C1s proteases. Incubation of nucleoli with C1 caused degradation of the nucleolar proteins nucleolin and nucleophosmin 1. This was inhibited by the C1 inhibitor. The nucleoli are abundant with autoantigens. C1q binding and C1r/C1s degradation of nucleolar antigens during cell apoptosis potentially reduces autoimmunity. These findings help us to understand why genetic C1q and C1r/C1s deficiencies cause systemic lupus erythematosus.
Introduction
The complement system consists of over 20 abundant serum proteins and numerous cell surface components (1, 2). In infection, complement can be activated through three pathways, although its discovery was made on the basis of the antibody-triggered classical pathway (3–5). The classical pathway is activated after C1 binds to antibodies congregated on microbial surfaces, and C1 is a pentameric complex between one C1q and four proteases (C1r2C1s2) (1, 2, 6). The complement system plays key roles in protecting hosts against microbial infections, and, therefore, deficiencies in one or more complement components can cause increased susceptibility to infection (7). In particular, genetic deficiencies in C1q, C1r/C1s, or C4 are strong causes for the autoimmune disease systemic lupus erythematosus (SLE)3 (8–10). The underlying pathogenic mechanism cannot be delineated directly on the basis of the infection model of complement activation.
A hallmark in SLE pathogenesis is the development of autoantibodies specific for DNA and other nuclear antigens, and these nuclear antigens surge in disease flare-ups, causing the formation of pathogenic immune complexes with the autoantibodies (11). In mice, injection of apoptotic cells induced antinuclear autoantibodies (12). Without a definitive preference, four mechanistic models have been documented regarding the relation between C1q and SLE pathogenesis. First, C1q binding to apoptotic cells enhances phagocytosis of cell debris by macrophages and dendritic cells (10, 13, 14). C1q deficiency may cause antinuclear autoantibodies because of impaired apoptotic cell clearance (1, 15). Second, apoptotic cell-bound C1q has been reported to induce tolerogenic properties in phagocytes that capture this debris (16).
A third C1q-related mechanism revolves around immune complex induction of IFN-α from plasmacytoid dendritic cells (17, 18). IFN-α production is persistent in SLE patients and is pathogenic, but C1q binding to these complexes strongly inhibits IFN-α induction from plasmacytoid dendritic cells (12, 19–21). Fourth, in a ligand-independent manner, C1q may directly induce tolerogenic properties in DCs (22, 23). Therefore, multiple mechanisms may exist for C1q to inhibit lupus pathogenesis. In contrast, there have not been any studies to delineate how deficiency of C1r/C1s, which are not known to participate in apoptotic cell phagocytosis or tolerance induction, causes SLE (24).
Besides being an autoantigen in lupus patients, DNA in its purified form is also a C1q ligand (25, 26). Because C1q binds to both DNA and apoptotic cells, we wondered whether it binds to exposed nuclear DNA in apoptotic cells. Therefore, we examined C1q binding at different stages of cell apoptosis. C1q bound to peripheral structures in early apoptotic cells, but it bound to the nucleolus at advanced stages. C1q also bound to isolated nucleoli. When nucleoli were incubated with the C1 complex, it caused C1r/C1s-dependent nucleolar antigen degradation. These results reveled an additional, novel mechanism by which C1q reacts to cell apoptosis. That is, besides enhancing phagocytosis, its binding to apoptotic cells also elicits the C1r/C1s-mediated degradation of apoptotic cellular antigens (27), adding a new dimension to the “waste disposal theory” (1, 15).
Experimental Procedures
Cell Culture and Reagents
Human cervical adenocarcinoma HeLa cells were cultured in DMEM containing 10% (v/v) HyClone FBS, 100 units/ml penicillin, 100 μg/ml streptomycin, and 2 mm l-glutamine at 37 °C and 5% CO2. The rabbit anti-nucleophosmin 1 (NPM1) antibody (catalog no. HPA11384) and purified C1q (catalog no. C1740) were obtained from Sigma-Aldrich (St. Louis, MO). The rabbit anti nucleolin (NCL) antibody (catalog no. ab22758) was purchased from Abcam (Cambridge, UK). A mouse anti-C1q antibody (catalog no. MCA2603) was purchased from Bio-Rad. A rabbit anti-human histone H3 antibody (catalog no. 9715), the C1 complex (catalog no. 204873), and the C1 inhibitor (catalog no. 204883) were purchased from Merck Millipore (Billerica, MA).
C1q Binding to Apoptotic Cells
HeLa cells were cultured overnight on 13-mm glass coverslips. After removing the medium, cells were washed with serum-free DMEM and maintained in the same medium (0.2 ml/well). Cells were UV-irradiated at 500 mJ/cm2 in a Spectroline Select XLE-1000 UV Crosslinker (254-nm lamp) and then cultured in serum-free medium for 1–24 h. As controls, cells were cultured in serum-free medium for 1 h without prior UV irradiation. Cells were washed in PBS and fixed for 30 min in 1% (w/v) paraformaldehyde (PFA). C1q was diluted in PBS containing 5% (w/v) BSA at 10 μg/ml, incubated with the cells for 1 h, and, after washing, incubated further with a mouse anti-C1q antibody for 1 h. Cells were then washed and incubated for 1 h with fluorescently conjugated goat anti-mouse IgG (Cy3). In some experiments, cells were incubated with both mouse anti-C1q and rabbit anti-NPM1 antibodies. Accordingly, goat anti-mouse (Cy3) and anti-rabbit (Alexa Fluor 488, AF488) IgG were used as secondary antibodies.
Confocal Microscopy
Stained cells were washed, mounted using VectorShield medium containing DAPI, and viewed using a FluoView FV1000 confocal microscope equipped with a ×100 oil objective (aperture 1.45) and Cool/SNAP HQ2 image acquisition camera (Olympus). Images were acquired with FV-ASW 1.6b software and analyzed using Imaris software (Bitplane AG).
Isolation of Nucleoli
HeLa cells were cultured to near confluence in 150-mm dishes, and harvested cells were, after washing, resuspended at 5 × 107 cells/ml in a cold 0.25 m sucrose buffer (10 mm Tris, 5.0 mm MgCl2, 0.25 m sucrose (pH 7.4)) supplemented with a mixture of protease inhibitors (Sigma-Aldrich). Cells were homogenized using an Isobiotec cell homogenizer at 10-μm clearance (Isobiotec Precision Engineering, Heidelberg, Germany). The homogenate was first centrifuged for 10 min at 600 × g, and the pellets were resuspended in 5 ml of a 2.2 m sucrose buffer (10 mm Tris, 5.0 mm MgCl2, and 2.2 m sucrose (pH 7.4)) and centrifuged for 30 min at 50,000 × g. The nuclei pellets were resuspended in 0.25 m sucrose buffer (2 ml) and were, in 0.2-ml aliquots, sonicated for 18 cycles in a UCD-200 Bioruptor (Diagenode s.a., Liege, Belgium) with the setting at maximal strength (15 s on and 45 s off per cycle). The nuclear homogenate was centrifuged for 5 min at 2500 × g and, after washing three times in a 0.88 m sucrose buffer (10 mm Tris, 5.0 MgCl2, and 0.88 m sucrose (pH 7.4)) by centrifuging for 5 min at 1000 × g, the crude nucleoli were resuspended in 0.25 m sucrose buffer (0.5 ml). It was then loaded on a 0.85–2.5 M sucrose gradient and centrifuged for 3 h at 33,000 rpm using a Himac CP 90WX preparative centrifuge and a P40ST rotor (Hitachi Ltd., Tokyo, Japan). The nucleolus pellets were resuspended in 0.25 m sucrose buffer (0.4 ml). The concentrations of the nucleoli slurries were estimated by NanoDrop (A280). Nucleoli obtained after the 0.88 m sucrose wash mostly read 1.5–3.0 optical density, and the final purified nucleoli read mostly between 1.0–2.0 optical density.
In some experiments, the crude nucleoli were, after washing in 0.88 m sucrose, incubated with purified C1q (50 μg/ml) for 30 min on ice and then centrifuged through the 0.85–2.5 m sucrose gradient. In incubation with C1q, crude nucleoli were diluted four times in the 0.25 m sucrose buffer, and 0.5 ml was used in the experiments. In these experiments, one fraction was collected for each gradient step, and nine fractions in total were collected, including the sample layer and the resuspended pellet fraction.
C1q Binding to Purified Nucleoli
Isolated nucleoli were incubated for 5 min on ice with glass coverslips and fixed for 30 min in 1% (w/v) PFA. After blocking for 30 min in PBS-BSA, coverslips were incubated with C1q (10 μg/ml) for 1 h on ice. Coverslips were then washed and incubated for 30 min with mouse anti-C1q and rabbit anti-NPM1 antibodies. This was followed by 30 min of incubation with goat anti-mouse (Cy3) and anti-rabbit (AF488) IgG. The nucleoli were then examined by confocal microscopy.
Nucleolus Digestion with C1
In a 50-μl reaction, 25 μl of purified nucleoli was incubated with 25 μl of diluted complement C1 (19.2 μg/ml) in TBS-Ca2+ (10 mm Tris, 150 mm NaCl, and 3.3 mm CaCl2 (pH 7.4)) at 37 °C for up to 2 h. Reactions were stopped by adding SDS-PAGE sample buffer and then analyzed by SDS-PAGE and Western blotting. In some experiments, C1 inhibitor was included at 160 μg/ml. In each 50-μl reaction, 12.5 μl of diluted C1 (19.2 μg/ml) was premixed with 12.5 μl of diluted C1 inhibitor (320 μg/ml) on ice and then combined with 25 μl of nucleoli before incubation for up to 2 h at 37 °C.
Western Blotting
The sucrose fractions or C1-digested nucleoli were separated on 12.5% (w/v) SDS-PAGE gels and analyzed by Western blotting. Briefly, the blots were blocked for 1 h in blocking buffer (50 mm Tris and 150 mm NaCl (pH 7.4)) containing 0.1% (v/v) Tween 20 and 5% (w/v) nonfat milk and incubated overnight at 4 °C with goat anti-C1q or rabbit anti-NPM1, anti-NCL, and anti-histone H3 antibodies in the blocking buffer. After washing, the blots were incubated for 2 h with horseradish peroxidase-conjugated secondary antibodies. Blots were probed sequentially after stripping for 20 min at 50 °C in 62.5 mm Tris (pH 6.8) containing 0.1 m 2-mecaptoethanol and 2% (w/v) SDS.
Results
In Detergent-permeabilized Cells, C1q Lacks Nuclear Binding
In separate experimental systems, C1q has been shown to bind to DNA and apoptotic cells, respectively (10, 13, 14, 25, 26). In this study, we began by surveying the subcellular regions in apoptotic cells that C1q recognizes, and we were particularly interested in whether C1q binds to the nuclear DNA. HeLa cells were UV-irradiated and subsequently cultured for 1, 3, 6, 12, and 24 h to obtain cells at different stages of apoptosis. As controls, non-irradiated cells were also evaluated for C1q binding. This was performed with or without Triton X-100 permeabilization.
As shown in Fig. 1, C1q lacked specific binding to intracellular structures without detergent permeabilization. Binding was noticed in extracellular spaces, and the signal levels were low. After permeabilization, intracellular C1q binding became prominent. C1q bound mainly to cortical skeletal and cytoplasmic regions and showed no obvious binding to the nuclear region. The interpretation at this point could be either the lack of C1q access to the nuclear region because it is a large 460-kDa protein (28) or the lack of C1q ligand(s) in the nucleus.
FIGURE 1.
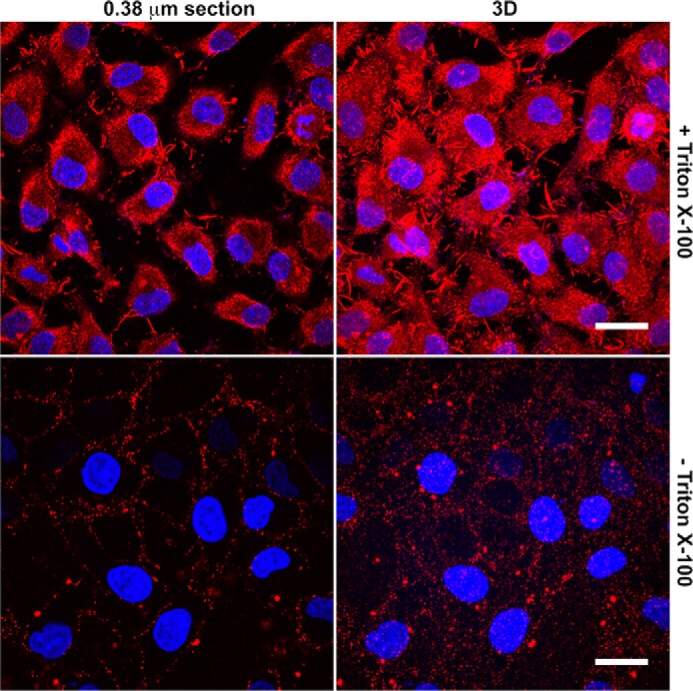
C1q lacks nuclear binding in permeabilized cells. HeLa cells on coverslips were fixed for 30 min in 1% (w/v) PFA and, with (top panels) or without (bottom panels) permeabilization for 1 h in 1% (v/v) Triton X-100, incubated with C1q (10 μg/ml) for 1 h. Bound C1q was detected by incubation with a mouse anti-C1q antibody, followed by goat anti-mouse IgG (Cy3, red). Coverslips were mounted using DAPI-containing VectorShield medium to visualize DNA chromatins (blue). Cells were analyzed using a FluoView FV1000 confocal microscope equipped with a ×100 oil objective, and 0.38-μm section (left panels) and reconstructed three-dimensional (3D, right panels) images were obtained using Imaris software. Scale bars = 20 μm.
In Early Apoptotic Cells, C1q Also Mainly Binds to Cortical and Cytoplasmic Regions
When C1q was incubated with UV-irradiated cells, its binding sites appeared progressively from the cell periphery to the nucleus following the progress of apoptosis (Fig. 2). One hour after UV irradiation, C1q showed prominent binding without detergent permeabilization, and its binding intensity differed among the cell surface, cytoplasmic, and nuclear regions. Strong binding was observed at the cell surface to structures resembling the filopodium type of cortical cytoskeleton (Fig. 2). In the cytoplasm, C1q binding sites were diffuse, appearing consistently networked. Because C1q has been reported previously to bind to monomeric actin (29), it is unclear whether the cortical and cytoplasmic binding by C1q were both contributed by actins. At this stage, binding to the nucleus was observed but remained low, and the bound structures were particulate in gaps between condensed chromatin regions (Fig. 2). The particulate nature of the C1q-bound nuclear structures were apparent on the basis of both 0.38-μm section images and reconstructed three-dimensional images. There was a clear lack of C1q binding to the large body of chromatins. By 3 h, similar binding patterns were maintained. Treatment of these cells with detergent did not significantly alter the C1q binding pattern (data not shown).
FIGURE 2.
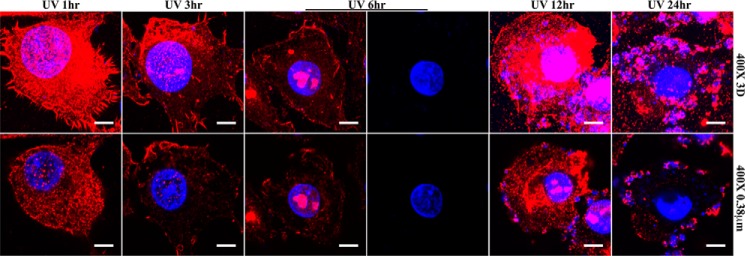
Temporal C1q binding to early and advanced apoptotic cells. HeLa cells on glass coverslips were UV-irradiated and cultured in serum-free medium for 1, 3, 6, 12, and 24 h and, after fixation in 1% (w/w) PFA for 30 min, incubated with C1q. Bound C1q was detected using a mouse anti-C1q antibody, followed by goat anti-mouse IgG (Cy3, red). Chromatins were visualized using DAPI-containing medium (blue). Images were captured using a ×100 oil objective under 4× zoom. Reconstructed three-dimensional (3D, top panels) and single 0.38-μm sections (bottom panels) are shown. With cells stained 6 h after UV irradiation, the same images were presented twice. In the left column, the two images show merged chromatin (DAPI) and C1q (Cy3) signals. In the right column, the C1q signals were omitted to show the morphology of the condensed chromatins. Scale bars = 5 μm.
C1q Binds Predominantly to the Nucleolus in Advanced Apoptotic Cells
Six hours after UV irradiation, both surface and cytoplasmic C1q binding subsided, but binding to the nucleus was intensified (Fig. 2). Furthermore, the pattern of C1q binding in the nucleus also changed from small particulate structures to one to two large congregated structures (Fig. 2). The C1q-bound subnuclear region was surrounded by a rim of chromatins, with sparse chromatin fragments being also found within the C1q-bound region. This C1q binding pattern is homogeneous among the cell population at 6 h. By 12 h, some cells began to exhibit nuclear fragmentation, and, in these cells, C1q binding was also found on the nuclear fragments (Fig. 2). More intensive nuclear fragmentation was observed by 24 h, and C1q continued to bind to these fragments. Meanwhile, the nuclear corpses remaining in the cell center began to lose C1q binding. Although C1q binding was generally near nuclear chromatins in advanced apoptotic cells, C1q probably bound to DNA-associated structures rather than DNA itself. The size and morphology of the C1q-bound nuclear structure resembled that of the nucleolus, as shown most clearly at 6 h (Fig. 2) (30, 31).
In Advanced Apoptotic Cells, the Major C1q-bound Nuclear Region Is the Nucleolus
To examine whether the C1q-bound subnuclear regions in the 6-h apoptotic cells were indeed nucleoli, cells were first incubated with C1q and then incubated with mouse anti-C1q and rabbit anti-NPM1 antibodies. NPM1 is a nuclear protein found predominantly in the granular component of the nucleolus (32). As shown in Fig. 3, the C1q-bound nuclear regions were strongly labeled by anti-NPM1 antibody (Fig. 3). At this stage, NPM1 also diffused broadly in the cytoplasm, where C1q binding was absent. It suggests that, although C1q binds to the nucleoli in late apoptotic cells, NPM1 is an unlikely C1q ligand. Even in the nucleolar region, C1q binding showed an incomplete overlap with NPM1 (Fig. 3D). Furthermore, C1q bound intensely in the cytoplasm 1 h after irradiation, when NPM1 was still restricted in the nucleus (Fig. 3, E–G). The lack of C1q binding to nucleoli in these early apoptotic cells may, again, be due to the lack of C1q access to the nucleolar regions.
FIGURE 3.

The C1q-bound structures in advanced apoptotic cells are nucleoli. After UV irradiation for 6 h (A–D) or 1 h (E–H), HeLa cells on coverslips were fixed with PFA. After incubation with C1q (10 μg/ml) for 1 h, cells were incubated further with mouse anti-C1q and rabbit anti-NPM1. This was followed by incubation with goat anti-mouse (Cy3, red) and anti-rabbit (AF488, green) IgG. Cells were mounted using DAPI-containing medium. A–C and E–G, reconstructed three-dimensional images. D and H, 0.38-μm section images. Scale bars = 5 μm.
The Nuclear Lamina Shows Impairment in Advanced Apoptotic Cells
At this point, the following two possibilities were considered: C1q ligands are only induced late in cell apoptosis, and the ligands exist constitutively in nucleoli, being inaccessible by C1q at earlier time points. In this part of the study, we evaluated the integrity of the nuclear lamina in apoptotic cells. Lamin B1 staining showed that apoptotic cell nuclei shrunk drastically in size 1–3 h after UV irradiation, but no apparent disruption was observed in the nuclear lamina (Fig. 4). By 6 h, the nuclear sizes were not reduced significantly further, but each nucleus typically developed a caved-in or dented area over which the layer of lamin B1 exhibited significant loss, suggesting impairment of the lamina layer. This is particularly obvious with the section images (Fig. 4). This may facilitate C1q access to inner nuclear regions.
FIGURE 4.

Size and integrity changes of nuclei during cell apoptosis. After UV irradiation, cells on coverslips were fixed at 1, 3, and 6 h and incubated with C1q. After washing, the cells were incubated with mouse anti-C1q and rabbit anti-lamin B1 (LB1) antibodies. The cells were then incubated with goat anti-mouse (Cy3, red) and anti-rabbit (AF488, green) IgG. Cells were mounted using DAPI-containing medium. Top panels, lamin B1 signal only. Bottom panels, overlapping lamin B1, C1q, and chromatin signals. Arrowheads indicate caved-in regions. Images for two different sections of 6-h apoptotic cells are presented. Scale bars = 5 μm. 3D, three-dimensional.
C1q Binds to Isolated Nucleoli from Non-irradiated Cells
To examine whether C1q binds to non-apoptotic nucleoli, a protocol was developed to isolate nucleoli from non-irradiated HeLa cells. After homogenization of the cells, nuclei were isolated from the homogenate by centrifugation, first in 0.25 m sucrose and then in 2.2 m sucrose (Fig. 5A). Nuclei were sonicated, and nucleoli were isolated from the homogenate by sequential centrifugation in 0.25 m and 0.88 m sucrose, followed by a 0.85–2.5 m sucrose gradient (Fig. 5A). Nucleoli were recovered in the pellets. The protein composition of isolated nucleoli showed progressive enrichment of NPM1 (Fig. 5B).
FIGURE 5.

Isolation of nucleoli. A, for the isolation of nucleoli, HeLa cells were homogenized, and nuclei were isolated through two sequential centrifugation steps in 0.25 m and 2.2 m sucrose. Nuclei were sonicated, and nucleoli were isolated from the nuclear homogenate through three sequential centrifugation steps in sucrose. Nucleoli in the pellet fraction were resuspended in 0.25 m sucrose (0.4 ml). The nuclear ghosts represent sequentially removed non-nucleolar chromatins. B, the protein composition of the progressively purified nucleoli and discarded fractions was analyzed by SDS-PAGE. NPM1 was identified by mass spectrometry.
In the first experiment, isolated nucleoli were attached to coverslips by incubation for 5 min on ice and then incubation with C1q. After fixation, bound C1q was detected simultaneously with the nucleolar protein NPM1. On the basis of the light images, the isolated nucleoli exhibited size heterogeneity (Fig. 6A). Inspection of individual fluorescent signals, i.e. NPM1, DNA, and bound C1q (Fig. 6, B–D), revealed a dense chromatin layer near the nucleolar surface. In some nucleoli, chromatin fragments were also found in inner regions. This was also observed in apoptotic cells (Fig. 2). NPM1 was detected as a continuous layer overlapping with the chromatin layer (Fig. 6E). C1q bound most intensely along the inner side of the chromatin layer (Fig. 6F). It was also found inside the nucleoli, but the intensity was much lower. Overall, nucleoli isolated from non-apoptotic cells contain C1q ligands, and C1q binding is largely confined close to the surface (Fig. 6, G and H).
FIGURE 6.

C1q binds to isolated nucleoli. Upon isolation, nucleoli were immobilized on coverslips by incubation for 5 min on ice and fixed in 1% (w/v) PFA. After incubation with C1q for 1 h (10 μg/ml), the coverslips were incubated with mouse anti-C1q and rabbit anti-NPM1 antibodies, followed by goat anti-mouse (Cy3, red) and anti-rabbit (AF488, green) IgG. A, reconstructed three-dimensional light image. B–H, 0.38-μm section images. B–D, single fluorescence images (NPM1, chromatin, and C1q, respectively). E–G, overlaps of two fluorescent signals. H, overlap of all three fluorescent signals. Scale bar = 3 μm.
In a separate experiment, C1q was incubated with crude nucleoli after washing in 0.88 m sucrose (Fig. 5). After incubation for 30 min on ice and centrifugation for 10 min at 600 × g, the nucleoli were resuspended in 0.25 m sucrose and applied to the 0.85–2.5 m sucrose gradient. After centrifugation, fractions were collected, one for each gradient step, and a total of nine fractions was obtained, including the sample layer and the resuspended pellet. By Western blotting, NPM1 and another nucleolar protein nucleolus (NCL) were found predominantly in the pellet fraction (Fig. 7A). Low levels of NPM1 and NCL were also detected in the 2.2 m and 2.5 m fractions, but none was detected in further lighter fractions. Without incubation with nucleoli, C1q was detected predominantly in the top two fractions, i.e. the loading layer and the 0.85 m fraction (Fig. 7B). The cosedimentation of C1q with the nucleolar marker proteins through a dense sucrose gradient again suggests C1q binding to these nucleoli. This shows that C1q ligands are constitutively present in the nucleoli, and the lack of C1q binding to nucleoli in permeabilized normal cells or early apoptotic cells probably reflected the inaccessibility of C1q to these subnuclear structures.
FIGURE 7.
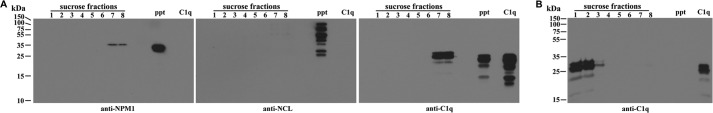
C1q binds to isolated nucleoli. A, after washing with 0.88 m sucrose, the crude nucleoli were diluted four times with 0.25 m sucrose buffer, and 0.5 ml was used to incubate with C1q (50 μg/ml) for 30 min on ice. This was then ultracentrifuged through the 0.85–2.5 m sucrose gradient for 3 h at 33,000 rpm. One fraction was collected for each gradient step, and nine fractions in total were obtained, including the top sample fraction and the resuspended pellet fraction. These fractions were subjected to SDS-PAGE and Western blot detection of C1q and the nucleolar proteins NPM1 and NCL. Purified C1q was included in the blot as a positive control (C1q). Lane 1, sample loading layer. Lanes 2–8, sucrose gradient steps: 0.85, 1.2, 1.5, 1.7, 2.0, 2.2, and 2.5 m. ppt, the pellet fraction. B, as a control, C1q was similarly loaded on the gradient without incubation with nucleoli and, after centrifugation, fractionated similarly. The fractions were analyzed similarly by Western blotting to detect C1q.
Nucleolar Proteins Are Degraded by C1 Proteases
Binding of cell debris by C1q has two known consequences. It may generally enhance phagocytosis, as reported previously (10, 13). Considering that in vivo C1q exists as the pentameric C1 complex (C1qC1r2C1s2), its binding to cell debris could also activate the C1r/C1s proteases and cause proteolytic degradation near the binding sites. C1r/C1s are historically considered highly specific for C4, C2, and the C1 inhibitor, but Kerr et al. (33) have discovered that C1s cleavage sites were predicted on many intracellular proteins. On the basis of the formula they developed, both NPM1 and NCL were each predicted to contain multiple C1s cleavage sites in open regions (Fig. 8, A–C). However, the predicted C1s cleavage sites in one of the histone H3 variant, H3.1, fell mostly in regions with secondary structures.
FIGURE 8.

Degradation of nucleolar antigens by complement C1 proteases. A—C, prediction of C1s cleavage sites. The amino acid sequences of NPM1, NCL, and histone H3.1 were analyzed using the Prediction of Protease Specificity program. Sites that were predicted with a score of >30.0 are indicated by an asterisk. The predicted secondary structures are indicated under the sequences: G, 3-turn helix (310 helix); E, extended strand, participates in β ladder; S, bend; H, α helix; T, hydrogen-bonded turn; B, residue in isolated β bridge. D, Western blot (WB). Isolated nucleoli (No) were incubated with C1 (9.6 μg/ml) for 2 h in the presence or absence of the C1 inhibitor (C1-inh, 160 μg/ml). The reactions were subjected to Western blotting with goat anti-C1s and rabbit antibodies for NPM1, NCL, or histone H3. Secondary antibodies were conjugates of horseradish peroxidase. Near complete inhibition could be achieved with 80 μg/ml C1 inhibitor.
To examine whether C1q binding to nucleoli could lead to C1s degradation of these nucleolar proteins, nucleoli were incubated with purified C1 for 2 h. By Western blotting, C1s, NPM1, NCL, and total histone H3 were probed on the blots (Fig. 8D). When nucleoli were incubated without C1, NPM1 and H3 degradation was undetectable (Fig. 8D). NCL exhibited multiple bands in nucleoli without incubation with C1. After nucleoli were incubated with C1, C1s exhibited its typical activated state of a 56-kDa heavy chain and a 27-kDa light chain (Fig. 8). In these reactions, NPM1 and NCL were completely degraded, but, with NPM1, a 13-kDa residual fragment remained detectable (Fig. 8). When the C1 inhibitor was included in the reaction, NPM1 degradation was almost completely inhibited. With NCL, degradation was also strongly inhibited.
After C1 treatment, a minor fraction of the 15-kDa histone H3 was cleaved in a limited way, and a minor 13-kDa fragment was produced (Fig. 8D). These results showed that NCL and NPM1, which contain predicted C1s cleavage sites, were indeed cleaved after nucleoli were treated with C1. It also showed that histone H3 was largely resistant to C1 cleavage. Because total H3 comprises many variants and prediction was only performed with the H3.1 variant (34), the minor fraction of H3 cleaved by C1 could also represent a separate, minor H3 variant.
C1 Rapidly Degrades Nucleolar NCL and NPM1
In view of the complete NPM1 and NCL degradation by C1 in a 2-h reaction, the incubation time was reduced to 30, 15, and 5 min. In these experiments, NPM1 degradation was noticeable after 5 min, as judged by the intensity reduction of the 36-kDa NPM1 band and the appearance of the 13-kDa NPM1 fragment (Fig. 9). By 30 min, the 36-kDa band was reduced markedly. When the C1 inhibitor was included, NPM1 degradation was blocked completely.
FIGURE 9.

Rapid nucleolar NPM1 and NCL degradation by C1. Nucleoli (No) were incubated with C1 (4.8 μg/ml) for 5, 15, or 30 min in the presence or absence of the C1 inhibitor (C1-inh, 80 μg/ml). The reactions were analyzed by Western blotting (WB) using antibodies for C1s, NPM1, and NCL. In each group of three lanes, as indicated at the top of the blots, the order of samples is 5, 15, and 30 min of digestion time.
NCL degradation was also obvious 5 min after incubation of nucleoli with C1, as shown by the disappearance of the higher molecular weight bands (Fig. 9). By 30 min, most of the high molecular weight bands were degraded. An intermediate fragment that was not detected after 2-h incubation with C1s remained after these shorter reactions. On the basis of these results, some nucleolar proteins are apparently sensitive to C1 degradation during cell apoptosis and, besides NPM1 and NCL, other nucleolar proteins may be cleaved by C1 the proteases that are expected to be recruited to the nucleoli by C1q. The nucleolus is highly immunogenic. Whether C1 cleaves and inactivates some of the nucleolar proteins that may be autoimmunogenic or facilitative to autoreactive B cell activation will be interesting to investigate.
Discussion
The host immune system is tasked to tolerate self-antigens as well as to respond to microbial infections. Cell apoptosis risks the exposure of otherwise intracellular antigens, some of which are autoantigens or damage-associated molecular patterns, and impaired apoptotic cell clearance may lead to autoreactive B and T cell activation and the development of autoimmunity (10, 15). Multiple mechanisms exist for apoptotic cell clearance, and C1q-mediated clearance has been investigated actively. Genetic C1q deficiency causes the production of antinuclear autoantibodies and SLE pathogenesis (8, 10).
C1q binding to apoptotic cells has been reported in several studies, but the nature of C1q binding, e.g. subcellular regions, molecular ligands, and temporal variations of C1q binding at different apoptotic stages, requires further investigations (13, 14, 16). Furthermore, C1r/C1s are naturally in complex with C1q in vivo, and, like C1q, genetic deficiency of these C1q-associated proteases also causes SLE (24). The question remains whether C1q binding to apoptotic cells causes significant C1r/C1s activation that may also impact the host response to this cell debris.
Our initial experiments aimed to monitor C1q binding to UV-irradiated cells in the course of progressive apoptosis and determine whether, at certain stages, C1q would bind to nuclear DNA in these cells. Purified DNA has been reported to be a C1q ligand, and it is also a pathogenic autoantigen in lupus patients (11, 25, 26). Initial results led to an extended study that, besides showing that nuclear DNA was not a primary C1q ligand, provided a series of novel data that help delineate C1q binding sites during cell apoptosis. The results also showed, for the first time, the novel effects of C1q-associated C1r/C1s proteases on apoptotic cellular antigens. The finding that the nucleolus was the dominant structure that C1q recognized in advanced apoptotic cells and that it was susceptible to C1r/C1s degradation is highly significant because the nucleolus is a highly autoimmunogenic nuclear structure (27).
C1q apparently binds to three subcellular regions or structures during the course of cell apoptosis. One hour after UV irradiation, it bound prominently to peripheral structures that resembled the cortical cytoskeleton (Fig. 2). Although the specific molecules that C1q recognized in these cytoskeleton-like regions were not characterized further, the binding pattern showed a similarity to that bound by Clec9A, a receptor on CD8+ DC that recognizes dead cells through binding to the exposed polymeric F-actin (35, 36). In this context, C1q has been reported to bind to purified monomeric actin (29). Clec9A is highly specific for CD8+ DC, a DC subset that is specialized for the cross presentation of captured antigens through MHC I to CD8+ T cells (37, 38). It would be interesting to know whether C1q and Clec9A share overlapping binding sites on the apoptotic cytoskeleton and whether C1q enhances or inhibits Clec9A-mediated uptake and cross-presentation of apoptotic antigens.
C1q also bound diffusely in the cytoplasm, which highlighted no specific cytoplasmic structure. The temporally dictated C1q binding to the nucleolus in advanced apoptotic cell was unexpected. C1q showed discrete binding to the nucleolus and selectively avoided binding to the large volume of chromatins in the vicinity of the nucleolus. The abundance of autoantigens in the nucleolus renders this finding particularly relevant to autoimmunity.
The consequence of an antigen binding to Clec9A would be its uptake by CD8+ DCs and cross-presentation to activate CD8+ T cells (39, 40). In addition, antigens targeted through Clec9A were also particularly potent in inducing antibody production (41). When the antigens that Clec9A recognizes are dead cells, this presents risks of autoimmunity. C1q binding to the cytoskeleton of the dead cell is also expected to promote phagocytosis, but this is expected to involve broader types of phagocytes. Besides phagocytic clearance, an additional effect after C1q binding to apoptotic cells is the activation of C1r/C1s proteases and proteolytic degradation of nucleolar antigens, as demonstrated in this study (Figs. 8 and 9). If Clec9A-mediated dead cell uptake is to induce immunity against viral or aberrant antigens born in the cell debris, then complementary C1q-elicited and C1r/C1s-mediated degradation of cellular antigens, as demonstrated, could inactivate some autoantigens and damage-associated molecular patterns and help reduce autoimmunity.
This C1q/C1r/C1s-mediated cellular antigen cleavage would be difficult to envisage on the basis of the historical norm that the C1r/C1s proteases are highly specific for the complement system. Besides C4, C2, and the C1 inhibitor, only three other molecules, all expressed extracellularly, have been reported to be cleaved by C1s: insulin-like growth factor binding protein 5, MHC class I, and the Wnt coreceptor low-density lipoprotein receptor-related protein 6 (42–44). However, on the basis of a new C1s substrate formula established using a library of peptides centered around the known C1s cleavage sites in C4, C2, and the C1 inhibitor, many intracellular proteins have been predicted to contain C1s cleavage sites (33). The prediction was largely made on the basis of sequence preference and local secondary structures, on the basis of which NCL and NPM1 were both predicted to contain multiple C1s cleavage sites (Fig. 8, A–C). After incubating the nucleolus with C1, NCL and NPM1 were found to be cleaved. The effective inhibition of the degradation by the C1 inhibitor supported the dominant roles of C1r/C1s proteases in the observed NPM1 and NCL degradation.
This finding raises the possibility that many cellular proteins near the C1q binding sites may be degraded in apoptotic cells, including autoreactive epitopes or damage-associated molecular patterns (Fig. 10). The collective outcome of this C1-dependent proteolytic mechanism during apoptosis can play important roles in reducing the host response to self-antigens to reduce the development of autoimmunity, an attractive hypothesis to explain their genetic deficiencies being causal to SLE pathogenesis. Deficiency in the C1 inhibitor, which causes angioedema (45), has also been reported to be associated with the production of autoantibodies and autoimmunity, including lupus-like conditions (46). The underlying mechanism is unclear, but it is possible, on the basis of our data, that, in C1 inhibitor deficiency, uncontrolled systemic C1r/C1s activation may occur that impairs targeted C1r/C1s trimming of antigens on cell debris, leading to autoantibody production. Besides this extracellularly installed “dead cell trimming” mechanism, apoptosis also activates intrinsic proteolytic breakdown of cellular antigens, including defined autoantigens (47). Dead cell trimming mechanisms may complement each other to delete different classes of antigens during apoptosis to preserve host tolerance to these self-antigens.
FIGURE 10.
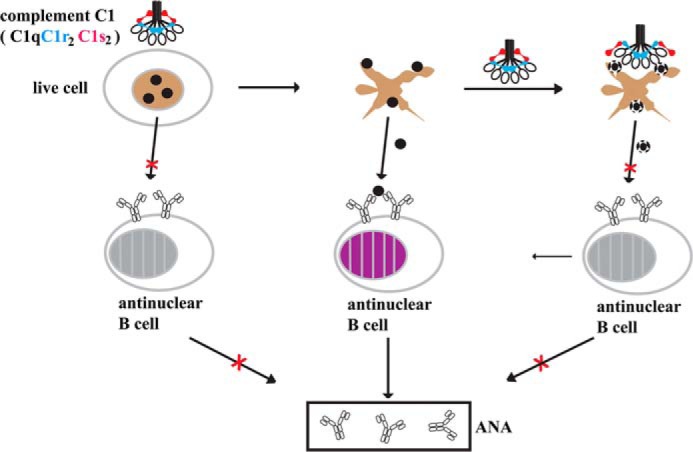
Schematic of the significance of C1q binding and C1 protease degradation of nucleolar antigens. In live cells, intracellular antigens, including nucleolar antigens, are privileged and are inaccessible for B cell and complement recognition. In advanced apoptotic cells, exposure of these intracellular antigens may activate B cells for autoantibody production. However, they are also susceptible to complement C1q recognition and C1r/C1s proteolytic inactivation. In C1q or C1r/C1s deficiency, some of these exposed intracellular proteins may remain intact and able to stimulate autoreactive B cells to produce autoantibodies and cause autoimmunity.
Author Contributions
J. L. conceived and coordinated the study, wrote the paper, generated data, and prepared Figs. 5 and 10. Y. C. performed the experiments, with assistance from B. H. D. T., for Figs. 1–4. Y. C. performed the experiments, with assistance from J. G. Y., for Figs. 7–9. Y. C. performed the experiments, with assistance from J. L., for Fig. 6.
Acknowledgments
We thank Shu Ying Lee and Weian Zhang for help in the National University of Singapore Yong Loo Lin School of Medicine Microscopy Unit.
This project was supported by Singapore National University Health System Seed Fund R-182-000-229-750 and Singapore Ministry of Education Tier 2 Grant MOE2012-T2-2-122). The authors declare that they have no conflict of interests with the contents of this article.
- SLE
- systemic lupus erythematosus
- DC
- dendritic cell
- NCL
- nucleolin
- PFA
- paraformaldehyde
- AF488
- Alexa Fluor 488.
References
- 1. Walport M. J. (2001) Complement: first of two parts. N. Engl. J. Med. 344, 1058–1066 [DOI] [PubMed] [Google Scholar]
- 2. Reid K. B. (1986) Activation and control of the complement system. Essays Biochem. 22, 27–68 [PubMed] [Google Scholar]
- 3. Bordet J., Gay F. P. (1906) Sur les relations des sensibilisatrices avec I'alexine. Ann. Inst. Pasteur 20, 467–498 [Google Scholar]
- 4. Kemper C., Atkinson J. P., Hourcade D. E. (2010) Properdin: emerging roles of a pattern-recognition molecule. Annu. Rev. Immunol. 28, 131–155 [DOI] [PubMed] [Google Scholar]
- 5. Fujita T. (2002) Evolution of the lectin-complement pathway and its role in innate immunity. Nat. Rev. Immunol. 2, 346–353 [DOI] [PubMed] [Google Scholar]
- 6. Lepow I. H., Naff G. B., Todd E. W., Pensky J., Hinz C. F. (1963) Chromatographic resolution of the first component of human complement into three activities. J. Exp. Med. 117, 983–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Skattum L., van Deuren M., van der Poll T., Truedsson L. (2011) Complement deficiency states and associated infections. Mol. Immunol. 48, 1643–1655 [DOI] [PubMed] [Google Scholar]
- 8. Lewis M. J., Botto M. (2006) Complement deficiencies in humans and animals: links to autoimmunity. Autoimmunity 39, 367–378 [DOI] [PubMed] [Google Scholar]
- 9. Moser K. L., Kelly J. A., Lessard C. J., Harley J. B. (2009) Recent insights into the genetic basis of systemic lupus erythematosus. Genes Immun. 10, 373–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Elkon K. B., Santer D. M. (2012) Complement, interferon and lupus. Curr. Opin. Immunol. 24, 665–670 [DOI] [PubMed] [Google Scholar]
- 11. Tan E. M. (2012) Autoantibodies, autoimmune disease, and the birth of immune diagnostics. J. Clin. Invest. 122, 3835–3836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mevorach D., Zhou J. L., Song X., Elkon K. B. (1998) Systemic exposure to irradiated apoptotic cells induces autoantibody production. J. Exp. Med. 188, 387–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ogden C. A., deCathelineau A., Hoffmann P. R., Bratton D., Ghebrehiwet B., Fadok V. A., Henson P. M. (2001) C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J. Exp. Med. 194, 781–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Korb L. C., Ahearn J. M. (1997) C1q binds directly and specifically to surface blebs of apoptotic human keratinocytes: complement deficiency and systemic lupus erythematosus revisited. J. Immunol. 158, 4525–4528 [PubMed] [Google Scholar]
- 15. Manderson A. P., Botto M., Walport M. J. (2004) The role of complement in the development of systemic lupus erythematosus. Annu. Rev. Immunol. 22, 431–456 [DOI] [PubMed] [Google Scholar]
- 16. Clarke E. V., Weist B. M., Walsh C. M., Tenner A. J. (2014) Complement protein C1q bound to apoptotic cells suppresses human macrophage and dendritic cell-mediated Th17 and Th1 T cell subset proliferation. J. Leukocyte Biol. 97, 147–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Båve U., Magnusson M., Eloranta M. L., Perers A., Alm G. V., Rönnblom L. (2003) Fc γ RIIa is expressed on natural IFN-α-producing cells (plasmacytoid dendritic cells) and is required for the IFN-α production induced by apoptotic cells combined with lupus IgG. J. Immunol. 171, 3296–3302 [DOI] [PubMed] [Google Scholar]
- 18. Means T. K., Latz E., Hayashi F., Murali M. R., Golenbock D. T., Luster A. D. (2005) Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. J. Clin. Invest. 115, 407–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Crow M. K. (2014) Type I interferon in the pathogenesis of lupus. J. Immunol. 192, 5459–5468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lood C., Gullstrand B., Truedsson L., Olin A. I., Alm G. V., Rönnblom L., Sturfelt G., Eloranta M. L., Bengtsson A. A. (2009) C1q inhibits immune complex-induced interferon-α production in plasmacytoid dendritic cells: a novel link between C1q deficiency and systemic lupus erythematosus pathogenesis. Arthritis Rheum. 60, 3081–3090 [DOI] [PubMed] [Google Scholar]
- 21. Santer D. M., Hall B. E., George T. C., Tangsombatvisit S., Liu C. L., Arkwright P. D., Elkon K. B. (2010) C1q deficiency leads to the defective suppression of IFN-α in response to nucleoprotein containing immune complexes. J. Immunol. 185, 4738–4749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Castellano G., Woltman A. M., Schlagwein N., Xu W., Schena F. P., Daha M. R., van Kooten C. (2007) Immune modulation of human dendritic cells by complement. Eur. J. Immunol. 37, 2803–2811 [DOI] [PubMed] [Google Scholar]
- 23. Teh B. K., Yeo J. G., Chern L. M., Lu J. (2011) C1q regulation of dendritic cell development from monocytes with distinct cytokine production and T cell stimulation. Mol. Immunol. 48, 1128–1138 [DOI] [PubMed] [Google Scholar]
- 24. Lipsker D., Hauptmann G. (2010) Cutaneous manifestations of complement deficiencies. Lupus 19, 1096–1106 [DOI] [PubMed] [Google Scholar]
- 25. Agnello V., Winchester R. J., Kunkel H. G. (1970) Precipitin reactions of the C1q component of complement with aggregated γ-globulin and immune complexes in gel diffusion. Immunology 19, 909–919 [PMC free article] [PubMed] [Google Scholar]
- 26. Jiang H., Cooper B., Robey F. A., Gewurz H. (1992) DNA binds and activates complement via residues 14–26 of the human C1q A chain. J. Biol. Chem. 267, 25597–25601 [PubMed] [Google Scholar]
- 27. Welting T. J., Raijmakers R., Pruijn G. J. (2003) Autoantigenicity of nucleolar complexes. Autoimmun. Rev. 2, 313–321 [DOI] [PubMed] [Google Scholar]
- 28. Reid K. B., Porter R. R. (1976) Subunit composition and structure of subcomponent C1q of the first component of human complement. Biochem. J. 155, 19–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nishioka M., Kobayashi K., Uchida M., Nakamura T. (1982) A binding activity of actin with human C1q. Biochem. Biophys. Res. Commun. 108, 1307–1312 [DOI] [PubMed] [Google Scholar]
- 30. Shaw P. J., Jordan E. G. (1995) The nucleolus. Annu. Rev. Cell Dev. Biol. 11, 93–121 [DOI] [PubMed] [Google Scholar]
- 31. Pederson T. (2011) The nucleolus. Cold Spring Harb. Perspect. Biol. 3, 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mamrack M. D., Olson M. O., Busch H. (1977) Negatively charged phosphopeptides of nucleolar nonhistone proteins from Novikoff hepatoma ascites cells. Biochem. Biophys. Res. Commun. 76, 150–157 [DOI] [PubMed] [Google Scholar]
- 33. Kerr F. K., O'Brien G., Quinsey N. S., Whisstock J. C., Boyd S., de la Banda M. G., Kaiserman D., Matthews A. Y., Bird P. I., Pike R. N. (2005) Elucidation of the substrate specificity of the C1s protease of the classical complement pathway. J. Biol. Chem. 280, 39510–39514 [DOI] [PubMed] [Google Scholar]
- 34. Maze I., Noh K. M., Soshnev A. A., Allis C. D. (2014) Every amino acid matters: essential contributions of histone variants to mammalian development and disease. Nat. Rev. Genet 15, 259–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang J. G., Czabotar P. E., Policheni A. N., Caminschi I., Wan S. S., Kitsoulis S., Tullett K. M., Robin A. Y., Brammananth R., van Delft M. F., Lu J., O'Reilly L. A., Josefsson E. C., Kile B. T., Chin W. J., Mintern J. D., Olshina M. A., Wong W., Baum J., Wright M. D., Huang D. C., Mohandas N., Coppel R. L., Colman P. M., Nicola N. A., Shortman K., Lahoud M. H. (2012) The dendritic cell receptor Clec9A binds damaged cells via exposed actin filaments. Immunity 36, 646–657 [DOI] [PubMed] [Google Scholar]
- 36. Ahrens S., Zelenay S., Sancho D., Hanč P., Kjær S., Feest C., Fletcher G., Durkin C., Postigo A., Skehel M., Batista F., Thompson B., Way M., Reis e Sousa C., Schulz O. (2012) F-actin is an evolutionarily conserved damage-associated molecular pattern recognized by DNGR-1, a receptor for dead cells. Immunity 36, 635–645 [DOI] [PubMed] [Google Scholar]
- 37. Brossart P., Bevan M. J. (1997) Presentation of exogenous protein antigens on major histocompatibility complex class I molecules by dendritic cells: pathway of presentation and regulation by cytokines. Blood 90, 1594–1599 [PMC free article] [PubMed] [Google Scholar]
- 38. Pooley J. L., Heath W. R., Shortman K. (2001) Cutting edge: intravenous soluble antigen is presented to CD4 T cells by CD8− dendritic cells, but cross-presented to CD8 T cells by CD8+ dendritic cells. J. Immunol. 166, 5327–5330 [DOI] [PubMed] [Google Scholar]
- 39. Geijtenbeek T. B. (2012) Actin' as a death signal. Immunity 36, 557–559 [DOI] [PubMed] [Google Scholar]
- 40. Brown G. D. (2012) Immunology: Actin' dangerously. Nature 485, 589–590 [DOI] [PubMed] [Google Scholar]
- 41. Caminschi I., Shortman K. (2012) Boosting antibody responses by targeting antigens to dendritic cells. Trends Immunol. 33, 71–77 [DOI] [PubMed] [Google Scholar]
- 42. Busby W. H. Jr., Nam T. J., Moralez A., Smith C., Jennings M., Clemmons D. R. (2000) The complement component C1s is the protease that accounts for cleavage of insulin-like growth factor-binding protein-5 in fibroblast medium. J. Biol. Chem. 275, 37638–37644 [DOI] [PubMed] [Google Scholar]
- 43. Eriksson H., Nissen M. H. (1990) Proteolysis of the heavy chain of major histocompatibility complex class I antigens by complement component C1s. Biochim. Biophys. Acta 1037, 209–215 [DOI] [PubMed] [Google Scholar]
- 44. Naito A. T., Sumida T., Nomura S., Liu M. L., Higo T., Nakagawa A., Okada K., Sakai T., Hashimoto A., Hara Y., Shimizu I., Zhu W., Toko H., Katada A., Akazawa H., Oka T., Lee J. K., Minamino T., Nagai T., Walsh K., Kikuchi A., Matsumoto M., Botto M., Shiojima I., Komuro I. (2012) Complement C1q activates canonical Wnt signaling and promotes aging-related phenotypes. Cell 149, 1298–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cicardi M., Johnston D. T. (2012) Hereditary and acquired complement component 1 esterase inhibitor deficiency: a review for the hematologist. Acta Haematol. 127, 208–220 [DOI] [PubMed] [Google Scholar]
- 46. Triggianese P., Chimenti M. S., Toubi E., Ballanti E., Guarino M. D., Perricone C., Perricone R. (2015) The autoimmune side of hereditary angioedema: insights on the pathogenesis. Autoimmun. Rev. 14, 665–669 [DOI] [PubMed] [Google Scholar]
- 47. Casiano C. A., Ochs R. L., Tan E. M. (1998) Distinct cleavage products of nuclear proteins in apoptosis and necrosis revealed by autoantibody probes. Cell Death Differ. 5, 183–190 [DOI] [PubMed] [Google Scholar]