

Background: The highly related small GTPases RalA and RalB exhibit distinct functions in cancer cell processes.
Results: Posttranslational modifications signaled by the C-terminal CAAX motif contribute to Ral isoform differences in subcellular localization, activation, and protein stability.
Conclusion: Modifications catalyzed by RCE1, ICMT, and protein palmitoyl acyltransferase cause Ral isoform-specific functional consequences.
Significance: Inhibitors of CAAX motif modifications will have complex consequences on Ral GTPase regulation.
Keywords: intracellular trafficking, plasma membrane, protein isoprenylation, protein palmitoylation, proteolytic enzyme, Ras protein, signal transduction, small GTPase, ICMT, RCE1
Abstract
The Ras-like small GTPases RalA and RalB are well validated effectors of RAS oncogene-driven human cancer growth, and pharmacologic inhibitors of Ral function may provide an effective anti-Ras therapeutic strategy. Intriguingly, although RalA and RalB share strong overall amino acid sequence identity, exhibit essentially identical structural and biochemical properties, and can utilize the same downstream effectors, they also exhibit divergent and sometimes opposing roles in the tumorigenic and metastatic growth of different cancer types. These distinct biological functions have been attributed largely to sequence divergence in their carboxyl-terminal hypervariable regions. However, the role of posttranslational modifications signaled by the hypervariable region carboxyl-terminal tetrapeptide CAAX motif (C = cysteine, A = aliphatic amino acid, X = terminal residue) in Ral isoform-selective functions has not been addressed. We determined that these modifications have distinct roles and consequences. Both RalA and RalB require Ras converting CAAX endopeptidase 1 (RCE1) for association with the plasma membrane, albeit not with endomembranes, and loss of RCE1 caused mislocalization as well as sustained activation of both RalA and RalB. In contrast, isoprenylcysteine carboxylmethyltransferase (ICMT) deficiency disrupted plasma membrane localization only of RalB, whereas RalA depended on ICMT for efficient endosomal localization. Furthermore, the absence of ICMT increased stability of RalB but not RalA protein. Finally, palmitoylation was critical for subcellular localization of RalB but not RalA. In summary, we have identified striking isoform-specific consequences of distinct CAAX-signaled posttranslational modifications that contribute to the divergent subcellular localization and activity of RalA and RalB.
Introduction
The Ras-like (Ral) small GTPases (RalA and RalB) are members of the Ras branch of the Ras superfamily of small GTPases (1–3). Like Ras, Ral small GTPases function as molecular on-off switches and cycle between inactive GDP- and active GTP-bound states. The cycle is catalyzed by Ral-selective guanine nucleotide exchange factors (RalGEFs)3 and GTPase-activating proteins (RalGAPs), which accelerate slow intrinsic exchange and GTPase activities, respectively (4). Ral GTPases are activated downstream of Ras oncoproteins. Activated Ras-GTP binds to and activates RalGEFs (e.g. RalGDS), stimulating formation of Ral-GTP. Active Ral-GTP binds preferentially to a spectrum of functionally diverse downstream effectors to regulate a diversity of cellular processes that include actin cytoskeletal organization, endocytosis and exocytosis, and mitochondrial function (5–8), as well as cell proliferation, survival, and autophagy (9, 10). The best validated effectors of Ral are the Sec5 and Exo84 subunits of the exocyst complex and RalBP1/RLIP76, a GAP for Rho family small GTPases (1–3).
RalA and RalB share significant overall sequence, structural, and biochemical identity (3). Their amino-terminal G-domains (amino acids 12–176) involved in GTP binding and hydrolysis, RalGEF and RalGAP regulation, and effector binding share 88% sequence identity. Remarkably, despite their regulation by shared GEFs and GAPs and interaction with shared effectors in vitro, RalA and RalB display divergent biological roles in normal and neoplastic cell growth. RalB was found to be specifically required for survival of tumor but not normal cells (11). In contrast, RalA was dispensable for survival, but was required for anchorage-independent proliferation. Differences in Ral isoform function have also been observed in various cancer types. Distinct aspects of cell migration were differentially regulated by RalA versus RalB in bladder cancer cells (12). RalA was essential for tumorigenic growth of pancreatic cancer cells, whereas RalB was required for invasive and metastatic growth (13). In contrast, RalA promoted whereas RalB antagonized anchorage-independent growth of colorectal carcinoma cells (14).
Like Ras, Ral proteins terminate in carboxyl-terminal CAAX tetrapeptide motifs (where C = cysteine, A = aliphatic amino acid, and X = terminal amino acid; Ral amino acids 203–206), which signal for a series of posttranslational modifications that facilitate intracellular trafficking and translocation to the plasma membrane. CAAX-signaled modifications begin with covalent addition of an isoprenoid lipid to the cysteine residue of the CAAX motif (15). The X residue determines protein prenyltransferase specificity (16). RalA and RalB proteins, both of which terminate in leucine, are modified by addition of a geranylgeranyl lipid, catalyzed by cytosolic geranylgeranyltransferase-I (GGTase I) (17, 18). Subsequent cleavage of the -AAX tripeptide by endoplasmic reticulum-associated Ras converting CAAX endopeptidase 1 (RCE1) leaves a lipid-modified terminal cysteine. This cysteine is then methylated by endoplasmic reticulum-associated isoprenylcysteine carboxylmethyltransferase (ICMT) (19).
The RalA and RalB functional differences identified to date have been attributed to the carboxyl-terminal sequences immediately adjacent to their CAAX motifs (7, 20). Termed the hypervariable region (HVR; amino acids 177–202), this sequence exhibits the greatest divergence between RalA and RalB (44% identity) and confers secondary membrane-targeting specificity. In particular, the RalA and RalB carboxyl termini possess phosphorylation sites recognized by distinct protein kinases, where phosphorylation causes translocation from the plasma membrane to endomembranes and a change in effector interaction (21–25). However, posttranslational modifications at other sites may also play an important role in Ral isoform functions.
A recent study of the brain-specific isoform of the Rho family small GTPase Cdc42 concluded that proteins terminating in a CA1A2X motif in which the A1 residue is a cysteine (CA1A2X = CCAX) can undergo an alternative processing pathway (26). Although a majority of Cdc42 undergoes the conventional post-prenyl modification pathway involving RCE1 and ICMT, a subset (5–20%) does not undergo RCE1-catalyzed removal of the AAX residues, and instead the A1 cysteine is covalently modified by palmitoylation. The resulting dually lipid-modified, nonmethylated pool of Cdc42 is no longer regulated by RhoGDIα, leading to enhanced plasma membrane association. The CAAX motif of RalA is CCIL, and that of RalB is CCLL (27), suggesting that, like Cdc42, Ral proteins may also undergo this alternative processing. However, unlike Cdc42, the membrane association of RalA and RalB is not regulated by RhoGDIα. Although both RalA and RalB were verified to undergo palmitoylation of the A1 cysteine of their CA1A2X motifs (26), the impact of dual lipid modification of the CAAX motif on Ral subcellular localization was not determined.
Additionally, the contributions of the CAAX-signaled RCE1 and ICMT modifications to RalA and RalB subcellular localization and membrane association have not been addressed. We therefore determined the consequences of RCE1 or ICMT deficiency on these properties of each Ral isoform. We found that both RalA and RalB required RCE1 for efficient plasma membrane targeting, whereas only RalB required ICMT for this association. Interestingly, whereas RalA could still concentrate at the plasma membrane in the absence of ICMT, it depended on ICMT for localization to recycling endosomes. We also found that RCE1 deficiency increased steady-state activity of both RalA and RalB, whereas ICMT deficiency increased steady-state levels of RalB but not RalA. Finally, we determined that CA1AX palmitoylation was essential for plasma membrane association of RalB, but not of RalA. Pharmacologic inhibitors of RCE1 and ICMT are being considered as anti-Ras therapeutic strategies (28). Because Ral GTPases are key drivers of Ras-dependent cancer growth, it will be critical to understand the complex and differential consequences of RCE1 and ICMT inhibition for Ral function.
Experimental Procedures
Plasmids and Cell Lines
cDNAs encoding green fluorescent protein (GFP)-tagged human RalA was described previously (22) and RalB was generated for this study as described for RalA. Site-directed mutagenesis was done to generate cysteine to serine missense mutants of the A1 cysteine residue of the Ral CCI/LL CA1A2X motif, which were then subcloned into pEGFP-C3. The pEGFP-RalA/B chimeric construct was provided by Dr. Christopher Counter (Duke University). The RalB/A chimeric cDNA was subcloned into pEGFP between the BamHI and XbaI sites. The pCDH-mCh lentivirus expression vector encoding mCherry-tagged RalA was previously described (21). All constructs were sequence-verified, and cloning details are available upon request. Mouse embryo fibroblasts (MEFs) deficient in RCE1 (Rce1−/−) or ICMT (Icmt−/−) were provided by Dr. Steven Young (UCLA) and cultured as described previously (29, 30).
Transfection, Transferrin Labeling, and Microscopy
Cells were plated, transfected, and imaged as described previously (31). Lipofectamine Plus was used for DNA transfections. Three h after transfection, cells were washed and cultured in phenol red-free Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum. Cells were labeled with transferrin following the manufacturer's protocol (Life Technologies). Cells were imaged in 35-mm culture dishes containing a 15-mm coverslip on the bottom made of number 1.5 glass (MatTek). Cells were examined with an inverted laser-scanning confocal microscope (Zeiss 710 LSM) using an oil immersion ×63 objective with a 1.4 numerical aperture. Images of GFP expression were captured by scanning with a 488-nm line of a solid-state laser using a BP 490–555 emission filter. Images of mCh and red fluorescent transferrin probes were captured by scanning with a 555-nm solid-state laser with the variable secondary dichroic set to 580 nm. Images were processed with ImageJ software, and thresholded Manders coefficients were calculated using the colocalization threshold plugin from the Wright Cell Imaging Facility.
GST Pulldown Assays
Endogenous activated RalA- and RalB-GTP were assayed as we described previously, by pulldown analyses using GST-Sec5-RBD (glutathione S-transferase fusion of the Ral-GTP binding domain of Sec5) (20). After SDS-PAGE and immunoblotting with the RalA- (BD Biosciences) or RalB (Millipore)-specific antibodies, the amount of active Ral-GTP was normalized to total Ral protein levels in total cellular lysates. Western blot analyses with anti-β-actin (Sigma) or anti-GAPDH (Sigma) were done to verify equivalent loading of total cellular protein.
Detection of Protein Palmitoylation
Palmitoylated proteins were purified using acyl-resin-assisted capture (acyl-RAC). Cells were lysed with RIPA buffer (20 mm Hepes-NaOH, pH 7.4, 100 mm NaCl, 3 mm MgCl2, 1% Triton X-100, 0.5% deoxycholate, and 0.1% SDS) supplemented with protease inhibitor. Cleared lysates were incubated in RIPA buffer containing 0.1% methyl methanethiosulfonate at 42 °C for 30 min with rotation. Proteins were precipitated with acetone, and then resuspended in binding buffer (100 mm Hepes-NaCl, pH 7.4, 1 mm EDTA, and 1% SDS). Samples were mixed with thiopropyl-Sepharose in the presence of 250 mm hydroxylamine to cleave thioester linkages. As a negative control, the same volume of NaCl was added instead of hydroxylamine. After a 3-h rotation at room temperature, the resins were washed four times with binding buffer, and samples were eluted in buffer containing 50 mm Tris-HCl, pH 6.8, 50 mm DTT, 10% glycerol, and 1% SDS. After immunoblotting with the RalA- and RalB-specific antibodies, the palmitoylation stoichiometry was calculated by a ratio of the precipitate intensity to the input intensity.
Quantification of Fluorescence
The statistics of fluorescence intensity were extracted from images of individual cells as a function of the distance from the cell edge. Cell masks (binary images) were obtained by thresholding with the MovThresh module of the CellGeo package (32). Each cell mask was then morphologically eroded using the MATLAB function “imerode” with a disk radius 1 to 50 pixels as a structural element. At each step of the morphological erosion, the boundary was traced and the intensity of the corresponding boundary pixels was determined from the original image. This allowed the mean, median, and standard deviation to be calculated for each cell as function of the distance 1 to 50 pixels from the cell edge. Finally, the intensity profiles for individual cells were normalized to their peak (max) values, and then averaged over all cells in a given genotype. These four processing steps constitute a single MATLAB code for batch processing.
Results
RalA and RalB Show Distinct Requirements for RCE1- and ICMT-mediated Posttranslational Modifications for Their Subcellular Localization and Plasma Membrane Association
We first addressed the importance of RCE1- and ICMT-catalyzed modifications in regulating RalA and RalB subcellular localization and membrane association. Because there are currently no potent and selective pharmacologic inhibitors of these enzymes, we utilized MEFs deficient in expression of either RCE1 or ICMT (29, 30). We transiently transfected wild-type (WT), Rce1−/−, or Icmt−/− MEFs with expression constructs encoding RalA or RalB, which were amino terminally tagged with GFP (Fig. 1A). To quantitate changes in Ral plasma membrane association, we developed a MATLAB image processing script to analyze fluorescence measurements for localization patterns in individual cells (Fig. 1, B and C). In particular, we quantified the GFP intensity as a function of the distance from the plasma membrane to the nucleus. As described previously (6, 7, 21), we found that RalA and RalB each exhibited both plasma membrane and endosome localization in WT MEFs (Fig. 1A). However, we found that in Rce1−/− MEFs, both RalA and RalB were mislocalized, with diminished plasma membrane association and increased diffuse cytoplasmic accumulation, although their endosome localization remained intact (Fig. 1A). These results were somewhat unexpected, because a previous study suggested that small GTPases modified by the hydrophobic geranylgeranyl lipid attached to Ral proteins would not be highly dependent on ICMT- and RCE1-catalyzed modifications for efficient plasma membrane association (33).
FIGURE 1.

Subcellular localization of Ral family proteins is differentially dependent on RCE1 and ICMT processing. Wild-type (WT), Rce1−/−, and Icmt−/− MEFs were transiently transfected with expression vectors encoding EGFP-tagged RalA or RalB. A, live cells were visualized using confocal microscopy. Images shown are representative of three independent experiments, each of which examined >40 cells. Images were analyzed using our MATLAB image processing script, for B, EGFP-RalA distribution, and C, EGFP-RalB distribution in WT, Rce1−/−, and Icmt−/− MEFs. Scale bar, 10 μm.
Because RCE1 deficiency also prevents the subsequent ICMT modification, we next determined the consequences of ICMT deficiency alone. Although the plasma membrane targeting of both Ral isoforms depended similarly on RCE1, surprisingly, we found a differential requirement for ICMT in regulating RalA versus RalB plasma membrane targeting. In Icmt−/− MEFs, we observed that RalA remained concentrated at the plasma membrane, similar to its localization in WT MEFs. Thus, the significant disruption of RalA plasma membrane association in Rce1−/− cells was caused by the absence of AAX proteolysis. In striking contrast, RalB association with the plasma membrane was reduced significantly in Icmt−/− MEFs, instead showing increased diffuse localization in the cytosol. Thus, plasma membrane association of RalB displays a greater requirement for ICMT compared with that of RalA.
We next investigated the basis for the different susceptibility of RalA and RalB to ICMT deficiency. Although their sequence divergence is greatest in the HVR (44% identity), there is also significant sequence divergence in the carboxyl-terminal portion of the G-domain (residues 91–153; 68% identity) (Fig. 2A). To distinguish between the importance of these two blocks of sequence divergence in ICMT-dependent Ral localization, we utilized chimeric RalA and RalB proteins that terminate in the reciprocal HVR (Fig. 2A). RalA/B is comprised of the RalA N terminus (residues 1–176), which includes RalA sequences involved in effector and regulatory protein binding, and the carboxyl-terminal HVR of RalB (residues 177–204) involved in membrane targeting. Conversely, RalB/A is comprised of the N terminus of RalB (residues 1–176) followed by the carboxyl-terminal HVR of RalA (residues 177–204). We transiently expressed GFP fusion proteins of RalA, RalB, RalA/B, or RalB/A in WT or Icmt−/− MEFs and observed their localization patterns. As expected, in WT MEFs the RalA/B and RalB/A chimeric proteins retained the predominantly plasma membrane localization patterns of full-length RalA and RalB (data not shown). However, in Icmt−/− MEFs, whereas RalB/A retained the strong plasma membrane subcellular localization of full-length RalA (Fig. 2, A and B), the RalA/B chimeric protein exhibited a loss of plasma membrane association similar to that seen with full-length RalB. We conclude that the distinct consequences of ICMT deficiency seen with RalA and RalB are due to sequence divergence in the HVR rather than in the G-domain.
FIGURE 2.
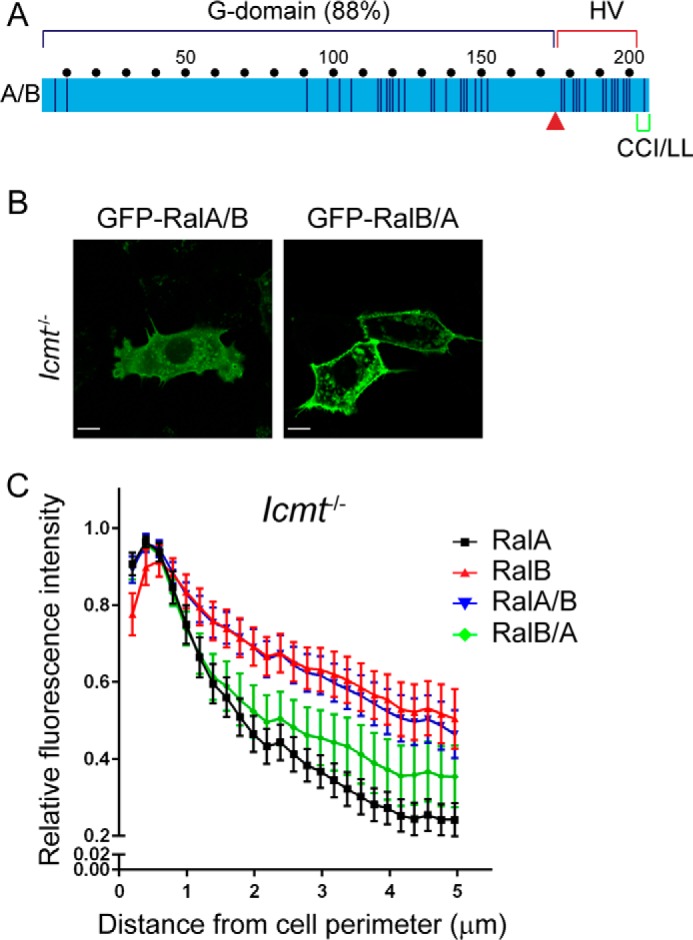
Hypervariable domain dictates Ral ICMT dependence. A, chimeric Ral proteins were constructed by a reciprocal switch of the carboxyl-terminal residues 176–206 (red arrow) comprised of the hypervariable domain and the CAAX motif. RalA/B is comprised of the RalA G-domain (residues 1–176) followed by RalB residues 177–206, whereas RalB/A is comprised of the RalB G-domain (residues 1–176) followed by RalA residues 177–206. WT, Rce1−/−, and Icmt−/− MEF cells were transiently transfected with expression constructs encoding EGFP-tagged Ral chimeras RalA/B or RalB/A. B, live cells were visualized by confocal microscopy. Images shown are representative of three independent experiments, each of which examined >40 cells. C, images from B were analyzed using our MATLAB image processing script. Scale bar, 10 μm.
Icmt Regulates RalA Localization to Recycling Endosomes
Although ICMT deficiency did not noticeably impact RalA plasma membrane targeting, we did observe a decreased level of endomembrane-associated RalA in Icmt−/− MEFs (Fig. 1A). Therefore, we used our MATLAB image processing script to quantify the spatial distribution of GFP-RalA. Our analysis revealed a decreased concentration of RalA closer to the nucleus in Icmt−/− MEFs. In contrast, RalB association with internal organelles was not impacted significantly by ICMT deficiency (Fig. 1B). Thus, RalA and RalB association with endomembranes exhibited different requirements for ICMT.
Because RalA is known to localize to recycling endosomes in various cell types (6, 7), we speculated that the RalA endomembrane localization that was disrupted in the absence of ICMT included this compartment. To address this possibility, we detected recycling endosomes by live cell microscopy using a fluorescent transferrin probe, and measured the colocalization of GFP-RalA with transferrin-loaded endosomes. Image analysis revealed a significant decrease in colocalization of RalA with transferrin in Icmt−/− MEFs compared with WT MEFs, indicating that RalA localization to recycling endosomes was impaired in the absence of ICMT (Fig. 3, A and B).
FIGURE 3.

RalA localization on recycling endosomes is impaired by loss of ICMT. A, WT and Icmt−/− MEF cells were transiently transfected with an expression construct encoding EGFP-RalA and incubated with pHrodo red transferrin conjugate for 30 min before visualization by confocal microscopy. The images were overlapped (Merge) to indicate the degree of colocalization on endosomes in WT cells compared with Icmt−/−. B, 25 images per condition were quantified. Values shown are means of Manders coefficients using thresholds ± S.E., and an unpaired t test was used to determine significance (p < 0.05). C, WT and Icmt−/− MEF cells were transiently transfected with expression constructs encoding EGFP-Rab9 and mCh-RalA. Cells were visualized by confocal microscopy. Images were merged as described for panel A, and in D were quantified as described in panel B. Scale bar, 10 μm.
To further validate this finding, we also measured RalA colocalization with a second marker of recycling endosomes, Rab9. For these analyses, we co-transfected expression vectors encoding mCherry-tagged RalA and GFP-Rab9 into WT or Icmt−/− MEFs. Colocalization analysis again showed a significant reduction in localization of RalA to Rab9-containing endosomes in Icmt−/− MEFs (Fig. 3, C and D). These results demonstrate that loss of ICMT significantly impaired RalA localization to recycling endosomes.
RCE1 Regulates Ral Activity
Because we previously observed that phosphorylation-mediated translocation of RalA or RalB from the plasma membrane to endosomes was associated with an increase in active GTP-bound protein (21, 22), we speculated that the altered subcellular localization of RalA and RalB seen in RCE1- or ICMT-deficient cells might also be accompanied by altered regulation of their GDP-GTP cycle and consequently of their activation state. To address this possibility, we first performed pulldown analyses to determine the level of activated, GTP-bound endogenous RalA and RalB in WT, Rce1−/−, and Icmt−/− MEFs. When normalized to total protein, we found that the steady-state levels of both endogenous RalA-GTP and RalB-GTP were elevated strikingly in Rce1−/− MEFs compared with WT MEFs (Fig. 4A). In contrast, there was limited increase in RalA-GTP and no increase in RalB-GTP levels in Icmt−/− MEFs, although RalB protein levels were markedly higher in these cells compared with WT MEFs.
FIGURE 4.

RCE1 deficiency increases activated Ral-GTP formed upon EGF stimulation. A, endogenous levels of RalA-GTP and RalB-GTP were compared in WT, Rce1−/−, and Icmt−/− MEFs by a pulldown assay using GST-Sec5 RBD. Western blot analyses were done with anti-RalA or anti-Ral antibody to determine levels of Ral-GTP and total Ral proteins. β-Actin was used to verify equivalent total protein loading. B, WT and Rce1−/− MEFs were serum-starved then stimulated with EGF. Cells were lysed at the indicated time points, followed by Western blot analyses as described in panel A. C, RalA-GTP, and D, RalB-GTP expression levels were quantified by densitometry of Western blot data in panel B and normalized to total RalA and RalB, respectively.
The elevated activity of the Ral GTPases observed in the absence of RCE1 when compared with WT cells suggests differential regulation of the mislocalized Ral proteins by their regulatory RalGAPs or RalGEFs. To address this possibility, we evaluated the kinetics of RalA-GTP formation in response to external stimuli. Growth factor-stimulated Ral activation is mediated through transient activation of Ras, leading to RalGEF activation and a rapid and transient increase in RalA-GTP (34), with RalGAP activity then terminating the activation. A previous study showed that, in cells depleted of RalGAP, EGF treatment resulted in an increased level of Ral activation and a decreased rate of termination of activation, leading to sustained Ral activity in stimulated cells (35). Therefore, we compared the kinetics of endogenous Ral-GTP formation in response to EGF in WT versus Rce1−/− MEFs.
We stimulated serum-starved WT and Rce1−/− MEFs with EGF and then monitored the GTP-bound levels of RalA and RalB over time by pulldown analyses (Fig. 4B). Although the activity of both RalA and RalB increased in response to EGF stimulation and returned to near basal levels at the same rate in both cell types, we did find that EGF stimulation caused a significantly greater initial increase in RalA-GTP and RalB-GTP activity in Rce1−/− MEFs compared with WT MEFs (Fig. 4, C and D). Thus, the Ral mislocalization that we observed in Rce1−/− MEFs is more likely to alter their regulation by one or more of the 6 RalGEFs than by RalGAPs (3).
Icmt Regulates RalB Stability
As described above, we observed that levels of endogenous RalB but not RalA proteins were stably elevated in Icmt−/− MEFs compared with WT MEFs (Fig. 4A). Previous studies demonstrated that RhoA protein was destabilized upon pharmacologic inhibition or genetic loss of ICMT, whereas Ras protein was stabilized (36, 37). Although we did find that the steady-state transcript levels of RalB were slightly greater in Icmt−/− MEFs compared with WT MEFs (Fig. 5A), this small change likely was an insufficient basis for the large difference in abundance of RalB protein in the two cell types (Fig. 5B). We therefore hypothesized that the increases in RalB protein in Icmt−/− MEFs were due to altered regulation of RalB stability in the absence of ICMT.
FIGURE 5.

ICMT deficiency increases RalB protein stability. A, RalB transcript levels were determined by TaqMan quantitative PCR analysis and normalized to GABPB1 in a triplicate set of assays ± S.E. Data displayed as relative to WT MEFs. B, endogenous RalB protein levels were quantified by densitometry of Western blot and normalized to total protein. Data are displayed as relative to WT MEFs. Experiment performed in triplicate. C, WT and Icmt−/− MEFs were treated with 25 μg/ml of cycloheximide and lysed at the indicated time points. Lysates were probed by Western blot for total RalB, c-Myc, and for GAPDH to verify equivalent total protein. Protein levels from blots in panel were quantified by densitometry and normalized to 0 h.
To investigate if RalB turnover was affected by loss of ICMT, we treated WT and Icmt−/− MEFs with cycloheximide to block protein synthesis and then measured changes in protein levels as the pre-existing RalB protein was degraded over time. We found that RalB exhibited a shorter half-life in WT MEFs compared with Icmt−/− MEFs, with only 20% RalB protein remaining in WT MEFs after 18 h of cycloheximide treatment compared with ∼60% in Icmt−/− MEFs (Fig. 5C). This difference indicates that ICMT regulates RalB protein stability and promotes its degradation.
Dual Lipid Modification of the Ral CAAX Motif Affects RalB Subcellular Localization
In addition to the conventional modification pathway involving RCE1 and ICMT, RalA and RalB can also undergo an alternative pathway, whereby the newly GGTase I-modified protein does not undergo subsequent RCE1-mediated proteolytic removal of the A1A2X residues but instead undergoes palmitoylation of the cysteine residue at the A1 position (26). Although the mechanism that regulates which pathway will be taken has not been elucidated, it was reported that the level of palmitoylation of RalA was increased in RCE1-deficient MEFs. Thus, impairment of one pathway can then favor a shift to the other pathway. We hypothesized that the altered subcellular localization of RalA and RalB that we observed in Rce1−/− MEFs (Fig. 1) might reflect not only loss of the RCE1- and ICMT-catalyzed modifications but additionally reflect alterations in dual lipidation by geranylgeranylation and palmitoylation of the CAAX motif.
To determine a role for this alternative dual lipid modification pathway in regulating RalA and RalB localization, we first generated the same palmitoylation-deficient Ral mutants utilized by Nishimura and Linder (26) in defining the pathway. To do so, we introduced a Cys to Ser amino acid substitution at the A1 position of each Ral isoform (C204S), designated RalA-CSIL and RalB-CSLL. This substitution ablates palmitoylation but not geranylgeranylation of the CAAX motif (26). Although we did not observe a significant alteration in RalA-CSIL subcellular localization compared with WT RalA, we did find reduced association of RalB-CSLL with the plasma membrane, along with increased cytosolic accumulation (Figs. 1A and 6A). To address the role of geranylgeranylation in Ral localization, we generated nonprenylatable RalB-SCLL and RalA-SCIL C203S mutants that are deficient in both the geranylgeranyl and palmitate modifications. As expected (18), we found that these mutants were completely disrupted in both plasma membrane and endomembrane localization, and showed a diffuse cytoplasmic distribution including nuclear accumulation (Fig. 6B). We used our MATLAB image processing script to quantify the spatial distribution of the nonpalmitoylated and nonprenylated mutant Ral proteins (Fig. 6C). We found that the alternative dual lipid modification site of the RalB but not the RalA CAAX motif affects subcellular localization to the plasma membrane. In support of this conclusion, analysis of palmitoylation of endogenous RalA and RalB revealed that the level of RalB palmitoylation is ∼2-fold greater than that of RalA (Fig. 6, D and E). An explanation of this result is that disruption of the palmitoylatable cysteine of RalB also disrupted prenylation. To address this, we inhibited geranylgeranylation of the palmitoylation-defective RalB-CSLL mutant with the pharmacological GGTase I inhibitor GGTI-2017. This treatment resulted in a localization similar to that of the nongeranylgeranylated mutant RalB-SCLL (Fig. 6F). We conclude that disruption of the palmitoylatable cysteine does not disrupt geranylgeranylation.
FIGURE 6.

CA1AX palmitoylation dictates RalB subcellular localization. WT MEFs were transiently transfected with an expression construct encoding either nonpalmitoylatable or nonprenylatable CAAX mutants of EGFP-RalA or EGFP-RalB, and then visualized by confocal microscopy. A, nonpalmitoylatable C204S CA1AX mutants (RalA-CSIL or RalB-CSLL) or B, nonprenylatable C203S SAAX mutant (RalA-SCIL or RalB-SCLL). C, images from A and B were analyzed using our MATLAB image processing script. D, hydroxylamine (NH2OH)-sensitive palmitoylated proteins from WT MEF lysates were immobilized on thiopropyl-Sepharose resin using resin assisted capture (acyl-RAC), and palmitoylation of RalA and RalB was detected by immunoblotting. E, palmitoylation stoichiometry was calculated by a ratio of the precipitate intensity to the input intensity. Data represent the mean ± S.E. (n = 6). F, WT MEFs were transiently transfected with the nonprenylatable C203S mutant EGFP-RalB-CSLL as in panel B, but then treated with 25 μm GGTI-2017 or dimethyl sulfoxide control for 24 h before visualization by confocal microscopy.
Discussion
Despite their significant sequence, structural, and biochemical similarities, RalA and RalB often serve divergent roles in normal and neoplastic cell functions (1–3, 38). In this study, we determined whether the CAAX-signaled posttranslational modifications of proteolysis, isoprenylcysteine carboxylmethylation, and CAAX motif palmitoylation could contribute to the distinct regulation of RalA and RalB. We found that both RalA and RalB require RCE1 function for association with the plasma membrane but not with endomembranes. In contrast, ICMT deficiency disrupted RalB association with the plasma membrane, whereas it disrupted RalA association with endosomes. Assessing the consequences for Ral regulation, we found that RCE1 deficiency increased the steady-state activation state of both RalA and RalB, whereas ICMT deficiency regulated protein stability of RalB, but not RalA. Finally, we determined that the A1 cysteine in the CA1A2X motif that leads to alternative modification by the fatty acid palmitate was more significant for RalB localization than for RalA. (Ral proteins do not contain the upstream palmitoylatable cysteines present in the HVR of many Ras family small GTPases (27).) We conclude that the differential roles of CAAX-signaled posttranslational modifications contribute significantly to the distinct regulation of RalA and RalB.
Previous reports have reached conflicting conclusions regarding the contribution of CAAX-signaled postprenyl modifications in promoting proper subcellular localization and membrane association of small GTPases. One study showed that both RCE1 and ICMT were critical for localization of farnesylated but not geranylgeranylated Ras and Rho proteins (33), whereas we found that loss of either RCE1 or ICMT did significantly impact the subcellular localization and membrane association of geranylgeranylated Rho GTPases (39). Although total membrane-bound RalA was shown previously to be unaffected by loss of ICMT, its differential membrane compartmentalization was not examined, and neither the membrane association nor localization of RalB was investigated (33). In the present study, we have demonstrated that the geranylgeranylated proteins RalA and RalB also require RCE1 and ICMT for proper subcellular localization. Thus, we conclude that the specific prenyl group modifying CAAX-containing proteins is not sufficient by itself to predict dependence on RCE1- and ICMT-catalyzed modifications. Because our studies utilized RCE1- and ICMT-deficient cells, we recognize that the consequences of their loss on Ral subcellular localization may be indirect and not due specifically to disrupted modification of Ral itself. However, because no known CAAX-terminating proteins have been described that interact with Ral, we feel that our results do likely demonstrate direct roles of these modifications in Ral function.
Although RalA and RalB both localize to the plasma membrane and to endosomes, they exhibited striking differences in the requirement of RCE1 and ICMT to facilitate their association with these two membrane compartments. Loss of RCE1 disrupted association of both RalA and RalB with the plasma membrane but not with endosomal membranes. In contrast, loss of ICMT disrupted RalB but not RalA plasma membrane association, whereas conversely disrupting endosome localization of RalA but not RalB. Utilizing chimeric Ral proteins, we determined that this differential regulation was due to sequence divergence in the HVR rather than in the G-domain. Both HVRs are largely lysine-rich sequences and contain no other obvious membrane targeting information. Therefore, how each HVR impacts the different consequences of ICMT deficiency is not clear. However, they do diverge in the location and sequence context of the serines that serve as phosphorylation sites for an isoform-specific variety of protein kinases (21–25). We conclude that inhibition of ICMT is a possible approach to blocking endosomal RalA function, and that this would provide a level of selectivity over inhibition of RCE1. Endosomal RalA function is important for cell processes including exocytosis (5, 40), cytokinesis (6), and cell polarity maintenance (7). Although it is unclear why prenylation of RalA followed by RCE1 processing alone is sufficient to take RalA to the plasma membrane, we previously found that the RhoB small GTPase also showed the same dependence (39). Thus, with some prenylated proteins, carboxylmethylation is dispensable for plasma membrane association. It is also possible that, because ICMT has additional substrates compared with RCE1, mislocalization of these other proteins may be affecting RalA and RalB differently.
In addition to altered subcellular localization, we also observed consequences for Ral activation in RCE1-deficient cells. We found that loss of RCE1 enhanced the steady-state activity of RalA and RalB. We speculated that this altered activity is a consequence of their altered subcellular localization and therefore of their interaction with RalGEFs or RalGAPs. Our analyses found that the kinetics of Ral-GTP formation upon EGF stimulation was consistent with increased activation by RalGEFs rather than decreased inactivation by RalGAPs. Because Ral proteins are displaced from the plasma membrane in Rce1−/− MEFs, there could be increased exposure of Ral to specific RalGEFs on internal membranes. For example, the RalGEF RGL2 is enriched on recycling endosomes (40). We conclude that RCE1 deficiency leads to aberrant RalA and RalB activation, and suggest that therapeutic intervention targeting RCE1 in Ras-dependent cancers could have pitfalls in unintentional Ral effector pathway activation.
Another consequence of the loss of CAAX-signaled modifications was the increased steady-state level of RalB protein but not RNA in Icmt−/− MEFs. Previous studies reported that ICMT-deficient, mutant K-Ras-transformed MEFs exhibited increased Ras protein stabilization but conversely a decreased half-life of RhoA protein but not RNA. However, no mechanistic basis for these observations was described (36). Although ubiquitination of Ral has been implicated in Ral-dependent lipid raft trafficking (41), a degradation mechanism for Ral has not been described. Perhaps RalB interaction with an E3 ubiquitin ligase controlling Ral stability is affected by mislocalization associated with loss of ICMT. Alternatively, the absence of the methylation modification, independent of altered localization, may alter RalB interaction with an E3 ligase. Consistent with this possibility, there is evidence that prenylcysteine methylation of CAAX-containing proteins can also play a role in regulating protein-protein interactions (42–45). In summary, our observations provide the first evidence that RalB protein stability may be regulated by a posttranslational mechanism.
Pharmacologic inhibition of CAAX-signaled modifications has been pursued extensively as an approach to inhibit Ras for cancer treatment. Initial efforts targeted the first step, catalyzed by farnesyltransferase. Unexpectedly, farnesyltransferase inhibitors were ineffective in blocking the membrane association of K-Ras and N-Ras, due to farnesyltransferase inhibitor-induced alternative prenylation by GGTase I (46, 47). Because blocking Ras membrane association remains an attractive approach (48), pharmacologic inhibition of RCE1 and ICMT has been considered. Initial cell-based studies using RCE1- or ICMT-deficient MEFs found that K-Ras-induced growth transformation was impaired (36, 49). However, subsequent mouse model studies indicate context-dependent consequences. In a mouse model of K-Ras-induced myeloproliferative disease, loss of ICMT ameliorated tumor growth, whereas loss of RCE1 accelerated it (50, 51). More recently, it was found that ICMT deficiency enhanced K-Ras-driven pancreatic cancer development (52). A logical explanation for these different consequences for Ras-driven oncogenesis is that ablation of RCE1 or ICMT expression alters the function of other CAAX-terminating prenylated proteins, including small GTPases such as Ral and Rac that function downstream of Ras effector signaling, or of Ras family proteins that can act as tumor suppressors (53). In conclusion, given the large number of other RCE1 and ICMT substrates, the usefulness of pharmacologic inhibitors of RCE1 and ICMT as anti-Ras drugs will likely be highly context-dependent.
Author Contributions
L. R. G. designed and performed experiments, analyzed the data, and wrote the manuscript. A. N. and M. N. performed experiments, analyzed the data, and contributed to the manuscript. A. D. C. provided reagents, analyzed the data, and made a substantial contribution to the manuscript writing. T. D. M. provided reagents and designed experiments. D. T. and T. C. E. analyzed results and contributed to the manuscript writing. C. J. D. directed the study, analyzed results, and wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Christopher Counter (Duke University) for Ral expression vectors and Steven Young (UCLA) for Rce1- and Icmt-deficient MEFs.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 CA042978 (to C. J. D.). The authors declare that they have no conflicts of interest with the contents of this article.
- RalGEF
- Ral-selective guanine nucleotide exchange factors
- RCE1
- Ras converting CAAX endopeptidase 1
- ICMT
- isoprenylcysteine carboxylmethyltransferase
- MEF
- mouse embryonic fibroblast
- mCh
- monomeric Cherry fluorescent protein
- CAAX
- cysteine-aliphatic aliphatic-any amino acid
- HVR
- hypervariable region.
References
- 1. Bodemann B. O., White M. A. (2008) Ral GTPases and cancer: linchpin support of the tumorigenic platform. Nat. Rev. Cancer 8, 133–140 [DOI] [PubMed] [Google Scholar]
- 2. Neel N. F., Martin T. D., Stratford J. K., Zand T. P., Reiner D. J., Der C. J. (2011) The RalGEF-Ral effector signaling network: the road less traveled for anti-Ras drug discovery. Genes Cancer 2, 275–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gentry L. R., Martin T. D., Reiner D. J., Der C. J. (2014) Ral small GTPase signaling and oncogenesis: more than just 15 minutes of fame. Biochim. Biophys. Acta 1843, 2976–2988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vigil D., Martin T. D., Williams F., Yeh J. J., Campbell S. L., Der C. J. (2010) Aberrant overexpression of the Rgl2 Ral small GTPase-specific guanine nucleotide exchange factor promotes pancreatic cancer growth through Ral-dependent and Ral-independent mechanisms. J. Biol. Chem. 285, 34729–34740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Balasubramanian N., Meier J. A., Scott D. W., Norambuena A., White M. A., Schwartz M. A. (2010) RalA-exocyst complex regulates integrin-dependent membrane raft exocytosis and growth signaling. Curr. Biol. 20, 75–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen X. W., Inoue M., Hsu S. C., Saltiel A. R. (2006) RalA-exocyst-dependent recycling endosome trafficking is required for the completion of cytokinesis. J. Biol. Chem. 281, 38609–38616 [DOI] [PubMed] [Google Scholar]
- 7. Shipitsin M., Feig L. A. (2004) RalA but not RalB enhances polarized delivery of membrane proteins to the basolateral surface of epithelial cells. Mol. Cell. Biol. 24, 5746–5756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kashatus D. F., Lim K. H., Brady D. C., Pershing N. L., Cox A. D., Counter C. M. (2011) RALA and RALBP1 regulate mitochondrial fission at mitosis. Nat. Cell. Biol. 13, 1108–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Martin T. D., Chen X. W., Kaplan R. E., Saltiel A. R., Walker C. L., Reiner D. J., Der C. J. (2014) Ral and Rheb GTPase activating proteins integrate mTOR and GTPase signaling in aging, autophagy, and tumor cell invasion. Mol. Cell 53, 209–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bodemann B. O., Orvedahl A., Cheng T., Ram R. R., Ou Y. H., Formstecher E., Maiti M., Hazelett C. C., Wauson E. M., Balakireva M., Camonis J. H., Yeaman C., Levine B., White M. A. (2011) RalB and the exocyst mediate the cellular starvation response by direct activation of autophagosome assembly. Cell 144, 253–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chien Y., White M. A. (2003) RAL GTPases are linchpin modulators of human tumour-cell proliferation and survival. EMBO Rep. 4, 800–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Oxford G., Owens C. R., Titus B. J., Foreman T. L., Herlevsen M. C., Smith S. C., Theodorescu D. (2005) RalA and RalB: antagonistic relatives in cancer cell migration. Cancer Res. 65, 7111–7120 [DOI] [PubMed] [Google Scholar]
- 13. Lim K. H., O'Hayer K., Adam S. J., Kendall S. D., Campbell P. M., Der C. J., Counter C. M. (2006) Divergent roles for RalA and RalB in malignant growth of human pancreatic carcinoma cells. Curr. Biol. 16, 2385–2394 [DOI] [PubMed] [Google Scholar]
- 14. Martin T. D., Samuel J. C., Routh E. D., Der C. J., Yeh J. J. (2011) Activation and involvement of Ral GTPases in colorectal cancer. Cancer Res. 71, 206–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Berndt N., Hamilton A. D., Sebti S. M. (2011) Targeting protein prenylation for cancer therapy. Nat. Rev. Cancer 11, 775–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Reid T. S., Terry K. L., Casey P. J., Beese L. S. (2004) Crystallographic analysis of CaaX prenyltransferases complexed with substrates defines rules of protein substrate selectivity. J. Mol. Biol. 343, 417–433 [DOI] [PubMed] [Google Scholar]
- 17. Kinsella B. T., Erdman R. A., Maltese W. A. (1991) Carboxyl-terminal isoprenylation of ras-related GTP-binding proteins encoded by rac1, rac2, and ralA. J. Biol. Chem. 266, 9786–9794 [PubMed] [Google Scholar]
- 18. Falsetti S. C., Wang D. A., Peng H., Carrico D., Cox A. D., Der C. J., Hamilton A. D., Sebti S. M. (2007) Geranylgeranyltransferase I inhibitors target RalB to inhibit anchorage-dependent growth and induce apoptosis and RalA to inhibit anchorage-independent growth. Mol. Cell. Biol. 27, 8003–8014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Winter-Vann A. M., Casey P. J. (2005) Post-prenylation-processing enzymes as new targets in oncogenesis. Nat. Rev. Cancer 5, 405–412 [DOI] [PubMed] [Google Scholar]
- 20. Lim K. H., Baines A. T., Fiordalisi J. J., Shipitsin M., Feig L. A., Cox A. D., Der C. J., Counter C. M. (2005) Activation of RalA is critical for Ras-induced tumorigenesis of human cells. Cancer Cell 7, 533–545 [DOI] [PubMed] [Google Scholar]
- 21. Martin T. D., Mitin N., Cox A. D., Yeh J. J., Der C. J. (2012) Phosphorylation by protein kinase Calpha regulates RalB small GTPase protein activation, subcellular localization, and effector utilization. J. Biol. Chem. 287, 14827–14836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lim K. H., Brady D. C., Kashatus D. F., Ancrile B. B., Der C. J., Cox A. D., Counter C. M. (2010) Aurora-A phosphorylates, activates, and relocalizes the small GTPase RalA. Mol. Cell. Biol. 30, 508–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang H., Owens C., Chandra N., Conaway M. R., Brautigan D. L., Theodorescu D. (2010) Phosphorylation of RalB is important for bladder cancer cell growth and metastasis. Cancer Res. 70, 8760–8769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sablina A. A., Chen W., Arroyo J. D., Corral L., Hector M., Bulmer S. E., DeCaprio J. A., Hahn W. C. (2007) The tumor suppressor PP2A Aβ regulates the RalA GTPase. Cell 129, 969–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wu J. C., Chen T. Y., Yu C. T., Tsai S. J., Hsu J. M., Tang M. J., Chou C. K., Lin W. J., Yuan C. J., Huang C. Y. (2005) Identification of V23RalA-Ser194 as a critical mediator for Aurora-A-induced cellular motility and transformation by small pool expression screening. J. Biol. Chem. 280, 9013–9022 [DOI] [PubMed] [Google Scholar]
- 26. Nishimura A., Linder M. E. (2013) Identification of a novel prenyl and palmitoyl modification at the CAAX motif of Cdc42 that regulates RhoGDI binding. Mol. Cell. Biol. 33, 1417–1429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chardin P., Tavitian A. (1989) Coding sequences of human ralA and ralB cDNAs. Nucleic Acids Res. 17, 4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cox A. D., Fesik S. W., Kimmelman A. C., Luo J., Der C. J. (2014) Drugging the undruggable RAS: mission possible? Nat. Rev. Drug Discov. 13, 828–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kim E., Ambroziak P., Otto J. C., Taylor B., Ashby M., Shannon K., Casey P. J., Young S. G. (1999) Disruption of the mouse Rce1 gene results in defective Ras processing and mislocalization of Ras within cells. J. Biol. Chem. 274, 8383–8390 [DOI] [PubMed] [Google Scholar]
- 30. Bergo M. O., Leung G. K., Ambroziak P., Otto J. C., Casey P. J., Gomes A. Q., Seabra M. C., Young S. G. (2001) Isoprenylcysteine carboxyl methyltransferase deficiency in mice. J. Biol. Chem. 276, 5841–5845 [DOI] [PubMed] [Google Scholar]
- 31. Mitin N., Roberts P. J., Chenette E. J., Der C. J. (2012) Posttranslational lipid modification of Rho family small GTPases. Methods Mol. Biol. 827, 87–95 [DOI] [PubMed] [Google Scholar]
- 32. Tsygankov D., Bilancia C. G., Vitriol E. A., Hahn K. M., Peifer M., Elston T. C. (2014) CellGeo: a computational platform for the analysis of shape changes in cells with complex geometries. J. Cell Biol. 204, 443–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Michaelson D., Ali W., Chiu V. K., Bergo M., Silletti J., Wright L., Young S. G., Philips M. (2005) Postprenylation CAAX processing is required for proper localization of Ras but not Rho GTPases. Mol. Biol. Cell 16, 1606–1616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wolthuis R. M., Zwartkruis F., Moen T. C., Bos J. L. (1998) Ras-dependent activation of the small GTPase Ral. Curr. Biol. 8, 471–474 [DOI] [PubMed] [Google Scholar]
- 35. Shirakawa R., Fukai S., Kawato M., Higashi T., Kondo H., Ikeda T., Nakayama E., Okawa K., Nureki O., Kimura T., Kita T., Horiuchi H. (2009) Tuberous sclerosis tumor suppressor complex-like complexes act as GTPase-activating proteins for Ral GTPases. J. Biol. Chem. 284, 21580–21588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bergo M. O., Gavino B. J., Hong C., Beigneux A. P., McMahon M., Casey P. J., Young S. G. (2004) Inactivation of Icmt inhibits transformation by oncogenic K-Ras and B-Raf. J. Clin. Invest. 113, 539–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Backlund P. S., Jr. (1997) Post-translational processing of RhoA: carboxyl methylation of the carboxyl-terminal prenylcysteine increases the half-life of RhoA. J. Biol. Chem. 272, 33175–33180 [DOI] [PubMed] [Google Scholar]
- 38. Kashatus D. F. (2013) Ral GTPases in tumorigenesis: emerging from the shadows. Exp. Cell Res. 319, 2337–2342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Roberts P. J., Mitin N., Keller P. J., Chenette E. J., Madigan J. P., Currin R. O., Cox A. D., Wilson O., Kirschmeier P., Der C. J. (2008) Rho Family GTPase modification and dependence on CAAX motif-signaled posttranslational modification. J. Biol. Chem. 283, 25150–25163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Takaya A., Kamio T., Masuda M., Mochizuki N., Sawa H., Sato M., Nagashima K., Mizutani A., Matsuno A., Kiyokawa E., Matsuda M. (2007) R-Ras regulates exocytosis by Rgl2/Rlf-mediated activation of RalA on endosomes. Mol. Biol. Cell 18, 1850–1860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Neyraud V., Aushev V. N., Hatzoglou A., Meunier B., Cascone I., Camonis J. (2012) RalA and RalB proteins are ubiquitinated GTPases, and ubiquitinated RalA increases lipid raft exposure at the plasma membrane. J. Biol. Chem. 287, 29397–29405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen Z., Otto J. C., Bergo M. O., Young S. G., Casey P. J. (2000) The C-terminal polylysine region and methylation of K-Ras are critical for the interaction between K-Ras and microtubules. J. Biol. Chem. 275, 41251–41257 [DOI] [PubMed] [Google Scholar]
- 43. Fukada Y., Matsuda T., Kokame K., Takao T., Shimonishi Y., Akino T., Yoshizawa T. (1994) Effects of carboxyl methylation of photoreceptor G protein gamma-subunit in visual transduction. J. Biol. Chem. 269, 5163–5170 [PubMed] [Google Scholar]
- 44. Maske C. P., Hollinshead M. S., Higbee N. C., Bergo M. O., Young S. G., Vaux D. J. (2003) A carboxyl-terminal interaction of lamin B1 is dependent on the CAAX endoprotease Rce1 and carboxymethylation. J. Cell Biol. 162, 1223–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Parish C. A., Smrcka A. V., Rando R. R. (1995) Functional significance of βγ-subunit carboxymethylation for the activation of phospholipase C and phosphoinositide 3-kinase. Biochemistry 34, 7722–7727 [DOI] [PubMed] [Google Scholar]
- 46. Rowell C. A., Kowalczyk J. J., Lewis M. D., Garcia A. M. (1997) Direct demonstration of geranylgeranylation and farnesylation of Ki-Ras in vivo. J. Biol. Chem. 272, 14093–14097 [DOI] [PubMed] [Google Scholar]
- 47. Whyte D. B., Kirschmeier P., Hockenberry T. N., Nunez-Oliva I., James L., Catino J. J., Bishop W. R., Pai J. K. (1997) K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J. Biol. Chem. 272, 14459–14464 [DOI] [PubMed] [Google Scholar]
- 48. Cox A. D., Philips M. R., Der C. J. (2015) Targeting RAS membrane association: back to the future for anti-RAS drug discovery? Clin. Cancer Res. 21, 1819–1827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bergo M. O., Ambroziak P., Gregory C., George A., Otto J. C., Kim E., Nagase H., Casey P. J., Balmain A., Young S. G. (2002) Absence of the CAAX endoprotease Rce1: effects on cell growth and transformation. Mol. Cell. Biol. 22, 171–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wahlstrom A. M., Cutts B. A., Karlsson C., Andersson K. M., Liu M., Sjogren A. K., Swolin B., Young S. G., Bergo M. O. (2007) Rce1 deficiency accelerates the development of K-RAS-induced myeloproliferative disease. Blood 109, 763–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wahlstrom A. M., Cutts B. A., Liu M., Lindskog A., Karlsson C., Sjogren A. K., Andersson K. M., Young S. G., Bergo M. O. (2008) Inactivating Icmt ameliorates K-RAS-induced myeloproliferative disease. Blood 112, 1357–1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Court H., Amoyel M., Hackman M., Lee K. E., Xu R., Miller G., Bar-Sagi D., Bach E. A., Bergö M. O., Philips M. R. (2013) Isoprenylcysteine carboxylmethyltransferase deficiency exacerbates KRAS-driven pancreatic neoplasia via Notch suppression. J. Clin. Invest. 123, 4681–4694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yu Y., Fujii S., Yuan J., Luo R. Z., Wang L., Bao J., Kadota M., Oshimura M., Dent S. R., Issa J. P., Bast R. C. Jr. (2003) Epigenetic regulation of ARHI in breast and ovarian cancer cells. Ann. N.Y. Acad. Sci. 983, 268–277 [DOI] [PubMed] [Google Scholar]