

Background: We studied how the endo/exonucleases EXO1, FAN1, and FEN1 process substrates resembling replication forks blocked by interstrand cross-links (ICLs).
Results: All three enzymes cleaved off the single-stranded 5′ flap, but FAN1 was also able to incise the substrate behind the ICL.
Conclusion: FAN1 can unhook ICLs.
Significance: In vivo, FAN1 may not require a 3′ flap nuclease to unhook ICLs.
Keywords: DNA damage, DNA endonuclease, DNA repair, DNA-protein interaction, enzyme mechanism, exonuclease, interstrand cross-links
Abstract
Cisplatin and its derivatives, nitrogen mustards and mitomycin C, are used widely in cancer chemotherapy. Their efficacy is linked primarily to their ability to generate DNA interstrand cross-links (ICLs), which effectively block the progression of transcription and replication machineries. Release of this block, referred to as unhooking, has been postulated to require endonucleases that incise one strand of the duplex on either side of the ICL. Here we investigated how the 5′ flap nucleases FANCD2-associated nuclease 1 (FAN1), exonuclease 1 (EXO1), and flap endonuclease 1 (FEN1) process a substrate reminiscent of a replication fork arrested at an ICL. We now show that EXO1 and FEN1 cleaved the substrate at the boundary between the single-stranded 5′ flap and the duplex, whereas FAN1 incised it three to four nucleotides in the double-stranded region. This affected the outcome of processing of a substrate containing a nitrogen mustard-like ICL two nucleotides in the duplex region because FAN1, unlike EXO1 and FEN1, incised the substrate predominantly beyond the ICL and, therefore, failed to release the 5′ flap. We also show that FAN1 was able to degrade a linear ICL substrate. This ability of FAN1 to traverse ICLs in DNA could help to elucidate its biological function, which is currently unknown.
Introduction
DNA interstrand cross-links (ICLs)2 are highly cytotoxic because of their propensity to block both transcription and replication (1). In all organisms studied to date, ICL repair involves protein constituents of the nucleotide excision repair pathway, translesion polymerases, and the machinery of homologous recombination (2). In higher eukaryotes, ICL processing also requires proteins of the FANC complementation group and their interactors. Correspondingly, cells lacking these proteins are exquisitely sensitive to ICL-inducing agents. This is of substantial clinical relevance. Mutations in genes encoding FANC polypeptides predispose to Fanconi anemia (FA), a severe genetic disorder characterized by bone marrow failure, congenital abnormalities and cancer predisposition, and cellular sensitivity to the cross-linking agents diepoxybutane or mitomycin C (MMC) is used in FA diagnosis (3).
Of the 17 FANC proteins identified to date, eight form the so-called core complex, which is activated at stalled replication forks by the ATR kinase. The FANCL subunit of the activated core complex monoubiquitylates the FANCD2/FANCI heterodimer that is believed to recruit to the cross-linked site in chromatin the polypeptides required for ICL removal (4). In the first step, these proteins have to separate the cross-linked strands in a process referred to as “unhooking,” in which one strand of the duplex is nucleolytically incised on either side of the cross-link. Studies carried out during the past three decades helped to identify several nuclease-encoding genes, the disruption of which results in sensitivity to ICL-inducing agents, and biochemical characterization of their respective gene products confirmed that these nucleases could indeed be involved in ICL unhooking. The primary candidates are SLX1-SLX4, XPF-ERCC1, MUS81-EME1, SNM1A, and FAN1. Despite some seminal mechanistic studies that have been described in the recent literature (reviewed in Ref. 5), our understanding of the possible functions of these enzymes in ICL unhooking is still rudimentary. The situation is further complicated by functional redundancies of several of the involved proteins, their interplay with other members of the ICL repair pathway, and differences in helical distortions induced by different types of ICLs (6).
FANCD2-associated nuclease 1 (FAN1) has been identified independently in four laboratories (7–10). It possesses a conserved PDXn(D/E)XK active site motif common to the restriction nuclease-like superfamily. This site is embedded in a VRR-nuclease domain, the structure of which has been solved recently (11). FAN1 has been implicated in ICL repair because its knockdown (7–10) or knockout (12, 13) caused sensitivity to the ICL-inducing agents MMC and cisplatin. The protein contains also a ubiquitin-binding zinc finger domain, which is necessary and sufficient for its recruitment to MMC-induced foci of the monoubiquitylated form of FANCD2/I (7–10). As a consequence, FAN1 was predicted to be a novel FANC protein. However, experiments with chicken DT40 knockout cell lines failed to yield evidence of an epistasis between FAN1 and FANCC or FANCJ (13), and no mutations in FAN1 were identified in unassigned FA patients. Instead, FAN1 loss has been linked to karyomegalic interstitial nephritis (14). Therefore, even though its involvement in ICL processing is beyond doubt, FAN1 does not appear to be a FANC protein. However, current evidence does not exclude the possibility that FAN1 does participate in the FANC pathway but that its deficiency does not lead to FA because of functional redundancy with other polypeptide(s).
In an attempt to address the latter point, we set out to compare how structures resembling ICL-blocked replication forks are addressed by three 5′ flap endonucleases and 5′ to 3′ exonucleases with known roles in DNA replication and repair: EXO1, FAN1, and FEN1. EXO1 (15) and FEN1 (16) belong to the same enzyme family and have been reported to incise 5′ flap structures primarily one nucleotide 3′ from the single-strand (ss)/double-strand (ds) junction (for a review, see Ref. 17). FAN1 has been shown to incise 5′ flap structures two to four nucleotides 3′ from the ss/ds junction (9). Given their similar substrate specificities, we wondered whether these enzymes could compensate for one another in ICL processing. We therefore synthesized a 5′ flap structure containing a single cross-link resembling an ICL induced by nitrogen mustards (NMs), positioned two nucleotides 3′ from the ss/ds junction (18). As controls, we also synthesized a linear duplex containing a similar ICL as well as several unmodified substrates. We now show that all three enzymes could cleave the 5′ flaps but that FAN1 was also able to traverse the cross-link on both ICL substrates. Furthermore, we show that FAN1 depletion leads to a reduction in the number of MMC-induced double-strand breaks (DSBs). This could imply that FAN1-dependent processing of ICLs leads to the generation of repairable DSBs, whereas FAN1 absence or malfunction may lead to ICL persistence and cytotoxicity.
Experimental Procedures
Antibodies
Sheep α-KIAA1018/MTMR15 (a gift from J. Rouse, 1:625), rabbit α-TFIIH (Ser-19, Santa Cruz Biotechnology, 1:1000), mouse α-MUS81 (Sigma, 1:2000), mouse α-Lamin B1 (Abcam, 1:100), rabbit α-γH2AX (Ser-139, Cell Signaling Technology, 1:1000), mouse α-RPA2 (Calbiochem, 1:50), mouse α-CtIP (Asp-4, Santa Cruz Biotechnology, 1:250), and rabbit α-pRPA2 (Ser-4/Ser-8, Bethyl Laboratories, 1:500) were used.
siRNA Treatments
The following siRNAs (Microsynth, Balgach, Switzerland) were used: siFAN1, GUAAGGCUCUUUCAACGUA; siLuciferase, CGUACGCGGAAUACUUCGA; and siMUS81, CACGCGCTTCGTATTTCAGAA and CGGGAGCACCTGAATCCTAAT. The siRNA treatments were carried out 1 day after seeding the cells at ∼30% confluency in 10-cm dishes. The transfection agent was Lipofectamine RNAiMAXTM (Invitrogen), which was used according to the instructions of the manufacturer.
Synthesis of Substrates
DNA ICLs were synthesized as described in Refs. 19, 20. Unmodified oligonucleotides were purchased from Microsynth. Sequences and graphical representation of the substrates can be found in Figs. 1A, 2A, 3A, and 4A.
FIGURE 1.

Comparison of 5′ flap endonuclease activities and the specificities of FAN1, EXO1, and FEN1. A, schematic representation of the DNA substrates used in this study. The unmodified flap substrates (left panel) were generated as described under “Experimental Procedures,” and the labeling of the flap substrates with a nitrogen mustard-like interstrand cross-link (right panel, cross-link shown in red) resulted primarily, but not exclusively, in the labeling of the I strand. The likely products generated by the above enzymes from these substrates are also shown. Red asterisks indicate the positions of the 32P-labeled 5′ phosphates. Fragments invisible on the autoradiograph are shown in gray. Dashed lines represent regions of exonucleolytic degradation. nt, nucleotides. B–D, product generated upon incubation of the substrates shown above the panels with FAN1 (B), EXO1 (C), and FEN1 (D). Aliquots were withdrawn at 0, 10 s, 2 min, 20 min, 40 min, and 80 min (B, lanes 1–6 and 7–12, respectively) or at 0, 10 s, 1 min, 10 min, 20 min, and 40 min (C and D, lanes 1–6 and 7–12, respectively). The protein-to-DNA ratios were 1:1 for FAN1 and EXO1 and 1:2 for FEN1. M, low molecular weight marker (Affymetrix). The oligonucleotide sizes are indicated on the left, and the position of the cross-link is indicated by a black arrowhead. The lowercase letters on the right correspond to the products in A. (Products b'–d' are seen solely in the reactions using the cross-linked substrate, in which both strands were labeled.) The black asterisk indicates the position of the non-cross-linked 35-mer oligonucleotide that was present in small amounts in the ICL substrate. The graphs below the autoradiographs of 20% denaturing polyacrylamide gels represent the quantification of either all product bands (total) or of only the bands indicated in red (arrowheads). The most prominent product bands in the reaction of FAN1 with the unmodified substrate resulted from incisions beyond the position of the ICL and are therefore not detectable in digestions of the cross-linked substrate. Error bars show mean ± S.D. (n = 3). E, comparative analysis of digestions of the indicated 5′ flap substrates by the three structure-specific endonucleases, including the nuclease-dead mutants of FAN1 (D960A) and EXO1 (D173N). DNA and proteins were all in equimolar ratios, and the reactions were carried out for 30 min at 37 °C.
FIGURE 2.

Comparison of 5′ to 3′ exonuclease activities and the specificities of FAN1, EXO1, and FEN1. A, schematic of the DNA substrates used in this study. The unmodified flap substrates (left panel) and the substrates with a nitrogen mustard-like interstrand cross-link (right panel, cross-link shown in red) were produced as described under “Experimental Procedures.” Schematics of products generated by the above enzymes are shown next to the corresponding substrates. Red asterisks indicate the positions of the 32P-labeled nucleotides. Fragments invisible on the autoradiograph are shown in gray. B–D, incubation of the indicated substrates with FAN1 (B), EXO1 (C), or FEN1 (D). Aliquots were withdrawn after 0, 10 s, 2 min, 20 min, 40 min, and 80 min (B, lanes 1–6 and 7–12, respectively) or 0, 10 s, 1 min, 20 min, and 40 min (C and D, lanes 1–6 and 7–12, respectively). Lowercase letters on the right correspond to the products in A. The protein-to-DNA ratios were FAN1 (10:1), EXO1 (3.5:1), and FEN1 (1:2). M, low molecular weight marker (Affymetrix). The oligonucleotide sizes are indicated on the left. The black asterisk indicates the position of the non-cross-linked 37-mer oligonucleotide that was present in small amounts in the ICL substrate. The graphs below the autoradiographs of 20% denaturing polyacrylamide gels show the quantification of bands a or b, respectively. b' was generated by incisions in the top strand. Error bars show mean ± S.D. (n = 3). E, comparative analysis of cleavage products generated on the 3′ side of the cross-link by the three nucleases as well as by the nuclease-dead mutants of FAN1 (D960A) and EXO1 (D173N). The reaction conditions were as in Fig. 1E.
FIGURE 3.

FAN1 can also traverse a cross-link on a linear DNA substrate. A, schematic of the linear DNA substrates labeled at the 3′ terminus of the upper strand by fill-in reactions with [α-32P]dTTP. A schematic of the expected products is shown next to the corresponding substrates (a, b, and a'–c'). Red asterisks indicate the positions of the 32P-labeled nucleotides. Fragments invisible on the autoradiograph are shown in gray. B–D, 5′ to 3′ exonuclease activities of FAN1 (B), EXO1 (C), and FEN1 (D) on the indicated substrates. Reaction conditions and protein concentrations were as in Fig. 2. Lowercase letters correspond to the products in A. M, low molecular weight marker (Affymetrix). The oligonucleotide sizes are indicated. The black asterisk indicates the position of the non-cross-linked 28-mer oligonucleotide that was present in small amounts in the ICL substrate. Bottom panels, quantifications of the degradation fragments. Error bars show mean ± S.D. (n = 3). E, comparative analysis of cleavage products generated on the 3′ side of the cross-link by the three nucleases as well as by the nuclease-dead mutants of FAN1 (D960A) and EXO1 (D173N). The reaction conditions were as in Fig. 1E. F, FAN1 WT and D960A activity on a recessed linear DNA substrate shown schematically above. Concentrations were as indicated, and samples were withdrawn after 1, 10, 30, or 60 min (WT, lanes 2–5) or after 1, 30, and 60 min (D960A, lanes 6–8). For the substrate sequence, see “Experimental Procedures.” Quantifications are as indicated above. Black asterisks indicate the non-cross-linked oligonucleotides present in the substrate preparation. The dashed line represents missing lanes.
FIGURE 4.
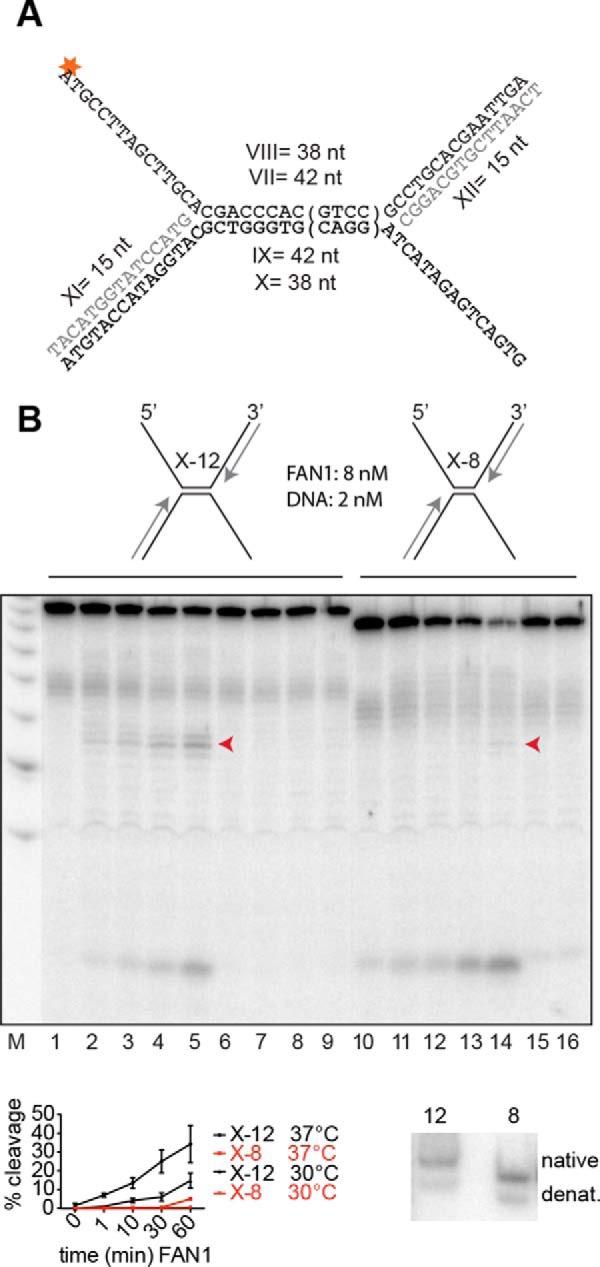
FAN1 cleavage requires ∼10 base pairs of dsDNA on both sides of the flap. A, schematic of the X-12 and X-8 substrates. The tetranucleotide sequence in parentheses is absent from the X-8 substrate. The asterisk indicates the 32P-labeled phosphate. B, the substrates were incubated with the enzymes at the indicated enzyme:substrate ratio at 37 °C for 1, 10, 30, and 60 min (FAN1-WT, lanes 1–5 and 10–14; FAN1 D960A, lanes 6–9) or 1 and 60 min (lanes 15 and 16). The X-8 substrate was extremely inefficiently processed even at 30 °C (bottom left panel), which indicates that the lack of flap cleavage was not caused by denaturation (denat) of the X structure. (Only the bands indicated by the arrowheads were quantified.) Bottom right panel, 10% native polyacrylamide gel of the two substrates, incubated at 37 °C for 30 min. This additional control experiment shows that both substrates were predominantly annealed under the conditions of the reaction.
Labeling and Annealing of Unmodified Substrates
The 5′ labeled flap substrate shown in Fig. 1A was generated by annealing oligonucleotide I labeled at its 5′ terminus with T4 polynucleotide kinase (New England Biolabs) and [γ-32P]ATP (Hartmann Analytic) to the unlabeled oligonucleotides II and III at 95 °C and cooling down to room temperature in annealing buffer (25 mm Hepes-KOH (pH 7.4) and 50 mm KCl).
The 5′ labeled flap substrates shown in Fig. 4A were generated by annealing either the 5′-labeled oligonucleotide VII with the unlabeled oligonucleotides IX, XI, and XII or the 5′-labeled oligonucleotide VIII with the unlabeled oligonucleotides X, XI, and XII. The 3′-labeled substrates were generated by first annealing oligonucleotides I and IV (Fig. 2A) or V and VI (Fig. 3A), and filling in the overhangs using Klenow polymerase (New England Biolabs) and [α-32P]dTTP (Hartmann Analytic) together with dATP, dCTP, and dGTP, followed where required by reannealing with oligonucleotide III (Fig. 2A).
Labeling and Annealing of ICL Substrates
The 5′ flap substrates were generated by labeling the 5′ termini of the cross-linked Y structure (oligonucleotides I and II, Fig. 1A) with T4 polynucleotide kinase (New England Biolabs) and [γ-32P]ATP and annealing with oligonucleotide III as described above. The 3′-labeled cross-linked substrates (Figs. 2A and 3A) were generated by filling in the overhangs using Klenow polymerase (New England Biolabs) and [α-32P]dTTP together with dATP, dCTP, and dGTP, followed by annealing with oligonucleotide III (Fig. 2A) as described above. The recessed substrate (5′ CCCTCTTCTXTCCTTCTTTC 3′/5′GAAAGAAGXACAGAAGAGGGTACCATCATAGAGTCAGTG 3′, where X represents the cross-linked guanines, Fig. 3F) was labeled at the ds 3′ end by cordycepin (PerkinElmer Life Sciences) and terminal transferase (Roche).
In Vitro Nuclease Assays
WT and mutant FAN1 were purified as described previously (9) and dialyzed against nuclease buffer (25 mm Hepes-KOH (pH 7.4, FAN1, or pH 7.8, EXO1 and FEN1), 25 mm KCl, 1 mm MgCl2, 0.5% PEG, and 0.05 mg/ml BSA) for 15 min at 4 °C before use. WT and mutant recombinant purified human EXO1 was a gift from Stephanie Bregenhorn (21). FEN1 was purchased from Gentaur (catalog no. C140). Unless stated otherwise, the nuclease assays were carried out by incubating the substrates for 30 min at 37 °C in the appropriate nuclease buffer (see above). Aliquots were withdrawn at the indicated time points, mixed with an equal volume of loading dye (80% formamide, 50 mm Tris-HCl (pH 8.3), and 1 mm EDTA), and loaded on 20% denaturing polyacrylamide gels (20 × 20 × 0.05 cm) that were run in TBE buffer for 1 h at 800 V. The gels were fixed, dried, and exposed to phospho screens, which were then scanned in a PhosphorImager (Typhoon 9400, GE Healthcare). Quantification of the bands was carried out using ImageJ, and the graphs were generated using GraphPad Prism. The overall efficiency of substrate processing was obtained by dividing the integral values of all the product bands by the integral value of all the bands. The cleavage efficiencies of the 5′ flaps in the unmodified and ICL substrates were calculated by dividing the integral values of the product bands by the integral value of all bands.
Pulsed Field Gel Electrophoresis
Pulsed field gel electrophoresis was performed as described previously (22) with minor modifications. 24 h after MMC treatment (3 μg/ml), cells were harvested, and agarose plugs containing 250,000 cells/plug were generated. Quantifications were done using ImageJ, and graphs were produced using GraphPad Prism.
5-Ethynyl-2′-deoxyuridine (EdU) Staining and FACS Analysis
After treating the cells with 10 μm EdU (Life Technologies) for 30 min, they were harvested, fixed in 1% formaldehyde, and labeled using the Click-iT® cell reaction buffer kit (Life Technologies) according to the instructions of the manufacturer. DNA was stained with 1 μg/ml DAPI, and the samples were analyzed on a Cyan ADP flow cytometer (Beckman Coulter) fitted with Summit software v4.3 (Beckman Coulter).
Clonogenic Survival Assay
Survival assays were performed as described previously (9). Eight days after treating the cells with MMC or carmustine (bromo-chloro-nitrosourea, BCNU) at the indicated dosages, the cells were fixed and stained with 0.5% crystal violet in 20% ethanol, and colonies containing more than 50 cells were counted. MMC-containing medium was replaced with fresh medium after 24 h.
Results
An NM-like ICL Does Not Block the Endonuclease Activity of FAN1 on a 5′ Flap Substrate
The ability of FAN1 to cleave substrates containing 5′ flaps, coupled to the sensitivity of FAN1-depleted cells to ICL-inducing agents (7–10), indicates that this protein might be involved in ICL unhooking. Experimental evidence implicating FAN1 in this process has, however, not been forthcoming. In Xenopus laevis egg extracts, FAN1 depletion caused no detectable defect in unhooking of a single ICL in a plasmid substrate, but this could be explained by functional redundancy with other nucleases in the extract (23). To learn with which nuclease FAN1 could be redundant, we set out to test its ability to process a 5′ flap substrate containing a single NM-like ICL (Fig. 1A) and compared it with those of two other 5′ flap endonucleases, EXO1 and FEN1. FAN1-catalyzed cleavage of the control, unmodified 5′ flap substrate labeled at the 5′ terminus of the flap gave rise to several species between 11 and 21 nucleotides in length, the most prominent of which was a 19-mer arising as a product of endonucleolytic cleavage of the lagging (flap-containing) strand four nucleotides 3′ from the ss/ds junction (Fig. 1B, lanes 1–6, products a).
EXO1 and FEN1 belong to the same nuclease family, possess highly conserved active sites, and have been reported to cleave 5′ flaps preferentially one nucleotide 3′ from the ss/ds junction (17). They were therefore both expected to release 16-mer flaps from the unmodified substrate (Fig. 1A). Somewhat surprisingly, the flap fragments generated by the two enzymes differed. The main product of EXO1-catalyzed cleavage was a 17-mer, whereas FEN1 generated predominantly a mixture of 15-mer and 16-mer fragments (cf. Fig. 1, C and D, lanes 1–6). Both enzymes are known to bind to the template strand and bend the DNA at the break. Our result suggests that the 90° bend introduced by EXO1 binding (17) partially melted the flap-containing strand to generate a single nucleotide gap between the 3′ terminus of the leading strand primer and the ss/ds boundary and subsequently cleaved the substrate 3′ from the first nucleotide of the duplex. The FEN1-generated products can also be explained by its modus operandi. It bends the substrate DNA by 100°, melts two base pairs downstream of the ss/ds boundary, and cleaves the strand between these two extruded nucleotides. This would explain the origin of the 16-mer fragment. The origin of the 15-mer can also be explained. FEN1 prefers to generate directly ligatable products and achieves this by binding and bending the DNA so that the 3′ end of the leading strand primer melts to give rise to a single-nucleotide 3′ flap. The ss/ds boundary in this structure is shifted by one nucleotide in the 5′ direction. Cleavage of this 5′ flap between the two extruded nucleotides would then liberate a 15-mer flap and leave behind a nick that can be ligated.
Analysis of products generated by the three nucleases from the 5′ phosphorylated cross-linked substrate was slightly complicated by the fact that the oligonucleotide had to be labeled by polynucleotide kinase after ICL synthesis, which means that both 5′ termini were labeled even though the ds end was phosphorylated less efficiently than the ss flap. This increased the number of possible products shown in Fig. 1A, right panel. FEN1 flap endonuclease activity was only weakly inhibited by the presence of the ICL (Fig. 1D, bottom panel). Because this enzyme lacks exonuclease activity on blunt-ended, double-stranded DNA, product b' was clearly detectable, as were the released flap products (a'), of which the 15-mer predominated (Fig. 1D, lanes 7–12). EXO1 activity on the ICL substrate was inhibited (Fig. 1C, bottom panel), as judged by the lower intensity of bands a' compared with bands a. Interestingly, EXO1 failed to release the 17-mer flap from the ICL substrate (Fig. 1C, lanes 7–12).
As shown in Fig. 1B, lanes 7–12, the abundance of flap fragments a/a' arising through cleavages 5′ from the ICL by FAN1 was almost comparable with the unmodified and cross-linked substrates, as shown by the intensity of the 15–17-mer bands (red arrowheads) relative to the full-length substrate (Fig. 1B, bottom panel). Because of the fact that the lower substrate strand was also labeled, products d' (Fig. 1A) were also detectable in the autoradiograph (Fig. 1B, lanes 7–12). Products c' were generated by a 5′-to-3′ degradation from the blunt-ended 5′ terminus of the ICL substrate, but the nature of band d' was uncertain. However, given that it migrated only slightly slower than the 37-mer, we postulated that it might correspond to species d', shown in Fig. 1A (see also below).
The most notable difference between the FAN1-generated products of the unmodified and the ICL substrate, however, was the absence of the 19-mer product in the latter reactions (Fig. 1B, cf. lanes 1–6 and 7–12). On the basis of the cleavage pattern of the unmodified flap substrate, this incision would have occurred on the 3′ side of the cross-link, and, therefore, the labeled flap fragment would not have been released from the substrate. In the experiments described above, we used recombinant FAN1 and EXO1 expressed in Sf9 cells and bacteria, respectively. To ensure that the observed enzymatic activities were indeed due to these polypeptides rather than contaminating nucleases, we also generated nuclease-dead variants of FAN1 (D960A) and EXO1 (D173N). As shown in Fig. 1E, the inactive variants generated no products comparable with those generated by the wild-type proteins.
To verify the incision on the 3′ side of the cross-link experimentally, we labeled the substrates at the 3′ termini of the upper strand (Fig. 2A). As anticipated, FAN1 (7–10), EXO1 (16), and FEN1 (24) degraded the unmodified substrate exonucleolytically from the sites of their respective endonucleolytic incisions in a 5′-to-3′ direction, with EXO1 (Fig. 2C, lanes 1–6) being more efficient than FAN1 (Fig. 2B, lanes 1–6), which was, in turn, more efficient than FEN1 (Fig. 2D, lanes 1–6). Most of the products detected in the FAN1-catalyzed degradation were longer than 11 nucleotides, which suggested that this enzyme might require a dsDNA stretch of ∼10 base pairs to which to bind. In contrast, EXO1 appeared to completely degrade the incised strand. FEN1 predominantly generated product a from the unmodified substrate. Incubation of the ICL substrate with EXO1 generated species c' as the primary product, and lower amounts of species a' and d' (Fig. 2C, lanes 7–12), which suggests that this enzyme may prefer a blunt-end terminus to a 5′ flap as a substrate (15). FEN1 predominantly yielded product a' (Fig. 2D, lanes 7–12). This indicated that both enzymes made their primary endonucleolytic incisions 5′ from the ICL at position 18 of the labeled strand and that their exonuclease activity failed to traverse the cross-link. In contrast, incubation of the unmodified and the ICL substrate with FAN1 yielded similar products (Fig. 2B, cf. products b and b', lanes 1–6 and lanes 7–12), which arose from incisions in the labeled strand on the 3′ side of nucleotide 18 (and, therefore, 3′ from the cross-link in the ICL substrate) and their subsequent 5′ to 3′ degradation. Fig. 2E shows again that the activities were due to the enzymatic activities of FAN1 and EXO1 because their mutants failed to generate the specific products.
FAN1 Can Degrade a Linear Duplex Containing an ICL
Our experiments carried out with the 5′-labeled substrates showed that the flap endonuclease of FAN1 was relatively promiscuous because it incised the structures both 5′ and 3′ of the ss/ds boundary. Moreover, its activity did not appear to be influenced by the presence of the cross-link. Given that the enzyme is also a robust 5′ to 3′ exonuclease, as seen with the 3′-labeled substrates, we wanted to test whether the presence of a cross-link affected the latter activity. We therefore generated linear duplex substrates, either unmodified or containing a single ICL (Fig. 3A). As shown in Fig. 3, B—D, lanes 1–6, all three enzymes were able to degrade the unmodified 3′-labeled substrate, albeit inefficiently. Unexpectedly, FAN1 digestion gave rise to a similar range of short oligonucleotide products as those seen with the unmodified substrate (Fig. 3B, cf. lanes 1–6 and 7–12), which indicated that the enzyme traversed the ICL. This result confirms the nature of product d' in Fig. 1B. In contrast, EXO1 and FEN1 generated a series of products b' from the ICL substrate (Fig. 3, C and D, lanes 7–12), which indicated that the enzyme was arrested by the ICL. As shown in Fig. 3E, the inactive variants generated no products comparable with those generated by the wild-type proteins.
We then tested FAN1 (WT and D960A) activity on a linear ICL substrate with a recessed 5′ terminus. Also on this substrate, the WT FAN1 protein was able to traverse the cross-link (Fig. 3F, lanes 1–5 (WT) and 6–8 (D960A)).
FAN1 Requires Regions of ∼10 Base Pairs on Either Side of the Flap for Cleavage
The experiments described above provided us with novel information concerning the biochemical properties of FAN1. Could these data help elucidate the role of FAN1 in ICL processing in vivo? That FAN1 is involved in ICL damage processing is beyond doubt, given that cells lacking the full complement of this protein are hypersensitive to cisplatin, MMC (7–10), and the nitrogen mustard BCNU (Fig. 5E). It is, however, unclear, which structures it addresses in genomic DNA and which products it generates. Evidence obtained from experiments carried out with an ICL-containing plasmid substrate and X. laevis egg extracts indicated that a replication fork encountering a cross-link stalls initially 20–40 base pairs from it because of the CDC45/MCM2–7/GINS replicative helicase remaining on the leading strand. This intermediate is not incised in this experimental system but persists until the arrival of the second fork from the opposite direction. Both helicase complexes are then unloaded, and the forks converge on the ICL. Unhooking was detected when the forks were just a single base pair from the cross-link (Ref. 25 and references therein). In this system, the nucleases primarily responsible for unhooking are believed to be SLX1/SLX4 on the 5′ side of the ICL and XPF/ERCC1 on the 3′ side (23). FAN1 has been shown not to be necessary (5, 23), and this finding is supported by our data, which suggest that FAN1 would not incise the X structure generated by the convergence of the two forks because it appears to require the ICL to be flanked by dsDNA on both sides of the flap, as witnessed by the fact that a Y junction is a poor FAN1 substrate (9) and that its exonuclease activity failed to generate fragments shorter than ∼10 nucleotides (Fig. 2B). This suggests that FAN1 requires at least this length of dsDNA for binding.
FIGURE 5.

FAN1- and/or MUS81-dependent DSB induction upon MMC treatment of human cells. A, top panel, FAN1 and/or MUS81 siRNA-mediated knockdown efficiencies assessed by Western blotting of total cell extracts of untreated (−) or MMC-treated (+) U2OS cells. TFIIH was used as a loading control. Quantification of the knockdown efficiencies shown in the bottom panel was carried out using ImageJ, and the graph was produced by GraphPad Prism (n = 3). The same extracts were probed for the markers of DSB metabolism RPA and CtIP. Center panel, Western blot analysis of the chromatin-enriched fraction of the above extracts probed for RPA, phospho-RPA, and γH2AX. Lamin was used as the loading control. B, time course of DSB formation assessed by pulsed field gel electrophoresis after MMC treatment (3 μg/ml) of U2OS cells, in which FAN1 or MUS81 were knocked down by siRNA. C, representative pulsed field gel electrophoresis image of DSBs induced by 24 h MMC (3 μg/ml) treatment of U2OS cells in which FAN1 and/or MUS81 were knocked down. The left panel shows a quantification of three independent experiments. siLUC was used as a control, and the ratio of DSBs of the MMC-treated samples divided by the untreated samples is shown for each siRNA condition. Error bars show mean ± S.E. D, quantifications of a FACS analysis of EdU-labeled cells pretreated with the indicated siRNAs and subsequently treated for 24 h with 3 μg/ml MMC. The knockdown did not affect cell viability during the course of the experiment. Error bars show mean ± S.E. (n = 3). E, clonogenic survival assay of U2OS cells treated with the indicated siRNAs and drugs. Colonies were counted 8 days after treatment, and MMC was washed out 24 h after treatment.
We decided to test this hypothesis directly by generating two X-like substrates reminiscent of two converging replication forks separated by 12 (X-12) or eight (X-8) nucleotides (Fig. 4A). As shown in Fig. 4B, the former substrate was processed more efficiently than the latter by FAN1 WT but not by the D960A mutant. This difference was not caused by the lower stability of the X-8 substrate. Both the X-12 and X-8 substrates were annealed efficiently under the assay conditions, as shown by native PAGE (Fig. 4B, bottom right panel). Therefore, coupled with the finding that the enzyme processes Y structures with only limited efficiency (9), the above result confirms our hypothesis that FAN1 requires regions of ∼10 base pairs on either side of the 5′ flap for cleavage.
MMC-induced DSBs Are Partially FAN1-dependent
If FAN1 is not involved in the resolution of replication forks converged at an ICL, then which structures does it address in vivo? This question is extremely difficult to answer, but it might be possible to gain some insights from the analysis of the products it generates from DNA treated with ICL-inducing agents. Processing of ICL-arrested replication forks by the FANC pathway (5) leads to lesion unhooking and to the generation of one or two DSBs that are subsequently repaired by homologous recombination (2). Some years ago, the MUS81 gene was also implicated in ICL repair. Like FAN1, MUS81 is not a known FANC gene. It encodes a protein that associates with EME1 to form a heterodimer that possesses 3′ flap endonuclease activity. Disruption of the Mus81 locus in the mouse resulted in sensitivity to ICL-inducing agents and to a decreased number of MMC-induced DSBs in ES cells in S phase, as shown by pulsed field gel electrophoresis (26). This suggested that Mus81 might be involved in the conversion of ICLs to DSBs, which are in turn repaired by recombination (22). We decided to make use of this assay to learn whether we could find evidence of similar processing of ICL-containing DNA by FAN1. We treated U2OS (osteosarcoma) cells with siRNAs targeting either luciferase (negative control), FAN1, and/or MUS81 and, 2 days later, exposed them to MMC. Western blot analysis of extracts isolated 24 h after MMC treatment revealed that FAN1 and/or MUS81 knockdown was associated with a reduction in phosphorylation of replication protein A (RPA), CtIP, and H2AX (Fig. 5A), indicative of less extensive resection of DSBs. This was confirmed by an analysis of genomic DNA 18, 24, and 30 h after MMC treatment. As shown in Fig. 5B, the number of DSBs was reduced in FAN1 or MUS81 knockdown cells compared with the control. Surprisingly, quantification of the 24-h time point (Fig. 5C) revealed that knockdown of both MUS81 and FAN1 gave rise to a similar number of DSBs as the FAN1 knockdown alone. This effect was not caused by a proliferative defect of the siRNA-treated cells, as confirmed by EdU incorporation and FACS analysis (Fig. 5D). The finding that knockdown of FAN1 or MUS81 resulted in a similar sensitivity to MMC or carmustine and that knockdown of both mRNAs failed to cause additional sensitization (Fig. 5E) suggested that the two proteins might act together in the processing of a subset of ICL substrates.
Discussion
The current hypotheses of ICL processing (5) posits that the repair machinery has to deal with either one of two distinct replication-dependent lesions: a single fork arrested at the ICL (Fig. 6A) or an X-shaped structure that arises at the cross-link through the convergence of two forks or through replication fork traverse through the ICL (Fig. 6B). Our findings suggest that FAN1 would be able to process the single arrested fork alone by first releasing the flap and then unhooking/traversing the ICL to leave behind a short oligonucleotide attached to the leading strand template that could be bypassed by translesion polymerases (Fig. 6C). However, this scenario would apply solely if FAN1 were able to incise the flap when the ICL was more than four nucleotides 3′ from the ss/ds boundary. If it were closer, FAN1 would fail to release the flap. FAN1 could also initiate the processing of an X-shaped structure, by releasing the lagging strand flap and, therefore, forming a substrate for a second nuclease, such as MUS81, which could release the 3′ flap formed by the leading strand of the second fork. This mechanism would only work if the two forks were at least 15 base pairs apart, with the ICL being positioned more than four nucleotides from the left ss/ds boundary (Fig. 6D) to provide FAN1 with a duplex platform to which it has to bind on either side of the flap (see above).
FIGURE 6.
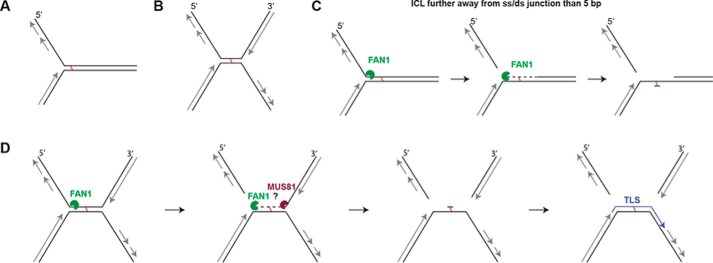
The putative mechanism of FAN1-dependent ICL unhooking. A, schematic of a single replication fork arrested at an ICL. B, the X-shaped structure arising through the convergence of two replication forks at an ICL. C, a single fork arrested at an ICL that is more than 5 bp from the ss/ds boundary would be incised by FAN1 5′ from the ICL, which would release the lagging strand. The enzyme would then degrade the nicked strand in a 5′-to-3′ direction to generate a substrate for translesion polymerases and subsequent repair by nucleotide excision repair. D, an X-shaped structure that contains a duplex longer than 10 bp where the lagging and leading strand flaps could be released by the action of FAN1 and, e.g., MUS81.
The hypothesis of ICL processing favoring the convergence of two forks arriving from opposite directions has recently received additional support (25). Of the known 5′ flap endonucleases, SLX1/SLX4 is emerging as the strongest candidate for introducing the first incision, with XPF/ERCC1 being the prime candidate for the 3′ incision. The involvement of FAN1 in the processing of a subset of these structures, where the forks remained some distance apart because of topological restraints, for example, cannot be excluded. But FAN1 does not necessarily have to be involved in ICL processing. We originally proposed that the enzyme might be involved downstream of the unhooking step, possibly during homologous recombination, because the enzyme processed a D loop structure with very high efficiency and because foci of γ-H2AX, RPA, and RAD51, which are generally believed to be markers of DSBs, appeared with similar kinetics in FAN1-proficient and knockdown cells but persisted longer in depleted cells (9), and depletion of MUS81 results in a similar phenotype (27). Interestingly, these foci also form after replication fork stalling (22, 28–30), and FAN1 has been reported to accumulate at replication forks stalled by aphidicolin (31). It is therefore possible that the persistent foci of γ-H2AX, RPA, and RAD51 represent stalled replication forks that collapsed because of the deficiency in enzymes able to mediate their restart and that are being processed by an alternative, less efficient mechanism.
Recently, the crystal structure of Pseudomonas aeruginosa FAN1 bound to a 5′ flap (32) revealed that the enzyme (lacking the ubiquitin-binding zinc finger domain) bends the DNA at the flap position by extensively interacting with the dsDNA regions on either side of the flap and that it cleaves the fourth phosphodiester moiety 3′ from the ss/dsDNA boundary. These observations fully agree with our findings. When our manuscript was in the final stages of preparation, two additional publications described the crystal structure of human FAN1 (also lacking the ubiquitin-binding zinc finger domain) bound to different flap substrates. In one study (33), FAN1 was shown to bind with a much greater affinity to substrates containing a 5′-phosphorylated flap of only one or two nucleotides, which it then incised in exonucleolytic steps of three nucleotides. In this way, it was able to unhook a triazole cross-link in a way analogous to that described in our study. The second study deployed a series of substrates in which the position of the ICL was moved further away from the ss/ds boundary of the flap (34). In the latter work, cleavage efficiency was seen to increase as the cross-link was moved 6, 12, or 16 base pairs away from the ss/ds boundary. This work predicted that, on the latter substrates, the enzyme would successfully traverse the ICL. In this scenario, it would be able to unhook the cross-link without the assistance of other enzymes. Taken together, our study and those of Wang et al. (33) and Zhao et al. (34) demonstrate that FAN1 can cleave long 5′ flaps such as those that would arise at blocked replication forks but that the enzyme may also process and unhook short flaps and nicks generated at ICLs by other nucleases that may not be able to bypass the ICL. In the latter reaction, FAN1 may be partially redundant with SNM1A, as is the case in Schizosaccharomyces pombe (35). Like FAN1, the latter enzyme has the ability to traverse cross-links, providing that a single-stranded nick 5′ from the ICL is present in the vicinity (36).
Because ICLs block the progression of transcription and replication machineries and because they can bring about different distortions in the helical structure of DNA (37), it is not surprising that nature has evolved several mechanisms of dealing with these extremely cytotoxic lesions. The investigation of which enzyme or pathway acts when and on which substrate(s) must await the outcome of genetic experiments that are currently in progress in several laboratories.
Author Contributions
J. P. helped design and carried out the experiments and cowrote the manuscript. S. M. synthesized the ICL oligonucleotides; O. D. S. helped design the experiments; and J. J. conceived the study, helped design the experiments, and wrote the manuscript.
Acknowledgments
We thank Anja Saxer for technical help. We also thank Stephanie Bregenhorn for the purified recombinant EXO1, Pavel Janscak for the MUS81 antibody, Alessandro Sartori for the CtIP antibody, and Svenja Kaden for helpful discussions and critical reading of the manuscript.
This work was supported by Swiss National Science Foundation Grants 310030B-133123 and 31003A-149989 and by Advanced ERC Grant MIRIAM (to J. J.). Funding for open access charge was from the University of Zurich. The authors declare that they have no conflicts of interest with the contents of this article.
- ICL
- interstrand cross-link
- FA
- Fanconi anemia
- MMC
- mitomycin C
- ss
- single-stranded
- ds
- double-stranded
- NM
- nitrogen mustard
- DSB
- double strand break.
References
- 1. Schärer O. D. (2005) DNA interstrand crosslinks: natural and drug-induced DNA adducts that induce unique cellular responses. ChemBioChem 6, 27–32 [DOI] [PubMed] [Google Scholar]
- 2. Hinz J. M. (2010) Role of homologous recombination in DNA interstrand crosslink repair. Environ. Mol. Mutagen. 51, 582–603 [DOI] [PubMed] [Google Scholar]
- 3. Auerbach A. D. (2009) Fanconi anemia and its diagnosis. Mutat. Res. 668, 4–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Crossan G. P., Patel K. J. (2012) The Fanconi anaemia pathway orchestrates incisions at sites of crosslinked DNA. J. Pathol. 226, 326–337 [DOI] [PubMed] [Google Scholar]
- 5. Zhang J., Walter J. C. (2014) Mechanism and regulation of incisions during DNA interstrand cross-link repair. DNA Repair 19, 135–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Clauson C., Schärer O. D., Niedernhofer L. (2013) Advances in understanding the complex mechanisms of DNA interstrand cross-link repair. Cold Spring Harb. Perspect. Biol. 5, a012732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liu T., Ghosal G., Yuan J., Chen J., Huang J. (2010) FAN1 acts with FANCI-FANCD2 to promote DNA interstrand cross-link repair. Science 329, 693–696 [DOI] [PubMed] [Google Scholar]
- 8. Smogorzewska A., Desetty R., Saito T. T., Schlabach M., Lach F. P., Sowa M. E., Clark A. B., Kunkel T. A., Harper J. W., Colaiácovo M. P., Elledge S. J. (2010) A genetic screen identifies FAN1, a Fanconi anemia-associated nuclease necessary for DNA interstrand crosslink repair. Mol. Cell 39, 36–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kratz K., Schöpf B., Kaden S., Sendoel A., Eberhard R., Lademann C., Cannavó E., Sartori A. A., Hengartner M. O., Jiricny J. (2010) Deficiency of FANCD2-associated nuclease KIAA1018/FAN1 sensitizes cells to interstrand crosslinking agents. Cell 142, 77–88 [DOI] [PubMed] [Google Scholar]
- 10. MacKay C., Déclais A. C., Lundin C., Agostinho A., Deans A. J., MacArtney T. J., Hofmann K., Gartner A., West S. C., Helleday T., Lilley D. M., Rouse J. (2010) Identification of KIAA1018/FAN1, a DNA repair nuclease recruited to DNA damage by monoubiquitinated FANCD2. Cell 142, 65–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pennell S., Déclais A. C., Li J., Haire L. F., Berg W., Saldanha J. W., Taylor I. A., Rouse J., Lilley D. M., Smerdon S. J. (2014) FAN1 activity on asymmetric repair intermediates is mediated by an atypical monomeric virus-type replication-repair nuclease domain. Cell Rep. 8, 84–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Munoz I. M., Szyniarowski P., Toth R., Rouse J., Lachaud C. (2014) Improved genome editing in human cell lines using the CRISPR method. PloS ONE 9, e109752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yoshikiyo K., Kratz K., Hirota K., Nishihara K., Takata M., Kurumizaka H., Horimoto S., Takeda S., Jiricny J. (2010) KIAA1018/FAN1 nuclease protects cells against genomic instability induced by interstrand cross-linking agents. Proc. Natl. Acad. Sci. U.S.A. 107, 21553–21557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhou W., Otto E. A., Cluckey A., Airik R., Hurd T. W., Chaki M., Diaz K., Lach F. P., Bennett G. R., Gee H. Y., Ghosh A. K., Natarajan S., Thongthip S., Veturi U., Allen S. J., Janssen S., Ramaswami G., Dixon J., Burkhalter F., Spoendlin M., Moch H., Mihatsch M. J., Verine J., Reade R., Soliman H., Godin M., Kiss D., Monga G., Mazzucco G., Amann K., Artunc F., Newland R. C., Wiech T., Zschiedrich S., Huber T. B., Friedl A., Slaats G. G., Joles J. A., Goldschmeding R., Washburn J., Giles R. H., Levy S., Smogorzewska A., Hildebrandt F. (2012) FAN1 mutations cause karyomegalic interstitial nephritis, linking chronic kidney failure to defective DNA damage repair. Nat. Genet. 44, 910–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Orans J., McSweeney E. A., Iyer R. R., Hast M. A., Hellinga H. W., Modrich P., Beese L. S. (2011) Structures of human exonuclease 1 DNA complexes suggest a unified mechanism for nuclease family. Cell 145, 212–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tsutakawa S. E., Classen S., Chapados B. R., Arvai A. S., Finger L. D., Guenther G., Tomlinson C. G., Thompson P., Sarker A. H., Shen B., Cooper P. K., Grasby J. A., Tainer J. A. (2011) Human flap endonuclease structures, DNA double-base flipping, and a unified understanding of the FEN1 superfamily. Cell 145, 198–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tsutakawa S. E., Lafrance-Vanasse J., Tainer J. A. (2014) The cutting edges in DNA repair, licensing, and fidelity: DNA and RNA repair nucleases sculpt DNA to measure twice, cut once. DNA Repair 19, 95–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mukherjee S., Guainazzi A., Schärer O. D. (2014) Synthesis of structurally diverse major groove DNA interstrand crosslinks using three different aldehyde precursors. Nucleic Acids Res. 42, 7429–7435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Guainazzi A., Campbell A. J., Angelov T., Simmerling C., Schärer O. D. (2010) Synthesis and molecular modeling of a nitrogen mustard DNA interstrand crosslink. Chemistry 16, 12100–12103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Angelov T., Guainazzi A., Schärer O. D. (2009) Generation of DNA interstrand cross-links by post-synthetic reductive amination. Org. Lett. 11, 661–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bregenhorn S., Jiricny J. (2014) Biochemical characterization of a cancer-associated E109K missense variant of human exonuclease 1. Nucleic Acids Res. 42, 7096–7103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hanada K., Budzowska M., Davies S. L., van Drunen E., Onizawa H., Beverloo H. B., Maas A., Essers J., Hickson I. D., Kanaar R. (2007) The structure-specific endonuclease Mus81 contributes to replication restart by generating double-strand DNA breaks. Nat. Struct. Mol. Biol. 14, 1096–1104 [DOI] [PubMed] [Google Scholar]
- 23. Klein Douwel D., Boonen R. A., Long D. T., Szypowska A. A., Räschle M., Walter J. C., Knipscheer P. (2014) XPF-ERCC1 acts in unhooking DNA interstrand crosslinks in cooperation with FANCD2 and FANCP/SLX4. Mol. Cell 54, 460–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Harrington J. J., Lieber M. R. (1994) Functional domains within FEN-1 and RAD2 define a family of structure-specific endonucleases: implications for nucleotide excision repair. Genes Dev. 8, 1344–1355 [DOI] [PubMed] [Google Scholar]
- 25. Zhang J., Dewar J. M., Budzowska M., Motnenko A., Cohn M. A., Walter J. C. (2015) DNA interstrand cross-link repair requires replication-fork convergence. Nat. Struct. Mol. Biol. 22, 242–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hanada K., Budzowska M., Modesti M., Maas A., Wyman C., Essers J., Kanaar R. (2006) The structure-specific endonuclease Mus81-Eme1 promotes conversion of interstrand DNA crosslinks into double-strands breaks. EMBO J. 25, 4921–4932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nair N., Castor D., Macartney T., Rouse J. (2014) Identification and characterization of MUS81 point mutations that abolish interaction with the SLX4 scaffold protein. DNA Repair 24, 131–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ward I. M., Chen J. (2001) Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J. Biol. Chem. 276, 47759–47762 [DOI] [PubMed] [Google Scholar]
- 29. Vassin V. M., Anantha R. W., Sokolova E., Kanner S., Borowiec J. A. (2009) Human RPA phosphorylation by ATR stimulates DNA synthesis and prevents ssDNA accumulation during DNA-replication stress. J. Cell Sci. 122, 4070–4080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Petermann E., Orta M. L., Issaeva N., Schultz N., Helleday T. (2010) Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol. Cell 37, 492–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chaudhury I., Stroik D. R., Sobeck A. (2014) FANCD2-controlled chromatin access of the Fanconi-associated nuclease FAN1 is crucial for the recovery of stalled replication forks. Mol. Cell. Biol. 34, 3939–3954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gwon G. H., Kim Y., Liu Y., Watson A. T., Jo A., Etheridge T. J., Yuan F., Zhang Y., Kim Y., Carr A. M., Cho Y. (2014) Crystal structure of a Fanconi anemia-associated nuclease homolog bound to 5′ flap DNA: basis of interstrand cross-link repair by FAN1. Genes Dev. 28, 2276–2290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang R., Persky N. S., Yoo B., Ouerfelli O., Smogorzewska A., Elledge S. J., Pavletich N. P. (2014) DNA repair. Mechanism of DNA interstrand cross-link processing by repair nuclease FAN1. Science 346, 1127–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhao Q., Xue X., Longerich S., Sung P., Xiong Y. (2014) Structural insights into 5′ flap DNA unwinding and incision by the human FAN1 dimer. Nat. Commun. 5, 5726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fontebasso Y., Etheridge T. J., Oliver A. W., Murray J. M., Carr A. M. (2013) The conserved Fanconi anemia nuclease Fan1 and the SUMO E3 ligase Pli1 act in two novel Pso2-independent pathways of DNA interstrand crosslink repair in yeast. DNA Repair 12, 1011–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang A. T., Sengerova B., Cattell E., Inagawa T., Hartley J. M., Kiakos K., Burgess-Brown N. A., Swift L. P., Enzlin J. H., Schofield C. J., Gileadi O., Hartley J. A., McHugh P. J. (2011) Human SNM1A and XPF-ERCC1 collaborate to initiate DNA interstrand cross-link repair. Genes Dev. 25, 1859–1870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Muniandy P. A., Liu J., Majumdar A., Liu S. T., Seidman M. M. (2010) DNA interstrand crosslink repair in mammalian cells: step by step. Crit. Rev. Biochem. Mol. Biol. 45, 23–49 [DOI] [PMC free article] [PubMed] [Google Scholar]