

Abstract
Statins or 3-hydroxy-methylglutaryl coenzyme A (HMG CoA) reductase inhibitors not only prevents the synthesis of cholesterol biosynthesis but also inhibits the synthesis of essential isoprenoid intermediates such as farnesyl pyrophosphate, geranylgeranyl pyrophosphate, isopentanyl adenosine, dolichols and polyisoprenoid side chains of ubiquinone, heme A, and nuclear lamins. These isoprenoid intermediates are required for activation of various intracellular/signaling proteins- small guanosine triphosphate bound protein Ras and Ras-like proteins like Rho, Rab, Rac, Ral, or Rap which plays an indispensible role in multiple cellular processes. Reduction of circulating isoprenoids intermediates as a result of HMG CoA reductase inhibition by statins prevents activation of these signalling proteins. Hence, the multiple effects of statins such as antiinflammatory effects, antioxidant effects, antiproliferative and immunomodulatory effects, plaque stability, normalization of sympathetic outflow, and prevention of platelet aggregation are due to reduction of circulating isoprenoids and hence inactivation of signalling proteins. These multiple lipid-independent effects of statins termed as statin pleiotropy would potentially open floodgates for research in multiple treatment domains catching attentions of researchers and clinician across the globe.
Keywords: Coronary artery disease, pleiotropic effects, rosuvastatin, statins
INTRODUCTION
Statins or 3-hydroxy-methylglutaryl coenzyme-A (HMG-CoA) reductase inhibitors are a potent class of cholesterol synthesis inhibitors and an established therapy for primary and secondary prevention of coronary artery diseases.[1]
Several large studies confirmed reduction in cardiac mortality and morbidity through the treatment with statins.[2,3,4,5,6,7,8] These clinical benefits were reported to be majorly due to lowering of low-density lipoprotein cholesterol (LDL-C). A linear relationship between LDL-C and cardiovascular (CV) event rates was first established by the Scandinavian Simvastatin Survival Study (4S).[2] The study showed landmark reduction in the rate of major coronary event by over 50% after use of statins. This reduction was associated with 2-mmol (78 mg/dl) reduction in LDL-C levels.[3] Later several other studies such as the cholesterol treatment trialists meta-analyses,[4] the treating to new target,[5] heart progression study (HPS),[6] cholesterol and recurrent events (CARE)[7] trial and Long-term intervention with pravastatin in ischaemic disease (LIPID) studies[8] also confirmed the findings of 4S.
Accumulating evidence have suggested that statins, in addition to LDL-C lowering, exhibited properties of plaque stabilization and endothelial homeostasis; anti-inflammatory, antioxidant, anti-proliferative and immunomodulatory effects; normalization of sympathetic outflow; and prevention of platelet aggregation. Such cholesterol-independent effects were called the pleiotropic effects of statins.[1,9]
“Pleiotropy” has originated from the Greek words “pleion,” which means more, and “tropy” meaning response or stimuli. Though pleiotropic effects are defined as a single gene affecting multiple systems or determining more than one phenotype, lately statin pleiotropy is referred as statins exerting multiple pharmacological activities.
The pleiotropic effect is the class effect of statins and is exhibited by all statins acting on non-hepatic tissues, including hydrophilic pravastatin, which penetrates poorly through the cell membranes of non-hepatic cells.[10,11]
STATIN PLEIOTROPY IN CLINICAL STUDIES
The rapidity with which statins produce beneficial effects has suggested involvement of mechanisms other than its lipid-lowering activities. Over the last more than 10 years, several clinical trials indicated that the benefits of statins extend beyond their effects on cholesterol, supporting the theory of “statin pleiotropy” [Table 1].[2,3,4,5,6,7,8,12,13,14,15,16,17,18,19,20,21,22,23,24,25]
Table 1.
Clinical studies elucidating statin pleiotropy
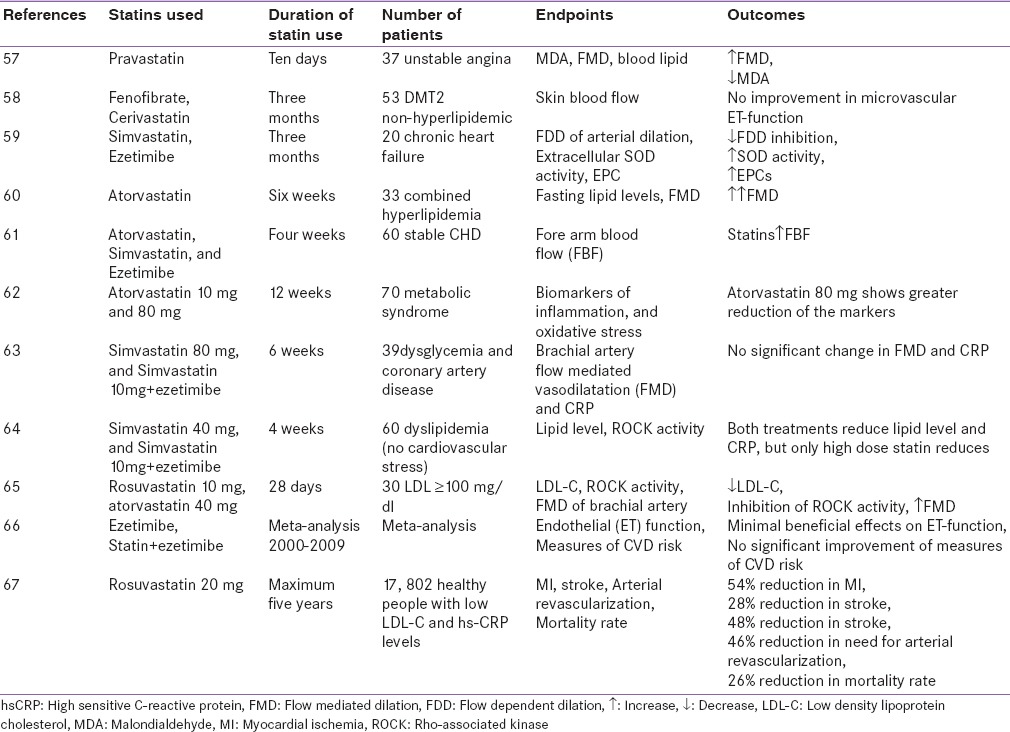
The program on the surgical control of the hyperlipidemias (POSCH) study[12] reported the benefits of cholesterol lowering after partial ileal bypass surgery after almost 10 years, whereas most statin trials showed benefits at much earlier time points (e.g., within 5 years). Comparing benefits after 5 years for all lipid-lowering trials, non-statin trials no longer were aligned to the same cholesterol:mortality risk reduction slope, whereas all statin trials aligned to this slope. In the POSCH study, that the 34% reduction in LDL-C within the first 3 months post ileal surgery was although earlier unnoticed could be explained by additional non-lipid effects of statins.[12]
In the west of scotland coronary prevention study study, Cox regression analyses of data highlighted that pravastatin treatment decreased LDL-C down to 24% and further reducing LDL-C levels did not show additive any benefit, and such phenomenal reduction in coronary heart disease risk was surprising in the pravastatin-treated group as such benefits could not be expected with decrease in LDL-C within this range. This strongly supported non-lipid-lowering theory of statins.[7,13] In another statin trial with 3000 subjects with unstable angina or non-Q-wave acute myocardial infarction, aggressive treatment with statins effectively reduced recurrent ischemic events within 16 weeks after acute coronary events, reduced serum LDL- by 40% and relative risk by 16%. According to the investigators, this period was too short to expect changes in lesion size and plaque stability of such magnitude.[14] Therefore, it was assumed that these early changes were due to the direct cellular effects on the vascular wall and improvement of endothelial function with statins.
Other studies that suggested the involvement of a non-lipid-lowering pleiotropic mechanism of statins were large prospective trials, such as the HPS and anglo-scandinavian cardiac outcomes (ASCOT) trials.[6,15] These studies demonstrated that relative risk reduction with statin treatment was independent of baseline lipid values. The HPS trial reported that absolute benefits were related directly to the individual vascular risk of the subjects. Patients with low HDL-C and high vascular risk, such as subjects with diabetes, significantly benefited from statin therapy possibly through lipid-independent mechanisms. In 2003, Sever et al.,[15] reported a randomised trial with hypertensive subjects (n = 10,305) with >3 CV risk factors. The subjects received either atorvastatin 10 mg/day or placebo and their total cholesterol level was measured to be <250 mg/dl. The study was halted after a median follow-up of 3.3 years as there was a 36% reduction in the risk of myocardial infarction and fatal coronary heart disease. These very early benefits with large reduction in coronary heart disease events were incredible and highlighted the involvement of mechanisms other than the lipid-lowering effects of stains. This was supported by earlier studies such as the myocardial ischaemia reduction with aggressive cholesterol lowering Study (MIRACL)[14] and the pravastatin or atorvastatin evaluation and infection therapy trial (PROVE-IT)[16] where early clinical benefits were seen in subjects with coronary heart disease. This rapid time course of event reduction in high-risk subjects with recurrent coronary ischemia suggested non-lipid-lowering effects.[17] In 2006, Vyas et al.,[18] reported over 35% reduced risk of sudden cardiac death or ventricular arrhythmia development due to the plaque-stabilizing effect of statins, increased coronary blood flow and microvascular myocardial perfusion.[18]
In 2004, Cannon et al.,[16] reported the PROVE-IT trial, comparing the effect of intensive therapy with atrovastatin (80 mg) daily versus moderate therapy with pravastatin (40 mg/day) in 4162 subjects with acute coronary syndrome. The authors reported that subjects were benefitted from intensive therapy than standard therapy, leading to separation of event curves within 3 weeks of treatment. Also, a decrease in hazard ratio was apparent as early as 6 months from treatment. In a sub-study of the PROVE-IT trial, Gibson et al.,[19] in the year 2009 reported the benefit of intensive statin therapy in clinical outcomes in subjects undergoing percutaneous coronary intervention for acute coronary syndrome. In 2007, Patti et al.,[20] reported a randomized trial where 12-h pretreatment with 40 mg/day atorvastatin prior to coronary angioplasty showed improvement in clinical outcomes in subjects with acute coronary syndrome. In this study, the early window of protection during which no reduction LDL-C in levels are commonly observed, suggested that the anti-inflammatory and pleiotropic properties of statins may be of clinical importance. In 2009, an observational study showed that statin users hospitalized for acute coronary syndrome were not only at a lower risk of developing in-hospital atrial fibrillation but also had a significantly lower risk of developing ventricular arrhythmias, cardiac arrest and/or death.[21] In these subjects, intensive statin therapy had reduced the risk of major adverse CV events compared with moderate-dose statin therapy. The reduction in the incidence of target vessel revascularization was independent of LDL-C and C-reactive protein (CRP) lowering, suggesting the statin pleiotropic effect of high-dose therapy. Scientists have been intrigued with the magnitude- and timing-reduced CV events in some clinical trials and asserted that these rapid effects mediated by statins could not be explained solely on the basis of the lipid-lowering mechanism of statins.
High-sensitivity C-reactive protein (hsCRP) is a non-specific marker of inflammatory diseases, and has been recognized as an independent risk predictor of cardiovascular diseases.
hsCRP has been correlated well with vascular risk independent of cholesterol levels.[9] Several studies have demonstrated lowering of hsCRP upon treatment with statins and this effect was independent of lowering of LDL-C. In patients with high CRP values, higher coronary deaths occurred in spite of low LDL-C levels. The Air Force/Texas Coronary Atherosclerosis Prevention Study reported lowering of CRP values through treatment with lovastatin, and improvement in cardiac outcomes was achieved with lowering of CRP levels independent of LDC-C levels.[23] In 2005, Nissen et al.,[24] reported the existence of an independent correlation between low levels of CRP and atheroma progression rate from a post-hoc analysis of data of a clinical study. According to the authors, regression of atheroma size was more rapid and significant with greater reduction in CRP levels. However, less regression in atheroma size was observed despite greater reduction in LDL-C levels.
In the primary prevention also treatment with statin benefitted patients with high hsCRP levels and low LDL-C levels and other CV risks. Subjects in the early or mild stages of heart failure benefitted from statins and the benefits were reported to be due to anti-inflammatory effects and improvement in endothelial function with statin treatment.[25] The presence of inflammatory components in mediating CV diseases were supported by a retrospective study, the CORONA (Controlled rosuvastatin multinational trial in heart failure) trial,[26] and patients benefitting from lowering of hsCRP levels suggested a non-lipid effect of rosuvastatin.[23,24] The finding was further confirmed by the multicenter COSMOS (coronary atherosclerosis Study measuring effects of rosuvastatin using intravascular ultrasound in Japanese subjects) trial. These trials with rosuvastatin showed significant reduction in plaque volume after treatment with rosuvastatin independent of LDL-C reduction, suggesting non-lipid-lowering effects.[27] In a double-blind study with 58 subjects with coronary artery disease, comparable reduction in LDL-C occurred through treatment with high- (80 mg/day) than low-dose atorvastatin (10 mg/day) plus ezetimibe (10 mg/day).[28]
MECHANISMS OF STATIN PLEIOTROPY
Statins are known to lower cholesterol by reversibly inhibiting HMG-CoA reductase, the well-known and widely established mechanism of action of statins. Inhibition of cholesterol synthesis occurs as a result of prevention of mevalonate from producing HMG-CoA, since mevalonate is not an immediate precursor of cholesterol synthesis and also acts as precursor for several other key molecules needed for normal functioning of cellular processes. In addition to cholesterol synthesis, mevalonate is required for production of non-steroidal isoprenoid intermediates such as the farnesyl pyrophosphate, geranylgeranyl pyrophosphate, isopentanyl adenosine, dolichols and polyisoprenoid side chains of ubiquinone and heme-A. These isoprenoid intermediates are integral to the post-translation modification and activation of several intracellular/signaling proteins such as the γ-subunit of heterotrimeric G-proteins; heme-A; nuclear lamins; and small GTP-bound protein Ras and Ras-like proteins such as Rho, Rab, Rac, Ral or Rap.[1,10,11] Post-translational isoprenylation, that is, attachment of isoprenoid chains helps these proteins in their covalent attachment, sub-cellular localization and intracellular trafficking. Both Ras and Rho proteins switch from their GDP-bound inactive state to the GTP-bound active state [Figure 1]. These signaling proteins play an indispensable role in multiple cellular processes – cell signaling, cell differentiation and proliferation, myelination, cytoskeleton dynamics and endocytotic/exocytotic transport [Figure 2].[11]
Figure 1.
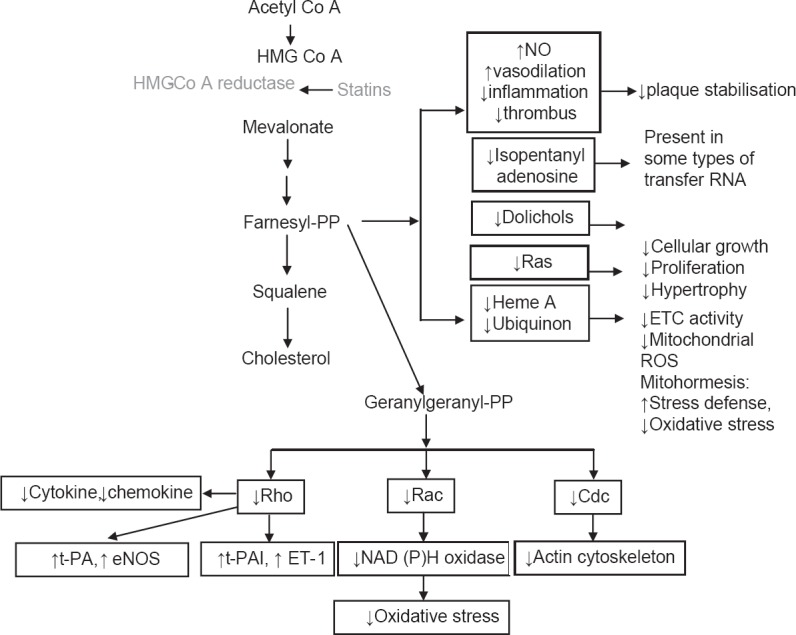
Regulation of the Rho GTPase cycle. Rho protein cycles between a cytosolic, inactive GDP-bound and an active, membrane, GTP-bound state. Inhibition of mevalonate synthesis by statins prevents membrane targeting of Rho and its subsequent activation of ROCK. This cycle is controlled by several cofactors, including guanine nucleotide exchange factors, GTPase-activating proteins, and guanine nucleotide dissociation inhibitors. An important step in the activation of Rho GTPases is posttranslational isoprenylation, which allows translocation of Rho to the cell membrane and subsequent activation
Figure 2.
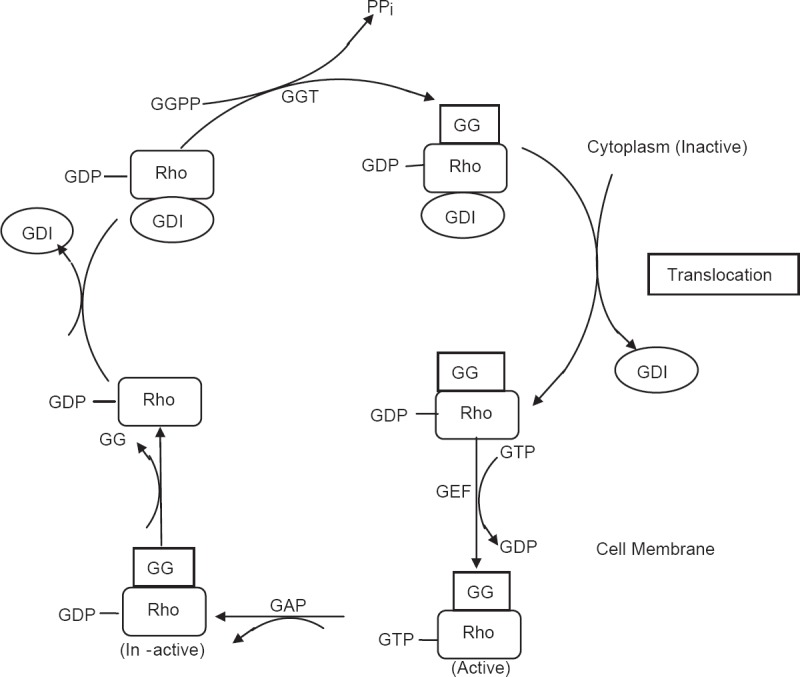
Mevalonate pathway for cholesterol biosynthesis showing the effects of inhibition of HMG-CoA reductase by statins. Statins decrease the isoprenylation of signaling molecules, which leads to modulation (↑[increase]/↓[decrease]) of various signaling pathways. Mitohormesis: Increased stress defense against oxidative damage to mitochondria and tissues. eNOS: Endothelial nitric oxide synthetase; ET-1: Endothelin-1; ETC: Electron transport chain; NADPH: Nicotinamide adenine dinucleotide phosphate (reduced); PAI-1: Plasminogen activator inhibitor-1; t-PA: Tissue-type plasminogen activator
Statins have been reported to prevent isoprenylation of Rho- and Rho-associated kinases (ROCK) at doses that were used to lower LDL-C levels.[6] Rho/ROCK are involved in controlling important functions such as regulation of contraction, migration and adhesion of vascular smooth muscle cells. Statins by inhibiting Rho/ROCK activation increase the bioavailability of nitric oxide (NO). The Rac protein has been involved in cardiac hypertrophy, actin cytoskeleton remodeling and generation of reactive oxygen species, and statins benefit by inhibiting Rac1.[10,29]
Inactivation of Rho by statins was reported to upregulate peroxisome proliferator-activated receptor (PPAR) expression and induce PPAR-γ transcription activity in macrophages, and inhibit lipopolysaccharide.[30] Matrix metalloproteinases (MMPs) are zinc-dependent endopeptidases that promote angiogenesis, intimal proliferation, vessel wall remodeling and extracellular matrix degradation through enhanced migration of vascular smooth muscle cells. The role of Rho/ROCK pathway in the secretion of MMP-2 through stimulation of endothelin-1 and angiotensin-II has been confirmed with use of small interfering RNA and the ROCK pathway inhibitor, Y-27632, which inhibited MMP-2 release.[31,32]
PLEIOTROPIC EFFECTS OF STATINS IN ATHEROSCLEROTIC VASCULAR DISORDERS
Statins decrease inflammatory cells in atherosclerotic plaques and increase plaque stability through combined reduction of lipids, macrophages and MMPs. Other cellular molecules inhibited by statins include nuclear factor-κb (NF-κB), monocyte chemoattractant protein-1 and hsCRP.[10,33] Under normal physiological conditions, NF-kB resides in the cytoplasm bound with inhibitor I-kB (IκB). In response to inflammatory stimuli, IkB is phosphorylated and degraded by the ubiquitin proteasome system releasing NF-kB. The NF-kB then translocates into the nucleus and induces the expression of inflammatory cytokines, monocyte chemoattractant protein-1 and intercellular adhesion molecule-1. Statins decrease the levels of reactive oxygen species and prevent dislocation of IkB from NF-kB, resulting in inactivation of NF-kB.[9] Statins have been reported to reduce the expression of monocyte chemoattractant protein-1, which lowers the interaction between monocytes and the vascular wall, monocyte chemotaxis, and growth and proliferation of macrophages.[1,9] Another anti-inflammatory mechanism of statins may be reduction of mRNA of cyclooxygenase-2.[1] Antigen-presenting cells express major histocompatibility complex-II constitutively, and its expression in human endothelial cells and monocytes increases in the presence of interferon-γ. Various statins reduce the expression of major histocompatibility complex-II on antigen-presenting cells, leading to decreased major histocompatibility complex-II-mediated activation of T-cells. In addition, statins reduce the production of various inflammatory markers such as tumor necrosis factor- α, interleukin-1β and chemotactic cytokines. Statins were reported to disrupt the oxidative stress/inflammatory cycles by decreasing lipid peroxidation and release of inflammatory mediators.[1,9,10,11,34]
Statins improve endothelial function by inhibiting the isoprenylation of Rac and Rho, and activation of the ROCK pathway. Inactivation of the ROCK pathway stabilizes the mRNA of endothelium-derived nitric oxide synthase (eNOS) and activates the Akt/PI3K cascade, leading to high eNOS activity, increased NO production and bioavailability. Protein kinase Akt has been associated with improvement of myocardial aerobic metabolism and increase in the levels of angiopoetine (protein growth factor), which accelerate the process of angiogenesis.[9,34] Inactivation of the Ras signaling pathway was associated with prevention or reversal of cardiac remodeling in humans.[35] In a preclinical study, atorvastatin reduced the synthesis of collagen, α-1-procollagen mRNA expression and expression of profibrotic peptide connective tissue growth factor in cell cultures of rat and human cardiac fibroblasts.[36] Rosuvastatin administration was reported to decrease the levels of TNF-α and MMP-2 and increased the circulating levels of bone marrow-derived stems cells.[37] Statins also have inhibitory effects on release of MMP-1 and MMP-3 from macrophages and endothelial cells, and modulate remodeling processes.[38] In endothelial cells, statins activate protein kinase Akt leading to stimulation of eNOS activity, increased NO synthesis and neoangiogenesis. In the central nervous system, statins regulate sympathetic and vagal outflow by increasing endothelial NO synthesis, and reduce the expression of angiotensin-II and endothelial receptors.[39] Statins by normalizing sympathetic outflow showed beneficial effects in hypertensive patients with a history of MI and cerebral ischemia. Statins attenuate the cytokine-mediated proliferation of vascular smooth muscle cells in coronary artery smooth muscle cells, arrest the cell cycle between G1–S phase transitions and inhibit smooth muscle cell proliferation through modulation of Ras and Rho activity. Atorvastatin inhibited serotonin-induced mitogenesis and migration by inhibiting GTP-RhoA formation in pulmonary artery vascular smooth muscle cells, which was reversed by geranylgeranyl pyrophosphate but not farnesyl pyrophosphate.[40,41] The anti-platelet and antithrombotic activities of statins have been attributed to reduction of thromboxane A2, tissue factor expression and PPARs.[41]
In heart failure, statins have been reported to benefit by mobilizing bone marrow cells. Statins activate phosphoinositide-3 kinase and Akt, and increase the circulating levels of bone marrow-derived endothelial progenitor cells, which may contribute to improved cardiac regeneration and/or repair.[42]
In 2012, Bouitbir et al., reported statins as a new activating factor of cardiac mitochondrial biogenesis and antioxidant capacities. However, they reported an opposite effect on skeletal muscles.[43] Mitohormesis is defined as a pre-conditioning or adaptive response of mitochondria to low levels of oxidative stress induced by physical exercise, reduced caloric uptake and glucose restriction.[43] This pleiotropic effect of statins was attributed to increased resistance/stress defense against higher levels of oxidative stress and damage to mitochondria and cardiac tissues. Use of antioxidants prevented statin-induced mitohormesis since antioxidant vitamins had decreased the clinical benefits of the simvastatin–niacin combination in subjects with coronary artery diseases and low levels of HDL-C in the study by Brown and co-workers.[22] Therefore, some of the cardioprotective mechanisms of statins could be attributed to statin-induced mitohormesis.
PLEIOTROPIC EFFECTS OF STATINS IN OTHER DISORDERS
Treatment with simvastatin has been reported to reverse the pulmonary vascular effects of cigarette smoke, including pulmonary hypertension, smoke-induced emphysema, smoke-induced small arterial remodeling and emphysema in guinea pigs exposed to cigarette smoke for 6 months.[44,45] In cancer, preclinical studies have shown anti-proliferative, pro-apoptotic, anti-invasive and radiosensitizing properties of statins.[46] Various lipophilic statins such as lovastatin, simvastatin and fluvastatin exerted direct anticancer activity in vitro by reducing proliferation and survival signals in susceptible breast cancer phenotypes, indicating potential use of statins in breast cancer.[46]
In 2008, Bifulco et al.,[47] suggested that statins may have potential to treat various other neurological disorders, including Alzheimer's disease, Parkinson's disease, multiple sclerosis and primary brain tumors as antioxidant, anti-inflammatory, anti-platelet activities and effects on the nitric oxide synthase system have been confirmed previously. In vitro studies, including a few animal studies, have suggested that statins may increase bone mass by enhancing bone morphogenetic protein-2-mediated osteoblast differentiation and activity.[48,49] In addition, statins also exerted beneficial effects in rheumatoid arthritis by inhibition and downregulation of these cytokines and chemokines.[50] In 2009, Glynn et al.,[51] reported an analysis of the JUPITER, which showed that rosuvastatin nearly halved the risk for symptomatic venous thromboembolism VTE in apparently healthy patients and the effects were ascribed to the pleiotropic effects of statins.[51] A recent meta-analyses including one randomized and nine observational trials was conducted on nearly 1 million people who were on statin therapy. The results showed a 32% decrease in the risk of VTE, 41% decrease in deep vein thrombosis and 30% decrease in pulmonary embolism.[52] In women with polycystic ovary syndrome, statins have been reported to directly or indirectly block oxidative stress-mediated increase in thecal interstitial cell proliferation, steroidogenesis and insulin resistance.[53,54] In seasonal and avian H5N1 influenza, administration of statins showed modulatory activities on various cytokines and increased the levels of other potential immunomodulatory molecules. The beneficial effect of statins in influenza was partly ascribed to their cardioprotective, anti-inflammatory and immunomodulatory effects.[55,56]
CONCLUSION
A global consensus on the concept of statin pleiotropy could not be reached as yet and remains a hot topic for debate. The pathophysiological implications of blockade of mevalonate production in the pathway leading to synthesis of cholesterol, that is, the mevalonate pathway suggested multiple mechanisms of statins. Over the years, analyses of several clinical studies, including the landmark HPS and ASCOT-LLA trial, reported findings with statins that were inexplicable with the lipid-lowering mechanism alone. The most probable mechanism of statin pleiotropy is prevention of isoprenoids synthesis as decrease in levels of circulating isoprenoids have been reported with treatment with statins.
ACKNOWLEDGEMENT
Danish Mahmood, Jr. Medical Writer, Max Neeman International, was responsible for the preparation of this review article.
Footnotes
Source of Support: AstraZeneca with respect to medical writing support
Conflict of Interest: None declared.
REFERENCES
- 1.Zhou Q, Liao JK. Statins and cardiovascular diseases: From cholesterol lowering to pleiotropy. Curr Pharm Des. 2009;15:467–78. doi: 10.2174/138161209787315684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Randomised clinical trial of cholesterol lowering in 4444 patients with coronary heart disease: The Scandinavian Simvastatin Survival Study (4S) Lancet. 1994;344:1383–9. [PubMed] [Google Scholar]
- 3.Pedersen TR, Olsson AG, Faergeman O, Kjekshus J, Wedel H, Berg K, et al. Lipoprotein changes and reduction in the incidence of major coronary heart disease events in the Scandinavian Simvastatin Survival Study (4S) Circulation. 1998;97:1453–60. doi: 10.1161/01.cir.97.15.1453. [DOI] [PubMed] [Google Scholar]
- 4.Baigent C, Keech A, Kearney PM, Blackwell L, Buck G, Pollicino C, et al. Efficacy and safety of cholesterol lowering treatment: Perspective meta-analyses of data from 90,056 participants in 14-randomised trials of statins. Lancet. 2005;366:1267–78. doi: 10.1016/S0140-6736(05)67394-1. [DOI] [PubMed] [Google Scholar]
- 5.LaRosa JC, Grundy SM, Waters DD, Shear C, Barter P, Fruchart JC, et al. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med. 2005;352:1425–35. doi: 10.1056/NEJMoa050461. [DOI] [PubMed] [Google Scholar]
- 6.Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high risk individuals: A randomised placebo-controlled trial. Lancet. 2002:3607–22. doi: 10.1016/S0140-6736(11)61125-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sacks FM, Pfeffer MA, Moye LA, Rouleau JL, Rutherford JD, Cole TG, et al. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events Trial investigators. N Engl J Med. 1996;335:1001–9. doi: 10.1056/NEJM199610033351401. [DOI] [PubMed] [Google Scholar]
- 8.Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. N Engl J Med. 1998;339:349–57. doi: 10.1056/NEJM199811053391902. [DOI] [PubMed] [Google Scholar]
- 9.Liao JK, Laufs U. Pleiotropic effects of statins. Annu Rev Pharmacol Toxicol. 2005;45:89–118. doi: 10.1146/annurev.pharmtox.45.120403.095748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang CY, Liu PY, Liao JK. Pleiotropic effects of statin therapy: Molecular mechanisms and clinical results. Trends Mol Med. 2008;14:37–44. doi: 10.1016/j.molmed.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou Q, Liao JK. Pleiotropic effects of statins: Basic research and clinical perspectives. Circ J. 2010;74:818–26. doi: 10.1253/circj.cj-10-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buchwald H, Varco RL, Matts JP, Long JM, Fitch LL, Campbell GS, et al. Effect of partial ileal bypass surgery on mortality and morbidity from coronary heart disease in patients with hypercholesterolemia. Report of the Program on the Surgical Control of the Hyperlipidemias (POSCH) N Engl J Med. 1990;323:946–5. doi: 10.1056/NEJM199010043231404. [DOI] [PubMed] [Google Scholar]
- 13.Shepherd J, Cobbe SM, Ford I, Isles CG, Lorimer AR, MacFarlane PW, et al. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. West of Scotland Coronary Prevention Study Group. N Engl J Med. 1995;333:1301–7. doi: 10.1056/NEJM199511163332001. [DOI] [PubMed] [Google Scholar]
- 14.Schwartz GG, Olsson AG, Ezekowitz MD, Ganz P, Oliver MF, Waters D, et al. Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes: The MIRACL study: A randomized controlled trial. JAMA. 2001;285:1711–8. doi: 10.1001/jama.285.13.1711. [DOI] [PubMed] [Google Scholar]
- 15.Sever PS, Dahlöf B, Poulter NR, Wedel H, Beevers G, Caulfield M, et al. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial – Lipid Lowering Arm (ASCOT-LLA): A multicentre randomised controlled trial. Lancet. 2003;361:1149–58. doi: 10.1016/S0140-6736(03)12948-0. [DOI] [PubMed] [Google Scholar]
- 16.Cannon CP, Braunwald E, McCabe CH, Rader DJ, Rouleau JL, Belder R, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med. 2004;350:1495–504. doi: 10.1056/NEJMoa040583. [DOI] [PubMed] [Google Scholar]
- 17.Sever PS, Chang CL, Gupta AK, Whitehouse A, Poulter NR. ASCOT Investigators. The Anglo-Scandinavian Cardiac Outcomes Trial: 11-year mortality follow-up of the lipid-lowering arm in the UK. Eur Heart J. 2011;32:2525–32. doi: 10.1093/eurheartj/ehr333. [DOI] [PubMed] [Google Scholar]
- 18.Vyas AK, Guo H, Moss AJ, Olshansky B, McNitt SA, Hall WJ, et al. Reduction in ventricular tachyarrhythmias with statins in the Multicenter Automatic Defibrillator Implantation Trial (MADIT)-II. J Am Coll Cardiol. 2006;47:769–73. doi: 10.1016/j.jacc.2005.09.053. [DOI] [PubMed] [Google Scholar]
- 19.Gibson CM, Pride YB, Hochberg CP, Sloan S, Sabatine MS, Cannon CP, et al. Effect of intensive statin therapy on clinical outcomes among patients undergoing percutaneous coronary intervention for acute coronary syndrome. PCI-PROVE IT: A PROVE IT-TIMI 22 (Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis In Myocardial Infarction 22) Substudy. J Am Coll Cardiol. 2009;54:2290–5. doi: 10.1016/j.jacc.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 20.Patti G, Nusca A, Chello M, Pasceri V, D’Ambrosio A, Vetrovec GW, et al. Usefulness of statin pretreatment to prevent contrast-induced nephropathy and to improve long-term outcome in patients undergoing percutaneous coronary intervention. Am J Cardiol. 2008;101:279–85. doi: 10.1016/j.amjcard.2007.08.030. [DOI] [PubMed] [Google Scholar]
- 21.Vedre A, Gurm H, Froehlich J, Kline-Rogers E, Montalescot G, Gore JM, et al. Impact of prior statin therapy on arrhythmic events in patients with acute coronary syndromes (from the Global Registry of Acute Coronary Events [GRACE]) Am J Cardiol. 2009;104:1613–17. doi: 10.1016/j.amjcard.2009.07.045. [DOI] [PubMed] [Google Scholar]
- 22.Ridker PM, Rifai N, Pfeffer MA, Sacks F, Braunwald E. Long term effects of pravastatin on plasma concentration of C-reactive protein. The Cholesterol and Recurrent Events (CARE) Investigators. Circulation. 1999;100:230–5. doi: 10.1161/01.cir.100.3.230. [DOI] [PubMed] [Google Scholar]
- 23.Ridker PM, Cannon CP, Morrow D, Rifai N, Rose LM, Mc-Cabe CH, et al. C-reactive protein levels and outcomes after statin therapy. N Engl J Med. 2005;352:20–8. doi: 10.1056/NEJMoa042378. [DOI] [PubMed] [Google Scholar]
- 24.Nissen SE, Tuzcu EM, Schoenhagen P, Crowe T, Sasiela WJ, Tsai J, et al. Statin therapy, LDL cholesterol, C-reactive protein, and coronary artery disease. N Engl J Med. 2005;352:29–38. doi: 10.1056/NEJMoa042000. [DOI] [PubMed] [Google Scholar]
- 25.Barrios V, Escobar E. Rosuvastatin along the cardiovascular continuum: From JUPITER to AURORA. Expert Rev Cardiovasc Ther. 2009;7:1317–27. doi: 10.1586/erc.09.119. [DOI] [PubMed] [Google Scholar]
- 26.McMurray JJ, Kjekshus J, Gullestad L, Dunselman P, Hjalmarson A, Wedel H, et al. Effects of statin therapy according to plasma high-sensitivity C-reactive protein concentration in the controlled rosuvastatin multinational trial in heart failure (CORONA): A retrospective analysis. Circulation. 2009;120:2188–96. doi: 10.1161/CIRCULATIONAHA.109.849117. [DOI] [PubMed] [Google Scholar]
- 27.Takayama T, Hiro T, Yamagishi M, Daida H, Hirayama A, Saito S, et al. Effect of rosuvastatin on coronary atheroma in stable coronary artery disease: Multicenter coronary atherosclerosis study measuring effects of rosuvastatin using intravascular ultrasound in Japanese subjects (COSMOS) Circ J. 2009;73:2110–17. doi: 10.1253/circj.cj-09-0358. [DOI] [PubMed] [Google Scholar]
- 28.Ostad MA, Eggeling S, Tschentscher P, Schwedhelm E, Boger R, Wenzel P, et al. Flow-mediated dilation in patients with coronary artery disease is enhanced by high dose atorvastatin compared to combined low dose atorvastatin and ezetimibe: Results of the CEZAR study. Atherosclerosis. 2009;205:227–32. doi: 10.1016/j.atherosclerosis.2008.11.032. [DOI] [PubMed] [Google Scholar]
- 29.Rashid M, Tawara S, Fukumoto Y, Seto M, Yano K, Shimokawa H. Importance of Rac1 signaling pathway inhibition in the pleiotropic effects of HMG-CoA reductase inhibitors. Circ J. 2009;73:361–70. doi: 10.1253/circj.cj-08-0817. [DOI] [PubMed] [Google Scholar]
- 30.Yano M, Matsumura T, Senokuchi T, Ishii N, Murata Y, Taketa K, et al. Statins activate peroxisome proliferator-activated receptor gamma through extracellular signal-regulated kinase 1/2 and p38 mitogen-activated protein kinase-dependent cyclooxygenase-2 expression in macrophages. Circ Res. 2007;100:1442–51. doi: 10.1161/01.RES.0000268411.49545.9c. [DOI] [PubMed] [Google Scholar]
- 31.Guo H, Shi Y, Liu L, Sun A, Xu F, Chi J. Rosuvastatin inhibits MMP-2 expression and limits the progression of atherosclerosis in LDLR-deficient mice. Arc Med Res. 2009;40:354–1. doi: 10.1016/j.arcmed.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 32.Ma S, Ma CC. Recent development in pleiotropic effects of statins on cardiovascular disease through regulation of transforming growth factor-beta superfamily. Cytokine Growth Factor Rev. 2011;22:167–75. doi: 10.1016/j.cytogfr.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 33.Mihl C, Dassen WR, Kuipers H. Cardiac remodelling: Concentric versus eccentric hypertrophy in strength and endurance athletes. Neth Heart J. 2008;16:129–33. doi: 10.1007/BF03086131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Inoue I, Goto S, Mizotani K, Awata T, Kawai S, Nakajima T, et al. Lipophilic HMG-CoA reductase inhibitor has an anti-inflammatory effect: Reduction of mRNA levels for interleukin-1beta, interleukin-6, cyclooxygenase-2, and p22phox by regulation of peroxisome proliferator-activated receptor alpha (PPARalpha) in primary endothelial cells. Life Sci. 2000;67:863–76. doi: 10.1016/s0024-3205(00)00680-9. [DOI] [PubMed] [Google Scholar]
- 35.Martin J, Denver R, Bailey M, Krum H. In vitro inhibitory effects of atorvastatin on cardiac fibroblasts: Implications for ventricular remodelling. Clin Exp Pharmacol Physiol. 2005;32:697–1. doi: 10.1111/j.1440-1681.2005.04256.x. [DOI] [PubMed] [Google Scholar]
- 36.Zacà V, Rastogi S, Imai M, Wang M, Sharov VG, Jiang A, et al. Chronic monotherapy with rosuvastatin prevents progressive left ventricular dysfunction and remodeling in dogs with heart failure. J Am Coll Cardiol. 2007;50:551–7. doi: 10.1016/j.jacc.2007.04.050. [DOI] [PubMed] [Google Scholar]
- 37.Kamio K, Liu XD, Sugiura H, Togo S, Kawasaki S, Wang X, et al. Statins inhibit matrix metalloproteinase release from human lung fibroblasts. Eur Respir J. 2010;35:637–46. doi: 10.1183/09031936.00134707. [DOI] [PubMed] [Google Scholar]
- 38.Malpas SC. Sympathetic nervous system overactivity and its role in the development of cardiovascular disease. Physiol Rev. 2010;90:513–57. doi: 10.1152/physrev.00007.2009. [DOI] [PubMed] [Google Scholar]
- 39.Bellosta S, Arnaboldi L, Gerosa L, Canavesi M, Parente R, Baetta R, et al. Statins effect on smooth muscle cell proliferation. Semin Vasc Med. 2004;4:347–56. doi: 10.1055/s-2004-869591. [DOI] [PubMed] [Google Scholar]
- 40.Li M, Liu Y, Dutt P, Fanburg BL, Toksoz D. Inhibition of serotonin-induced mitogenesis, migration, and ERK MAPK nuclear translocation in vascular smooth muscle cells by atorvastatin. Am J Physiol Lung Cell Mol Physiol. 2007;293:L463–71. doi: 10.1152/ajplung.00133.2007. [DOI] [PubMed] [Google Scholar]
- 41.Ali FY, Armstrong PC, Dhanji AR, Tucker AT, Paul-Clark MJ, Mitchell JA, et al. Antiplatelet actions of statins and fibrates are mediated by PPARs. Arterioscler Thromb Vasc Biol. 2009;29:706–11. doi: 10.1161/ATVBAHA.108.183160. [DOI] [PubMed] [Google Scholar]
- 42.Mathur N, Ramasubbu K, Mann DL. Spectrum of pleiotropic effects of statins in heart failure. Heart Fail Clin. 2008;4:153–1. doi: 10.1016/j.hfc.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bouitbir J, Charles AL, Echaniz-Laguna A, Kindo M, Daussin F, Auwerx J, et al. Opposite effects of statins on mitochondria of cardiac and skeletal muscles: A ‘mitohormesis’ mechanism involving reactive oxygen species and PGC-1. Eur Heart J. 2012;33:1397–407. doi: 10.1093/eurheartj/ehr224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wright JL, Zhou S, Preobrazhenska O, Marshall C, Sin DD, Laher I, et al. Statin reverses smoke-induced pulmonary hypertension and prevents emphysema but not airway remodeling. Am J Respir Crit Care Med. 2011;183:50–8. doi: 10.1164/rccm.201003-0399OC. [DOI] [PubMed] [Google Scholar]
- 45.Campbell MJ, Esserman LJ, Zhou Y, Shoemaker M, Lobo M, Borman E, et al. Breast cancer growth prevention by statins. Cancer Res. 2006;66:8707–14. doi: 10.1158/0008-5472.CAN-05-4061. [DOI] [PubMed] [Google Scholar]
- 46.Bifulco M, Malfitano AM, Marasco G. Potential therapeutic role of statins in neurological disorders. Exp Rev Neurother. 2008;8:827–37. doi: 10.1586/14737175.8.5.827. [DOI] [PubMed] [Google Scholar]
- 47.Uzzan B, Cohen R, Nicolas P, Cucherat M, Perret GY. Effects of statins on bone mineral density: A meta-analysis of clinical studies. Bone. 2007;40:1581–7. doi: 10.1016/j.bone.2007.02.019. [DOI] [PubMed] [Google Scholar]
- 48.Schlienger RG, Meier CR. Effect of selective serotonin reuptake inhibitors on platelet activation: Can they prevent acute myocardial infarction? Am J Cardiovasc Drugs. 2003;3:149–62. doi: 10.2165/00129784-200303030-00001. [DOI] [PubMed] [Google Scholar]
- 49.Blaschke S, Brandt P, Wessels JT, Müller GA. Expression and function of the C-class chemokine lymphotactin (XCL1) in Wegener's granulomatosis. J Rheumatol. 2009;36:2491–500. doi: 10.3899/jrheum.090244. [DOI] [PubMed] [Google Scholar]
- 50.Agarwal V, Phung OJ, Tongbram V, Bhardwaj A, Coleman CI. Statin use and the prevention of venous thromboembolism: A meta-analysis. Int J Clin Pract. 2010;64:1375–83. doi: 10.1111/j.1742-1241.2010.02439.x. [DOI] [PubMed] [Google Scholar]
- 51.Glynn RJ, Danielson E, Fonseca FA, Genest J, Gotto AM, Jr, Kastelein JJ, et al. A randomized trial of rosuvastatin in the prevention of venous thromboembolism. N Engl J Med. 2009;360:1851–61. doi: 10.1056/NEJMoa0900241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Khemasuwan D, Divietro ML, Tangdhanakanond K, Pomerantz SC, Eiger G. Statins decrease the occurrence of venous thromboembolism in patients with cancer. Am J Med. 2010;123:60–5. doi: 10.1016/j.amjmed.2009.05.025. [DOI] [PubMed] [Google Scholar]
- 53.Kodaman PH, Duleba AJ. Statins in the treatment of polycystic ovary syndrome. Semin Reprod Med. 2008;26:127–38. doi: 10.1055/s-2007-992933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rzepczynska IJ, Piotrowski PC, Wong D, Cress AB, Villanueva J, Duleba AJ. Role of isoprenylation in simvastatin-induced inhibition of ovarian theca-interstitial growth in the rat. Biol Reprod. 2009;81:850–5. doi: 10.1095/biolreprod.109.078667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fedson DS. Pandemic influenza: A potential role for statins in treatment and prophylaxis. Clin Infect Dis. 2006;43:199–205. doi: 10.1086/505116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Leren P, Askevold EM, Foss OP, Froili A, Grymyr D, Helgeland A, et al. The Oslo study. Cardiovascular disease in middle age-aged and young Oslo men. Acta Med Scand Suppl. 1975;588:1–38. [PubMed] [Google Scholar]
- 57.Karatzis E, Lekakis J, Papamichael C, Andreadou I, Cimponeriu A, Aznaouridis K, et al. Rapid effect of pravastatin on endothelial function and lipid peroxidation in unstable angina. Int J Cardiol. 2005;101:65–70. doi: 10.1016/j.ijcard.2004.03.060. [DOI] [PubMed] [Google Scholar]
- 58.Fegan PG, Shore AC, Mawson D, Tooke JE, MacLeod KM. Microvascular endothelial function in subjects with type 2 diabetes and the effect of lipid-lowering therapy. Diabet Med. 2005;22:1670–6. doi: 10.1111/j.1464-5491.2005.01719.x. [DOI] [PubMed] [Google Scholar]
- 59.Landmesser U, Bahlmann F, Mueller M, Spiekermann S, Kirchhoff N, Schulz S, et al. Simvastatin versus ezetimibe: Pleiotropic and lipid-lowering effects on endothelial function in humans. Circulation. 2005;111:2356–63. doi: 10.1161/01.CIR.0000164260.82417.3F. [DOI] [PubMed] [Google Scholar]
- 60.Taneva E, Borucki K, Wiens L, Makarova R, Schmidt-Lucke C, Luley C, et al. Early effects on endothelial function of atorvastatin 40 mg twice daily and its withdrawal. Am J Cardiol. 2006;97:1002–6. doi: 10.1016/j.amjcard.2005.10.032. [DOI] [PubMed] [Google Scholar]
- 61.Fichtlscherer S, Schmidt-Lucke C, Bojunga S, Rössig L, Heeschen C, Dimmeler S, et al. Differential effects of short-term lipid lowering with ezetimibe and statins on endothelial function in patients with CAD: Clinical evidence for ‘pleiotropic’ functions of statin therapy. Eur Heart J. 2006;27:1182–90. doi: 10.1093/eurheartj/ehi881. [DOI] [PubMed] [Google Scholar]
- 62.Singh U, Devaraj S, Jialal I, Siegel D. Comparison effect of atorvastatin (10 versus 80 mg) on biomarkers of inflammation and oxidative stress in subjects with metabolic syndrome. Am J Cardiol. 2008;102:321–5. doi: 10.1016/j.amjcard.2008.03.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Settergren M, Böhm F, Rydén L, Pernow J. Cholesterol lowering is more important than pleiotropic effects of statins for endothelial function in patients with dysglycaemia and coronary artery disease. Eur Heart J. 2008;29:1753–60. doi: 10.1093/eurheartj/ehn166. [DOI] [PubMed] [Google Scholar]
- 64.Liu PY, Liu YW, Lin LJ, Chen JH, Liao JK. Evidence for statin pleiotropy in humans: Differential effects of statins and ezetimibe on rho-associated coiled-coil containing protein kinase activity, endothelial function, and inflammation. Circulation. 2009;119:131–8. doi: 10.1161/CIRCULATIONAHA.108.813311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rawlings R, Nohria A, Liu PY, Donnelly J, Creager MA, Ganz P, et al. Comparison of effects of rosuvastatin (10 mg) versus atorvastatin (40 mg) on rho kinase activity in Caucasian men with a previous atherosclerotic event. Am J Cardiol. 2009;103:437–41. doi: 10.1016/j.amjcard.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bass A, Hinderliter AL, Lee CR. The impact of ezetimibe on endothelial function and other markers of cardiovascular risk. Ann Pharmacother. 2009;43:2021–30. doi: 10.1345/aph.1M302. [DOI] [PubMed] [Google Scholar]
- 67.Everett BM, Glynn RJ, MacFadyen JG, Ridker PM. Rosuvastatin in the prevention of stroke among men and women with elevated levels of C-reactive protein: Justification for the use of statins in prevention: An Intervention Trial Evaluating Rosuvastatin (JUPITER) Circulation. 2010;121:143–50. doi: 10.1161/CIRCULATIONAHA.109.874834. [DOI] [PubMed] [Google Scholar]