

Background: Different snake venom three-finger toxins interact with various receptors, channels, and membranes.
Results: Here, we demonstrate that GABAA receptors are inhibited by α-cobratoxin, other long chain α-neurotoxins, nonconventional toxin from Naja kaouthia, and α-conotoxin ImI.
Conclusion: Some toxin blockers of nicotinic acetylcholine receptors also inhibit GABAA receptors.
Significance: Three-finger toxins offer new scaffolds for the design of GABAA receptor effectors.
Keywords: Cys-loop receptor, electrophysiology, fluorescence, molecular dynamics, molecular modeling, neurotoxin, snake venom, GABAA receptor, three finger toxin
Abstract
Ionotropic receptors of γ-aminobutyric acid (GABAAR) regulate neuronal inhibition and are targeted by benzodiazepines and general anesthetics. We show that a fluorescent derivative of α-cobratoxin (α-Ctx), belonging to the family of three-finger toxins from snake venoms, specifically stained the α1β3γ2 receptor; and at 10 μm α-Ctx completely blocked GABA-induced currents in this receptor expressed in Xenopus oocytes (IC50 = 236 nm) and less potently inhibited α1β2γ2 ≈ α2β2γ2 > α5β2γ2 > α2β3γ2 and α1β3δ GABAARs. The α1β3γ2 receptor was also inhibited by some other three-finger toxins, long α-neurotoxin Ls III and nonconventional toxin WTX. α-Conotoxin ImI displayed inhibitory activity as well. Electrophysiology experiments showed mixed competitive and noncompetitive α-Ctx action. Fluorescent α-Ctx, however, could be displaced by muscimol indicating that most of the α-Ctx-binding sites overlap with the orthosteric sites at the β/α subunit interface. Modeling and molecular dynamic studies indicated that α-Ctx or α-bungarotoxin seem to interact with GABAAR in a way similar to their interaction with the acetylcholine-binding protein or the ligand-binding domain of nicotinic receptors. This was supported by mutagenesis studies and experiments with α-conotoxin ImI and a chimeric Naja oxiana α-neurotoxin indicating that the major role in α-Ctx binding to GABAAR is played by the tip of its central loop II accommodating under loop C of the receptors.
Introduction
Type A γ-aminobutyric acid receptor (GABAAR)4 is abundantly expressed across the nervous system. Most of the GABAARs in the brain are constructed from α1–6 and β1–3 subunits co-assembled with γ1–3 or δ/ϵ/π/θ subunits. Interestingly, some β subunits, usually found in heteropentameric GABAARs could also form constitutively active homopentameric ligand-gated ion channels. Channels with such properties were found in cultured neurons (1), and in fact the β3-homopentamer at the moment is the only GABAAR with known three-dimensional structure (2).
GABAARs are targeted by diverse toxins of low molecular weight, such as the channel blocker picrotoxin (3), a polyacetylenic compound oenanthotoxin (4), the GABA site antagonist bicuculline (5), and by GABA site agonists such as ibotenic acid and muscimol. Benzodiazepines are positive allosteric modulators of GABAAR and were the world's most prescribed drugs in the 1980s (6). It is almost impossible to introduce fluorescent labels into such molecules, and therefore, they cannot be used in live cell imaging studies. In contrast, peptide toxins can easily be tagged with fluorescent or radioactive labels. Many of them, for example α-conotoxins from Conus snails and three-finger toxins (TFTs) from snake venoms, target nicotinic acetylcholine receptors (nAChRs) and show high levels of selectivity between the different nAChR types (7). For example, α-bungarotoxin (α-Bgt) from multibanded krait venom binds with nanomolar affinity to the muscle-type and α7 receptors but not to the heteropentameric neuronal nAChRs (8, 9). Such peptide toxins also could be quite helpful in structural studies. The x-ray crystal structure of the extracellular domain of α1 (10) and α9 (11) nAChR subunits were obtained in a complex with α-Bgt.
TFTs form an abundant group of proteins found in Colubridae, Elapidae, and Psammophiinae snake venoms. Members of this group possess no enzymatic activity and bind to receptors from different groups as follow: nAChRs (short and long chain α-neurotoxins), muscarinic acetylcholine receptors (African mamba-derived muscarinic toxins), L-type calcium channels (calciseptine from black mamba venom); and ASIC1, acid-sensitive ion channel (mambalgines). There are also three-finger cytotoxins (cardiotoxins) that interact with the cell plasma membrane (12).
Typical members of the long-chain group of TFTs, such as α-Bgt from Bungarus multicinctus as well as α-cobratoxin (α-Ctx) from Naja kaouthia cobra, neurotoxin NT I from Naja oxiana, and LsIII from marine krait Laticauda semifasciata, have an additional disulfide bond in loop II (Fig. 1, bottom left). This disulfide has been identified as a crucial feature for binding at the neuronal nAChR intersubunit sites (13, 14). Short neurotoxins, such as Naja oxiana neurotoxin II (NT II), acting exclusively on muscle nAChRs, do not have this additional disulfide (Fig. 1, bottom center). Another group, closely related to α-neurotoxins, is the group of so-called nonconventional or weak neurotoxins. In contrast to the previous two groups, weak toxins have an additional disulfide bond in the first loop (Fig. 1, bottom right), and show both weaker affinity toward nAChRs and some cross-reactions with other receptors (15). The toxins listed above block nAChRs with affinities that range from nanomoles to tens of micromoles/liter, and α-Bgt is the most active (Table 1). Interestingly, evidence for α-Ctx inhibition of T-type calcium channels through muscarinic receptors was recently provided (16), which may indicate that α-Ctx also has biological targets other than nAChRs.
FIGURE 1.

Structural diversity of three-finger neurotoxins. Top set represents sequence alignments. All TFTs have three loops stabilized by four disulfides, but some neurotoxins have one additional disulfide bond. Long (names higlighted green) and weak or nonconventional (names highlighted pink) neurotoxins have additional disulfide bonds in the second and the first loops, respectively, although short neurotoxins (name highlighted yellow) do not have additional disulfides. Bottom set represents typical spatial structures of three toxin groups as follows: long neurotoxin α-Ctx (PDB code 1CTX), short neurotoxin NT II (PDB code 1NOR), and nonconventional or weak toxin, here represented by the published structure of candoxin (PDB code 1JGK) that shares homology with WTX (N. kaouthia nonconventional toxin) and OWT (N. oxiana nonconventional toxin). Sequence alignments were produced via Clustal W algorithm; signal peptide sequences were removed manually, and top line shows numbering of α-Ctx residues. Backbones are shown in gray; β-strands are shown as arrows; cysteine residues, forming disulfide bonds are shown as yellow sticks on structures and highlighted orange in sequences, loop II arginine and valine residues in sequences are shown in blue and green, respectively.
TABLE 1.
Affinities of different three-finger toxins toward nAChRs and acetylcholine-binding proteins
α7-nAChR is the human neuronal homopentameric nAChR, Tca-nAChR is the Torpedo californica muscle-type nAChR; Ac- and Ls-AChBP are L. stagnalis and Aplysia californica acetylcholine-binding proteins, respectively. ND means not determined.
Fluorescent α-Bgt is widely used in applications concerning nAChR location and expression (17–19). Moreover, the “bungarotoxin-binding site” comprising a fragment of the α1 nAChR could be incorporated into the extracellular loops of different receptors allowing imaging of the receptors on the surface of living cells with the fluorescently labeled α-Bgt, which in particular also has been done for the GABAAR (20, 21). Using fluorophore-conjugated α-Bgt, about a decade ago the first evidence of α-Bgt binding to GABAAR was obtained (22). However, it was suggested to be limited to the peculiar β3/β3-binding sites, whose existence in vivo in the wild-type organisms has not been unambiguously demonstrated. This work aroused our interest in searching for novel GABAAR ligands among snake venom components structurally similar to α-Bgt, namely among various TFTs, as well as among α-conotoxins, another class of nAChR inhibitors.
Here, we report specific labeling of the α1β3γ2 subtype of GABAAR with a fluorescent derivative of α-Ctx, as well as inhibition of GABA-induced currents in a series of GABAARs by well known TFTs belonging to different structural groups and exhibiting anticholinergic properties; interaction of α-conotoxin ImI with α1β3γ2 GABAAR was found as well.
Experimental Procedures
Three-finger Toxin Preparations
Ls III was from Latoxan (Valence, France). WTX, α-Ctx, and NT II were obtained as described previously (23–25) and OWT as described previously (26). NT I was obtained as described previously (27) with additional purification by reversed phase HPLC. α-Bgt was purified from B. multicinctus venom by combination of gel filtration, ion-exchange, and reverse-phase HPLC, and α-Bgt Val-31 analogue was used in this work. The structures of toxins obtained were confirmed by mass spectrometry. Chimeric NT II/I toxin with the tip of the second loop from NT I grafted to the NT II structure was obtained as described previously (14).
Peptide Chemical Synthesis
Peptide WCDAFCSIRGKR from the α-Ctx loop II and α-conotoxin ImI was synthesized as described previously (28).
Electrophysiology
Oocytes were obtained from healthy mature (>10 cm) Xenopus frogs. After manual separation, oocytes were injected with 2–3 ng of laboratory PCI vector containing DNA sequences encoding mouse GABAAR subunits. Oocytes were incubated at 18 °C in ND96 solution (5 mm HEPES/NaOH, pH 7.6, 96 mm NaCl, 2 mm KCl, 1.8 mm CaCl2, 2 mm MgCl2) supplemented with ampicillin and kanamycin. Recordings were performed 36–72 h after injections using turbo TEC-03X setup (npi electronic GmbH, Germany). During a typical experiment, agonist (γ-aminobutyric acid) was applied every 5 min. To evaluate the activity of the tested TFTs, they were pre-applied before the agonist and then co-applied with it. Incorporation of the γ2 subunit was confirmed by application of diazepam. Incorporation of δ subunit was confirmed using DS2 compound as described previously (29). Data are presented as mean ± S.E. and/or 95% confidence interval in parentheses.
Fluorescent α-Ctx
Coupling of Alexa Fluor 546 NHS reagent (Invitrogen) to α-Ctx was conducted according to the manufacturer's protocol. This fluorophore has been chosen due to similar spectral properties to commercially available Alexa Fluor 555 α-Bgt (Invitrogen) also used in this work.
Live Cell Imaging with Fluorescent Ligand
Mouse neuroblastoma Neuro2a cells grown in 96-well plates were transfected with laboratory PCI plasmids encoding GABAAR subunits and GFP-encoding plasmid (Evrogen, Russia) following the Lipofectamine transfection protocol (Invitrogen). Transfected cells were grown in DMEM (Paneco, Russia) supplemented with 10% fetal bovine serum (PAA Laboratories, Austria) at 37 °C in a CO2 incubator for 48 h. The growth medium was removed; cells were rinsed with a buffer (20 mm HEPES, 140 mm NaCl, 2.8 mm KCl, 2 mm CaCl2, 1 mm MgCl2, 10 mm glucose, pH 7.4) and incubated with 50 nm Alexa Fluor 555-labeled α-Bgt (Invitrogen) or Alexa Fluor 546-labeled α-Ctx for 20 min at room temperature. In control experiments, 50-fold molar excess of nonlabeled α-Ctx was added to the incubation medium. The cells were then washed twice with the buffer and were visualized by an epifluorescent microscope (Olympus, Japan) using the appropriate filter combination. In competition experiments, 1–4000 μm muscimol or 400 μm diazepam were added to the incubation medium (140 mm NaCl, 2 mm CaCl2, 2.8 mm KCl, 4 mm MgCl2, 20 mm HEPES, 10 mm glucose, 1% BSA, pH 7.4) as described previously (19). Data were analyzed using the ImageJ program with the standard set of plugins (30). Fluorescence was quantified as follows: field of view was photographed in two channels as follows: one to detect fluorescent ligand and another to detect GFP. Background of images was subtracted, and maximum points were found on the GFP image and selected. Selection was enlarged to fit cells on the image and transferred to an image containing fluorescent ligand detection (same field of view); then the mean fluorescence was measured and normalized to the difference between total fluorescent binding and unstained cells fluorescence. In each well, 2–4 random fields of view were photographed, and each individual experiment included several wells; at least three independent experiments were performed.
Molecular Modeling
The extracellular domains of α and β subunits forming the β+α− interface of GABAAR were modeled using the atomic coordinates of the β3 extracellular domain taken from the recently published β3-homopentameric x-ray structure (2) and the extracellular domain of the α1 subunit, which was constructed via the Swiss model homology modeling service using the crystal structure of the β3 subunit from homo-oligomeric receptor as a template (31). GABAAR subunits share some homology with the Lymnaea stagnalis acetylcholine-binding protein (AChBP). Assuming that α-Ctx binds to GABAARs in a way similar to the crystal structure of the α-Ctx-bound AChBP, we constructed models of α-Ctx complexes with GABAAR extracellular domains by aligning the β3 subunit extracellular domain with the principal AChBP subunit and α1 subunit extracellular domain with the complementary AChBP subunit from its x-ray structure with α-Ctx (PDB 1YI5) using UCSF Chimera (32). Resulting structures were energy-minimized with GROMACS 5.0 (33) and then treated with molecular dynamics procedure using OpenMM libraries (34) with the aid of AMBER force field parameters (35) and GBSA implicit solvent model to simulate 100 ns of complex behavior. Resulting coordinates were analyzed with VMD (36) and UCSF Chimera software.
Single-point Mutagenesis
The S67K and K67S mutations (in the α1 and α2 subunit, respectively) were obtained by PCR amplification of α1 and α2 subunits in the vector with the following primers: forward primer 5-gtgtttttccgccaaaagtggaaggatgaaagattaaaattc-3 and reverse primer 5-catccttccagctttggcggaaaaacacatctattg-3 (for S67K); forward primer 5-gctcgtctttccagctttgtcgaaagaaaacatctattg-3 and reverse primer 5-ctttcgacaaagctggaaagacgagcggttaaaatttaaag-3 for (K67S) by using Phusion High-Fidelity DNA polymerase. Amplification product was analyzed after 25 cycles of the following thermal steps: 2 min of initial denaturation at 98 °C; 20 s of denaturation at 98 °C; 1 min of annealing at 65–70 °C; 3 min of extension at 72 °C, and 10 min of final extension at 72 °C. The product obtained was incubated with DpnI enzyme to restrict the methylated DNA and then transformed into XL1-Blue supercompetent cells. The mutations were confirmed by DNA sequencing.
Results
Long-chain α-Ctx and Naja oxiana α-Neurotoxin NT I Compete with Fluorescent α-Bgt for GABAAR-binding Site
First of all, we decided to check whether α-Ctx could displace a fluorescent α-Bgt derivative from α1β3 GABAAR. We reproduced (Fig. 2A) fluorescent Alexa Fluor 555 α-Bgt binding to (presumably) the β3/β3-interface of α1β3 receptors first reported by McCann et al. (22). α-Ctx at 5 μm concentration competed with fluorescent α-Bgt and displaced it almost completely from the receptor. Interestingly, Naja oxiana NT I (see Fig. 1 for sequence alignments) was also an effective competitor; it diminished Alexa Fluor 555 α-Bgt binding by 91 ± 2% (experiments not shown).
FIGURE 2.

Alexa Fluor 555 α-Bgt and Alexa Fluor 546 α-Ctx bind to the surface of cells expressing α1β3 or α1β3γ2 receptors and binding is differentially inhibited by α-Bgt and α-Ctx. Alexa Fluor 555 α-Bgt (A and C) and Alexa Fluor 546 α-Ctx (B and D) binding to Neuro2a cells transfected with α1β3 (A and B) and α1β3γ2 (C and D) GABAAR subunits (scale bar, 50 μm). Bars represent remaining fluorescence in the presence of α-Bgt or α-Ctx of α1β3 cells stained by Alexa Fluor 555 α-Bgt (E) or Alexa Fluor 546 α-Ctx (F). Note that Alexa Fluor 546 α-Ctx binding to α1β3-transfected cells was significantly (p < 0.05) better blocked by α-Ctx, whereas Alexa Fluor 555 α-Bgt binding was blocked with equal efficiency by both toxins. However, Alexa Fluor 555 α-Bgt (G) and Alexa Fluor 546 α-Ctx (H) binding to cells transfected with α1β3γ2 were blocked more effectively by α-Ctx (p < 0.05). Concentrations of fluorescent derivatives were 50 nm; the mixtures were incubated for 15 min, and then unbound ligand was washed out with buffer. Competition experiments using fluorescent derivatives and native α-Ctx and α-Bgt were performed by incubation of cells with unlabeled toxins at 5 μm along with addition of labeled analogues. The data were analyzed with ImageJ. Note that data are normalized to differences between mean stained and unstained cell fluorescence. Three independent experiments were performed for each data point. Data are presented as mean ± 95% confidence interval; differences were interpreted as significant (p < 0.05, asterisk) if intervals are not intersecting (if intervals intersect, difference was interpreted as nonsignificant, n.s.).
Fluorescently Labeled α-Ctx Can Be Used for Imaging of Live Cells Expressing GABAAR
To study α-Ctx competition with α-Bgt in detail, we tested the applicability of a α-Ctx fluorescent derivative in cytochemical experiments for imaging of living cells heterologously expressing α1β3 and α1β3γ2 GABAAR. α-Ctx bearing Alexa Fluor 546 fluorophore has been chosen due to similar spectral properties to commercially available Alexa Fluor 555 α-Bgt. It should be noted that we had no intention of registering simultaneously Alexa Fluor 555 α-Bgt and Alexa Fluor 546 α-Ctx binding. Both ligands (Alexa Fluor 555 α-Bgt and Alexa Fluor 546 α-Ctx) brightly stained cells expressing either α1β3 (Fig. 2, A and B) or α1β3γ2 subunit combinations (Fig. 2, C and D). The actual expression of the γ2-subunit in α1β3γ2 receptors was confirmed in patch clamp experiments with diazepam as discussed below (Fig. 4A). Binding of fluorescent toxins was then blocked by unlabeled toxins (α-Ctx or α-Bgt) to determine specificity of staining. Interestingly, only for α1β3-expressing cells stained by Alexa Fluor 555 α-Bgt (Fig. 2E), we detected no difference in efficiency of fluorescence block by 5 μm toxins (α-Bgt or α-Ctx). Fluorescence of α1β3-expressing cells stained by Alexa Fluor 546 α-Ctx, as well as α1β3γ2-expressing cells stained by either Alexa Fluor 555 α-Bgt or Alexa Fluor 546 α-Ctx, was more effectively blocked by 5 μm α-Ctx (Fig. 2, F–H).
FIGURE 4.

Inhibition of different GABAAR subtypes by α-Ctx and inhibition of Alexa Fluor 546 α-Ctx binding on α1β3γ2 by muscimol. A, inhibition of α1β3 and α1β3γ2 GABAARs by α-Ctx. The presence of γ subunit is confirmed by the effect of the benzodiazepine 1 μm diazepam (BZD) and differential sensitivity to 50 μm Zn2+ (see insets). Absence of a γ subunit influence on α-Ctx inhibition is consistent with the assumption that the α-Ctx-binding site is located at an interface between α and β subunits. B, this toxin manifests modest subtype selectivity; it inhibits α1β3γ2 receptor five times more potently than α2β3γ2. The potency of α-Ctx against diverse sets of GABAAR subunits was examined using the two-electrode voltage clamp method. To measure the effect of different α-Ctx concentration, α-Ctx was applied alone to the oocyte for 5 min before control GABA application and then was co-applied with GABA (10 μm, EC20–50). Current amplitudes obtained in the presence of α-Ctx were expressed as percentage of control amplitudes and plotted against logarithm of toxin molar concentration. C, GABA dose-response curve at α1β3γ2 receptors without α-Ctx (upper curve) and in the presence of 250 nm α-Ctx (lower curve). GABA EC50 is shifted to the right and the Hill slope is reduced, suggesting noncompetitive or some mixed competitive/noncompetitive type of inhibition. D, muscimol competes with Alexa Fluor 546 α-Ctx on α1β3γ2 GABAAR suggesting α-Ctx-binding sites overlap with orthosteric sites located at the β/α interface. Fluorescence data were normalized to the difference between mean stained cell fluorescence and mean cell fluorescence blocked by 10 μm α-Ctx (i.e. data represent specific α-Ctx binding). Data were fitted to a dose-response equation using the Origin 7.5 software.
Snake Venom Neurotoxins from Different Structural Groups Inhibit GABAAR
To test whether α-Ctx and other toxins could inhibit α1β3γ2 GABAAR, we performed two-electrode voltage clamp experiments. It was found that α-Ctx, as well as Ls III (long-chain α-neurotoxin from Laticauda semifasciata), and nonconventional toxin WTX from Naja kaouthia venom inhibit the GABAARs in a dose-dependent manner (Fig. 3). Given at 10 μm Ls III and WTX blocked GABA-evoked response by 83 ± 12 and 31 ± 8%, respectively. Long-chain NT I and α-Bgt at 10 μm inhibited α1β3γ2 receptors only by 17 ± 5 and 19 ± 3%, respectively. No inhibition was observed for short-chain neurotoxin NT II and nonconventional toxin OWT, both from N. oxiana venom, at concentrations of 10 μm. Interesting, α-Ctx (5 μm) also inhibited α1β3δ receptor (Fig. 3).
FIGURE 3.

Effects of different three-finger toxins on GABA-evoked currents. Electrophysiological recordings demonstrate that three-finger toxins can inhibit GABAAR. GABA was applied alone or co-applied with the indicated toxin to oocytes injected with cDNA encoding α1, β3, and γ2 subunits. First peak of each set represents control (contr.) with a 1-s application of 10 μm GABA alone, corresponding to EC20–50; the concentration of co-applied toxin is indicated above the recording. The traces marked as wash represent the response to GABA after 5 min of oocyte washing with the buffer solution. The lower right inset shows inhibition of α1β3δ receptor by α-Ctx at 5 μm.
To characterize more broadly the α-Ctx potency and selectivity, we tested it on GABAAR with different subunit combinations. First of all, we investigated the potency of α-Ctx for inhibition of GABA-induced currents at α1β3 and α1β3γ2 receptors. As expected, GABA-induced currents at α1β3 receptors were completely inhibited by 50 μm Zn2+ in contrast to those at α1β3γ2 receptors (Fig. 4A, insets). The incorporation of a γ2 subunit in α1β3γ2 receptors was further demonstrated by their sensitivity to 1 μm diazepam. Interestingly, α-Ctx was able to completely inhibit diazepam-stimulated GABA currents, again demonstrating that α-Ctx is able to also block the actions of α1β3γ2 receptors (Fig. 4A, insets). As shown in Fig. 4A, α-Ctx exhibited a comparable potency for inhibition of these two receptor subtypes, indicating that the γ2 subunit does not influence the potency of α-Ctx.
Then we investigated the effects of α-Ctx at α1β2γ2, α1β3γ2, α2β2γ2, α2β3γ2, and α5β2γ2 receptors (Fig. 4B). α-Ctx exhibited a differential potency for inhibition of GABA-induced currents at the individual GABAA receptor subtypes. IC50 values for each of them are shown in Table 2. The potency of α-Ctx depended on the types of α and β subunits present in the receptor, supporting the conclusion that the α-Ctx-binding site is located at an interface between α and β subunits. In contrast to the strong inhibition of α1β3γ2, long-chain Ls III (10 μm) inhibited α5β2γ2 and α2β2γ2 only by 13 ± 5 and 39 ± 9%, respectively (experiments not shown).
TABLE 2.
EC50 values for GABA and IC50 values for α-Ctx at different combinations of GABAAR subunits
| Subunit combination | EC50 ± S.E. (95% confidence interval) | IC50 ± S.E. (95% confidence interval) |
|---|---|---|
| μm | nm | |
| α1β2γ2 | 35 ± 1 (33, 36) | 469 ± 23 (427, 515) |
| α1β3γ2 | 29 ± 7 (19, 44) | 236 ± 7 (223, 250) |
| α2β2γ2 | 45 ± 1 (43, 47) | 485 ± 39 (417, 564) |
| α2β3γ2 | 12 ± 1 (11, 13) | 1099 ± 57 (1016, 1088) |
| α5β3γ2 | 20 ± 2 (17, 24) | 635 ± 91 (489, 825) |
Electrophysiology Measurements Indicate That α-Ctx Inhibits GABAAR in a Mixed Competitive/Noncompetitive Manner
To shed light on the α-Ctx mode of inhibition, we compared the GABA dose-response dependence (for α1β3γ2 subtype) in the presence of 250 nm α-Ctx, a concentration that is sufficient to inhibit receptor response to 10 μm GABA by about 50% as compared with control (Fig. 4C). GABA EC50 value was shifted from 29 ± 7 to 128 ± 25 μm in the presence of α-Ctx. After looking at the individual data points, however, it seems that α-Ctx caused a parallel shift of the GABA dose-response curve in at least two steps. Such behavior would be consistent with a competitive inhibition of GABA binding by α-Ctx, when binding of α-Ctx to one of the two orthosteric sites caused an allosteric reduction of the GABA affinity for the second orthosteric site.
At 1 mm GABA, however, the current induced in the presence of toxin is 40% smaller than in its absence (Fig. 4C). Although the data do not allow a firm conclusion because the GABA concentration-response curve in the presence of toxin is not yet saturated at this concentration, they are consistent with an additional noncompetitive inhibition of GABA currents by α-Ctx (see below).
Muscimol but Not Diazepam Competes with Alexa Fluor 546 α-Ctx
We investigated whether diazepam, a classical benzodiazepine that binds at the α/γ interface (37), or the agonist muscimol that binds at the β/α interface can compete with Alexa Fluor 546 α-Ctx for binding to Neuro2a cells expressing α1β3γ2 GABAAR. Indeed, 200 μm muscimol reduced fluorescence by about 80% (Fig. 4D), whereas diazepam had no effect at concentrations up to 400 μm (experiments not shown). These data support the conclusion that Alexa Fluor 546 α-Ctx to a large extent binds to the GABA-binding site of GABAA receptors. The remaining 20% of bound Alexa Fluor 546 α-Ctx cannot be displaced by muscimol, suggesting that this binding of α-Ctx occurs via a site different from the orthosteric binding site.
α-Cobratoxin Loop II Peptide (WCDAFCSIRGKR) and α-Conotoxin ImI Selective for α7 nAChR but Not the α-Conotoxins SIA and MII Selective for Muscle Type nAChR and Heteromeric Neuronal nAChRs, Respectively, Inhibit α1β3γ2 GABAAR
We hypothesized that other polypeptide nAChR ligands could also inhibit GABAAR and chose α-conotoxin ImI because it was reported previously that ImI shares some structural features of the α-Ctx loop II (28). ImI binds efficiently to α7 nAChR, i.e. to neuronal homopentameric nAChR, whereas α-Ctx in addition to α7 nAChR also binds to muscle type nAChR. We decided to test two other conotoxins: SIA that binds to muscle nAChR and MII that binds to neuronal non α7 nAChRs. α-Conotoxin ImI (see Fig. 5A for structure) inhibited the α1β3γ2 GABAAR at 100 μm by 45 ± 5% in terms of GABA-evoked peak current (Fig. 5B). α-Conotoxins SIA and MII failed to show inhibition of GABAAR at 100 μm (Fig. 5B, bottom). This experiment also proves specificity of inhibition, and because 100 μm is a rather high concentration, we had to make some control experiments with peptides chemically similar to ImI at such a high concentration.
FIGURE 5.
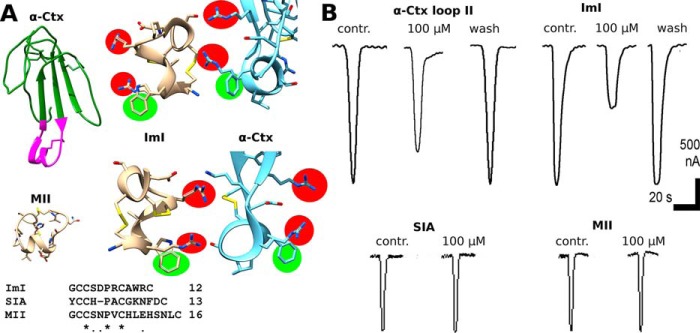
A, structural similarity of α-conotoxin ImI, selective for α7 nAChR, to the tip of the α-Ctx loop II (magenta). Note that two positively charged arginines (highlighted red) are exposed in a very similar manner with respect to aromatic residues (tryptophan or phenylalanine, highlighted green) in both ImI (shown wheat) and loop II of α-Ctx (shown blue). Two different views (1st and 2nd rows) are provided for convenience. Amino acid sequences of α-conotoxins SIA and MII differ greatly from that of ImI (bottom). B, chemically synthesized WCDAFCSIRGKR peptide that mimics tip of α-Ctx loop II and α-conotoxin ImI at 100 μm inhibits 10 μm GABA-evoked current (upper left and right traces, respectively). SIA specific for muscle type nAChR and MII specific for heteromeric neuronal nAChR have no effect on the currents at 100 μm.
We also performed experiments with the synthetic peptide WCDAFCSIRGKR comprising the tip of α-Ctx loop II. Synthetic fragment (see Fig. 5A for structure) inhibited the α1β3γ2 GABAAR at 100 μm by 28 ± 10% in terms of GABA-evoked peak current, respectively (Fig. 5B). The above results suggest that new peptide ligands of GABAARs could be found among toxins sharing common structural features with α-Ctx loop II.
α-Cobratoxin at Concentrations of Up to 40 μm Does Not Inhibit Homopentameric Glycine Receptor
Glycine receptor is another example of abundantly expressed Cl−-permeable Cys-loop receptors. It also shares very high sequence homology with GABAAR subunits. To check whether α-Ctx possesses some activity against this receptor, we tested its activity against the human homopentameric α1 glycine receptor. At concentrations up to 40 μm, α-Ctx failed to inhibit glycine-evoked currents (data not shown). Thus, α-Ctx blocks GABAARs but not the glycine receptor. However, it should be noticed that there are other subsets of glycine receptor (α2–3 and β) that were not tested.
Model for GABAAR·α-Ctx Complex
GABAAR spatial structure (2) has a very high degree of similarity to structures of AChBPs and extracellular nAChR domains for which the α-neurotoxin-binding sites were defined. Based on these data, we assumed that a complex of α-Ctx with the GABAAR might resemble published structures of α-neurotoxins bound to AChBP or to the extracellular nAChR domain. Under this assumption, we suggested that loop II of α-Ctx might find its place under the loop C of the GABAAR receptor subunit. To get a picture of such a hypothetical intersubunit binding site, we constructed homology models of extracellular domains of α1 GABAAR subunits using the recently published structure of GABAAR β3-homopentamer (2) as a template. Then a molecular model of the β3/α1 dimer with α-Ctx introduced under loop C of the β3 subunit was constructed by superimposition and alignment of the β3 extracellular domain structure with that of the AChBP protomer (PDB code 1YI5). The structure obtained was subjected to 100 ns of molecular dynamics. The last 40-ns complex was stable, and its backbone root mean square deviation did not exceed 0.1 nm. By the end of simulation, the complex retained the overall “TFT to Cys-loop” binding mode known from the x-ray structures (Fig. 6A) with the toxin loop II buried under loop C of the receptor. Interestingly, α-Ctx residues Arg-33 and Arg-36 form a stable network of salt bridges with both Asp-27 and β3 Glu-155 (Fig. 6B), which was present during all simulation times. The toxin C-terminal region and tips of loop I and loop II also formed contacts with both dimer subunits.
FIGURE 6.

Molecular modeling of α-Ctx binding to the GABAAR. A, side and top view of the molecular model of complex formed by α-Ctx with the orthosteric site of the GABAAR. Toxin (green) rests under loop C of the β3 (plum) subunit. GABAAR α1 subunit is colored coral. B, Arg-33 and Arg-36 from the α-Ctx loop II form a network of hydrogen bonds (shown as light blue sticks) with both α-Ctx Asp-27 and Glu-155 of the β3 subunit.
Electrophysiological Analysis of Chimeric NT II/I
To verify the model, we decided to test whether the loop II of the toxins plays a crucial role in functional inhibition of GABAAR. In previous studies, short-chain toxin NT II with grafted loop II from long neurotoxin I was used to study the role of loop II in α-neurotoxin interaction with neuronal nAChRs (14). We used the same chimera in electrophysiological experiments on GABAAR. Short-chain toxin NT II at 10 μm did not show any inhibition of the receptor but gained the inhibitory activity upon the grafting of loop II from long neurotoxin NT I (Fig. 7A). Chimeric NT II/I toxin and NT I generated a comparable inhibition of α1β3γ2 receptors (16 ± 3 and 19 ± 3%, respectively, Fig. 7B). Molecular dynamics simulations indicated that the model of NT II was unable to form a complex with the GABAAR orthosteric intersubunit site in the manner similar to α-Ctx (Fig. 7C). In contrast, the model of NT II/I formed a stable complex with loop II resting under the C-loop of β3 extracellular domain model (Fig. 7D).
FIGURE 7.

Delineation of the active site in TFTs interacting with GABAARs. A, loop II peptide fragment CDAWCGS of NT I was grafted to NT II structure, replacing the corresponding SDH sequence (see Fig. 1 for sequence alignment). B, normalized GABA-evoked currents (10 μm of GABA, α1β3γ2 GABAAR) in the presence of wild-type NT I, NT II, and chimeric NT II/I at 10 μm. Wild-type NT II does not inhibit the receptor, whereas both wild-type NT I and NT II/I chimeric toxin show current inhibition by 19 ± 3 and 16 ± 3%, respectively. Asterisks indicate significant inhibition (p < 0.01, n = 6, paired t test). C, last frame of a 100-ns run of molecular dynamics simulation of NT II complex with β3/α1 extracellular domains. NT II failed to reproduce mode of binding shown by α-Ctx model. D, molecular model of chimeric NT II/I toxin complex with β3/α1 extracellular domains. Note that the tip of the loop II (which was grafted to the short toxin NT II molecule from long NT I toxin) rests under the C loop of β3 subunit.
Single Point Mutations α1(S67K) and α2(K67S) Located at the Complementary Surface of the Respective α-Subunit Change Inhibition by α-Ctx
Because we observed modest α-Ctx selectivity toward α1β3γ2 (Table 2), we decided to test our model by introduction of α1→α2 and α2→α1 point mutations. Loop II of the toxin carries a significant positive charge, and we hypothesized that addition or deletion of a positive charge at position 67 of α1 and α2 subunits, located in close vicinity to the hypothetical toxin-binding site and occupied by a serine residue (Fig. 8A), might diminish affinity to α-Ctx in the α1(S67K) mutant and increase affinity toward α2(K67S) mutant. Indeed, we found that the α-Ctx IC50 value for α1(S67K)β3γ2 GABAAR (703 ± 39 nm) is closer to the α2-containing receptor (1099 ± 57 nm, Table 2) than the α1-containing receptor (236 ± 7 nm, Table 2), and vice versa the α-Ctx IC50 value for α2(K67S)β3γ2 GABAAR (442 ± 54 nm) is closer to the α1-containing receptor than the α2-containing receptor (Fig. 8B). At the same time GABA EC50 values were not affected by these mutations.
FIGURE 8.
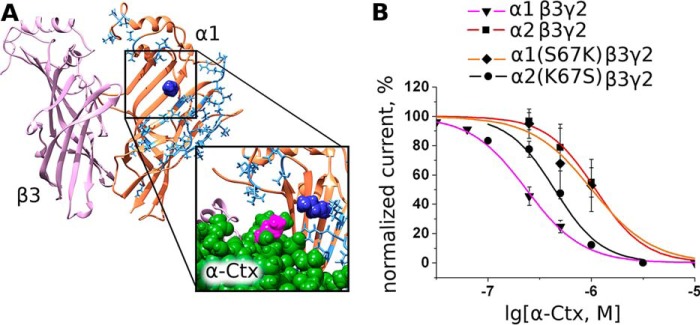
Influence of GABAAR single point mutation on α-Ctx binding. A, extracellular domains of subunits forming β3/α1 interface. Residues that are different in α1 and α2 subunits are shown blue (stick representation), whereas α1 structure conserved between these two subunits is colored orange. Note that one of the closest to agonist-binding site residues is Ser-67 (dark blue). Inset shows that, according to our molecular modeling, by being mutated to lysine (as in α2 subunit) this residue comes close to the lysine of the α-Ctx loop II (magenta, space-filling model). B, inhibition of mutant receptor α1(S67K)β3γ2 by α-Ctx is characterized by a higher IC50 = 703 nm (632, 781 nm) value than wild-type α1β3γ2. However, inhibition of mutant receptor α2(K67S)β3γ2 by α-Ctx is characterized by a lower IC50 = 442 nm (352, 554 nm) value than wild-type α2β3γ2 suggesting the importance of β/α interface. GABA was given at 10 μm, which is slightly less than EC50. Data points are expressed as percentage of control amplitudes (mean peak current ± S.E.).
Discussion
There has been considerable progress in understanding the structure and function of GABAARs (38). Notwithstanding, there are still significant gaps in our knowledge on peptide and proteins interacting with GABAARs. In general, polypeptide ligands manifest higher selectivity to particular receptor subtypes as compared with low molecular weight organics. Several polypeptides interacting with GABAAR have been reported earlier that include α-Bgt (22), diazepam-binding inhibitor protein, and its processing fragments such as the octadecaneuropeptide (39, 40). Given a plethora of different GABAAR subunits, it seems surprising that there is such a paucity of known proteins interacting with the receptor. For example, a great deal of polypeptide toxins is known for nAChR, closely related to GABAAR. Here, for a series of GABAAR subtypes, we report specific fluorescent labeling and inhibition of GABA-induced ion currents by several protein toxins from snake venoms.
As a starting point, we reproduced α-Bgt binding to the α1β3 GABAAR reported in Ref. 22 to obtain a verified test system of binding. As expected, long-chain α-neurotoxin α-Ctx, closely related to α-Bgt, showed competition with α-Bgt binding. We then investigated whether a fluorescently labeled α-Ctx, could stain cells expressing α1β3γ2 or α1β3 GABAAR. We found that Alexa Fluor 546 α-Ctx, as well as Alexa Fluor 555 α-Bgt, were able to stain both types of cells (Fig. 2, A–D). Whereas α-Ctx blocks Alexa Fluor 555 α-Bgt binding at α1β3-transfected cells (Fig. 2E) as effectively as α-Bgt, Alexa Fluor 555 α-Bgt or Alexa Fluor 546 α-Ctx staining of α1β3- or α1β3γ2-transfected cells was blocked more effectively by α-Ctx (Fig. 2, F–H). This observation suggests that GABAARs with different subunit combinations bear binding sites with slightly different properties.
To investigate how α-Ctx and other anticholinergic TFTs might influence GABAAR function, we performed electrophysiology studies of α1β2γ2, α1β3γ2, α2β2γ2, α2β3γ2, α5β2γ2, and α1β3δ GABAAR subunit sets expressed in Xenopus oocytes. It was found that α-Ctx blocks α1β3γ2 with a relatively high potency (IC50 = 236 nm, Fig. 4, A and B) and other receptor subtypes with lower potencies, namely in submicromolar to low micromolar range (Fig. 4B). It is worth noting that α-Ctx manifests some selectivity toward α1 as compared with the α2 subunit only in the presence of the β3 subunit, whereas in the presence of the β2 subunit it does not show noticeable selectivity to any α subunit investigated. This suggests that the α-Ctx-binding site might be located at the interface between the β and α subunit. Other tested TFTs were less active, and their potencies were not measured. However, it is clear that Ls III and WTX are active in the micromolar range, whereas NT II and OWT do not show any inhibition of α1β3γ2 receptor at 10 μm.
Electrophysiology measurements indicated that α-Ctx (for the α1β3γ2 subtype) shifted GABA dose-response curve to the right, at the same time changes in the slope and maximally achieved current were observed (Fig. 4C). Such a picture is typical of noncompetitive antagonists. However, we found that muscimol, acting as agonist of GABAAR, but not the allosteric modulator diazepam, displaced Alexa Fluor 546 α-Ctx from α1β3γ2-transfected cells (Fig. 4D) to the level of ∼20%. So a large part of α-Ctx action seems to be competitive and to be mediated via binding to the GABA-binding sites of GABAA receptors. Because occupation of only one orthosteric site is sufficient for receptor activation, the α-Ctx could bind either to one orthosteric site (as a competitive inhibitor) and allosterically inhibit receptor activation through the second site or α-Ctx could bind to both orthosteric and allosteric types of sites, shifting GABA dose-response curve and diminishing maximal current amplitude. The second possibility is supported by the finding that about 20% of bound Alexa Fluor 546 α-Ctx could not be displaced by muscimol, supporting an additional interaction of Alexa Fluor 546 α-Ctx with a nonorthosteric binding site.
The tip of loop II is the most important part of α-Ctx and other TFTs for binding to AChBP and nAChRs (41). To test whether this region is also involved in the interaction with GABAAR, we applied mutant NT II with loop II grafted from NT I. This chimeric toxin (14) seems to be a suitable candidate because we have not detected any inhibition of GABAAR by NT II at 10 μm, but we observed some inhibition by NT I at the same concentration. Upon grafting of the NT I loop II fragment bearing a disulfide loop to the short neurotoxin NT II, the latter became functionally active against the receptor, strongly supporting the key role of loop II in TFTs in the interaction with GABAAR. Thus, we conclude that the tip of the loop II of TFTs plays a crucial role in GABAAR binding and inhibition.
Previously, it was suggested that some structural similarity (see Fig. 5A) of α-conotoxin ImI to the α-Ctx loop II may contribute to its binding to the neuronal nAChRs (28). Electrophysiology revealed that ImI inhibited GABAAR at 100 μm (Fig. 5B), whereas muscle nAChR-specific SIA and heteromeric nAChR-specific MII, lacking α-Ctx-similar features, were ineffective (Fig. 5B, bottom). Therefore, even molecules with structural similarity to the α-Ctx loop II could inhibit GABAAR. These data indicated that α-conotoxin ImI represents another type of polypeptide compounds able to interact with GABAAR. Moreover, synthetic peptide representing the sequence WCDAFCSIRGKR of the α-Ctx loop II tip also inhibited GABAAR (Fig. 5B). Despite the fact that we observe only weak inhibition at a relatively high concentration, short peptide GABAAR ligands on the base of loop II peptide or ImI could represent a good starting point for rational design of more specific ligands.
Our findings suggest that α-Ctx binds at the β/α interface and the loop II tip plays a crucial role in its binding. Given that, we constructed a molecular model of α-Ctx bound to the β3/α1 dimer of extracellular domains and performed 100-ns molecular dynamics studies in implicit solvent. Loop II residues 25–36 rested stably at the site under the loop C of the β3 subunit (Fig. 6A). With respect to β3 subunit backbone, root mean square deviation of these residues have not exceeded 0.15 nm, suggesting that such a complex might exist in principle. Interestingly, we found that both Arg-33 and Arg-36 residues in α-Ctx form salt bridges with α-Ctx Asp-27 and β3 Glu-155 (Fig. 6B). Arg-36 residue is unique to α-Ctx among all tested toxins, and its substitution by hydrophobic residues in other toxins (Fig. 1) could explain their weaker GABAAR inhibiting activity. Molecular modeling of NT II interaction with the β3/α1 dimer revealed inability of this short toxin to form a complex with the orthosteric site of GABAAR (Fig. 7C). In contrast, a molecular model of the NT II/I chimeric toxin showed properties very close to that of α-Ctx complex model with the β3/α1 dimer. In particular, loop II, grafted to the NT II scaffold from NT I, formed stable contacts with the C-loop of the β3 subunit extracellular domain (Fig. 7D).
It should be noted that previously we demonstrated that upon modification of α-Ctx with NHS ethers, the major product is the one with the label attached at Lys-23 (42). This lysine residue is in loop II, which according to our model takes part in receptor binding. However, it is not one of the deeply buried residues, and its modification does not necessarily disrupt α-Ctx binding, as indicated by the interaction of modified α-Ctx with the nAChRs (42).
In electrophysiological studies, we observed a somewhat more potent inhibition of α1β3γ2 over α2β3γ2 GABAAR. Because both α1 and α2 subunits share extremely high sequence homology, the respective receptors should differ only slightly in their extracellular parts. To test the above-described model, as a candidate for single-point mutagenesis we chose the Ser-67 residue (numbering of residues is given according to the homology model of extracellular domain) situated at the β/α1 interface. It is substituted by a lysine residue in the α2 subunit, and this positively charged residue could confer the difference in the α-Ctx affinity toward these two receptors, because α-Ctx loop II bears significant positive charge itself. This mutation was done, and the α-Ctx potency at α1(S67K)β3γ2 GABAAR was diminished, approaching that at α2β3γ2 (Fig. 8).
When our manuscript was ready for submission, two papers (43, 44) appeared, clearly demonstrating the perspective of TFTs for research on GABAARs. In Ref. 43, the authors analyzed in detail the interactions of α-Bgt and its fluorescent derivative with GABAAR in hippocampal neurons and with recombinant GABAAR subtypes, comparing their effects on ion currents and staining patterns in the presence of diverse ligands (agonists and antagonists) both with GABAARs and nAChRs. Their conclusion was that α-Bgt inhibits various functional GABAAR subtypes with different efficiency, with the α2β2γ2 receptor being inhibited most potently. Here, we show that the preferred target of α-Ctx is the α1β3γ2 receptor, indicating that diverse α-neurotoxins may have different affinity for distinct GABAAR subtypes and might be able to distinguish between various GABAAR subtypes. Concerning the mode of α-Bgt binding, Hannan et al. (43) described it as “a mixed inhibitory manner.” It is quite clear that further work is necessary to obtain a high resolution picture for recognition of GABAAR by both α-Bgt and α-Ctx.
Interestingly, in Ref. 43 it was also found that α-Bgt inhibited spontaneous channel openings in GABAAR composed of α4 and δ subunits, but in the presence of GABA the toxin behaved as a positive allosteric modulator. Such effects were not observed in this study, supporting our observation (Fig. 2) that different toxins, dependent on the receptor composition, might exhibit different effects. However, α-Bgt was found to bind at the orthosteric site (43). It is thus possible that binding of toxins to one of two orthosteric sites could lead to an allosteric change in the conformation of the second site and that the functional consequences of these allosteric interactions might be influenced by the type of additional (in this case δ and γ) subunits in the receptor (45).
The second above-mentioned paper (44) also deals with GABAAR potentiators by describing two TFTs from the coral snake venom that potentiate the GABAAR activity at low nanomolar concentrations. An undisputable advantage of these toxins is that they do not act on nAChRs and are the first, strictly speaking, GABAAR-specific TFTs. These toxins (MmTX1 and MmTX2) are not α-neurotoxins (like α-Ctx or α-Bgt) but belong to the nonconventional neurotoxins (additional 5th disulfide in loop I) like the GABAA receptor inhibiting toxin WTX identified in our study (Fig. 3). We and the authors of Ref. 44 hypothesized that TFTs should bind to GABAAR in a similar manner as α-neurotoxins bind to nAChRs. This is supported by their finding that the toxin interaction is decreased upon the H33S mutation in the central loop II of MmTX2 or upon double mutation G228E/Q231K in the receptor loop C. Concerning the binding interface of the toxin, this single mutation agrees with the conclusions we made based on the activity of the chimeric TFT. Our S67K and K67S mutations in the α1 and α2 subunits of GABAAR, respectively, although not being in the loop C, also affect binding, and it is clear that mapping of the toxin-binding interfaces in the GABAAR is a challenging task.
In conclusion, even such extensively studied objects as α-Ctx, which has been investigated since the 1970s, could hide some secrets, as demonstrated by its earlier overlooked inhibition of GABAARs. We showed that α-Ctx and NT I compete with α-Bgt for binding site(s) at the α1β3 receptor, and we found that α-Ctx, LsIII, and WTX inhibit the functional activity of α1β3γ2 GABAAR in the submicromolar to micromolar concentration ranges. In addition, the first peptide toxin α-conotoxin ImI inhibiting GABAAR was identified. These facts together with previously published observations demonstrate that GABAARs are the target for α-conotoxin and diverse three-finger toxins.
Author Contributions
V. I. T., W. S., and Y. N. U. made study concepts and design. D. S. K., I. V. S., L. V. S., L. O. O., E. V. K., E. N. L., D. A. D., M. N. Z., I. A. I., I. E. K., V. G. S. and J. R. designed, performed, and analyzed the experiments and interpreted the data. D. S. K., I. V. S., E. N. L., L. O. O., and E. V. K. wrote the manuscript. M. N. Z., I. A. I., and V. G. S. provided technical assistance. W. S., V. I. T,. and Y. N. U. revised the manuscript critically for important intellectual content and made a final approval of the version to be published. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgment
We thank Dr. Christoph Methfessel for help with electrophysiology measurements.
This work was supported by Russian Science Foundation Grant 14-24-00118. The authors declare that they have no conflicts of interest with the contents of this article.
- GABAAR
- type A γ-aminobutyric acid receptor
- AChBP
- acetylcholine-binding protein
- α-Bgt
- α-bungarotoxin
- α-Ctx
- α-cobratoxin
- GlyR
- glycine receptor
- nAChR
- nicotinic acetylcholine receptor
- TFT
- three-finger toxin
- PDB
- Protein Data Bank
- NT II
- neurotoxin II
- Ls III
- α-elapitoxin-Ls2a.
References
- 1. Krishek B. J., Moss S. J., Smart T. G. (1996) Homomeric β1 γ-aminobutyric acid A receptor-ion channels: evaluation of pharmacological and physiological properties. Mol. Pharmacol. 49, 494–504 [PubMed] [Google Scholar]
- 2. Miller P. S., Aricescu A. R. (2014) Crystal structure of a human GABAA receptor. Nature 512, 270–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Olsen R. W. (2006) Picrotoxin-like channel blockers of GABAA receptors. Proc. Natl. Acad. Sci. U.S.A. 103, 6081–6082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wyrembek P., Negri R., Appendino G., Mozrzymas J. W. (2012) Inhibitory effects of oenanthotoxin analogues on GABAergic currents in cultured rat hippocampal neurons depend on the polyacetylenes' polarity. Eur. J. Pharmacol. 683, 35–42 [DOI] [PubMed] [Google Scholar]
- 5. Razet R., Thomet U., Furtmüller R., Jursky F., Sigel E., Sieghart W., Dodd R. H. (2000) Use of bicuculline, a GABA antagonist, as a template for the development of a new class of ligands showing positive allosteric modulation of the GABAA receptor. Bioorg. Med. Chem. Lett. 10, 2579–2583 [DOI] [PubMed] [Google Scholar]
- 6. Ashton H. (2005) The diagnosis and management of benzodiazepine dependence. Curr. Opin. Psychiatry 18, 249–255 [DOI] [PubMed] [Google Scholar]
- 7. Kasheverov I. E., Utkin Y. N., Tsetlin V. I. (2009) Naturally occurring and synthetic peptides acting on nicotinic acetylcholine receptors. Curr. Pharm. Des. 15, 2430–2452 [DOI] [PubMed] [Google Scholar]
- 8. Berg D. K., Kelly R. B., Sargent P. B., Williamson P., Hall Z. W. (1972) Binding of α-bungarotoxin to acetylcholine receptors in mammalian muscle. Proc. Natl. Acad. Sci. U.S.A. 69, 147–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Clarke P. B., Schwartz R. D., Paul S. M., Pert C. B., Pert A. (1985) Nicotinic binding in rat brain: autoradiographic comparison of [3H]acetylcholine, [3H]nicotine, and [125I]α-bungarotoxin. J. Neurosci. 5, 1307–1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dellisanti C. D., Yao Y., Stroud J. C., Wang Z.-Z., Chen L. (2007) Crystal structure of the extracellular domain of nAChR α1 bound to α-bungarotoxin at 1.94 A resolution. Nat. Neurosci. 10, 953–962 [DOI] [PubMed] [Google Scholar]
- 11. Zouridakis M., Giastas P., Zarkadas E., Chroni-Tzartou D., Bregestovski P., Tzartos S. J. (2014) Crystal structures of free and antagonist-bound states of human α9 nicotinic receptor extracellular domain. Nat. Struct. Mol. Biol. 21, 976–980 [DOI] [PubMed] [Google Scholar]
- 12. Dubovskii P. V., Konshina A. G., Efremov R. G. (2014) Cobra cardiotoxins: membrane interactions and pharmacological potential. Curr. Med. Chem. 21, 270–287 [DOI] [PubMed] [Google Scholar]
- 13. Antil-Delbeke S., Gaillard C., Tamiya T., Corringer P. J., Changeux J. P., Servent D., Ménez A. (2000) Molecular determinants by which a long chain toxin from snake venom interacts with the neuronal α7-nicotinic acetylcholine receptor. J. Biol. Chem. 275, 29594–29601 [DOI] [PubMed] [Google Scholar]
- 14. Lyukmanova E. N., Shenkarev Z. O., Schulga A. A., Ermolyuk Y. S., Mordvintsev D. Y., Utkin Y. N., Shoulepko M. A., Hogg R. C., Bertrand D., Dolgikh D. A., Tsetlin V. I., Kirpichnikov M. P. (2007) Bacterial expression, NMR, and electrophysiology analysis of chimeric short/long-chain α-neurotoxins acting on neuronal nicotinic receptors. J. Biol. Chem. 282, 24784–24791 [DOI] [PubMed] [Google Scholar]
- 15. Mordvintsev D. Y., Polyak Y. L., Rodionov D. I., Jakubik J., Dolezal V., Karlsson E., Tsetlin V. I., Utkin Y. N. (2009) Weak toxin WTX from Naja kaouthia cobra venom interacts with both nicotinic and muscarinic acetylcholine receptors. FEBS J. 276, 5065–5075 [DOI] [PubMed] [Google Scholar]
- 16. Zhang L., Zhang Y., Jiang D., Reid P. F., Jiang X., Qin Z., Tao J. (2012) Alpha-cobratoxin inhibits T-type calcium currents through muscarinic M4 receptor and Gο-protein βγ subunits-dependent protein kinase A pathway in dorsal root ganglion neurons. Neuropharmacology 62, 1062–1072 [DOI] [PubMed] [Google Scholar]
- 17. Gergalova G., Lykhmus O., Kalashnyk O., Koval L., Chernyshov V., Kryukova E., Tsetlin V., Komisarenko S., Skok M. (2012) Mitochondria express α7 nicotinic acetylcholine receptors to regulate Ca2+ accumulation and cytochrome c release: study on isolated mitochondria. PLoS ONE 7, e31361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shelukhina I. V., Kryukova E. V., Lips K. S., Tsetlin V. I., Kummer W. (2009) Presence of α7 nicotinic acetylcholine receptors on dorsal root ganglion neurons proved using knockout mice and selective α-neurotoxins in histochemistry. J. Neurochem. 109, 1087–1095 [DOI] [PubMed] [Google Scholar]
- 19. Tsetlin V., Shelukhina I., Kryukova E., Burbaeva G., Starodubtseva L., Skok M., Volpina O., Utkin Y. (2007) Detection of α7 nicotinic acetylcholine receptors with the aid of antibodies and toxins. Life Sci. 80, 2202–2205 [DOI] [PubMed] [Google Scholar]
- 20. Bogdanov Y., Michels G., Armstrong-Gold C., Haydon P. G., Lindstrom J., Pangalos M., Moss S. J. (2006) Synaptic GABAA receptors are directly recruited from their extrasynaptic counterparts. EMBO J. 25, 4381–4389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Joshi S., Keith K. J., Ilyas A., Kapur J. (2013) GABAA receptor membrane insertion rates are specified by their subunit composition. Mol. Cell. Neurosci. 56, 201–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McCann C. M., Bracamontes J., Steinbach J. H., Sanes J. R. (2006) The cholinergic antagonist α-bungarotoxin also binds and blocks a subset of GABA receptors. Proc. Natl. Acad. Sci. U.S.A. 103, 5149–5154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Utkin Y. N., Kukhtina V. V., Kryukova E. V., Chiodini F., Bertrand D., Methfessel C., Tsetlin V. I. (2001) “Weak toxin” from Naja kaouthia is a nontoxic antagonist of α7 and muscle-type nicotinic acetylcholine receptors. J. Biol. Chem. 276, 15810–15815 [DOI] [PubMed] [Google Scholar]
- 24. Osipov A. V., Rucktooa P., Kasheverov I. E., Filkin S. Y., Starkov V. G., Andreeva T. V., Sixma T. K., Bertrand D., Utkin Y. N., Tsetlin V. I. (2012) Dimeric α-cobratoxin x-ray structure: localization of intermolecular disulfides and possible mode of binding to nicotinic acetylcholine receptors. J. Biol. Chem. 287, 6725–6734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tsetlin V. I., Karlsson E., Arseniev A. S., Utkin Y. N., Surin A. M., Pashkov V. S., Pluzhnikov K. A., Ivanov V. T., Bystrov V. F., Ovchinnikov Y. A. (1979) EPR and fluorescence study of interaction of Naja naja oxiana neurotoxin II and its derivatives with acetylcholine receptor protein from Torpedo marmorata. FEBS Lett. 106, 47–52 [DOI] [PubMed] [Google Scholar]
- 26. Starkov V. G., Polyak Y. L., Vulfius E. A., Kryukova E. V., Tsetlin V. I., Utkin Y. N. (2009) New weak toxins from the cobra venom. Russ. J. Bioorganic Chem. 35, 10–18 [DOI] [PubMed] [Google Scholar]
- 27. Grishin E. V., Sukhikh A. P., Slobodyan L. N., Ovchinnikov YuA, Sorokin V. M. (1974) Amino acid sequence of neurotoxin I from Naja naja oxiana venom. FEBS Lett. 45, 118–121 [DOI] [PubMed] [Google Scholar]
- 28. Maslennikov I. V., Shenkarev Z. O., Zhmak M. N., Ivanov V. T., Methfessel C., Tsetlin V. I., Arseniev A. S. (1999) NMR spatial structure of α-conotoxin ImI reveals a common scaffold in snail and snake toxins recognizing neuronal nicotinic acetylcholine receptors. FEBS Lett. 444, 275–280 [DOI] [PubMed] [Google Scholar]
- 29. Mirheydari P., Ramerstorfer J., Varagic Z., Scholze P., Wimmer L., Mihovilovic M. M., Sieghart W., Ernst M. (2014) Unexpected properties of δ-containing GABAA receptors in response to ligands interacting with the α+ β− site. Neurochem. Res. 39, 1057–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Abràmoff M. D., Magalhães P. J., Ram S. J. (2004) Image processing with ImageJ. Biophotonics Int. 11, 36–41 [Google Scholar]
- 31. Guex N., Peitsch M. C., Schwede T. (2009) Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: a historical perspective. Electrophoresis 30, S162-S173 [DOI] [PubMed] [Google Scholar]
- 32. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 33. Berendsen H. J., van der Spoel D., van Drunen R. (1995) GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 91, 43–56 [Google Scholar]
- 34. Eastman P., Pande V. (2015) OpenMM: a hardware-independent framework for molecular simulations. Comput. Sci. Eng. 12, 34–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cornell W. D., Cieplak P., Bayly C. I., Gould I. R., Merz K. M. Jr., Ferguson D. M., Spellmeyer D. C., Fox T., Caldwell J. W., Kollman P. A. (1995) A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 117, 5179–5197 [Google Scholar]
- 36. Humphrey W., Dalke A., Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38 [DOI] [PubMed] [Google Scholar]
- 37. Bergmann R., Kongsbak K., Sørensen P. L., Sander T., Balle T. (2013) A unified model of the GABAA receptor comprising agonist and benzodiazepine binding sites. PLoS ONE 8, e52323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sigel E., Steinmann M. E. (2012) Structure, function, and modulation of GABAA receptors. J. Biol. Chem. 287, 40224–40231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Christian C. A., Herbert A. G., Holt R. L., Peng K., Sherwood K. D., Pangratz-Fuehrer S., Rudolph U., Huguenard J. R. (2013) Endogenous positive allosteric modulation of GABAA receptors by diazepam binding inhibitor. Neuron 78, 1063–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Alfonso J., Le Magueresse C., Zuccotti A., Khodosevich K., Monyer H. (2012) Diazepam binding inhibitor promotes progenitor proliferation in the postnatal SVZ by reducing GABA signaling. Cell Stem Cell 10, 76–87 [DOI] [PubMed] [Google Scholar]
- 41. Bourne Y., Talley T. T., Hansen S. B., Taylor P., Marchot P. (2005) Crystal structure of a Cbtx-AChBP complex reveals essential interactions between snake α-neurotoxins and nicotinic receptors. EMBO J. 24, 1512–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Utkin Y. N., Krivoshein A. V., Davydov V. L., Kasheverov I. E., Franke P., Maslennikov I. V., Arseniev A. S., Hucho F., Tsetlin V. I. (1998) Labeling of Torpedo californica nicotinic acetylcholine receptor subunits by cobratoxin derivatives with photoactivatable groups of different chemical nature at Lys23. Eur. J. Biochem. 253, 229–235 [DOI] [PubMed] [Google Scholar]
- 43. Hannan S., Mortensen M., Smart T. G. (2015) Snake neurotoxin α-bungarotoxin is an antagonist at native GABAA receptors. Neuropharmacology 93, 28–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rosso J.-P., Schwarz J. R., Diaz-Bustamante M., Céard B., Gutiérrez J. M., Kneussel M., Pongs O., Bosmans F., Bougis P. E. (2015) MmTX1 and MmTX2 from coral snake venom potently modulate GABAA receptor activity. Proc. Natl. Acad. Sci. U.S.A. 112, E891–E900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sieghart W. (2015) Allosteric modulation of GABAA receptors via multiple drug-binding sites. Adv. Pharmacol. 72, 53–96 [DOI] [PubMed] [Google Scholar]
- 46. Hansen S. B., Talley T. T., Radic Z., Taylor P. (2004) Structural and ligand recognition characteristics of an acetylcholine-binding protein from Aplysia californica. J. Biol. Chem. 279, 24197–24202 [DOI] [PMC free article] [PubMed] [Google Scholar]