

Background: Axon regeneration relies on HDAC5-dependent tubulin deacetylation at the site of injury.
Results: Filamin A interacts with HDAC5 and is required for injury-induced tubulin deacetylation and axon regeneration.
Conclusion: Filamin A is required in injured axons for HDAC5 activity and axon regeneration.
Significance: Revealing the mechanisms involved in the spatial control of HDAC5 activity along the axon length is important to understand regenerative mechanisms in peripheral neurons.
Keywords: axon, filamin, histone deacetylase (HDAC), regeneration, tubulin, deacetylation
Abstract
Microtubule dynamics are important for axon growth during development as well as axon regeneration after injury. We have previously identified HDAC5 as an injury-regulated tubulin deacetylase that functions at the injury site to promote axon regeneration. However, the mechanisms involved in the spatial control of HDAC5 activity remain poorly understood. Here we reveal that HDAC5 interacts with the actin binding protein filamin A via its C-terminal domain. Filamin A plays critical roles in HDAC5-dependent tubulin deacetylation because, in cells lacking filamin A, the levels of acetylated tubulin are elevated markedly. We found that nerve injury increases filamin A axonal expression in a protein synthesis-dependent manner. Reducing filamin A levels or interfering with the interaction between HDAC5 and filamin A prevents injury-induced tubulin deacetylation as well as HDAC5 localization at the injured axon tips. In addition, neurons lacking filamin A display reduced axon regeneration. Our findings suggest a model in which filamin A local translation following axon injury controls localized HDAC5 activity to promote axon regeneration.
Introduction
The microtubule cytoskeleton plays an important role in axon growth during development and axon regeneration after injury. A number of studies have shown that a precise balance between stable and dynamic microtubules is required for growth cone formation and axon regeneration (1–3). Several signaling pathways converge onto microtubules to promote axon regeneration in the mammalian system (4). The GSK3-β pathway regulates the activity of microtubule-binding proteins such as cytoplasmic linker associated protein (CLASP), adenomatous polyposis coli, and CRMP2 to coordinate the stability, growth speed, and configuration of microtubules in growth cones and regulates axon growth (5, 6). In addition to microtubule-binding proteins, posttranslational modifications of tubulin are believed to correlate with the dynamic properties of microtubules (7). For example, acetylated tubulin is enriched in stable microtubules along the axon shaft, correlating with microtubule stability (8). We have previously demonstrated a role for histone deacetylase 5 (HDAC5)3 in the control of microtubule dynamics in injured sensory neurons (9). HDAC5 tubulin deacetylase activity increases following axon injury in a calcium and PKC-dependent manner and is required for growth cone dynamics and axon regeneration (9). Axon injury elicits the formation of a gradient of tubulin deacetylation, with low levels of acetylated tubulin in proximity of the injury site (9). Although a gradient of HDAC5 correlates with the gradient of deacetylated tubulin, the precise mechanisms involved in the spatial control of HDAC5 activity along the axon length remain poorly understood.
An important aspect of axon growth is the functional interplay between actin and the microtubule cytoskeleton. Indeed, extending neurites contain bundled microtubules surrounded by an actin-rich growth cone (10). Recent studies have demonstrated that actin turnover and organization mediated by actin depolymerizing factor (ADF)/cofilin is necessary for microtubule penetration and coalescence (11). In the absence of ADF/cofilin, pharmacological depolymerization of actin filaments enables microtubules to protrude toward the cell periphery and allow neurite formation (11). Another study revealed a link between the actin network and microtubules mediated by a protein complex containing the Rho guanine nucleotide exchange factor TRIO and the plus-end tracking protein Navigator 1 (NAV1) (12). Capture of this complex by EB1-labeled microtubule dynamic ends localizes Rac1 activity and promotes neurite extension (12). Although these studies reveal that, with a permissive actin network, microtubules can extend and lead to neurite formation, how microtubule dynamics are precisely controlled at the microtubule/actin network interface has not been explored.
Filamin A is a large homodimeric protein that organizes the actin filaments into an orthogonal network by its ability to cross-link actin filaments (13–17). Filamin A has been implicated in numerous aspects of cell motility and cell migration by providing a scaffolding platform for cytoskeletal and signaling proteins and by mediating membrane-cytoskeleton interactions (17, 18). Filamin A is composed of an amino-terminal actin-binding domain and 24 IgG-like domains that can bind more than 70 diverse proteins (19). Human filamin A loss-of-function mutations cause periventricular heterotopia, a neuronal migration disorder in which many neurons destined for the cerebral cortex fail to migrate (20, 21). In mice, complete loss of filamin A results in embryonic lethality and causes cardiovascular defects (22) as well as neuronal migration defects (23). Filamin A-deficient mouse embryonic fibroblasts display minimal motility defects (22), whereas filamin A-deficient M2 melanoma cells have extensive motility-related defects (16, 17). The absence of filamin A in mouse embryonic fibroblasts has been shown to cause a defect in microtubule extension toward the cell periphery (24), but whether this is a result of defects in the surrounding actin network or a more direct effect of filamin A on microtubules has not been explored.
Here we report that filamin A binds to HDAC5 and is required for HDAC5 deacetylase activity toward tubulin in injured axons. Following axon injury, filamin A is translated locally, suggesting a role for filamin A in injury-induced control of local HDAC5 activity. Accordingly, reducing filamin A levels impairs injury-induced tubulin deacetylation as well as HDAC5 localization at the injured axon tips. Sensory neurons with reduced filamin A levels displayed impaired axon regeneration. Our findings reveal that local translation of filamin A following axon injury is required for the localized HDAC5 deacetylase activity toward tubulin and contributes to promote axon regeneration.
Experimental Procedures
Antibodies, Constructs, Lentiviruses, and Cell Lines
Antibodies used in this study were as follows: anti-HDAC5 (Sigma, catalog no. H4538), anti-filamin A (Santa Cruz Biotechnology, catalog no. SC-28284), anti-FLAG (Sigma, catalog no. F1804), anti-GFP (Santa Cruz Biotechnology, catalog no. SC-9996), anti-PKCμ (Cell Signaling Technology, catalog no. 2052), anti-p-PKCμ (Cell Signaling Technology, catalog no. 2054), anti-α-tubulin (Abcam, catalog no. 15246), anti-acetylated tubulin (Sigma, catalog no. T7451), anti-tyrosinated tubulin (Sigma, catalog no. T9028), and anti-βIII tubulin (Covance, catalog no. MMS-435P). To knock down filamin A, two pLKO shRNA plasmids from Sigma (TRCN0000239209 and TRC0000239210) were used. Lentiviruses were produced as described previously (9). FLAG-tagged human HDAC5 was a gift from Eric Verdin (Addgene, plasmid 13822) (25). HDAC5 fragments were generated by PCR from amino acids 1–680, 1–1040, or 1040–1122 and cloned into pEGFP-C1 (Clontech) in-frame. The FUGW lentiviral expression vector was used to produce the GFP-Cterm (HDAC51040–1122)-overexpressing lentivirus as described previously (9). HEK293T cells were used for coimmunoprecipitation and virus production. Filamin A-deficient human melanoma M2 cells were provided by Dr. Thomas P. Stossel (Harvard Medical School). The filamin A-recovered control cell line A7 was purchased from the ATCC (catalog no. CRL-2500). The following chemicals were used in cultured cells: ingenol-3-angelate (I3A, 1 μm, Adipogen) and Scriptaid (5 μm, Tocris).
Protein Extraction and Coimmunoprecipitation
To extract protein lysates from cultured cells, lysis buffer (Cell Signaling Technology, catalog no. 9803) supplemented with protease inhibitor mixture and phosphatase inhibitor mixture (Roche, catalog nos. 11873580001 and 04906845001) was used. To screen for HDAC5-interacting proteins, HEK293T cells overexpressing FLAG-HDAC5 were lysed with buffer A (50 mm HEPES (pH 7.4), 1% Nonidet P-40, 0.1% SDS, 300 mm NaCl, and 1 mm EDTA supplemented with protease and phosphatase inhibitor mixtures). A total of 10 mg of FLAG- or FLAG-HDAC5-overexpressing lysates was precleared, loaded onto an anti-FLAG antibody-agarose resin column (Sigma, catalog no. A2220), and washed 10 times with 10 bed volumes of washing buffer (buffer A containing 500 mm NaCl). Proteins were eluted with 1 bed volume of elution buffer (washing buffer containing 100 mg/ml FLAG peptide (Sigma, F3290)) five times. Eluted proteins were pooled and precipitated using the TCA precipitation method (26). Proteins were separated by SDS-PAGE and detected by silver staining (Life Technologies). Proteins were identified as described previously (27). Briefly, protein bands were excised, destained, and trypsinized. Peptide mass fingerprints were obtained using a mass spectrometer (Voyager DE-TM STR biospectrometry workstation). The database search was performed using the MS-FIT proteomic tool.
For immunoprecipitation experiments from cultured DRG neurons, protein extract from DIV7 cultured DRG neurons were prepared with lysis buffer (Cell Signaling Technology, catalog no. 9803) supplemented with protease inhibitor mixture and phosphatase inhibitor mixture (Roche, catalog nos. 11873580001 and 04906845001), and immunoprecipitation was performed with control IgG, anti-HDAC5, or anti-filamin A antibodies. HDAC5 nuclear export was induced by treating DIV7 cultured DRG neurons with I3A (1 μm) for 3 h before cell lysis. HDAC enzymatic activity was blocked by treating DIV7 cultured DRG neurons with the HDAC inhibitor Scriptaid (5 μm) for 16 h before cell lysis.
Embryonic DRG Culture, Immunofluorescence Imaging, and in Vitro Axotomy
Mouse embryonic DRG culture was performed as described previously (9). Briefly, for spot culture, DRG from embryonic day 13.5 embryos were dissociated, and 104 cells/2.5 μl were plated. For the in vitro regeneration assay, DRG neurons were axotomized at DIV7 using a blade and stained 40 h later for the regenerative marker SCG10 (28). Fluorescence images were obtained using an Epi-fluorescence microscope (Nikon, TE2000E) and a confocal microscope (Nikon, Eclipse C1). Intensity and the in vitro regeneration index were plotted using ImageJ (9). For the DRG replating assay (29), DRG neurons were infected with control or filamin A shRNA lentivirus at DIV1, trypsinized (0.025% trypsin-EDTA, Life Technologies), and replated on poly-d-lysine-laminin-coated culture plastic dishes at DIV4. Replated neurons were fixed and stained with TUJ1 antibody 16 h after replating.
Animal Surgeries, Chemical Treatments, and Tissue Sample Preparations
All surgical procedures were approved by the Washington University in St. Louis School of Medicine Animal Studies Committee. Mouse tissue samples for Western blot analysis and immunohistochemistry were prepared as described previously (9). For the Western blot analysis, mouse sciatic nerves were dissected and lysed in lysis buffer (Cell Signaling Technology, catalog no. 9803) supplemented with protease and phosphatase inhibitor mixtures (Roche, catalog nos. 11873580001 and 04906845001). For immunohistochemistry, sciatic nerves were dissected, fixed in 4% paraformaldehyde in PBS for 1 h, incubated in 30% sucrose in PBS for 24 h, embedded in OCT solution (Tissue-Tek), and frozen in dry ice-cooled methylbutane. For sciatic nerve delivery of chemical compounds, the chemicals cycloheximide (1 mg/kg, Sigma), EGTA (10 mm, Life Technologies), Gö6976 (1 mg/kg, Tocris), and H-89 (1 mg/kg, Tocris) were dissolved in dimethyl sulfoxide. Surgifoam (Johnson and Johnson) was soaked with 100 μl of dimethyl sulfoxide alone as a control or dimethyl sulfoxide containing the indicated chemical and applied directly to the sciatic nerve 30 min prior to injury.
Results
HDAC5 Interacts with Filamin A via Its C-terminal Domain
We have shown previously that HDAC5 functions as a tubulin deacetylase in injured axons to promote axon regeneration (9). Our studies revealed that axon injury enhanced microtubule deacetylation in close proximity but not further away from the injury site. Because HDAC5 is transported anterogradely along axons (9), it is important to understand how its activity is controlled to allow tubulin deacetylation at the axon tip but not along the axon shaft. To understand the mechanisms regulating local HDAC5 activity, we first aimed to identify HDAC5 interacting partners. FLAG-tagged HDAC5 was expressed in HEK293T cells, and the immunoprecipitated FLAG-HDAC5 was analyzed by SDS-PAGE and silver staining. Non-transfected cells were used as a negative control. A specific band was detected in the high molecular weight range and identified by mass spectrometry as the actin-binding protein filamin A (Fig. 1A). To define the regions of HDAC5 responsible for filamin A binding, GFP-tagged HDAC5 full-length or HDAC5 mutants lacking the C-terminal domain (Cterm, HDAC51040) or lacking both the deacetylase domain and the Cterm (HDAC5680) were expressed in HEK293T cells (Fig. 1B). Filamin A was immunoprecipitated, and the binding of HDAC5 was probed for GFP. HDAC5 full-length, but neither of the deletion mutants, was bound to filamin A (Fig. 1C), indicating that the Cterm of HDAC5 is required for the association with filamin A. To test whether the Cterm of HDAC5 is sufficient for filamin A binding, the GFP-tagged Cterm of HDAC5 (HDAC51040–1122, hereafter referred to as GFP-Cterm) was expressed in HEK293T cells and immunoprecipitated with anti-GFP antibodies. Filamin A efficiently coprecipitated with GFP-Cterm (Fig. 1D). The interaction between the Cterm of HDAC5 and filamin A was further confirmed in GST pulldown experiments (Fig. 1E). We then tested whether GFP-Cterm could interfere with the interaction between endogenous filamin A and HDAC5. We performed a competition assay by expressing GFP-Cterm or GFP as a control in HEK293T cells and examined, by immunoprecipitation of filamin A, whether the interaction with HDAC5 was affected. We observed that expression of GFP-Cterm, but not GFP, disrupted the interaction of endogenous filamin A with HDAC5 (Fig. 1F). This experiment confirmed the specificity of the interaction between HDAC5 and filamin A.
FIGURE 1.

Filamin A is a novel binding partner for HDAC5. A, FLAG-HDAC5 overexpressed in HEK293T cells was immunopurified using anti-FLAG antibody-agarose resin. The immunoprecipitates were analyzed by SDS-PAGE and silver staining. A high molecular weight protein coimmunoprecipitated with FLAG-HDAC5 and was identified as filamin A by mass spectrometry analysis. IP, immunoprecipitation. B, schematic of HDAC5 constructs. The amino acid numbering refers to human HDAC5. C, filamin A was immunoprecipitated from HEK293T cells transiently expressing the indicated GFP-HDAC5 constructs. The HDAC5 Cterm (residues 1040–1122) is required for HDAC5 interaction with filamin A. D, GFP-Cterm (HDAC51040–1122) was expressed in HEK293T cells and immunoprecipitated using GFP antibody, and the immunoprecipitated material was analyzed by Western blot for GFP and filamin A. TCL, total cell lysate. E, purified GST-Cterm (HDAC51040–1122) was incubated with HEK293T cell lysates, and pulldown material was analyzed by Western blot. Ponceau S staining showed that equal amounts of GST and GST-Cterm were used. The HDAC5 C-terminal domain (residues 1040–1122) is sufficient for HDAC5 interaction with filamin A. F, GFP or GFP-Cterm was overexpressed in HEK293T cells, and endogenous filamin A was immunoprecipitated. Overexpression of GFP-Cterm disrupted the interaction between endogenous filamin A and HDAC5. G, HDAC5 was immunoprecipitated from cultured DRG neurons treated with or without I3A, and immunoprecipitated material was analyzed by Western blot. p-PKC and p-HDAC5 were used as controls for the effectiveness of I3A. H, cultured DRG neurons were treated with the HDAC inhibitor Scriptaid or left untreated, and filamin A was immunoprecipitated. Immunoprecipitated material was analyzed by Western blot for HDAC5 and filamin A. Acetylated histone H3 (Ac-H3) was used as a control for the effectiveness of HDAC inhibition by Scriptaid.
Next we performed coimmunoprecipitation of endogenous protein to test whether HDAC5 forms a complex with filamin A in cultured DRG neurons. We observed that filamin A coimmunoprecipitated with HDAC5 (Fig. 1G). Treatment of cultured DRG neurons with the PKC activator I3A, which, as we have shown previously, stimulates HDAC5 nuclear export (30), enhanced HDAC5 interaction with filamin A (Fig. 1G). HDAC5 interaction with filamin A did not require HDAC5 deacetylase enzymatic activity because treatment of cultured DRG neurons with the HDAC inhibitor Scriptaid (9) did not affect the ability of filamin A to coimmunoprecipitate with HDAC5 (Fig. 1H). Acetylated histone in the lysate was used as a control for the effectiveness of HDAC inhibition. These results demonstrate that HDAC5 interacts with filamin A via its Cterm and that the interaction between HDAC5 and filamin A occurs in DRG neurons.
Filamin A Is Required for HDAC5-dependent Tubulin Deacetylation
To examine whether filamin A regulates the tubulin deacetylase activity of HDAC5, we investigated the level of α-tubulin acetylation in filamin A-null cells, M2 cells (17). We found that α-tubulin is highly acetylated in filamin A-null M2 cells, whereas, in filamin A-recovered A7 cells (17), α-tubulin is mostly deacetylated (Fig. 2A). Tubulin tyrosination and total levels of tubulin showed no difference in both cell lines, indicating that filamin A specifically affects tubulin acetylation. We also tested whether a reduction in filamin A levels in DRG neurons alters the levels of acetylated tubulin. For this experiment, cultured DRG neurons were infected with a lentivirus encoding scrambled shRNA control (Control) or filamin A shRNA (FLNA KD). Western blot analysis showed that the knockdown was effective and resulted in increased levels of α-tubulin acetylation (Fig. 2B). Next we asked whether filamin A is required for HDAC5-mediated tubulin deacetylation. GFP-HDAC5 was overexpressed in M2 cells. Under vehicle treatment conditions, GFP-HDAC5 was mostly nuclear, whereas, upon stimulation of the PKC pathway with I3A, GFP-HDAC5 relocalized to the cytoplasm (Fig. 2C), as expected (30, 31). However, the levels of acetylated tubulin were not altered by increased cytoplasmic localization of HDAC5 in filamin A-null M2 cells (Fig. 2, C–E). In filamin A-recovered A7 cells, GFP-HDAC5 also relocalized to the cytoplasm following I3A treatment (Fig. 2F), as observed in M2 cells. Interestingly, in contrast to M2 cells, increased cytoplasmic localization of HDAC5 in A7 cells resulted in tubulin deacetylation (Fig. 2, F–H). Tubulin deacetylation in I3A-treated A7 cells was blocked by the presence of the HDAC inhibitor Scriptaid (Fig. 2F). These results indicate that filamin A is required for HDAC5-dependent tubulin deacetylation but not for the PKC-mediated cytoplasmic translocation of HDAC5.
FIGURE 2.

Filamin A is required for HDAC5-mediated tubulin deacetylation. A, cell lysates from filamin A-deficient human melanoma cells (M2) and filamin A-recovered M2 cells (A7) were analyzed for tubulin acetylation by Western blot. The levels of acetylated α-tubulin (Ac-tub) were drastically higher in M2 cells compared with A7 cells, whereas the levels of tyrosinated α-tubulin (Tyr-tub), total α- or β-tubulin, and actin were similar in both cell lines. B, cultured DRG neurons were infected with control shRNA lentivirus or FLNA KD lentivirus and analyzed by Western blot. Filamin A knockdown increased the level of acetylated tubulin in DRG neurons. C, M2 cells expressing GFP-HDAC5 were treated with the PKC activator I3A or left untreated, and the cellular localization of HDAC5 and the level of Ac-tub in the cells was analyzed by immunocytochemistry. I3A-induced cytoplasmic localization of GFP-HDAC5 did not alter Ac-tub levels. Scale bar = 20 μm. D, cell lysates from M2 cells expressing GFP-HDAC5 were analyzed for tubulin acetylation by Western blot. I3A treatment led to the expected phosphorylation of PKCμ and HDAC5 but did not change Ac-tub levels. E, quantification of the Ac-tub levels in C. Data are mean ± S.E.; n = 3; ns, not significant by analysis of variance. F, A7 cells expressing GFP-HDAC5 were treated with I3A alone or with I3A together with the HDAC inhibitor Scriptaid, and the cellular localization of HDAC5 and the levels of Ac-tub in the cells were analyzed by immunocytochemistry. I3A induced translocation of GFP-HDAC5 from the nucleus to the cytoplasm, accompanied by a decrease in the level of Ac-tub. Scriptaid blocked the effect of I3A on the level of Ac-tub without affecting HDAC5 translocation. Scale bar = 20 μm. G, cell lysates from A7 cells expressing GFP-HDAC5 were analyzed for tubulin acetylation by Western blot. I3A treatment induced phosphorylation of PKCμ and HDAC5 and decreased Ac-tub levels. H, quantification of the Ac-tub levels in F. Data are mean ± S.E.; n = 3; **, p < 0.01 by analysis of variance.
Filamin A Levels Are Increased in Injured Axons
To test whether HDAC5 activity in injured axons requires filamin A, we first examined filamin A protein levels in sciatic nerve axons. We found that, in uninjured nerves, very little filamin A is expressed, whereas, 24 h following sciatic nerve injury, filamin A levels were up-regulated dramatically in the 3-mm segment of nerve proximal to the injury site (Figs. 3, A–C). Immunofluorescence of a section of nerve within the 3-mm segment proximal to the injury site showed that the injury-induced filamin A colocalized with TUJ1, an axonal marker, suggesting that the increased expression of filamin A occurs in axons (Fig. 3D). To further determine whether filamin A expression was localized in close proximity to the injury site, we performed immunofluorescence of a 2-mm longitudinal segment of nerve, which revealed that filamin A expression was enriched within the first millimeter nerve segment immediately proximal to the injury site (Fig. 3, E and F). To further confirm whether the increased filamin A levels after injury occurs in axons, we performed immunostaining on sciatic nerve cross-sections. Injury clearly induced filamin A expression in axons which were labeled with the βIII tubulin axonal marker (TUJ1 antibody) and surrounded by the Schwann cell marker S100β (Fig. 3, G and H).
FIGURE 3.

Filamin A levels increase in axons following injury. A, schematic of the injured sciatic nerve segment used for analysis. B, representative Western blot of control sciatic nerve (−Ax) and sciatic nerve 24 h after axotomy (+Ax). C, quantification of the filamin A levels in A. Data are mean ± S.E.; n = 8; **, p < 0.01 by Student's t test. D, longitudinal sections of a control uninjured nerve and an injured segment of the sciatic nerve within the 3-mm proximal to the injury site, stained for filamin A and the axon marker βIII tubulin (TUJ1 antibody). Scale bar = 100 μm. E, representative image of a 2-mm longitudinal section of injured sciatic nerve stained with filamin A and TUJ1 antibody. Scale bar = 500 μm. Dotted boxes (a and b) indicate the magnified regions 1 mm adjacent to the injury site (a) or 2 mm away from the injury site (b). Scale bar = 50 μm. The dotted arrows indicate the injury site. F, quantification of filamin A fluorescence intensity in unit area a and b in E. AU, arbitrary unit per square micrometer. Data are mean ± S.E. n = 15 for 1 mm and 2 mm from the injury site; ***, p < 0.001 by Student's t test. G, cross-sections of control and injured sciatic nerves stained for filamin A and Schwann cell marker S100β antibodies. Scale bar = 10 μm. H, cross-sections of control and injured sciatic nerves stained with filamin A and TUJ1 antibodies. Scale bar = 10 μm.
Filamin A is Translated Locally in Injured Axons
To test whether filamin A accumulation is due to local protein synthesis, sciatic nerves were treated locally with cycloheximide for 30 min prior to injury to block local protein synthesis. The result showed that cycloheximide treatment prevented filamin A up-regulation after injury (Fig. 4, A and B). Because axonal calcium influx and PKC activation are known to induce local protein synthesis in axons (32), we next tested whether filamin A expression requires calcium and PKC activity. We found that blocking calcium influx with EGTA or inhibiting PKC activity with the PKC inhibitor Gö6976 locally at the site of nerve injury abolishes filamin A accumulation (Fig. 4, C–F). As a control, we used the PKA inhibitor H-89, which failed to block injury-induced filamin A accumulation (Fig. 4, G and H). Together with the fact that filamin A mRNA is present in DRG axons (33), these results indicate that filamin A levels in injured nerves are regulated by local protein translation.
FIGURE 4.
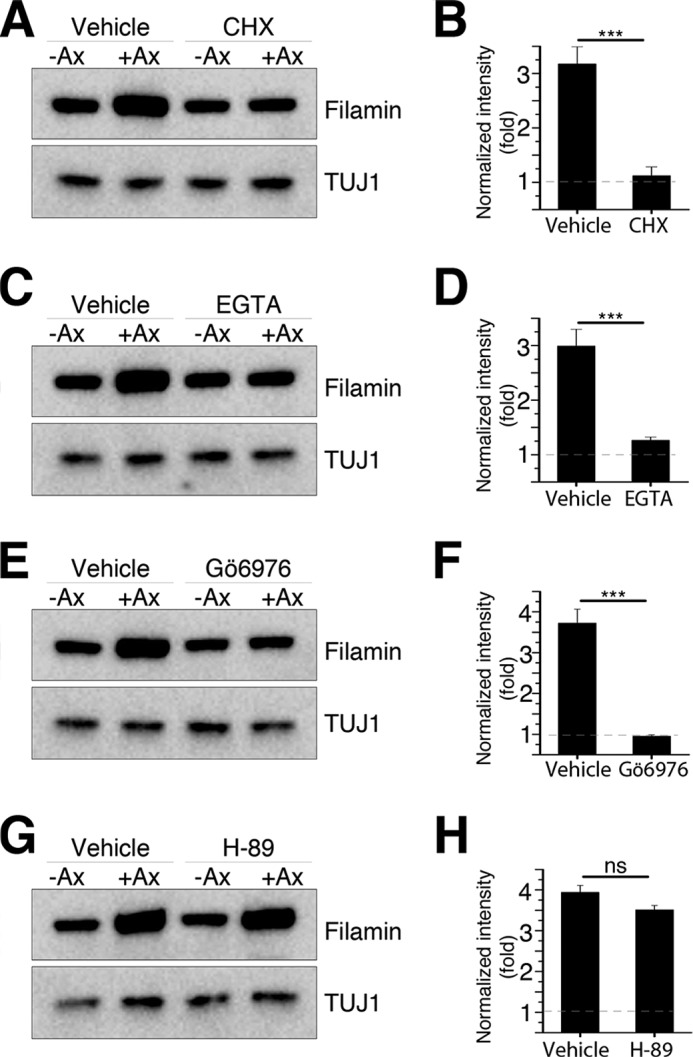
Filamin A is locally translated in injured axons. A, C, E, and G, Western blots of control (−Ax) and axotomized (+Ax) sciatic nerves treated with vehicle or cycloheximide (CHX, A), EGTA (C), Gö6976 (E), and H-89 (G). B, D, F, and H, quantifications of filamin A levels in A, C, E, and G. Data are mean ± S.E. n = 3; ***, p < 0.001 by Student's t test; ns, not significant.
Filamin A Is Required for HDAC5-mediated Tubulin Deacetylation in Injured Axons
Localized filamin A translation may represent a mechanism to locally regulate HDAC5 activity at newly formed axons tips. To test this hypothesis, we first examined whether filamin A colocalizes with HDAC5 in growth cones. We observed that, in hippocampal neurons, filamin A colocalizes with HDAC5 at axon tips, where the level of acetylated tubulin is relatively low compared with the axon shaft (Fig. 5A). In injured DRG neurons, HDAC5 localization with TUJ1-labeled microtubules in growth cones was impaired by knockdown of filamin A (Fig. 5B). These results suggest that filamin A is important for HDAC5 function toward tubulin as well as HDAC5 localization in injured axon tips.
FIGURE 5.

Filamin A is required for HDAC5 localization in growth cones. A, representative images of hippocampal neurons fixed at DIV1 and stained for filamin A, HDAC5, and Ac-tub. Filamin A and HDAC5 colocalized in the growth cone, where the level of acetylated tubulin is low. Scale bar = 5 μm. B, representative images of control or FLNA-KD DRG neurons fixed 3 h after in vitro axotomy and stained for HDAC5 and βIII-tubulin. Knockdown of filamin A reduces HDAC5 levels in the βIII-tubulin-labeled growth cone. Scale bar = 5 μm.
To further test whether reduction in filamin A levels impairs injury-induced tubulin deacetylation, as we have reported previously (9), we quantified tubulin deacetylation following axon injury in control and FLNA KD cultured DRG neurons. In control neurons, axotomy induced tubulin deacetylation at the injured axon tips (Fig. 6, A–C), as reported previously (9). In contrast, axotomy-induced tubulin deacetylation at the injured axon tips was impaired in FLNA KD neurons (Fig. 6, A–C). To test whether the impaired injury-induced tubulin deacetylation results from a reduced interaction between HDAC5 and filamin A, we expressed GFP Cterm (Fig. 1, D and F) in cultured DRG neurons. In the presence of GFP-Cterm, but not GFP as a control, injury failed to induce tubulin deacetylation (Fig. 6, D–F). The levels of HDAC5 at the injured axon tips were also reduced in both FLNA KD and GFP-Cterm-expressing DRG neurons (Fig. 6, G and H).
FIGURE 6.

Filamin A is required for tubulin deacetylation after axon injury. A, control or FLNA-KD DRG neurons were axotomized, fixed 3 h later, and stained for Ac-tub and total α-tubulin. Scale bar = 100 μm. B, average intensity plot of Ac-tub in A. The intensity of Ac-tub was normalized to α-tubulin and plotted in function of distance toward the cell body, with 0 referring to the axotomy site (n = 6 for control and 7 for FLNA KD). Data are mean ± S.E. C, average slopes of the Ac-tub ratio calculated from the plots in B (n = 6 for control and 7 for FLNA KD). Data are mean ± S.E. ***, p < 0.001 by Student's t test. D, control (FUGW lentivirus) or Cterm-overexpressing (FUGW-GFP-Cterm lentivirus) DRG neurons were axotomized, fixed 3 h later, and stained for Ac-tub and total α-tubulin. Scale bar = 100 μm. E, average intensity plot of Ac-tub in D (n = 5 for control and 5 for GFP-Cterm). Data are mean ± S.E. F, average slopes of the Ac-tub ratio calculated from the plots in B (n = 5 for control and 5 for GFP-Cterm). Data are mean ± S.E. ***, p < 0.001 by Student's t test. G, control, FLNA-KD, or GFP-Cterm-overexpressing (FUGW-Cterm lentivirus) DRG neurons were axotomized, fixed 3 h later, and stained for HDAC5 and βIII tubulin (TUJ1 antibody). Scale bar = 100 μm. H, normalized intensity of HDAC5 from G (n = 6, 8, and 8 for control, FLNA KD, and GFP-Cterm, respectively). Data are mean ± S.E. ***, p < 0.001 by analysis of variance).
Filamin A Contributes to Promote Axon Regeneration
We next asked whether filamin A is required for axon regeneration. Control or FLNA KD DRG spot cultures were axotomized at DIV7, and axon regrowth was visualized by staining DRG neurons with SCG10, a regenerative marker (28). To assess the regenerative capacity of injured axons, a regeneration index was calculated from the images acquired 40 h after axotomy. Axotomized axons displayed robust regeneration, whereas filamin A knockdown significantly impaired axon regeneration (Fig. 7, A–C). Disruption of the interaction between HDAC5 and filamin A with the expression of GFP-Cterm also impaired axon regeneration (Fig. 7, D–F).
FIGURE 7.

Filamin A contributes to the regulation of axon regeneration. A, representative images of the in vitro regeneration assay of control or FLNA-KD DRG neurons. Scale bar = 100 μm. B, schematic of the experiment time line. DRG neurons were infected with control or shFLNA lentivirus (shLenti) at DIV3, axotomized at DIV7 to start the in vitro regeneration assay, and stained 40 h later for SCG10. C, average regeneration index obtained from regeneration assays (n = 14 for control and 17 for FLNA KD). Data are mean ± S.E. ***, p < 0.001. D, representative images of in vitro regeneration assay of control (FUGW lentivirus) or GFP-Cterm-overexpressing (FUGW-Cterm lentivirus) DRG neurons. Scale bar = 100 μm. E, schematic of the experiment time line. DRG neurons were infected with control or GFP-Cterm lentivirus at DIV3, axotomized at DIV7 to start the in vitro regeneration assay, and stained 40 h later for SCG10. F, average regeneration index obtained from regeneration assays (n = 8 for each condition). Data are mean ± S.E. **, p < 0.01. G, representative images of the neuron replating assay of control or FLNA-KD DRG neurons. Scale bar = 20 μm. H, schematic of the experiment time line. DRG neurons were infected with control or shFLNA lentivirus at DIV1, replated at DIV4, and fixed at 12 h after replating. I, average of maximum axon length obtained from the neuron replating assays (n = 60 for control and 49 for FLNA KD). Data are mean ± S.E. ***, p < 0.001.
We next employed a replating assay (29) to assess the role of filamin A in axon regeneration. In this assay, neurons were plated, infected with lentivirus at DIV1, trypsinized, and replated at DIV4 (Fig. 7H). This assay is more stringent than the in vitro axotomy assay described above because neurons need to reconstruct their axons after complete axonal loss. We observed that control DRG neurons successfully extended axons, whereas filamin A knockdown DRG neurons failed to extend long axons (Fig. 7, G and I). These results indicate that filamin A promotes initial axon growth from the cell body as well as regeneration of injured axons.
Discussion
Neurite extension and axon growth require the coordinated actions of the actin and microtubule cytoskeleton network (10). Neurites and axons contain bundled microtubules, which are surrounded in the growth cone by an actin-rich network. A dynamic actin network allows microtubules to advance toward the growth cone periphery. Specifically, a constitutive retrograde flow of actin has been shown to allow microtubules to protrude and generate a neurite from a spherical neuron (11). Actin dynamics are therefore believed to drive growth cone protrusion and motility (10). However, growth cone steering and migration can also be guided by microtubules (34, 35). Our data reveal a novel link between the actin cytoskeleton and microtubule dynamics, with the actin-associated protein filamin A regulating the activity of HDAC5 toward tubulin at the tip of injured axons to promote axon regeneration.
Our studies indicate that filamin A regulates axon regeneration in sensory neurons in part via regulating HDAC5-dependent tubulin deacetylation at axon tips. Our previous studies have shown that tubulin deacetylation following axon injury correlate with increased microtubule dynamics (9). The absence of filamin A may therefore limit microtubule dynamics in newly formed growth cones. We found that expression of the HDAC5 Cterm exerts dominant negative functions, indicating that the C-terminal region of HDAC5 is responsible for its association with filamin A. This interaction is likely to be specific for HDAC5 and not shared with the other tubulin deacetylase, HDAC6 (36), because the Cterm of HDAC5 has no significant sequence homology with the C-terminal domain of HDAC6 (37). The filamin A-dependent regulation of HDAC5 may also underlie the different functions HDAC5 and HDAC6 play in injured axons. Indeed, in contrast to HDAC5 inhibition, which blocks axon regeneration (9), inhibiting HDAC6 heightens the level of tubulin acetylation and increases axon regeneration in the presence of inhibitory factors found in the damaged CNS (38). The apparent opposite roles of HDAC5 and HDAC6 may result, beyond the different environment, from different substrate specificity and different interacting partners, such as filamin A.
Consistent with the notion that filamin A may regulate microtubule dynamic properties at the actin interface, Lynch et al. (24) have observed that, in filamin-depleted mouse embryonic fibroblast, microtubules exhibited lower growth rates than controls and did not extend to the cell edge during early stages of cell spreading. Interestingly, a decrease in microtubule extension rates does not result from peripheral actin flow, which is also decreased in the filamin A-depleted system (24), suggesting a more direct role of filamin A depletion in microtubules. Interestingly, a recent study revealed that the plus-end tracking protein NAV1 forms a complex with the Rho guanine nucleotide exchange factor TRIO and localizes to microtubule dynamic ends to promote neurite extension (12). TRIO also interacts with filamin (39), further supporting the notion of filamin A being a scaffold for the spatial organization of signaling pathways at the junction between actin and the microtubule cytoskeleton.
These functions of filamin A in regulating microtubule dynamics may be more important for axon regeneration after injury than for normal axon growth. Indeed, filamin A-null ES cells or neural progenitor cells successfully differentiate in vitro into neurons bearing long axons and normal growth cones (22). F-actin levels are not significantly altered in filamin-null fibroblast, neurons, and endothelia cells, and filamin-null fibroblasts show normal motility and membrane ruffling (22, 40). Compensation from filamin B, which has high homology with filamin A, may be responsible for the lack of phenotype in these settings. Alternatively, HDAC5-mediated tubulin deacetylation is not essential for normal axon outgrowth, as supported by the fact that mice lacking HDAC5 mice develop normally (41).
The ability of axons to regenerate depends in part on their capacity to locally synthesize new proteins (32). Newly synthesized proteins that contribute to the regeneration process include cytoskeletal proteins (42) and membrane-associated proteins (43, 44). Here we reveal that local translation of filamin A contributes to control of the spatial activity of HDAC5 in injured axons. The function of filamin A may also be regulated posttranslationally in injured axons. Indeed, filamin A participates in signaling events by interacting with numerous protein kinases. One of them is protein kinase C (45), which is implicated in actin reorganization, cell adhesion, and cell spreading. Filamin A not only serves as a scaffolding protein of PKC, it is also a substrate of PKC (45). Given that PKC and PKCμ are activated locally after injury to control HDAC5 phosphorylation and activity (9), PKC may also regulate the function of filamin A in injured axons.
Filamin A may play other roles in regenerating axons beyond regulating HDAC5 activity. Indeed, filamin A is required for vimentin-mediated cell adhesion and spreading (46) and interacts with vimentin in vitro (47). Vimentin is also locally translated at the axon lesion side and plays a role in injury signaling (48). Locally translated filamin A may also promote more efficient recycling of integrins, which enhance sensory axon regeneration by being transported in distinct recycling endosomal cargoes (49–51). In the absence of filamin A, β1 integrin surface expression is reduced in the initial phase of cell matrix contact (47). The absence of filamin A alters the trafficking of adhesion receptors such as b1 integrin, leads to cell spreading and migration defects (47), and may also affect growth cone migration.
The importance of actin network reorganization (11) and dynamic microtubules (12) for neurite formation and extension correlates well with the requirement for filamin A at the intersection of the microtubule and actin cytoskeletal elements in the growth cone and in axon regeneration. Future studies will elucidate the roles filamin A may play to guide axon regeneration and whether filamin A, or its absence, may play a role in the disorganized microtubule network observed in retraction bulbs in central nervous system axons.
Author Contributions
Y. C. conceived, designed, and performed the experiments and analyzed the data. Y. C., D. P., and V. C. conceived the experiments and wrote the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant NS082446 (to V. C.). This work was also supported by National Research Foundation of Korea Grants NRF-2012R1A6A3A03039290 (to Y. C.) and M103KV010013-04K2201-01310 (to D. P.). The authors declare that they have no conflicts of interest with the contents of this article.
- HDAC
- histone deacetylase
- I3A
- ingenol-3- angelate
- DRG
- dorsal root ganglion
- DIV
- day(s) in vitro
- Cterm
- C-terminal domain
- Ac-tub
- acetylated α-tubulin.
References
- 1. Ertürk A., Hellal F., Enes J., Bradke F. (2007) Disorganized microtubules underlie the formation of retraction bulbs and the failure of axonal regeneration. J. Neurosci. 27, 9169–9180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hellal F., Hurtado A., Ruschel J., Flynn K. C., Laskowski C. J., Umlauf M., Kapitein L. C., Strikis D., Lemmon V., Bixby J., Hoogenraad C. C., Bradke F. (2011) Microtubule stabilization reduces scarring and causes axon regeneration after spinal cord injury. Science 331, 928–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sengottuvel V., Leibinger M., Pfreimer M., Andreadaki A., Fischer D. (2011) Taxol facilitates axon regeneration in the mature CNS. J. Neurosci. 31, 2688–2699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Saijilafu, Zhang B. Y., Zhou F. Q. (2013) Signaling pathways that regulate axon regeneration. Neurosci. Bull. 29, 411–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hur E. M., Saijilafu, Lee B. D., Kim S. J., Xu W. L., Zhou F. Q. (2011) GSK3 controls axon growth via CLASP-mediated regulation of growth cone microtubules. Genes Dev. 25, 1968–1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liz M. A., Mar F. M., Santos T. E., Pimentel H. I., Marques A. M., Morgado M. M., Vieira S., Sousa V. F., Pemble H., Wittmann T., Sutherland C., Woodgett J. R., Sousa M. M. (2014) Neuronal deletion of GSK3β increases microtubule speed in the growth cone and enhances axon regeneration via CRMP-2 and independently of MAP1B and CLASP2. BMC Biol. 12, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hammond J. W., Cai D., Verhey K. J. (2008) Tubulin modifications and their cellular functions. Curr. Opin. Cell Biol. 20, 71–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Janke C., Kneussel M. (2010) Tubulin post-translational modifications: encoding functions on the neuronal microtubule cytoskeleton. Trends Neurosci. 33, 362–372 [DOI] [PubMed] [Google Scholar]
- 9. Cho Y., Cavalli V. (2012) HDAC5 is a novel injury-regulated tubulin deacetylase controlling axon regeneration. EMBO J. 31, 3063–3078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dent E. W., Gertler F. B. (2003) Cytoskeletal dynamics and transport in growth cone motility and axon guidance. Neuron 40, 209–227 [DOI] [PubMed] [Google Scholar]
- 11. Flynn K. C., Hellal F., Neukirchen D., Jacob S., Tahirovic S., Dupraz S., Stern S., Garvalov B. K., Gurniak C., Shaw A. E., Meyn L., Wedlich-Söldner R., Bamburg J. R., Small J. V., Witke W., Bradke F. (2012) ADF/cofilin-mediated actin retrograde flow directs neurite formation in the developing brain. Neuron 76, 1091–1107 [DOI] [PubMed] [Google Scholar]
- 12. van Haren J., Boudeau J., Schmidt S., Basu S., Liu Z., Lammers D., Demmers J., Benhari J., Grosveld F., Debant A., Galjart N. (2014) Dynamic microtubules catalyze formation of Navigator-TRIO complexes to regulate neurite extension. Curr. Biol. 24, 1778–1785 [DOI] [PubMed] [Google Scholar]
- 13. Hartwig J. H., Stossel T. P. (1975) Isolation and properties of actin, myosin, and a new actin binding protein in rabbit alveolar macrophages. J. Biol. Chem. 250, 5696–5705 [PubMed] [Google Scholar]
- 14. Hartwig J. H., Tyler J., Stossel T. P. (1980) Actin-binding protein promotes the bipolar and perpendicular branching of actin filaments. J. Cell Biol. 87, 841–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gorlin J. B., Yamin R., Egan S., Stewart M., Stossel T. P., Kwiatkowski D. J., Hartwig J. H. (1990) Human endothelial actin-binding protein (ABP-280, nonmuscle filamin): a molecular leaf spring. J. Cell Biol. 111, 1089–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Flanagan L. A., Chou J., Falet H., Neujahr R., Hartwig J. H., Stossel T. P. (2001) Filamin A, the Arp2/3 complex, and the morphology and function of cortical actin filaments in human melanoma cells. J. Cell Biol. 155, 511–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cunningham C. C., Gorlin J. B., Kwiatkowski D. J., Hartwig J. H., Janmey P. A., Byers H. R., Stossel T. P. (1992) Actin-binding protein requirement for cortical stability and efficient locomotion. Science 255, 325–327 [DOI] [PubMed] [Google Scholar]
- 18. Kim H., McCulloch C. A. (2011) Filamin A mediates interactions between cytoskeletal proteins that control cell adhesion. FEBS Lett. 585, 18–22 [DOI] [PubMed] [Google Scholar]
- 19. Zhou A. X., Hartwig J. H., Akyürek L. M. (2010) Filamins in cell signaling, transcription and organ development. Trends Cell Biol. 20, 113–123 [DOI] [PubMed] [Google Scholar]
- 20. Fox J. W., Lamperti E. D., Ekşioğlu Y. Z., Hong S. E., Feng Y., Graham D. A., Scheffer I. E., Dobyns W. B., Hirsch B. A., Radtke R. A., Berkovic S. F., Huttenlocher P. R., Walsh C. A. (1998) Mutations in filamin 1 prevent migration of cerebral cortical neurons in human periventricular heterotopia. Neuron 21, 1315–1325 [DOI] [PubMed] [Google Scholar]
- 21. Sheen V. L., Dixon P. H., Fox J. W., Hong S. E., Kinton L., Sisodiya S. M., Duncan J. S., Dubeau F., Scheffer I. E., Schachter S. C., Wilner A., Henchy R., Crino P., Kamuro K., DiMario F., Berg M., Kuzniecky R., Cole A. J., Bromfield E., Biber M., Schomer D., Wheless J., Silver K., Mochida G. H., Berkovic S. F., Andermann F., Andermann E., Dobyns W. B., Wood N. W., Walsh C. A. (2001) Mutations in the X-linked filamin 1 gene cause periventricular nodular heterotopia in males as well as in females. Hum. Mol. Genet. 10, 1775–1783 [DOI] [PubMed] [Google Scholar]
- 22. Feng Y., Chen M. H., Moskowitz I. P., Mendonza A. M., Vidali L., Nakamura F., Kwiatkowski D. J., Walsh C. A. (2006) Filamin A (FLNA) is required for cell-cell contact in vascular development and cardiac morphogenesis. Proc. Natl. Acad. Sci. U.S.A. 103, 19836–19841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang J., Neal J., Lian G., Hu J., Lu J., Sheen V. (2013) Filamin A regulates neuronal migration through brefeldin A-inhibited guanine exchange factor 2-dependent Arf1 activation. J. Neurosci. 33, 15735–15746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lynch C. D., Gauthier N. C., Biais N., Lazar A. M., Roca-Cusachs P., Yu C. H., Sheetz M. P. (2011) Filamin depletion blocks endoplasmic spreading and destabilizes force-bearing adhesions. Mol. Biol. Cell 22, 1263–1273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fischle W., Emiliani S., Hendzel M. J., Nagase T., Nomura N., Voelter W., Verdin E. (1999) A new family of human histone deacetylases related to Saccharomyces cerevisiae HDA1p. J. Biol. Chem. 274, 11713–11720 [DOI] [PubMed] [Google Scholar]
- 26. Kim C., Cho Y., Kang C. H., Kim M. G., Lee H., Cho E. G., Park D. (2005) Filamin is essential for shedding of the transmembrane serine protease, epithin. EMBO Rep. 6, 1045–1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim C., Lee H. S., Lee D., Lee S. D., Cho E. G., Yang S. J., Kim S. B., Park D., Kim M. G. (2011) Epithin/PRSS14 proteolytically regulates angiopoietin receptor Tie2 during transendothelial migration. Blood 117, 1415–1424 [DOI] [PubMed] [Google Scholar]
- 28. Shin J. E., Geisler S., DiAntonio A. (2014) Dynamic regulation of SCG10 in regenerating axons after injury. Exp. Neurol. 252, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zou H., Ho C., Wong K., Tessier-Lavigne M. (2009) Axotomy-induced Smad1 activation promotes axonal growth in adult sensory neurons. J. Neurosci. 29, 7116–7123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cho Y., Sloutsky R., Naegle K. M., Cavalli V. (2013) Injury-induced HDAC5 nuclear export is essential for axon regeneration. Cell 155, 894–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vega R. B., Harrison B. C., Meadows E., Roberts C. R., Papst P. J., Olson E. N., McKinsey T. A. (2004) Protein kinases C and D mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5. Mol. Cell. Biol. 24, 8374–8385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gumy L. F., Tan C. L., Fawcett J. W. (2010) The role of local protein synthesis and degradation in axon regeneration. Exp. Neurol. 223, 28–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gumy L. F., Yeo G. S., Tung Y. C., Zivraj K. H., Willis D., Coppola G., Lam B. Y., Twiss J. L., Holt C. E., Fawcett J. W. (2011) Transcriptome analysis of embryonic and adult sensory axons reveals changes in mRNA repertoire localization. RNA 17, 85–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dent E. W., Callaway J. L., Szebenyi G., Baas P. W., Kalil K. (1999) Reorganization and movement of microtubules in axonal growth cones and developing interstitial branches. J. Neurosci. 19, 8894–8908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Williamson T., Gordon-Weeks P. R., Schachner M., Taylor J. (1996) Microtubule reorganization is obligatory for growth cone turning. Proc. Natl. Acad. Sci. U.S.A. 93, 15221–15226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hubbert C., Guardiola A., Shao R., Kawaguchi Y., Ito A., Nixon A., Yoshida M., Wang X. F., Yao T. P. (2002) HDAC6 is a microtubule-associated deacetylase. Nature 417, 455–458 [DOI] [PubMed] [Google Scholar]
- 37. Yang X. J., Grégoire S. (2005) Class II histone deacetylases: from sequence to function, regulation, and clinical implication. Mol. Cell. Biol. 25, 2873–2884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rivieccio M. A., Brochier C., Willis D. E., Walker B. A., D'Annibale M. A., McLaughlin K., Siddiq A., Kozikowski A. P., Jaffrey S. R., Twiss J. L., Ratan R. R., Langley B. (2009) HDAC6 is a target for protection and regeneration following injury in the nervous system. Proc. Natl. Acad. Sci. U.S.A. 106, 19599–19604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bellanger J. M., Astier C., Sardet C., Ohta Y., Stossel T. P., Debant A. (2000) The Rac1- and RhoG-specific GEF domain of Trio targets filamin to remodel cytoskeletal actin. Nat. Cell Biol. 2, 888–892 [DOI] [PubMed] [Google Scholar]
- 40. Hart A. W., Morgan J. E., Schneider J., West K., McKie L., Bhattacharya S., Jackson I. J., Cross S. H. (2006) Cardiac malformations and midline skeletal defects in mice lacking filamin A. Hum. Mol. Genet. 15, 2457–2467 [DOI] [PubMed] [Google Scholar]
- 41. Chang S., McKinsey T. A., Zhang C. L., Richardson J. A., Hill J. A., Olson E. N. (2004) Histone deacetylases 5 and 9 govern responsiveness of the heart to a subset of stress signals and play redundant roles in heart development. Mol. Cell. Biol. 24, 8467–8476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Donnelly C. J., Park M., Spillane M., Yoo S., Pacheco A., Gomes C., Vuppalanchi D., McDonald M., Kim H. H., Kim H. K., Merianda T. T., Gallo G., Twiss J. L. (2013) Axonally synthesized β-actin and GAP-43 proteins support distinct modes of axonal growth. J. Neurosci. 33, 3311–3322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cho Y., Di Liberto V., Carlin D., Abe N., Li K. H., Burlingame A. L., Guan S., Michaelevski I., Cavalli V. (2014) Syntaxin13 expression is regulated by mammalian target of rapamycin (mTOR) in injured neurons to promote axon regeneration. J. Biol. Chem. 289, 15820–15832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Merianda T. T., Vuppalanchi D., Yoo S., Blesch A., Twiss J. L. (2013) Axonal transport of neural membrane protein 35 mRNA increases axon growth. J. Cell Sci. 126, 90–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tigges U., Koch B., Wissing J., Jockusch B. M., Ziegler W. H. (2003) The F-actin cross-linking and focal adhesion protein filamin A is a ligand and in vivo substrate for protein kinase C α. J. Biol. Chem. 278, 23561–23569 [DOI] [PubMed] [Google Scholar]
- 46. Kim H., Nakamura F., Lee W., Hong C., Pérez-Sala D., McCulloch C. A. (2010) Regulation of cell adhesion to collagen via β1 integrins is dependent on interactions of filamin A with vimentin and protein kinase C ϵ. Exp. Cell Res. 316, 1829–1844 [DOI] [PubMed] [Google Scholar]
- 47. Kim H., Nakamura F., Lee W., Shifrin Y., Arora P., McCulloch C. A. (2010) Filamin A is required for vimentin-mediated cell adhesion and spreading. Am. J. Physiol. Cell Physiol. 298, C221–C236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Perlson E., Hanz S., Ben-Yaakov K., Segal-Ruder Y., Seger R., Fainzilber M. (2005) Vimentin-dependent spatial translocation of an activated MAP kinase in injured nerve. Neuron 45, 715–726 [DOI] [PubMed] [Google Scholar]
- 49. Eva R., Crisp S., Marland J. R., Norman J. C., Kanamarlapudi V., ffrench-Constant C., Fawcett J. W. (2012) ARF6 directs axon transport and traffic of integrins and regulates axon growth in adult DRG neurons. J. Neurosci. 32, 10352–10364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Eva R., Dassie E., Caswell P. T., Dick G., ffrench-Constant C., Norman J. C., Fawcett J. W. (2010) Rab11 and its effector Rab coupling protein contribute to the trafficking of β 1 integrins during axon growth in adult dorsal root ganglion neurons and PC12 cells. J. Neurosci. 30, 11654–11669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Andrews M. R., Czvitkovich S., Dassie E., Vogelaar C. F., Faissner A., Blits B., Gage F. H., ffrench-Constant C., Fawcett J. W. (2009) α9 integrin promotes neurite outgrowth on tenascin-C and enhances sensory axon regeneration. J Neurosci. 29, 5546–5557 [DOI] [PMC free article] [PubMed] [Google Scholar]