

Background: MDM2 and p53 form a negative feedback loop in regulation.
Results: HBXIP up-regulates MDM2 by recruiting p300 to activate transcription factor p53.
Conclusion: HBXIP is involved in the MDM2/p53 feedback loop to promote the growth of breast cancer.
Significance: Our finding provides a new insight into the mechanism of MDM2/p53 circuit involved in HBXIP in the development of breast cancer.
Keywords: breast cancer, mouse double minute 2 homolog (MDM2), oncogene, p53, tumor, HBXIP, p300
Abstract
MDM2 and p53 form a negative feedback loop, in which p53 as a transcription factor positively regulates MDM2 and MDM2 negatively regulates tumor suppressor p53 through promoting its degradation. However, the mechanism of the feedback loop is poorly understood in cancers. We had reported previously that the oncoprotein hepatitis B X-interacting protein (HBXIP) is a key oncoprotein in the development of cancer. Thus, we supposed that HBXIP might be involved in the event. Here, we observed that the expression levels of HBXIP were positively correlated to those of MDM2 in clinical breast cancer tissues. Interestingly, HBXIP was able to up-regulate MDM2 at the levels of mRNA and protein in MCF-7 breast cancer cells. Mechanically, HBXIP increased the promoter activities of MDM2 through directly binding to p53 in the P2 promoter of MDM2. Strikingly, we identified that the acetyltransferase p300 was recruited by HBXIP to p53 in the promoter of MDM2. Moreover, we validated that HBXIP enhanced the p53 degradation mediated by MDM2. Functionally, the knockdown of HBXIP or/and p300 inhibited the proliferation of breast cancer cells in vitro, and the depletion of MDM2 or overexpression of p53 significantly blocked the HBXIP-promoted growth of breast cancer in vitro and in vivo. Thus, we concluded that highly expressed HBXIP accelerates the MDM2-mediated degradation of p53 in breast cancer through modulating the feedback loop of MDM2/p53, resulting in the fast growth of breast cancer cells. Our findings provide new insights into the mechanism of the acceleration of the MDM2/p53 feedback loop in the development of cancer.
Introduction
Tumor suppressor p53, a transcription factor that activates or represses the expression of multiple genes, has been documented in a large number of biological processes and plays a critical role in cell cycle arrest, senescence, apoptosis, and metabolism (1, 2). Activation of p53-dependent apoptosis can lead to mitochondrial apoptotic changes through the intrinsic and extrinsic pathways triggering cell death execution, most notably by release of cytochrome c and activation of the caspase cascade (3).
MDM2 is overexpressed in a variety of human cancers, including melanoma, non-small cell lung cancer, breast cancer, esophageal cancer, leukemia, non-Hodgkin lymphoma, and sarcoma (4). MDM2 is able to negatively regulate the levels of p53 in cells by acting as an E3 ligase for the proteasome degradation of p53, which involves interaction with the N-terminal hydrophobic pocket and the acidic domain of MDM2, to promote cancer growth (5–9). MDM2 can interfere with p53-mediated apoptosis and growth arrest of tumor; this is the major oncogenic activity of MDM2 (10). The levels of MDM2 are transcriptionally up-regulated in response to a variety of oncogenic and tumor suppressor pathways and are closely related to the degree of tumor malignancy in cancers (11). Many studies have indicated that the MDM2 expression is elevated at mRNA levels in a majority of tumor tissues (12), and the transcription of MDM2 is controlled by two distinct promoters, P1 and P2 (13). It has been reported that p53 protein as a transcription factor can up-regulate MDM2 at the transcription levels on the P2 promoter (11, 14). However, the regulatory mechanism of the MDM2/p53 feedback loop, especially the transcription of MDM2 mediated by p53, remains undefined.
Hepatitis B X-interacting protein (HBXIP),3 a cellular 18-kDa protein originally identified by its interaction with the C terminus of hepatitis B virus X protein (15), regulates centrosome duplication in HeLa cells (16, 17) and may form a complex with survivin, an antiapoptotic protein, resulting in the suppression of cell apoptosis via the mitochondrial/cytochrome pathway (18). The current study demonstrates that HBXIP is required for amino acid sensing by the mTORC1 pathway in cell growth (19). Our group has reported that HBXIP plays a critical role in promoting the proliferation and migration of breast cancer cells by acting as a co-activator of transcription factors such as E2F1, TFIID, CREB (cAMP-response element-binding protein), and Sp1 (20–25).
In this study, the role of HBXIP in modulating the MDM2/p53 pathway in breast cancer drew our attention. Interestingly, we found that the oncoprotein HBXIP as a co-activator is involved in the transcription of MDM2 mediated by p53, in which the acetyltransferase p300 is recruited to p53 by HBXIP. Moreover, HBXIP enhances MDM2-mediated p53 degradation, leading to the growth of breast cancer. Our finding provides a new insight into the mechanism by which the oncoprotein HBXIP joins the MDM2/p53 feedback loop in breast cancer.
Experimental Procedures
Cell Culture and Treatment
Breast cancer cell lines MCF-7, LM-MCF-7 (a metastatic subclone of MCF-7 breast cancer cell line), and MCF-7-HBXIP were cultured in RPMI 1640 (Gibco). Human kidney epithelial (HEK) 293T cells and human colon cancer cell lines HCT116 p53+/+ and HCT116 p53−/− were maintained in Dulbecco's modified Eagle's medium (Gibco). All cell lines were supplemented with heat-inactivated 10% FBS (Gibco), 100 units/ml penicillin, and 100 mg/ml streptomycin and grown at 5% CO2 and 37°C. Cells were collected and seeded in 6-, 24-, or 96-well plates for 24 h and then transfected with plasmid or siRNA. All transfections were performed using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's protocol.
Immunohistochemistry Staining
A breast cancer tissue array (C0048), comprising duplicates of 95 cases of infiltrating duct carcinoma tissue samples, was purchased from Xi'an Aomei Biotechnology (Xi'an, China). Immunohistochemistry staining was performed using rabbit anti-HBXIP (or rabbit anti-MDM2) antibody as described previously (18).
Total RNA Isolation, RT-PCR, and Real-time PCR
Total RNA was isolated from cells using TRIzol reagent (Invitrogen) according to the manufacturer's instructions. First-strand cDNA was synthesized with the PrimeScript reverse transcriptase kit (TaKaRa Bio, Shiga, Japan). RT-PCR and real-time PCR were performed as described previously (23). The primers of HBXIP, p53, MDM2, p21, PUMA, and GAPDH cDNA used were described previously (21, 23, 26–28).
Plasmid Construction and Small Interfering RNA
RNA (siRNA), pCMV-tag2B, pGL3-Basic, and pGL3-Control vectors (Promega, Madison, WI) and pCMV-HBXIP were kept in our laboratory. The complete human MDM2 (GenBankTM accession number NG_016708.1) cDNA or p53 (GenBankTM accession number NG_017013.2) cDNA was subcloned into the pCMV-tag2B vector to generate the pCMV-MDM2 or pCMV-p53 construct, respectively. The nucleotide sequence of the MDM2 promoter region with or without the p53-binding site (29, 30) was cloned into the pGL3-Basic vector to generate the pGL3-MDM2 (P1/P2) construct. Primers for pGL3-P2-mut vector were 5′-GGG GTA CCG TGT TTA GGT TGA CTC AGC TTT-3′ and 5′-CCC AAG CTT TCC GTT TTC ACA GGT CTA CCC TCC A-3′. The nucleotide sequence of siRNA for MDM2 was cloned into the pSilencer 3.1-H1 neo vector to generate the pSilencer-MDM2 construct. The 2′-O-methyl interfering RNAs directly against HBXIP 3′-UTR, p53, MDM2, and p300 and the control siRNAs were purchased from the Riobio Co. (Guangzhou, China). The sequences of siRNAs for MDM2, HBXIP, p53, and p300 were described previously (16, 31–34).
Western Blot Analysis
Western blot analysis was carried out with the standard protocols (18). Some of the results were quantified by using Quantity One software (Bio-Rad). The primary antibodies used were rabbit anti-p53 (Proteintech Group), rabbit anti-MDM2 (Wuhan Boster Biological Technology Ltd., Wuhan, China), anti-HBXIP antibody (Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-p21 (Proteintech Group), rabbit anti-PUMA (Proteintech Group), rabbit anti-ubiquitin (Proteintech Group), rabbit anti-p300 (Santa Cruz Biotechnology), rabbit anti-acetylated lysine antibody (Cell Signaling Technology, Beverly, MA), rabbit anti-histone H3 (Proteintech Group), and mouse anti-β-actin (Sigma-Aldrich).
Luciferase Reporter Gene Assays
Adherent cells were seeded into 24-well plates and co-transfected with pGL3-MDM2 (P1/P2), pGL3-Basic, or pGL3-P2-mut and the pRL-TK plasmid (Promega), which is used for internal normalization. Cell extracts were harvested after 36 h and lysed using lysis buffer (Promega). Luciferase reporter gene assays were implemented using the Dual-Luciferase reporter gene assay system (Promega) according to the manufacturer's instructions. All experiments were performed at least three times.
Co-immunoprecipitation Assays
The co-immunoprecipitation (co-IP) protocol was described in detail in previously published articles (35). The lysates harvested from MCF-7 cells were incubated with antibodies and protein G-conjugated agarose beads at 4 °C for 2 h. The precipitates were washed eight times with ice-cold lysis buffer, resuspended in the PBS, and resolved by SDS-PAGE followed by Western blot analysis.
GST Pulldown Assays
The cDNA of HBXIP was cloned into the pGEX-4T1 expression vector (pGEX-HBXIP), and the cDNA of p53 was cloned into the pET-28a expression vector (pET-p53); the construct vectors pGEX-HBXIP, pGEX-4T1, and pET-p53, were transformed into Escherichia coli strains BL21(DE3). The detailed procedure was performed according to published protocols (36). Glutathione beads were recovered by a brief centrifugation and washed eight times with lysis buffer. The final wash used PBS followed by resolution by SDS-PAGE and detection by Western blot analysis using the anti-p53 antibody.
Chromatin Immunoprecipitation (ChIP) Assays
The ChIP assays were performed using an EpiQuikTM chromatin immunoprecipitation kit from Epigentek Group Inc. (Brooklyn, NY). Protein-DNA complexes were immunoprecipitated with anti-HBXIP or anti-p300 antibody. DNA from these samples was then subjected to PCR analysis. Primers sets for the MDM2 promoter were 5′-GTGGGCAGGTTGACTCAG-3′ and 5′-GGTCTACCCTCCAATCGC-3′ followed by sequencing.
Electrophoretic Mobility Shift Assays (EMSA)
Nuclear protein extracts were prepared from MCF-7 cells. Probes were generated by annealing single-strand oligonucleotides containing the MDM2 or MDM2-mut promoter and labeling the ends with [γ-32P]ATP using T4 polynucleotide kinase (TaKaRa Bio). Binding reactions were performed at 4 °C for 1 h in 10-μl mixtures containing 20 mm Hepes, 50 mm KCl, 0.05 mm DTT, 0.05 mm EDTA, 1 mm MgCl2, 5% glycerol, 0.05 μg/μl poly(dI·dC), 0.05% Nonidet P-40, 50 ng of probe, and 1.5 μg of nuclear extracts. The specificity of the HBXIP-DNA or p53-DNA interaction was confirmed by competition or supershift with HBXIP or p53 antibody, respectively. For the antibody competitive or supershift experiment, 1 μg of HBXIP or p53 antibody was added into the reaction mixture and incubated at 4 °C for 30 min before the DNA probe was added. For the competitive binding experiment, 500 ng of unlabeled DNA was added after the initial incubation for an additional 30 min. The binding mixtures were then resolved on a native 6% polyacrylamide gel in 0.5× TBE at 4 °C. The gel was dried and exposed to x-ray film for autoradiography. An analogous experiment was repeated with purified HBXIP and p53. The primers for the MDM2 promoter DNA fragment were 5′-GTG GGC AGG TTG ACT CAG CTT T-3′ and 5′-TCC GTG CCC ACA GGT CTA CCC TCC A-3′ and for the mutant MDM2 promoter fragment were 5′-GTG TTT AGG TTG ACT CAG CTT T-3′ and 5-TCC GTT TTC ACA GGT CTA CCC TCC A-3′.
Preparation of Recombinant Proteins
The plasmid pET-28a-HBXIP or pET-28a-p53 was used to express recombinant His-HBXIP or His-p53 in E. coli strain BL21. The recombinant proteins were expressed by induction with 0.5 mm isopropyl β-d-1-thiogalactopyranoside at 16°C for 20 h and purified by Ni2+-nitrilotriacetic acid affinity chromatography according to standard procedures.
In Vivo Ubiquitination Assays
HEK293T or MCF-7 cells were transfected as indicated and pretreated with 40 μm MG-132. Ten mm N-ethylmaleimide and 25 μm MG-132 were added into the lysis buffer. The ubiquitination level of p53 was examined by co-IP and Western blot analysis.
MTT Assays
Cell proliferation was determined by MTT (Sigma) assay as described previously (37).
Colony Formation Assays
For clonogenicity analysis, 48 h after transfection, 1000 viable transfected cells were placed in 6-well plates and maintained in complete medium for 2 weeks. Colonies were fixed with methanol and stained with methylene blue.
Animal Transplantation
Four groups of 4-week-old female BALB/c athymic nude mice (Experiment Animal Center of Peking, China; each group, n = 5) were housed and treated according to guidelines established by the National Institutes of Health Guide for the Care and Use of Laboratory Animals. According to the report (38), each nude mouse was implanted each with 1.7 mg of a 17β-estradiol pellet (90 days release; Beijing Solarbio Science and Technology Co., China) to supplement the estrogen requirements for MCF-7 proliferation. A week after implantation of the 17β-estradiol pellet, MCF-7-pCMV-shControl, MCF-7-HBXIP-shControl, MCF-7-HBXIP-shMDM2 (MCF-7-HBXIP cells were transfected with 7.5 μg of pSilencer-MDM2), and MCF-7-HBXIP-p53 (MCF-7-HBXIP cells were transfected with 7.5 μg of pCMV-p53) cells were harvested, suspended at 5× 107 cells/ml with phosphate-buffered saline, and then injected subcutaneously into mouse mammary fat pads with 0.2 ml of the cell suspensions. Beginning 5 days after injection, the tumor growth was measured every 3 days. Tumor volume (V), monitored by measuring the length (L) and width (W) with calipers, was calculated with the formula (L × W2) × 0.5. After 30 days, four groups of the mice were sacrificed, and the tumors were excised and measured.
Statistical Analysis
Each experiment was repeated at least three times. Statistical significance, assessed by comparing mean values ± S.D. using a Student's t test for independent groups, was assumed for p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***). Pearson's correlation coefficient was used to determine the expression correlation of genes in clinical breast cancer tissues. Statistical analysis of the tissue array was performed with the chi-square test or Kruskal-Wallis test using the SPSS software program (SPSS Inc., Chicago).
Results
The Expression Levels of HBXIP Are Positively Associated with Those of MDM2 in Clinical Breast Cancer Tissues, and HBXIP Up-regulates MDM2 in Breast Cancer Cells
Previously, we reported that a high level HBXIP is closely related to carcinogenesis in breast cancer (22, 39). Many studies have demonstrated that MDM2 is well characterized as an oncoprotein in breast cancer (40) that negatively regulates the levels of p53 in the cells by acting as an E3 ligase for proteasome degradation of p53 (7). Accordingly, we theorized that HBXIP might be associated with MDM2 in carcinogenesis. Then, we examined the expression relationship between HBXIP and MDM2 via immunohistochemistry staining using tissue arrays that were from the same tissue paraffin block. Our data indicated that the positive rate of HBXIP was 82.1% (78 of 95) in breast cancer tissues, in which 2.1% were cytoplasm-positive, 13.7% were nucleus-positive, and 66.3% were positive for both cytoplasm and nucleus. Also, the positive rate of MDM2 was 91% in 78 cases of HBXIP-positive samples of breast cancer. The cytoplasm-positive rate for MDM2 was 14.7%, the nucleus-positive rate was 30.5%, and the positive rate for both cytoplasm and nucleus was 31.6% (Table 1 and Fig. 1A). Moreover, the expression relationship between HBXIP and MDM2 was validated by real-time PCR at mRNA levels in 30 clinical breast cancer tissue samples (Fig. 1B), indicating that the expression of HBXIP was positively related to MDM2 in breast cancer. Our data further showed that HBXIP was able to up-regulate the expression of MDM2 at the levels of mRNA and protein in MCF-7 cells in a dose-dependent manner as indicated by RT-PCR and Western blot analysis (Fig. 1C). However, knockdown of HBXIP showed the opposite results in LM-MCF-7 cells, a metastatic subclone of the MCF-7 breast cancer cell line expressing a high level of HBXIP (22) (Fig. 1D). Then, the core regions of the MDM2 promoter, P1 and P2 (41), were cloned into the pGL3-Basic plasmid (Fig. 2A). We determined that the promoter activities of P2, rather than P1, were markedly increased by HBXIP in MCF-7 cells (Fig. 2B); this was validated in a dose-dependent manner in MCF-7 and 293T cells. The silencing of HBXIP attenuated the activities of the P2 promoter in LM-MCF-7 cells (Fig. 2, C–E). Thus, the above data suggest that the expression levels of HBXIP are positively associated with those of MDM2 in clinical breast cancer tissues and HBXIP up-regulates MDM2 through activating the P2 promoter activity of MDM2 in breast cancer cells.
TABLE 1.
Cross-tabulation analysis of HBXIP and MDM2 in breast cancer tissues
χ2 = 74.582, p < 0.001, n = 95.
| Breast cancer | MDM2 |
Total | ||||
|---|---|---|---|---|---|---|
| − | + | ++ | ||||
| HBXIP | − | Count | 15 | 2 | 0 | 17 |
| % within MDM2 | 88.2 | 11.8 | 0 | 100 | ||
| + | Count | 5 | 33 | 8 | 46 | |
| % within MDM2 | 10.9 | 71.7 | 17.4 | 100 | ||
| ++ | Count | 2 | 8 | 22 | 32 | |
| % within MDM2 | 6.2 | 25 | 68.8 | 100 | ||
| Total | Count | 22 | 43 | 30 | 95 | |
| % within MDM2 | 23.1 | 45.3 | 31.6 | 100 | ||
FIGURE 1.
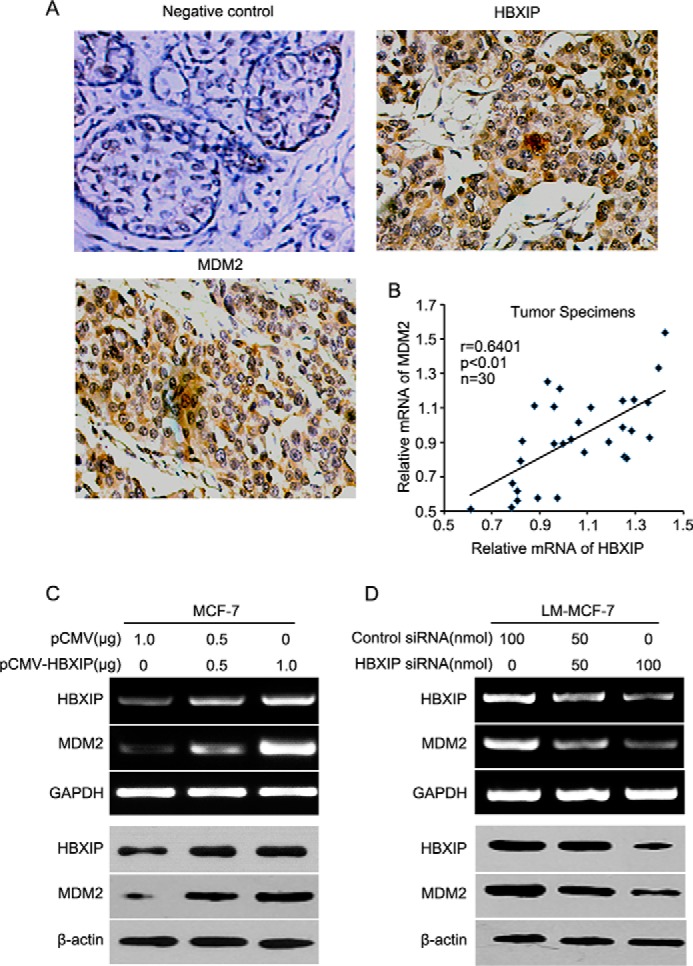
The expression levels of HBXIP are positively associated with those of MDM2 in breast cancer. A, the expression of HBXIP and MDM2 was determined by immunohistochemical staining in breast cancer tissues using tissue microarray. B, the relative expression levels of HBXIP and MDM2 were detected by real-time PCR in 30 clinical breast cancer tissues (p < 0.01, r = 0.6401, Pearson's correlation). C and D, the expression of HBXIP and MDM2 at the levels of mRNA and protein was examined by RT-PCR and Western blot analysis in MCF-7 and LM-MCF-7 cells, respectively. Each experiment was repeated at least three times.
FIGURE 2.

HBXIP activates the P2 promoter of MDM2 in breast cancer cells. A, locations of P1 and P2 of the MDM2 promoter are shown. B, the promoter (P1 and P2) activities of MDM2 were examined by luciferase reporter gene assays in MCF-7 cells transfected with pCMV-HBXIP plasmids. NS, not significant. C and D, the promoter (P2) activities of MDM2 were examined by luciferase reporter gene assays in MCF-7 and HEK293T cells transfected with pCMV-HBXIP plasmids. E, effect of HBXIP siRNA on promoter (P2) activities of MDM2 was detected by luciferase reporter gene assays in LM-MCF-7 cells. Each experiment was repeated at least three times. Student's t test; *, p < 0.05; **, p < 0.01.
HBXIP Activates the MDM2 Promoter by Stimulating Transcription Factor P53
Next, we sought to investigate the underlying mechanism by which HBXIP enhances MDM2. It has been reported that the P2 promoter is regulated by a variety of transcription factors, including transcription factor p53 (11, 29). Therefore, we hypothesized that HBXIP might act as a co-activator of p53 to stimulate the MDM2 promoter in breast cancer cells. Thus, we cloned the construct of P2 promoter with a mutated p53 element (termed pGL3-P2-mut) (Fig. 3A) and observed that HBXIP failed to promote the activity of the mutant P2 promoter (Fig. 3B). Moreover, p53 knockdown markedly abolished the increased activities of P2 promoter mediated by HBXIP (Fig. 3, C and D). Furthermore, luciferase reporter gene assays showed that HBXIP increased P2 promoter activity in HCT116 p53+/+ cells but failed to work in HCT116 p53−/− cells (Fig. 3E). In addition, we observed that overexpression of HBXIP or/and p53 increased P2 promoter activities in MCF-7 and 293T cells (Fig. 3, F and G). These results suggest that HBXIP may co-activate p53 to stimulate the P2 promoter in breast cancer cells.
FIGURE 3.

HBXIP up-regulates MDM2 involving the p53 element in breast cancer cells. A, the mutation of the p53 element is shown in the P2 promoter of MDM2. B, the luciferase activities were examined by luciferase reporter gene assays in the MDM2 promoter (P2) with wild type or mutant binding sites of p53 in MCF-7 cells. C and D, the luciferase activities of the MDM2 promoter (P2) were detected by luciferase reporter gene assays in MCF-7 and HEK293T cells. The efficiency of p53 siRNA was determined using Western blot analysis. E, the promoter (P2) activities of MDM2 were examined by luciferase reporter gene assays in HCT116 p53−/− and HCT116 p53+/+ cells transfected with pCMV-HBXIP plasmids. NS, not significant. F and G, the promoter (P2) activities of MDM2 were examined by luciferase reporter gene assays in MCF-7 and HEK293T cells transfected with pCMV-HBXIP and pCMV-p53 plasmids. Each experiment was repeated at least three times. Student's t test; *, p < 0.05, **, p < 0.01.
Next, co-IP assays revealed that HBXIP and p53 bound to each other in MCF-7 cells (Fig. 4A, left panel). GST pulldown assays showed that the p53 protein with His tag could be pulled down by GST-HBXIP but not by the control GST alone (Fig. 4A, right panel), suggesting that HBXIP directly bound to p53 protein in the cells. Importantly, ChIP assays revealed that HBXIP was able to interact with the MDM2 promoter through p53 (Fig. 4B). The interference efficiency of p53 siRNA was validated by Western blot analysis in MCF-7 cells (Fig. 4C). EMSAs demonstrated that the MDM2 promoter probe with a mutated p53 binding site failed to interact with the nuclear extracts. Moreover, the anti-p53 (or anti-HBXIP) antibody disrupted the interaction between the complex of p53-HBXIP and the MDM2 promoter (Fig. 4D). We further validated that the purified p53 and HBXIP protein together resulted in a supershift compared with p53 protein only (Fig. 4E). The results further support the idea that HBXIP is able to occupy the MDM2 promoter through binding to p53. Luciferase reporter gene assays validated that p53 and HBXIP dependently activated the P2 promoter of MDM2 in breast cancer cells (Fig. 4, F and G). Thus, we concluded that HBXIP activates the MDM2 promoter through binding to and activating transcription factor p53.
FIGURE 4.

HBXIP activates the MDM2 promoter through stimulating transcription factor p53. A, the interaction between HBXIP and p53 was detected by co-IP assays in MCF-7 cells in vivo. The direct interaction between recombinant GST-HBXIP and His-p53 proteins was detected by GST pulldown assays followed by Western blot analysis (WB) in vitro. B, MCF-7 cells were transfected with p53 siRNA. ChIP assays were conducted 24 h after transfection. C, the interference efficiency of p53 siRNA was detected by Western blot analysis in MCF-7 cells. D, the interaction between nuclear proteins from MCF-7 cells and MDM2 promoter (P2) probe with wild type or mutant binding sites of p53 was determined by EMSA. E, the interaction between MDM2 promoter (P2) probe and HBXIP was tested by EMSA using purified HBXIP and p53. F, the promoter activities of MDM2 (P2) were measured by luciferase reporter gene assays in MCF-7 cells. G, the luciferase activities of the MDM2 promoter (P2) were detected by luciferase reporter gene assays in HEK293T cells transfected with pCMV-p53 and HBXIP siRNAs. Each experiment was repeated at least three times. Student's t test; **, p < 0.01.
HBXIP Recruits P300 to Transcription Factor P53 in Activation of the MDM2 Promoter
Given that p300 histone acetylatransferase acts as a co-activator of p53 to enhance the transcription activity of p53 (42) and is able to promote the acetylation of histone H3 (43), we questioned whether p300 was involved in the MDM2 transcription induced by HBXIP. Luciferase reporter gene assays showed that overexpression of p300 could increase the promoter activities of P2 in MCF-7 cells in a dose-dependent manner, but p300 siRNA resulted in the decrease of promoter activities of P2 in the cells (Fig. 5A). Moreover, we observed that p300 siRNA abolished the HBXIP-enhanced promoter activities of P2 in MCF-7 cells in a dose-dependent manner (Fig. 5B). Interestingly, HBXIP siRNA impaired the p300-increased activities of P2 promoter in the cells (Fig. 5C), suggesting that p300 was involved in the activation of P2 mediated by HBXIP. Furthermore, co-IP assays showed that the silencing of HBXIP could attenuate the interaction of p300 with p53 in MCF-7 cells, whereas the overexpression of HBXIP enhanced the interaction of p300 with p53 in the cells (Fig. 5D). Meanwhile, ChIP assays confirmed that HBXIP siRNA disrupted the interaction of p300 with MDM2 promoter in MCF-7 cells using p300 antibody (Fig. 5E), suggesting that HBXIP could recruit p300 to the MDM2 promoter in breast cancer cells. The interference efficiency of p300 siRNA was tested in MCF-7 cells (Fig. 5F). Moreover, our data indicated that HBXIP siRNA abolished the p300-induced acetylation of histone H3 (Fig. 5G), and silencing p300 also blocked the effect of HBXIP in the cells (Fig. 5H), confirming that HBXIP recruited p300 to the MDM2 promoter in breast cancer cells, forming a transcriptional complex of HBXIP-p53-p300. Taking these findings together, we concluded that HBXIP recruits p300 to transcription factor p53 to activate the MDM2 promoter.
FIGURE 5.

HBXIP recruits p300 to transcription factor p53 to activate MDM2 promoter. A, P2 promoter activities of MDM2 were tested by luciferase reporter gene assays in MCF-7 cells. B, P2 promoter activities of MDM2 were examined by luciferase reporter gene assays in MCF-7 cells. C, P2 promoter activities of MDM2 were measured by luciferase reporter gene assays in MCF-7 cells. The efficiency of HBXIP siRNA was determined using Western blot analysis (WB). D, the interaction of p300 with p53 was detected by co-IP assays in MCF-7 cells. E, MCF-7 cells were transfected with HBXIP siRNA. ChIP assays were measured 24 h after transfection. F, the interference efficiency of p300 siRNA was tested in MCF-7 cells. G and H, the acetylation levels of histone H3 were examined by co-IP assays in MCF-7 cells with different treatments. Each experiment was repeated at least three times. Student's t test; *, p < 0.05; **, p < 0.01.
HBXIP Up-regulates MDM2, Leading to the Further Degradation of P53
Growing evidence has revealed that MDM2 is a negative regulator of tumor suppressor p53 and is able to regulate the level of p53 in cells by acting as an E3 ligase for the ubiquitination and proteasomal degradation of p53 (5, 6, 11). Therefore, we further validated the effect of HBXIP on MDM2-mediated p53 degradation. As expected, RT-PCR revealed that the overexpression of HBXIP was able to down-regulate the target genes of p53, such as p21 and PUMA, but not p53, at the mRNA levels in MCF-7 cells (Fig. 6A). Also, the knockdown of HBXIP by siRNA failed to influence p53 at the mRNA levels in LM-MCF-7 cells (Fig. 6B). Moreover, Western blot analysis indicated that the overexpression of HBXIP significantly decreased the level of p53 and down-regulated its target genes at the level of protein in MCF-7 cells in a dose-dependent manner (Fig. 6C). The opposite results were observed in LM-MCF-7 cells mediated by HBXIP siRNA (Fig. 6D). Following treatment with the proteasome inhibitor MG-132, we found that p53 had significantly accumulated in MCF-7 cells, but no accumulation of p53 was observed in the cells treated with siHBXIP (Fig. 6E). The half-life assays demonstrated that the knockdown of HBXIP could suppress the degradation of p53 and increase the half-life of p53 from 39.3 to 82.6 min (Fig. 6F), suggesting that HBXIP contributed to the degradation of p53 in breast cancer cells. Furthermore, the in vivo ubiquitination assays validated that HBXIP increased the ubiquitination levels of p53 (Fig. 6G), whereas the siRNA of MDM2 abolished the ubiquitination levels of p53 enhanced by HBXIP in MCF-7 cells (Fig. 6H). Moreover, the depletion of MDM2 abolished the degradation of p53 mediated by HBXIP in the cells (Fig. 6I). The above data show that HBXIP enhances MDM2-mediated p53 degradation and reduces the expression of p53 target genes. Therefore, we concluded that HBXIP up-regulates MDM2, leading to the further degradation of p53 in breast cancer cells.
FIGURE 6.

HBXIP up-regulates MDM2, leading to the further degradation of p53. A and B, the expression levels of HBXIP, p53, and p53 target genes such as p21 and PUMA were detected by RT-PCR in MCF-7 and LM-MCF-7 cells. C and D, the expression levels of HBXIP, p53, and p53 target genes such as p21 and PUMA were detected by Western blot analysis in MCF-7 and LM-MCF-7 cells. E, MCF-7 cells were treated with 25 mm MG-132 for 5 h, as indicated, followed by Western blot analysis. F, upper lane, MCF-7 cells were treated with 100 mg/ml cycloheximide and harvested using SDS lysis buffer at the time points indicated. The levels of p53 were analyzed by Western blot analysis, and the results were quantified using Quantity One software (Bio-Rad) as shown. Lower lane, summary of three independent experiments. G, the in vivo ubiquitination assays were performed by Western blot analysis in HEK293T cells transfected with pCMV-HBXIP plasmids. H, the in vivo ubiquitination assays were performed by Western blot analysis in MCF-7 cells transfected with pCMV-MDM2, pCMV-HBXIP plasmids, and MDM2 siRNA. I, effect of MDM2 siRNA on p53 was examined by Western blot analysis in MCF-7 cells when HBXIP was overexpressed. Each experiment was repeated at least three times.
P300 Is Involved in HBXIP-modulated Tumorigenesis, and HBXIP Enhances the Growth of Breast Cancer through Modulating the MDM2/P53 Circuit
Next, we examined the effect of HBXIP and p300 on cell proliferation. MTT assays and colony formation assays revealed that the knockdown of HBXIP or/and p300 could significantly inhibit the cell proliferation of LM-MCF-7 cells (*, p < 0.05; **, p < 0.01, Student's t test; Fig. 7, A and B), suggesting that p300 was involved in the HBXIP-modulated tumorigenesis. Then we evaluated the effect of HBXIP on the MDM2/p53 pathway in breast cancer cells in vitro and in vivo. It has been reported that elevated MDM2 protein levels or poor wild type p53 expression levels can promote the progression of breast cancer (44). Interestingly, we observed by MTT assays that treatment with MDM2 siRNA or the overexpression of p53 could abolish the HBXIP-enhanced proliferation of MCF-7 cells in a time course-dependent manner (Fig. 7C), suggesting that HBXIP was involved in the MDM2/p53 pathway in promoting the proliferation of breast cancer cells. We further validated that MDM2 knockdown or overexpression of p53 significantly blocked the growth of MCF-7-HBXIP cells in mice (Fig. 7, D–F, **, p < 0.01, Student's t test), showing that HBXIP accelerated tumor growth by enhancing MDM2-mediated degradation of p53 in breast cancer cells. Meanwhile, we confirmed the expression levels of HBXIP, MDM2, and p53 by Western blot analysis in tumor tissues from mice (Fig. 7G). Therefore, we concluded that HBXIP enhances the growth of breast cancer through modulating the MDM2/p53 pathway in vitro and in vivo.
FIGURE 7.

HBXIP enhances the growth of breast cancer through modulating MDM2/p53 circuit. A and B, the effect of HBXIP siRNA or/and p300 siRNA on cell proliferation was measured by MTT and colony formation assays in LM-MCF-7 cells. C, the effect of MDM2 siRNA or overexpression of p53 on HBXIP-enhanced cell proliferation was measured by MTT in MCF-7 cells. D, growth curve of tumors in nude mice. E, average weight of tumors. F, tumor images. G, the relative expression levels of HBXIP, MDM2, and p53 in the tumor tissues from mice were detected by Western blot analysis. The data presented are from three independent experiments; error bars represent S.E. Student's t test; *, p < 0.05; **, p < 0.01.
Discussion
MDM2 has proved to be a key player in human cancer and thus an important target for therapeutic intervention (11, 40). p53 and MDM2 form a negative feedback loop in which p53 positively regulates MDM2 by activating MDM2 transcription (45, 46) and MDM2 negatively regulates p53 by promoting its ubiquitination and degradation (5, 7). The MDM2/p53 circuit plays a crucial role in breast cancer (47, 48). Our findings show that HBXIP, acting as a co-activator of transcription factors, is an important oncoprotein (18, 20, 24, 49, 50). In our current study, we were interested in whether the oncoprotein HBXIP is involved in the regulation of the MDM2/p53 feedback loop in breast cancer.
In this study, our results showed that the expression of HBXIP was significantly correlated with that of MDM2 in clinical breast cancer tissues. In addition, HBXIP was able to up-regulate MDM2 at the levels of mRNA, protein, and promoter in breast cancer cells. It has been reported that p53 as a transcription factor can up-regulate MDM2 at the level of transcription (29). Because HBXIP acts as a co-activator of transcription factors, we speculated that HBXIP might activate p53 to stimulate MDM2. As expected, our data validated that HBXIP increased MDM2 promoter activity through direct interaction with p53. The transcription of MDM2 is controlled by a variety of transcription factors, including AP-1, the Ets family, and Smad2/3, as well as p53 (11). Here we found that HBXIP displaying a co-activator of transcription factor p53 modulated the MDM2 transcription in breast cancer cells. It has been reported that p53 is bound and inactivated by large T-antigen in HEK293T cells (9). Indeed, we validated that p53 knockdown alone did not affect P2 promoter activity in HEK293T cells (data not shown). Therefore, we concluded that the reason for the increase in P2 promoter activity mediated by p53 in the cells is related to HBXIP overexpression.
Thus, we aimed to detect the mechanism by which HBXIP cooperated with p53 to activate the MDM2 promoter. The highly conserved co-activator histone acetyltransferase p300 has been reported to interact with a number of DNA-binding transcriptional activators to mediate the recruitment of basal transcription machinery to the promoter (51, 52). Previous studies report that the functions of p53 require the participation of p300 (53). However, it is elusive whether p300 is required for the transcription of MDM2 mediated by p53. Therefore, we were interested in whether HBXIP was involved in the p53-p300 complex. Our results revealed that HBXIP was able to enhance the MDM2 promoter activity coordinating with p300. It has been reported that the histone can be acetylated by p300, which contributes to chromatin disassembly during transcriptional activation (43, 54). Interestingly, our data further demonstrated that HBXIP could recruit p300 to p53, forming a transcriptional complex of HBXIP-p53-p300, to promote the acetylation level of histone H3 in breast cancer cells. This event leads to the enhancement of transcription of the MDM2 gene. Therefore, we determined whether the recruitment of p300 by HBXIP also modulates other p300-dependent H3 acetylation for multiple genes. We observed that HBXIP was able to up-regulate other p300 target genes such as MMD, NR4A1, EGR1, and TPM1 (55, 56) but not p300 (data not shown). The p53 protein as a transcription factor has been reported to enhance the transcription levels of MDM2 on the P2 promoter (11), but the regulatory mechanism remains unclear. In this study we report that HBXIP is required for the activation of MDM2 mediated by transcription factor p53 to recruit p300 in breast cancer.
Growing evidence has revealed that the oncoprotein MDM2, which is an E3 ubiquitin ligase that targets p53 for proteasome-dependent degradation, plays an essential yet non-redundant role in the inactivation of tumor suppressor p53 protein levels (5, 6). Because the above data have revealed that HBXIP has a remarkable function in the up-regulation of MDM2, we further validated the effect of HBXIP on p53 through up-regulating MDM2 in the cells. As expected, HBXIP was able to decrease the levels of p53 and its target genes at the level of protein in breast cancer cells rather than at the level of mRNA. Treatment with the proteasome inhibitor MG-132 and half-life assays further showed that HBXIP was involved in the degradation of p53 in breast cancer cells. Thus, we identified that HBXIP could increase the ubiquitination levels of p53 and that the effect of HBXIP on p53 was attenuated by MDM2 siRNA in the cells. Many studies have demonstrated that the levels of p53 are decreased by a variety of oncoproteins through up-regulating MDM2 in cancers (57, 58). Here we found that HBXIP enhanced the degradation of p53 through up-regulating MDM2. Functionally, we validated that p300 was involved in the HBXIP-modulated tumorigenesis. It has been reported that elevated MDM2 protein levels or poor wild type p53 expression levels are associated with the progression of breast cancer (44). Next, we provided evidence that HBXIP promoted the growth of breast cancer via MDM2 and MDM2-mediated p53 degradation in vivo and in vitro. Therapeutically, HBXIP may serve as a target in breast cancer.
The main mechanism for p53 stabilization in response to stress signals is a reduction in its interaction with MDM2, causing an increase in p53 levels (59). However, little is known about the mechanism of promoting p53 ubiquitination and proteasomal degradation in cancer. In this study, we presented a model showing that highly expressed HBXIP can promote a MDM2/p53 feedback loop and breast cancer growth through accelerating the up-regulation of MDM2 to keep p53 at a lower level in the development of breast cancer (shown in Fig. 8). Our finding illuminates the concept that HBXIP up-regulates MDM2 through activating transcription factor p53, in which acetyltransferase p300 is recruited to p53 by HBXIP, forming a transcriptional complex of HBXIP-p53-p300 in breast cancer cells. Highly expressed HBXIP accelerates the MDM2-mediated degradation of p53 in breast cancer cells through modulating the feedback loop of MDM2/p53, resulting in the fast growth of breast cancer cells. Thus, our finding provides new insights into the MDM2/p53 feedback loop, especially the mechanism by which p53 positively regulates MDM2 in breast cancer.
FIGURE 8.
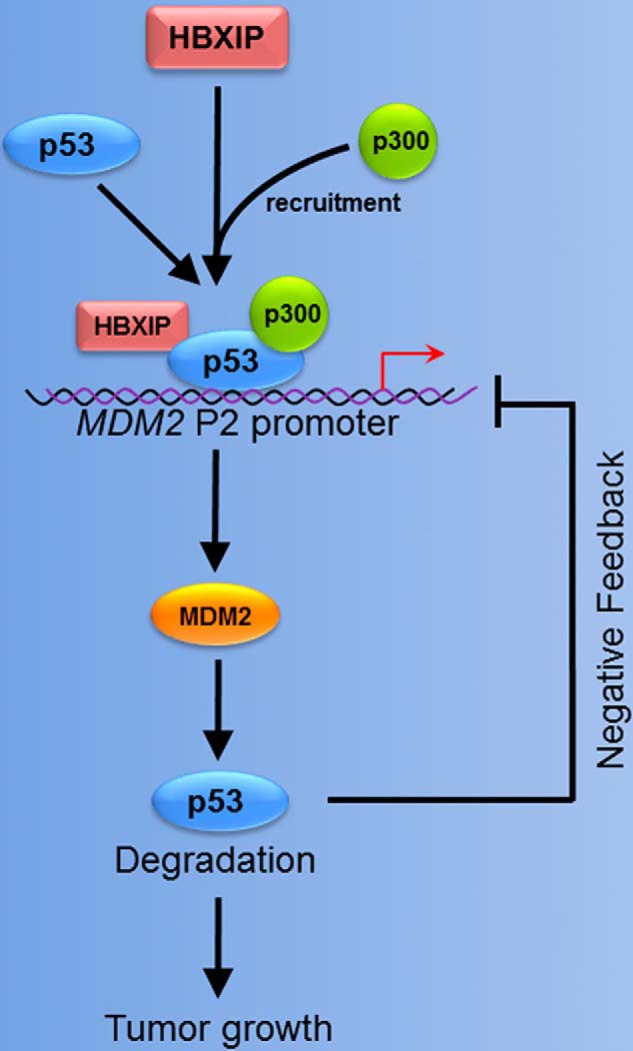
A model showing that HBXIP modulates the feedback loop of MDM2/p53 in breast cancer. HBXIP acting as a co-activator up-regulates MDM2 through binding to the promoter of MDM2 and activating transcription factor p53. HBXIP recruits acetyltransferase p300 to p53 forming a transcriptional complex of HBXIP-p53-p300. The HBXIP-enhanced MDM2 accelerates the degradation of p53 in breast cancer cells. Thus, HBXIP promotes the growth of breast cancer through modulation of the MDM2/p53 feedback loop.
Author Contributions
L. Y., X. Z., and H. L. designed the study and wrote the paper. Q. L. and Z. W. performed the experiments shown in Figs. 1, 6, and 7. Y. S. and X. C. performed the experiments shown in Figs. 2 and 3. Y. G. and Y. L. performed the experiments shown in Figs. 4 and 5. X. Z., H. L., and R. F. provided technical assistance and contributed to the preparation of the figures. L. Y. and X. Z. analyzed the experiments shown in all of the figures. All authors reviewed the results and approved the final version of the manuscript.
This work was supported by Grant 2015CB553905 from the National Basic Research Program of China (973 Program), Grants 81372186, 81272217, and 81272218 from the National Natural Science Foundation of China, and Grant 2014ZX0002002-005 from the Project for Prevention and Treatment of Key Infectious Diseases. The authors declare that they have no conflicts of interest with the contents of this article.
- HBXIP
- hepatitis B X-interacting protein
- co-IP
- co-immunoprecipitation
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.
References
- 1. Amaral J. D., Castro R. E., Solá S., Steer C. J., Rodrigues C. M. (2007) p53 is a key molecular target of ursodeoxycholic acid in regulating apoptosis. J. Biol. Chem. 282, 34250–34259 [DOI] [PubMed] [Google Scholar]
- 2. Volonte D., Zou H., Bartholomew J. N., Liu Z., Morel P. A., Galbiati F. (2015) Oxidative stress-induced inhibition of Sirt1 by caveolin-1 promotes p53-dependent premature senescence and stimulates the secretion of interleukin 6 (IL-6). J. Biol. Chem. 290, 4202–4214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Okita N., Ishikawa N., Mizunoe Y., Oku M., Nagai W., Suzuki Y., Matsushima S., Mikami K., Okado H., Sasaki T., Higami Y. (2014) Inhibitory effect of p53 on mitochondrial content and function during adipogenesis. Biochem. Biophys. Res. Commun. 446, 91–97 [DOI] [PubMed] [Google Scholar]
- 4. Onel K., Cordon-Cardo C. (2004) MDM2 and prognosis. Mol. Cancer Res. 2, 1–8 [PubMed] [Google Scholar]
- 5. Kubbutat M. H., Jones S. N., Vousden K. H. (1997) Regulation of p53 stability by Mdm2. Nature 387, 299–303 [DOI] [PubMed] [Google Scholar]
- 6. Sczaniecka M., Gladstone K., Pettersson S., McLaren L., Huart A. S., Wallace M. (2012) MDM2 protein-mediated ubiquitination of numb protein: identification of a second physiological substrate of MDM2 that employs a dual-site docking mechanism. J. Biol. Chem. 287, 14052–14068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Haupt Y., Maya R., Kazaz A., Oren M. (1997) Mdm2 promotes the rapid degradation of p53. Nature 387, 296–299 [DOI] [PubMed] [Google Scholar]
- 8. D'Arcy P., Ryan B. A., Brodin B. (2009) Reactivation of p53 function in synovial sarcoma cells by inhibition of p53-HDM2 interaction. Cancer Lett. 275, 285–292 [DOI] [PubMed] [Google Scholar]
- 9. Wang X., Wang J., Jiang X. (2011) MdmX protein is essential for Mdm2 protein-mediated p53 polyubiquitination. J. Biol. Chem. 286, 23725–23734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang Z., Zhang R. (2005) p53-independent activities of MDM2 and their relevance to cancer therapy. Curr. Cancer Drug Targets 5, 9–20 [DOI] [PubMed] [Google Scholar]
- 11. Manfredi J. J. (2010) The Mdm2-p53 relationship evolves: Mdm2 swings both ways as an oncogene and a tumor suppressor. Genes Dev. 24, 1580–1589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Miyajima K., Tamiya S., Oda Y., Adachi T., Konomoto T., Toyoshiba H., Masuda K., Tsuneyoshi M. (2001) Relative quantitation of p53 and MDM2 gene expression in leiomyosarcoma; real-time semi-quantitative reverse transcription-polymerase chain reaction. Cancer Lett. 164, 177–188 [DOI] [PubMed] [Google Scholar]
- 13. Barak Y., Gottlieb E., Juven-Gershon T., Oren M. (1994) Regulation of mdm2 expression by p53: alternative promoters produce transcripts with nonidentical translation potential. Genes Dev. 8, 1739–1749 [DOI] [PubMed] [Google Scholar]
- 14. Phelps M., Darley M., Primrose J. N., Blaydes J. P. (2003) p53-independent activation of the hdm2-P2 promoter through multiple transcription factor response elements results in elevated hdm2 expression in estrogen receptor alpha-positive breast cancer cells. Cancer Res. 63, 2616–2623 [PubMed] [Google Scholar]
- 15. Melegari M., Scaglioni P. P., Wands J. R. (1998) Cloning and characterization of a novel hepatitis B virus x binding protein that inhibits viral replication. J. Virol. 72, 1737–1743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fujii R., Zhu C., Wen Y., Marusawa H., Bailly-Maitre B., Matsuzawa S., Zhang H., Kim Y., Bennett C. F., Jiang W., Reed J. C. (2006) HBXIP, cellular target of hepatitis B virus oncoprotein, is a regulator of centrosome dynamics and cytokinesis. Cancer Res. 66, 9099–9107 [DOI] [PubMed] [Google Scholar]
- 17. Wen Y., Golubkov V. S., Strongin A. Y., Jiang W., Reed J. C. (2008) Interaction of hepatitis B viral oncoprotein with cellular target HBXIP dysregulates centrosome dynamics and mitotic spindle formation. J. Biol. Chem. 283, 2793–2803 [DOI] [PubMed] [Google Scholar]
- 18. Liu S., Li L., Zhang Y., Zhang Y., Zhao Y., You X., Lin Z., Zhang X., Ye L. (2012) The oncoprotein HBXIP uses two pathways to up-regulate S100A4 in promotion of growth and migration of breast cancer cells. J. Biol. Chem. 287, 30228–30239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bar-Peled L., Schweitzer L. D., Zoncu R., Sabatini D. M. (2012) Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell 150, 1196–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang Y., Zhao Y., Li H., Li Y., Cai X., Shen Y., Shi H., Li L., Liu Q., Zhang X., Ye L. (2013) The nuclear import of oncoprotein hepatitis B X-interacting protein depends on interacting with c-Fos and phosphorylation of both proteins in breast cancer cells. J. Biol. Chem. 288, 18961–18974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xu F., You X., Liu F., Shen X., Yao Y., Ye L., Zhang X. (2013) The oncoprotein HBXIP up-regulates Skp2 via activating transcription factor E2F1 to promote proliferation of breast cancer cells. Cancer Lett. 333, 124–132 [DOI] [PubMed] [Google Scholar]
- 22. Liu Q., Bai X., Li H., Zhang Y., Zhao Y., Zhang X., Ye L. (2013) The oncoprotein HBXIP upregulates Lin28B via activating TF II D to promote proliferation of breast cancer cells. Int. J. Cancer 133, 1310–1322 [DOI] [PubMed] [Google Scholar]
- 23. Hu N., Zhang J., Cui W., Kong G., Zhang S., Yue L., Bai X., Zhang Z., Zhang W., Zhang X., Ye L. (2011) miR-520b regulates migration of breast cancer cells by targeting hepatitis B X-interacting protein and interleukin-8. J. Biol. Chem. 286, 13714–13722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu F., You X., Wang Y., Liu Q., Liu Y., Zhang S., Chen L., Zhang X., Ye L. (2014) The oncoprotein HBXIP enhances angiogenesis and growth of breast cancer through modulating FGF8 and VEGF. Carcinogenesis 35, 1144–1153 [DOI] [PubMed] [Google Scholar]
- 25. Xu F., Zhu X., Han T., You X., Liu F., Ye L., Zhang X., Wang X., Yao Y. (2014) The oncoprotein hepatitis B X-interacting protein promotes the migration of ovarian cancer cells through the upregulation of S-phase kinase-associated protein 2 by Sp1. Int. J. Oncol. 45, 255–263 [DOI] [PubMed] [Google Scholar]
- 26. Phan R. T., Dalla-Favera R. (2004) The BCL6 proto-oncogene suppresses p53 expression in germinal-centre B cells. Nature 432, 635–639 [DOI] [PubMed] [Google Scholar]
- 27. Li S., Lao J., Chen B. P., Li Y. S., Zhao Y., Chu J., Chen K. D., Tsou T. C., Peck K., Chien S. (2003) Genomic analysis of smooth muscle cells in 3-dimensional collagen matrix. FASEB J. 17, 97–99 [DOI] [PubMed] [Google Scholar]
- 28. Villunger A., Michalak E. M., Coultas L., Müllauer F., Böck G., Ausserlechner M. J., Adams J. M., Strasser A. (2003) p53- and drug-induced apoptotic responses mediated by BH3-only proteins Puma and Noxa. Science 302, 1036–1038 [DOI] [PubMed] [Google Scholar]
- 29. Juven T., Barak Y., Zauberman A., George D. L., Oren M. (1993) Wild type p53 can mediate sequence-specific transactivation of an internal promoter within the mdm2 gene. Oncogene 8, 3411–3416 [PubMed] [Google Scholar]
- 30. Chang C. J., Freeman D. J., Wu H. (2004) PTEN regulates Mdm2 expression through the P1 promoter. J. Biol. Chem. 279, 29841–29848 [DOI] [PubMed] [Google Scholar]
- 31. Chen H., Ma X., Li Z., Shi Q., Zheng W., Liu Y., Wang P. (2012) Functionalization of single-walled carbon nanotubes enables efficient intracellular delivery of siRNA targeting MDM2 to inhibit breast cancer cells growth. Biomed. Pharmacother. 66, 334–338 [DOI] [PubMed] [Google Scholar]
- 32. Li S. D., Chono S., Huang L. (2008) Efficient oncogene silencing and metastasis inhibition via systemic delivery of siRNA. Mol. Ther. 16, 942–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shi X., Kachirskaia I., Yamaguchi H., West L. E., Wen H., Wang E. W., Dutta S., Appella E., Gozani O. (2007) Modulation of p53 function by SET8-mediated methylation at lysine 382. Mol. Cell 27, 636–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Debes J. D., Schmidt L. J., Huang H., Tindall D. J. (2002) p300 mediates androgen-independent transactivation of the androgen receptor by interleukin 6. Cancer Res. 62, 5632–5636 [PubMed] [Google Scholar]
- 35. Marusawa H., Matsuzawa S., Welsh K., Zou H., Armstrong R., Tamm I., Reed J. C. (2003) HBXIP functions as a cofactor of survivin in apoptosis suppression. EMBO J. 22, 2729–2740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vikis H. G., Guan K. L. (2004) Glutathione S-transferase fusion-based assays for studying protein-protein interactions. Methods Mol. Biol. 261, 175–186 [DOI] [PubMed] [Google Scholar]
- 37. Shan C., Xu F., Zhang S., You J., You X., Qiu L., Zheng J., Ye L., Zhang X. (2010) Hepatitis B virus X protein promotes liver cell proliferation via a positive cascade loop involving arachidonic acid metabolism and p-ERK1/2. Cell Res. 20, 563–575 [DOI] [PubMed] [Google Scholar]
- 38. González L., Agulló-Ortuño M. T., García-Martínez J. M., Calcabrini A., Gamallo C., Palacios J., Aranda A., Martín-Pérez J. (2006) Role of c-Src in human MCF7 breast cancer cell tumorigenesis. J. Biol. Chem. 281, 20851–20864 [DOI] [PubMed] [Google Scholar]
- 39. Wang F. Z., Sha L., Ye L. H., Zhang X. D. (2008) Promotion of cell proliferation by HBXIP via upregulation of human telomerase reverse transcriptase in human mesenchymal stem cells. Acta Pharmacol. Sin. 29, 83–89 [DOI] [PubMed] [Google Scholar]
- 40. Turbin D. A., Cheang M. C., Bajdik C. D., Gelmon K. A., Yorida E., De Luca A., Nielsen T. O., Huntsman D. G., Gilks C. B. (2006) MDM2 protein expression is a negative prognostic marker in breast carcinoma. Mod. Pathol. 19, 69–74 [DOI] [PubMed] [Google Scholar]
- 41. Zauberman A., Flusberg D., Haupt Y., Barak Y., Oren M. (1995) A functional p53-responsive intronic promoter is contained within the human mdm2 gene. Nucleic Acids Res. 23, 2584–2592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Miller Jenkins L. M., Feng H., Durell S. R., Tagad H. D., Mazur S. J., Tropea J. E., Bai Y., Appella E. (2015) Characterization of the p300 Taz2-p53 TAD2 complex and comparison with the p300 Taz2-p53 TAD1 complex. Biochemistry 54, 2001–2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Das C., Lucia M. S., Hansen K. C., Tyler J. K. (2009) CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature 459, 113–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Maglic D., Zhu S., Fry E. A., Taneja P., Kai F., Kendig R. D., Sugiyama T., Miller L. D., Willingham M. C., Inoue K. (2013) Prognostic value of the hDMP1-ARF-Hdm2-p53 pathway in breast cancer. Oncogene 32, 4120–4129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Barak Y., Juven T., Haffner R., Oren M. (1993) mdm2 expression is induced by wild type p53 activity. EMBO J. 12, 461–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wu X., Bayle J. H., Olson D., Levine A. J. (1993) The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 7, 1126–1132 [DOI] [PubMed] [Google Scholar]
- 47. Lahav G. (2008) Oscillations by the p53-Mdm2 feedback loop. Adv. Exp. Med. Biol. 641, 28–38 [DOI] [PubMed] [Google Scholar]
- 48. Ren Z. J., Nong X. Y., Lv Y. R., Sun H. H., An P. P., Wang F., Li X., Liu M., Tang H. (2014) Mir-509–5p joins the Mdm2/p53 feedback loop and regulates cancer cell growth. Cell Death Dis. 5, e1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li Y., Zhang Z., Zhou X., Li L., Liu Q., Wang Z., Bai X., Zhao Y., Shi H., Zhang X., Ye L. (2014) The oncoprotein HBXIP enhances migration of breast cancer cells through increasing filopodia formation involving MEKK2/ERK1/2/Capn4 signaling. Cancer Lett. 355, 288–296 [DOI] [PubMed] [Google Scholar]
- 50. Zhang Y., Zhao Y., Li L., Shen Y., Cai X., Zhang X., Ye L. (2013) The oncoprotein HBXIP upregulates PDGFB via activating transcription factor Sp1 to promote the proliferation of breast cancer cells. Biochem. Biophys. Res. Commun. 434, 305–310 [DOI] [PubMed] [Google Scholar]
- 51. Wang F., Marshall C. B., Ikura M. (2013) Transcriptional/epigenetic regulator CBP/p300 in tumorigenesis: structural and functional versatility in target recognition. Cell Mol. Life Sci. 70, 3989–4008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Garbati M. R., Alço G., Gilmore T. D. (2010) Histone acetyltransferase p300 is a coactivator for transcription factor REL and is C-terminally truncated in the human diffuse large B-cell lymphoma cell line RC-K8. Cancer Lett. 291, 237–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sebti S., Prébois C., Pérez-Gracia E., Bauvy C., Desmots F., Pirot N., Gongora C., Bach A. S., Hubberstey A. V., Palissot V., Berchem G., Codogno P., Linares L. K., Liaudet-Coopman E., Pattingre S. (2014) BAT3 modulates p300-dependent acetylation of p53 and autophagy-related protein 7 (ATG7) during autophagy. Proc. Natl. Acad. Sci. U.S.A. 111, 4115–4120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ogiwara H., Ui A., Otsuka A., Satoh H., Yokomi I., Nakajima S., Yasui A., Yokota J., Kohno T. (2011) Histone acetylation by CBP and p300 at double-strand break sites facilitates SWI/SNF chromatin remodeling and the recruitment of non-homologous end joining factors. Oncogene 30, 2135–2146 [DOI] [PubMed] [Google Scholar]
- 55. Ramos Y. F., Hestand M. S., Verlaan M., Krabbendam E., Ariyurek Y., van Galen M., van Dam H., van Ommen G. J., den Dunnen J. T., Zantema A., 't Hoen P. A. (2010) Genome-wide assessment of differential roles for p300 and CBP in transcription regulation. Nucleic Acids Res. 38, 5396–5408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Stossi F., Madak-Erdogan Z., Katzenellenbogen B. S. (2009) Estrogen receptor alpha represses transcription of early target genes via p300 and CtBP1. Mol. Cell. Biol. 29, 1749–1759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yu H., Yue X., Zhao Y., Li X., Wu L., Zhang C., Liu Z., Lin K., Xu-Monette Z. Y., Young K. H., Liu J., Shen Z., Feng Z., Hu W. (2014) LIF negatively regulates tumour-suppressor p53 through Stat3/ID1/MDM2 in colorectal cancers. Nat. Commun. 5, 5218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhang X., Zhang Z., Cheng J., Li M., Wang W., Xu W., Wang H., Zhang R. (2012) Transcription factor NFAT1 activates the mdm2 oncogene independent of p53. J. Biol. Chem. 287, 30468–30476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Michael D., Oren M. (2003) The p53-Mdm2 module and the ubiquitin system. Semin. Cancer Biol. 13, 49–58 [DOI] [PubMed] [Google Scholar]