

1. Introduction
Since the dawn of the 20th century, the average life expectancy at birth has nearly doubled due to advances in science and medicine. As medical records grew more reliable, it became apparent that, while overall life span was increasing, there were significant epidemiological differences between geographical regions, with people from countries such as Japan, France, and Switzerland enjoying longer life expectancy as compared to other regions of the world. The causative factors for these disparities in both mortality and morbidity have been attributed not only to genetics but also to the contributions of highly variable lifestyle and environmental influences.1 Barring differences in modernization and public health policy, diet is the most significant epidemiological factor cross-culturally and is thought to be a profoundly important contributor to health and overall well-being.
Prior to the early 1990s, there was a general consensus in support of the belief that moderate alcohol consumption significantly lowered the risk of coronary heart disease (CHD).2 However, it was not clear whether this was true of all alcoholic beverages or which constituents in the drinks were responsible for these beneficial properties.3 In 1992, Siemann and Creasy4 reported that resveratrol (1) was present in significant concentrations in red wine, drawing attention to the fact that it was also an active principle found in Japanese and Chinese folk medicines used to treat ailments related to the liver, skin, heart, and lipid metabolism.5 The discovery of resveratrol in wine was particularly timely; although the correlation between high fat and cholesterol consumption and coronary heart disease was widely accepted,6 certain populations, namely the French, had a low incidence of CHD mortality despite a diet and lifestyle that exposed these individuals to elevated risk factors.7 The cardioprotective ability of resveratrol, its oligomers, along with other phytochemicals present in wine, appeared to resolve this “French Paradox” and advanced the notion that a chemical constituent of one’s diet could be beneficial to health.8 Although the reality of the French Paradox remains contentious,9,10 research into the health benefits of resveratrol has exploded as a result. The reported biological activities of resveratrol are numerous, including antioxidant,11 anticancer,12 antidiabetic,13,14 cardioprotective,15 and even antiaging properties,16 to name a few. The body of literature regarding the biological activity of resveratrol is expansive and beyond of the scope of this review. We invite the readers to the following resources for background on this exciting and ongoing field of research.17−21
Despite its enormous popularity, resveratrol is but a single compound in a large, structurally diverse class of oligomeric stilbenoids that are present in the wines and foods that we consume.22,23 Until recently, access to these oligomeric natural products was limited to isolation from natural sources, which severely hampered their biological evaluation. The scientific interest in resveratrol has undoubtedly instigated the renewed attention in these compounds by the chemical community. Advances in the characterization, isolation techniques, and synthesis of these natural products have significantly enriched our understanding of their chemistry and biology. Our review seeks to document the history and recent progress in the isolation, chemical synthesis, and biology of this fascinating class of phytochemicals, while providing new insight on the biosynthesis and future prospects for the field as a whole.
2. Resveratrol Biosynthesis and Biological Role in the Plant
Resveratrol, among many natural stilbenoids, shares a common biosynthetic pathway responsible for the production of cinnamic acids and flavonoids (e.g. naringenin, Scheme 1) through the phenylpropanoid pathway.24 Phenylalanine, a product of the shikimate pathway, undergoes a series of enzymatic reactions to produce the linear tetraketide 2. This intermediate is critical for the synthesis of flavonoids through constitutively expressed chalcone synthase.25 The gene encoding stilbene synthase, on the other hand, is transcribed only when induced by stimuli such as pathogenic invasion, physical trauma, or UV irradiation.26 It is important to consider any externalities imposed on the plant by this diversion of biosynthetic resources. Though resveratrol and its derivatives are found in lignified stem tissue, they are deliberately not produced in photosynthetic tissues where they have been shown to interfere with ion transport and associated redox processes.27 These cellular effects are partially responsible for the cytotoxicity of resveratrol and its derivatives against invasive pathogens, and have implications for the treatment of human disease (see section 8).28
Scheme 1. Resveratrol (A) Biosynthesis and (B) Post-Synthetic Modification and Derivatization.

Resveratrol can undergo several structural modifications after its biosynthesis. These derivatizations can increase its antifungal activity and alter the antioxidant properties of the stilbene, rendering its oxidation by laccase enzymes more facile.29 The glycosylation of 1, as seen in the resveratrol adduct piceid (5), is thought to aid in the storage and translocation of resveratrol within the cell tissues as well as protect resveratrol from oxidation.30 Alternatively, the resveratrol monomer can be oligomerized to form complex polyphenolic secondary metabolites composed of 2–8 resveratrol units. Collectively, these natural products are referred to as the “resveratrol oligomers” and are the subject of this review.
Resveratrol oligomers, like many secondary metabolites, are chiefly expressed as biological defense compounds and occur as dimers, trimers, tetramers, and higher-order oligomers in plants. The biological role of resveratrol and its oligomers within plants was first described in the 1970s by Langcake and Pryce.31 They identified resveratrol as the compound responsible for the formation of fluorescent lesions on the leaves of Vitis vinifera upon infection with the plant pathogens Botrytis cinerea (gray mold) and Plasmopara viticola (powdery mildew). Although resveratrol itself did not possess particularly potent antifungal properties, its expression during pathogenic invasion was the first indication of such a role in V. vinifera. This was further supported by the observation that resveratrol expression was inducible by fungal inoculation and ultraviolet light irradiation, and that the local concentration of 1 increased nearer to the site of the lesion.32
A more comprehensive comparison between susceptible and resistant species of grape vines revealed that the fungal lesions on the leaves of resistant species contained relatively low quantities of resveratrol but rather high concentrations of the oligomeric resveratrol natural products α-viniferin (6) and ε-viniferin (7) (see section 3.2).33 Unlike resveratrol, these compounds exhibited potent antifungal activity and were present in high concentrations in fungal-resistant cultivars of the grape vine. Even at this early stage, Langcake, Pryce, and others34 had speculated that these molecules were the products of the oxidative oligomerization of resveratrol. Drawing on the known chemistry of related lignan phytoalexins such as licarin A,35 they demonstrated that 1 could be dimerized to produce more potent antifungal compounds, indicating that resveratrol was a precursor to the actual phytoalexins in the plant.36,37 It is now believed that while most oligomers are produced endogenously, some may in fact be formed by the action of oxidase enzymes that are part of the invasive fungus.38 Whether this is part of a “detoxifying” process of fungal origin that enables pathogenesis or a plant-designed defense mechanism that exploits the invading species’ metabolic machinery for its own demise is currently unknown.
3. Isolation and Characterization of Resveratrol Oligomers
The first reported resveratrol oligomer was characterized 50 years ago by single-crystal X-ray diffraction (XRD) analysis.34,39,40 Over the next quarter-century, approximately 25 structurally related compounds were identified. As the number of natural product reports increased, structural patterns emerged, including the presence of dihydrobenzofuran and indane moieties, as well as bicyclic [3.2.1] and [3.3.0] ring systems. Today, >300 resveratrol oligomers have been characterized, largely due to the advent of advanced NMR and chromatographic technologies.41 Additionally, the search for these natural products has recently been incentivized by a series of reports lauding the broad spectrum biological activities of resveratrol.12,14−17 In the plant kingdom, resveratrol oligomers have now been isolated from the following nine families: Dipterocarpaceae, Vitaceae, Cyperaceae, Gnetaceae, Fabaceae (Leguminosae), Paeoniaceae, Apiaceae (Umbelliferae), Haemodoraceae, and Musaceae. In this section, five representative case studies are presented which we feel speak to the beauty of these molecules and highlight dedicated efforts required to elucidate their complex architectures.
The structural elucidation of these natural products has been accomplished using a combination of mass spectrometry (MS) with UV–vis, infrared (IR), and NMR spectroscopy. Although the connectivity of these compounds can typically be deciphered through detailed analysis of the 2D NMR spectra (COSY, HSQC, HMBC), the relative and absolute configurations have often been difficult to resolve. Notably, vicinal coupling constants are notoriously poor predictors of relative configuration within the five- and seven-membered ring systems which are prevalent in this class of natural products,42 leading to a number of structural misassignments43 and revisions.44,45 Modern NOE(SY) and ROESY experiments have enabled assignment of relative configuration to a reasonable level of accuracy, although the determination of absolute configuration remains a difficult task. Typically only derivatives of the natural products are capable of forming crystals suitable for XRD analysis, which introduces concerns about compound stability and propensity for skeletal rearrangement. The development of circular dichroism (CD) has helped to address this challenge, but suffers from a dependence on analogy to previous reports. In this approach, the absolute configurations are assigned on the basis of observed Cotton effects46−48 in the CD spectra and can be inferred by comparison to the spectral signatures of analogous structures whose absolute configurations were previously determined. The risk in this approach is apparent: an error in assignment can propagate through the literature as spectroscopists rely on the original report as their reference spectrum. Nonetheless, the assignments for the resveratrol oligomers are reasonably well-supported.
The most relevant example of this is the assignment of the absolute configuration of (−)-ε-viniferin (7) by Kurihara and co-workers.49 This assignment has been highly influential as a large number of resveratrol oligomers are believed to derive from 7. Indeed, the absolute configurations of several higher-order oligomers have been deduced on the basis of biomimetic transformations of optically pure (−)-ε-viniferin.50−52 Kurihara et al. prepared permethylated derivatives of the syn and anti diastereomers of 7 in order to determine the relative configuration of the natural sample. As expected, the anti diastereomer was found to be consistent with the NMR data for the enantiopure authentic sample. The final assignment of (−)-ε-viniferin as 7a(R), 8a(R) was made by comparison of the CD spectrum of a hydrogenated sample of 7 to the CD spectrum that had been used for the assignment of gnetin F (8) 5 years prior (Figure 1).53 The stereochemical assignment of 8, in turn, had been established by comparison to the CD spectra of benzofuranoid neolignans whose absolute configurations were determined by X-ray crystallography.54
Figure 1.

Establishing the absolute configuration of (−)-ε-viniferin.
3.1. Hopeaphenol
The first resveratrol oligomer ever characterized was hopeaphenol (9, Figure 2), a resveratrol tetramer isolated by Coggon et al.34,39,40 Originally isolated in 1951 from the heartwood of Hopea odorata and Balanocarpus heimii, two members of the Dipterocarpaceae family, another 15 years passed before the structure was solved. Ultimately, the isolation and characterization of this natural product were made possible by its poor solubility, enabling its isolation by crystallization and eventually its structural determination by single-crystal XRD of a dibromo-decamethyl ether derivative. It was known that exposure of the permethyl ether compound to electrophilic halide sources did not induce skeletal rearrangement, and therefore analysis of this derivative enabled the assignment of the absolute structure of hopeaphenol itself. Interestingly, stilbenes had not yet been reported as occurring in any Dipterocarpaceous plants at this time, but the researchers correctly assumed that the biosynthesis occurs through the oligomerization of resveratrol (1), which had been identified some 20 years earlier.55 However, it was not until a decade later that ε-viniferin (7), the building block for hopeaphenol (and nearly all higher-order oligomers), was identified. Indeed, the absolute configuration of (−)-ε-viniferin (7), as determined by Kurihara and co-workers in 1990, was consistent with the absolute configuration of the dihydrobenzofurans in (−)-9, the structure reported by Coggon and co-workers.
Figure 2.
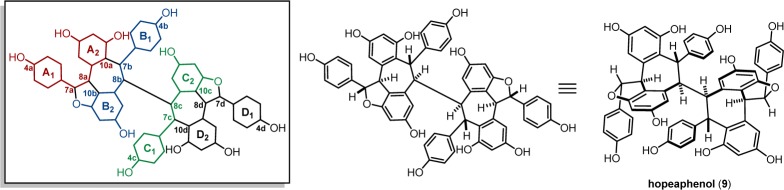
Numbering scheme for hopeaphenol.
In 1992, Kawabata et al. were able to confirm the structure of hopeaphenol using advanced NMR spectroscopic methods that were unavailable at the time of its initial isolation.56 Despite the complex polycyclic scaffold of hopeaphenol (9), its C2-axis of symmetry halves the number of resonances observed in the NMR spectra. Determining such structures by NMR spectroscopic methods is confounded by difficulty in distinguishing between equivalent nuclei. For equivalent, hydrogen-bearing carbons that are covalently bonded, it is possible to ascertain the connectivity using isotopomeric asymmetry. In hopeaphenol there are three possible isotopic combinations for 8b and 8c: 12C–12C (97.8%), 12C–13C (2.18%), and 13C–13C (0.01%). Although the latter is below detection limits, the broken symmetry of the 12C–13C species can be exploited. Using INEPT (insensitive nuclei enhancement through polarization transfer), LSPD (long-range selective proton decoupling), COLOC (correlation through long range coupling), and HMBC, these researchers were able to unequivocally establish the 8b–8c connectivity through detection of the 1J(C–H) and 2J(C–H) coupling constants across the H–12C–13C–H spin system.
3.2. Viniferins
In 1976, Ingham and co-workers discovered that groundnut hypocotyls accumulate a mixture of cis- and trans-resveratrol in response to infection by the nonpathogenic fungus, Helminthosporium carbonum, representing the first time that resveratrol had been implicated as a phytoalexin.57 Shortly thereafter, Langcake and Pryce discovered that resveratrol also accumulates in the leaves and lignified stem tissue of grapevines that have been infected with fungus or exposed to UV irradiation.37 Resveratrol, although not constitutively expressed in the leaves, was present at a significant concentration (50–400 μg/g fresh weight in leaves and 700 μg/g in stem tissue). In search of other resveratrol-derived materials, they extracted vine leaves from the grapevine Vitis vinifera (Vitaceae family) that had been infected with the necrotrophic fungus, Botrytis cinerea. Structural elucidation using a combination of derivatization, MS, UV–vis, and NMR studies, enabled the identification of ε-viniferin (7), a trans-dehydrodimer of resveratrol, as well as α-viniferin (6), a resveratrol cyclotrimer (Figure 3). Due to the relatively low antifungal activity of resveratrol as compared to the viniferin oligomers, these researchers proposed that resveratrol should not be considered a phytoalexin. Rather, they proposed that resveratrol serves as a biosynthetic precursor to the more active viniferins, drawing analogy to the oxidative oligomerization of cinnamyl alcohols (monolignols) during the biosynthesis of phenylpropanoid lignans. This insightful hypothesis was experimentally supported by the oxidative dimerization of resveratrol upon treatment with horseradish peroxidase (HRP) and H2O2, although the regioisomeric trans-dehydrodimer, δ-viniferin (10), was isolated (Figure 3).36
Figure 3.

Identification of α- and ε-viniferin, phytoalexin constituents of Vitis vinifera.
While the structure of α-viniferin (6) was proposed in their prior communication, limited structural evidence was presented.37 In a follow-up paper, these authors shared their efforts toward elucidating the structure of this resveratrol cyclotrimer.58 Impressively, the structural assignment was made using only 1H NMR spectroscopy, as there was not enough material to obtain a 13C NMR spectrum. It was assumed that each of the 2,3-dihydrobenzofurans were trans because this was the only relative configuration that had been found thus far in natural isolates. Next, the relative disposition of the three dihydrobenzofurans about the 9-membered ring was assigned using circular dichroism and 1H NMR spectroscopy. Because the compound was optically active, the cyclotrimer could not have a C3-axis of symmetry, and the 1H NMR data were consistent with this observation. Using a Dreiding model to rationalize observed coupling constants based on the Karplus curve, α-viniferin (6) was proposed to have a trans, cisoid, trans, transoid, trans stereostructure, as depicted in Figure 3.
In 1990, the proposed structure of α-viniferin was confirmed by Kitanaka and co-workers using a combination of single- (HETCOR) and multiple- (COLOC) bond heteronuclear correlation NMR experiments, which today have been supplanted by HSQC and HMBC, respectively.59 NOESY experiments supported the relative configuration proposed by Langcake and Pryce. Kitanaka et al. found that the Cotton effects of the CD spectrum obtained on their sample from Caragana chamlagu (Fabaceae) showed opposite curves to those previously reported, and (+)-α-viniferin was therefore deemed antipodal. This supports the hypothesis that these oligomers are synthesized from ε-viniferin (7), as Vitaceous plants are known to produce the (+)-isomer of this resveratrol dimer while nearly all other resveratrol-producing plant families synthesize the enantiomer. Notably, α-viniferin has since demonstrated a wide range of interesting biological activities (see section 8.2).
3.3. Vaticaffinol
Fifteen years after the milestone discovery of the structure of hopeaphenol (9), Sultanbawa and Bladon reported the isolation (from Vatica affinis) and structural elucidation of a second resveratrol tetramer, vaticaffinol (11) (Figure 4).60 Unlike hopeaphenol, in which the vicinal 7/8b, 7/8c and magnetically equivalent 8b/8c protons do not couple (Figure 2), decoupling experiments on vaticaffinol revealed vicinal coupling through 7a–8a–8c–7c at the core of the molecule (Figure 4). Hopeaphenol has a C2-axis of symmetry, resulting in just four observable aliphatic proton signals, whereas the 1H NMR spectrum of vaticaffinol has eight distinct proton resonances. Additionally, the physical properties of 11 (mp =280 °C, [α]D = −22.5°) were found to be distinct from those of 9 (mp = 350 °C, [α]D = −407°). It is worth noting that ε-viniferin (7) was isolated from the same extract, representing the first report of the dimer from the Dipterocarpaceae, and lending credence to the hypothesis that tetramers such as 9 and 11 are formed via the oxidative coupling of 7 (see section 5.4.2).
Figure 4.
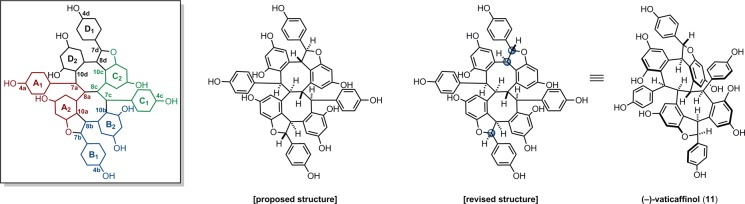
Reassignment of the structure of vaticaffinol.
Several years after these initial reports, Sotheeswaran and Kraus revised the stereochemical assignment of 11 using a sample isolated from Stemonoporus canaliculatus.44 Using NOE and 1H-homodecoupling experiments, the relative configuration at 7b/8b was revised from trans to cis, and an NOE correlation between 7a, 8a, and 8d led them to invert the stereogenic centers at 7d and 8d so as to place 8d cisoid to 7a and 8a (Figure 4). This stereochemical revision has important implications with respect to the biogenesis of these compounds. The previously proposed diastereomer of vaticaffinol would have required the crossed coupling of (+)- and (−)-ε-viniferin for this to be true, whereas in the revised structure, the nonepimerizable C-3 stereogenic centers of each of the dihydrobenzofurans are of the same absolute configuration.
3.4. Gnetin A and Kobophenol B
In 1982, a team of researchers discovered and reported the structure of gnetin A (12) (Figure 5), a bicyclo-[3.2.1]-octen-dione based resveratrol dimer isolated from Gnetum leyboldii (Gnetaceae).61 The connectivity about the bicyclic core could be deduced through a combination of 13C NMR chemical shift and 1H NMR coupling analyses, and the substitution pattern and relative configuration of the vicinal arenes were deduced through evaluation of 3-bond coupling constants and homodecoupling experiments. An identical structure was later isolated from the roots of Vitis thunbergii (Vitaceae) by Chen and co-workers and given a second name, vitisinol D.62
Figure 5.
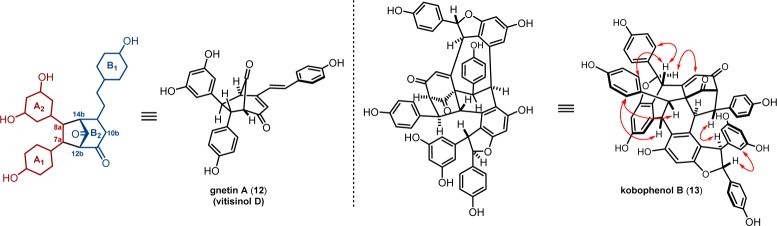
Resveratrol oligomers with a dearomatized A2 resorcinol ring.
In 1991, Kawabata et al. elucidated the structure of kobophenol B (13) (Figure 5), a highly complex resveratrol tetramer isolated from the subterranean parts of Carex pumila.63 Mass spectra of the natural product and its peralkyl and peracetyl derivatives demonstrated that it was a resveratrol tetramer. IR spectroscopy indicated that the molecule contained both conjugated (1640 cm–1) and isolated (1740 cm–1) carbonyl groups, and the presence of 13C resonances at δ 194.0 and 203.4 was consistent with this observation. The previous report of the structure of gnetin A validated the proposal of a bicyclo-[3.2.1]-octen-dione. The remaining connectivity was determined through extensive HETCOR and COLOC correlation NMR experiments. Relative stereochemical assignments were made using NOESY, and selected examples of these enhancements are highlighted in Figure 5. As with the foregoing tetramers, kobophenol B (13) is proposed to arise from the oxidative coupling of two molecules of ε-viniferin (7), which has been isolated from the same plant source.
3.5. Shoreaketone
In 2005, Ito and co-workers reported the structure of shoreaketone (14) (Scheme 2), a resveratrol tetramer that was isolated from three species of Dipterocarpaceous plants: Shorea uliginosa, Shorea hemsleyana, and Vateria indica.64,65 While gnetin A (12) and kobophenol B (13) each contained dearomatized resorcinol rings, shoreaketone is characterized by a dearomatized phenol. This molecule represents the first example of an atropisomeric resveratrol oligomer, although hindered rotation had been observed previously at reduced temperatures for other oligomers.66,67 At ambient temperature, each of the rotational isomers of 14 were configurationally stable. Using variable-temperature (VT) NMR, these researchers were able to observe interconversion of the isomers by the variance in signal enhancement of the NOE between H-8b and H-14b (Scheme 2) and via anisotropic effects on the chemical shift of several protons.
Scheme 2. Atropisomerism of Shoreaketone and Its Acid-Mediated Interconversion to Isohopeaphenol Monomethyl Ether.

Once the connectivity had been established through extensive COSY, HSQC, and HMBC experiments, the relative configuration and ring conformations of 14 were determined using ROESY. Selected dipolar couplings are highlighted in Scheme 2. The relative proportions of the two conformers were dependent on solvent polarity, viscosity, and temperature. This equilibrium could be shifted to populate predominantly one rotamer when the spectra were recorded with increasing concentrations of deuterium oxide (D2O) in acetone-d6. Interestingly, subjection of 14 to either trifluoromethanesulfonic acid or sulfuric acid in methanol resulted in its skeletal rearrangement to the monomethyl ether derivative 15a of (+)-isohopeaphenol (Scheme 2). As the stereochemical information at each of the dihydrobenzofurans should be unchanged by this rearrangement, the authors proposed the absolute configuration for shoreaketone based on a previously reported assignment for (−)-isohopeaphenol (15, see Scheme 17, B).44,65
Scheme 17. Studies of the Biosynthesis of 8–8′ Trimers (A) and Tetramers (B).

4. The Biosynthesis of Resveratrol Dimers
The resveratrol oligomers are a highly diverse class of natural products that are produced by a small set of phylogenetically distant plant families.68 Although the number of individual resveratrol-based natural products is >300, there are many characteristic structural motifs that are conserved across the oligostilbene producing plants. There have been several attempts to phenotypically classify the resveratrol oligomers based on the presence of these conserved structural motifs, a position which has waned in popularity as the field matured. Instead, as Cichewicz and Kouzi posited, a genotypic approach to oligomer classification that presents the oligomers based on their biological source is more useful, since it provides a link between taxonomy and chemical structure.42 However, this approach is still imperfect and does not account for the possibility of biosynthetic pathways that are conserved between plant families. In this review we will classify the natural products on the basis of the regioisomeric mode of their dimerization, and will employ the numbering scheme favored by Kawabata and co-workers (Scheme 3),49 which conveniently standardizes the arenes of each resveratrol subunit as A1/A2 for the phenol and resorcinol rings, respectively. In the literature, the resveratrol unit designated “A” is usually arbitrarily assigned and each successive resveratrol subunit is then assigned the next letter in the alphabet (e.g., “B”, “C”, ...).
Scheme 3. Regioisomeric Modes of Resveratrol Dimerization.

Resveratrol oligomerization appears to proceed via the coupling of oxidatively generated phenoxyl radicals (Scheme 3, A–D) as originally proposed by Langcake and Pryce.37 The dimerization typically occurs through three regioisomeric modes: the 8–10′ coupling (as found in ε-viniferin (7) and ampelopsin F (16)69), 8–8′ coupling (quadrangularin A (17)70,71 and pallidol (18)72), and 3–8′ coupling (e.g., δ-viniferin (10), Scheme 3). Several exotic regioisomers, like the 8–12′ (gnetin C (19))61 or the 12–12′ coupling (amurensin M),73 have also been identified, but they are relatively uncommon. Following dimerization, highly reactive para-quinone methides such as 20–23 can undergo numerous regiodivergent Friedel–Crafts reactions, nucleophilic trappings, or tautomerizations. Although the divergent reactivity of these intermediates quickly generates an impressive number of complex architectures, nearly all of the resveratrol natural products can be reduced to these initial bond disconnections.
In the section below, we will outline the biosynthesis of the resveratrol natural products and present as much of the skeletal diversity as possible. Our objective is not to provide a comprehensive list of natural products (for this the reader is directed to refs (41, 42)), but rather to methodically delineate a plausible biogenic relationship between compounds possessing very different topologies. These “proposed” biosyntheses are strongly supported by empirical studies on the biogenesis of these natural products, with several of the ensuing schemes representing a compilation of many years of work and, at times, dozens of papers. Where appropriate, experimental data will be included to supplement the biosynthetic relationships as presented. In the following reaction schemes, each of the natural products is depicted as a single stereoisomer. However, due to the existence of antipodal resveratrol oligomers that derive from enantiomeric starting materials, there will sometimes be apparent changes in the molecule’s configuration with respect to these starting materials.74 In these instances, we do not mean to imply a biosynthetic epimerization, but rather only to depict the structure in the reported configuration.
4.1. Biosynthesis of 8–10′ Dimers
The 8–10′ connectivity is by far the most prevalent found among resveratrol-derived oligomeric natural products.42 The parent molecule of 8–10′ oligomers, ε-viniferin (7), has the relatively uncommon distinction of being a natural product that is found naturally in both enantiomeric forms.75 (−)-ε-Viniferin is found in several plant families such as Dipterocarpaceae, Gnetaceae, Cyperaceae, and Fabaceae (Leguminosae), while its enantiomer, (+)-ε-viniferin, is exclusively found in plants from the family Vitaceae (Scheme 4).41 Upon oxidation of 1 and 8–10′ dimerization, hypothetical para-quinone methide intermediate 21 can follow divergent cyclization pathways to generate either ε-viniferin via an oxa-conjugate addition (Scheme 4, path A), or para-quinone methide 24 through a vinylogous Friedel–Crafts reaction (path B). From here, a second intramolecular cyclization yields the bridged [3.2.1] bicyclic dimer ampelopsin F (16)69 (path C), while tautomerization yields the indane isomer ampelopsin D (25) (path D).76 The existence of the intermediate para-quinone methide 24 is strongly supported by the isolation of viniferethers A/B (26),77 the products of the nucleophilic trapping of 24 by methanol (path E). Caraphenol B (27) likely follows a similar biosynthesis: hydration of 24 with water, followed by oxidation forms the benzylic ketone.43 Oxidative cleavage of the olefin of ampelopsin D (25) results in the formation of the natural product pauciflorol F (28),78 which was inadvertently synthesized by Oshima and co-workers 11 years before its eventual isolation.76
Scheme 4. Proposed Biosynthesis of the 8–10′ Dimers.

Given the ubiquity of the 8–10′ dihydrobenzofuran motif in the resveratrol oligomers, it appears that ε-viniferin plays a central role in the biosynthesis of the entire class of 8–10′ dimers and oligomers. The biogenic relationship of 7 to ampelopsins F (16), D (25), and B (29) was delineated by Niwa and co-workers50 through a series of biomimetic Brønsted-acid-mediated rearrangements of natural (+)-ε-viniferin (Scheme 5, A). Additionally, they were able to convert peracetyl 7 to ampelopsin A (30) through an epoxidation/fragmentation/cyclization sequence in 55% overall yield (Scheme 5, B). The optical rotations of each of these synthetically derived compounds were compared to reported values for the isolated natural products, verifying that (+)-ε-viniferin was their biogenic precursor and by analogy, confirming their absolute configuration.
Scheme 5. Niwa’s Brønsted-Acid-Mediated Conversion of ε-Viniferin to Various 8–10′ Dimers.

4.2. Biosynthesis of Oxidized 8–10′ Dimers
Plants found in the Dipterocarpaceae family synthesize an array of highly oxidized and structurally rearranged 8–10′ dibenzocycloheptane dimers that are unique to this plant family. Although no unified biosynthesis of these rearranged 8–10′ dimers has been proposed, the biogenic relationship between 7 and hopeanol (31),79 for example, can be inferred from the isolation of several natural products of intermediate levels of oxidation and rearrangement that ultimately lead to 31. The efforts of the Tan and Ito groups in the isolation and detailed characterization of these fascinating (and surprisingly stable) natural products provide key insight into the biogenic relationships of these compounds.
Following epoxidation of the para-hydroxy styrene of 7, trans-stilbene oxide 32 can undergo a dearomative epoxide fragmentation to generate intermediate 33, which is prone to an intramolecular 7-exo trig Friedel–Crafts cyclization to afford (+)-balanocarpol (34)80 or its epimers (−)-ampelopsin A (30)81,82 and acuminatol (35)83 (Scheme 6). The epimeric [3.2.2] oxabicyclic natural products heimiol A (37)84 and hopeahainol D (38)85 possibly derive from an intramolecular etherification of the C7b–O7a bond upon an acid-mediated formation of para-quinone methide 36 (path A). A direct benzylic alcohol oxidation of 30 or 35 to the ketone affords pauciflorol E 39 (path B).78
Scheme 6. Proposed Biogenic Relationship Between ε-Viniferin and the 8–10′ Dibenzocycloheptane Dimers.

The biogenic relationship between the (+)-balanocarpol (34), hopeanol (31), and the hopeahainols A/B (40/41)86 can be deduced from the isolation of several oxidized intermediary natural products. The oxidation of 34 may occur through the hypothetical intermediate 42, since the natural product hopeahainanphenol (43)87 would represent a product of its oxidative dearomatization. Dehydration of intermediate 42 may lead to the formation of (−)-malibatol A (44),88 a plausible biosynthetic precursor to the several other oxidized 8–10′ dimers. The oxidation of benzylic alcohol 44 yields the natural product shoreaphenol (45)89 (also known as hopeafuran),90 although biosynthesis via a formal dehydrogenation of pauciflorol E (39) cannot be ruled out. The biogenic relationship between (−)-malibatol A (44) and the hopeanols/hopeahainols is supported by the isolation of the oxidized 8–10′ dimers vaticahainols B (46) and A (47).91 Vaticahainol B (46) is presumably derived from a diastereoselective oxidative dearomatization of 44, perhaps through an epoxidation. A stereoselective 1,2 aryl migration from 46 yields vaticahainol A (47). Subsequent C8a oxidation and oxidative dearomatization of phenol A1 would yield the natural product hopeahainol A (40). The biogenic relationships between hopeanol (31),79 hopeanol B (48),86 and hopeahainols A (40) and B (41) are supported by total synthesis (see sections 7.1.4–7.1.5).92 Upon trapping with water, vaticahainol B (46)91 can undergo an oxidative cleavage of its C7b–C8b bond followed by hydrolysis of 4-hydroxybenzoate to hemsleyanol E (49)93 (or its epimer) and diptoindonesin D (50),94 which differ only in the oxidation state at the C8a benzylic position.
The isolation of the rearranged dibenzocycloheptane natural products demonstrates another level of chemical diversity that is available to Dipterocarpaceous plants. These transformations also provide insight to how other plant families may perform oxidative modifications to other dimers, as in the case of the conversion of 7 to viniferifuran (51) (also known as amurensin H)95 (Scheme 6) or ampelopsin D (25) to pauciflorol F (28) (Scheme 4).
4.3. Biosynthesis of 8–8′ Dimers
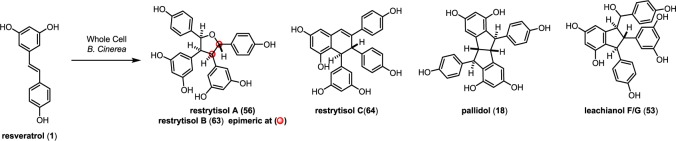
The natural products pallidol (18)71 and quadrangularin A (17) (formerly known as cyphostemmin B)70,71 were the first 8–8′ dimers isolated from Vitaceae. The 8–8′ dimers are relatively uncommon regioisomers and have been isolated from a limited number of plant families, primarily from Vitaceae. The proposed 8–8′ dimeric intermediate 22 has two para-quinone methides and two vicinal stereogenic centers, which promote unusual diversification reactions when compared to the regioisomeric 8–10′ and 3–8′ dimers. The relative configuration of these vicinal stereocenters has important consequences from a product determination standpoint. The C2-symmetric diastereomer of 22 has the correct relative configuration to undergo the sequential Friedel–Crafts cyclizations necessary to form the [3.3.0] bicyclic core of pallidol (18) (Scheme 7, path A). After the first cyclization reaction of meso22, however, the anti,anti-configuration of the resultant quinone methide 52 would prevent a second cyclization reaction due to the thermodynamically unfavorable formation of a trans-fused bicyclo[3.3.0]octane (Scheme 7, path B).96 Tautomerization of para-quinone methide intermediate 52 to the E-alkene provides (−)-quadrangularin A (17). There are several natural products that presumably derive from the nucleophilic trapping of quinone methide indane 52 (path C) including leachinols F/G (53),97 parthenostilbenins A/B (54),98 and quadrangularins B/C (55).71 It is probable that these natural products are actually isolation artefacts, since methanol and ethanol were used as extraction solvents for 54 and 55, respectively. The isomeric furan dimers restrytisol A (56)38 and tricuspidatol A (57),99 which derive from the double addition of water across the para-quinone methide stereoisomers of 22 (path D), provide indirect evidence for the existence of both the meso- and C2-symmetric diastereomers. Amurensin A (59)100 is the product of trapping of water onto the linear mono-para-quinone methide 58, a tautomer of 22 (Scheme 7, path E).
Scheme 7. Proposed Biosynthesis of the 8–8′ Dimers.

So far, quadrangularin A (17) has only been isolated from Vitaceae as the (−) optical isomer, but the oxidized derivative caraphenol C (60),43 isolated from Fabaceae, appears to derive from (+)-quadrangularin A, or intermediate 52, suggesting that other plant families are capable of biosynthesizing the opposite enantiomer of 17. However, this has yet to be substantiated (Scheme 8). Quadrangularin A (17) can undergo an alkene isomerization, generating its Z-isomer parthenocissin A (61).101 An oxidative cyclization reaction, perhaps through a 6π electrocyclic ring closure followed by dehydrogenation, yields laetevirenol A (62).102 It is unknown whether the interconversion of 17 to 61 and 62 is mediated by enzymes or if they are simply the result of background isomerization/oxidation. For a related discussion on the proposed biosynthesis of these oxidized 8–8′ dimers, see section 5.5.1.
Scheme 8. Oxidized Derivatives of 8–8′ Dimeric Natural Products.
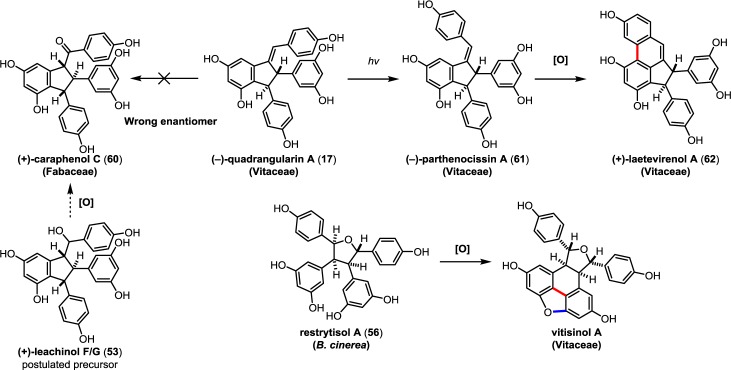
In 2000, Cichewicz and Kouzi demonstrated that the grape pathogen B. cinerea could exogenously produce resveratrol dimers when supplemented with resveratrol in its fermentation broth.38 They isolated three natural products: two 8–8′ dimers, restrytisols A/B (56/63) and the dihydronaphthylene restrytisol C (64), which were isolated as optically active compounds. In addition, they isolated two other 8–8′ resveratrol dimers (Scheme 9), pallidol (18) and leachinols F/G (53), indicating that B. cinerea is capable of producing an array of 8–8′ dimers and that some fraction of these dimers can be formed through uncontrolled oxidation. The authors suggest that fungi not only induce the expression of resveratrol in plants, but may also produce oligomers by fungal laccase catalysis. They further speculate that fungi utilize this as a mechanism for detoxifying resveratrol and other chemical defenses of the plant and that the presence of certain resveratrol oligomers in plant extracts can be attributed in part to nonplant based pathogens.
Scheme 9. Whole Cell B. Cinerea-Mediated Dimerization of 1 to 8–8′ Dimers.
4.4. Biosynthesis of 3–8′ Dimers
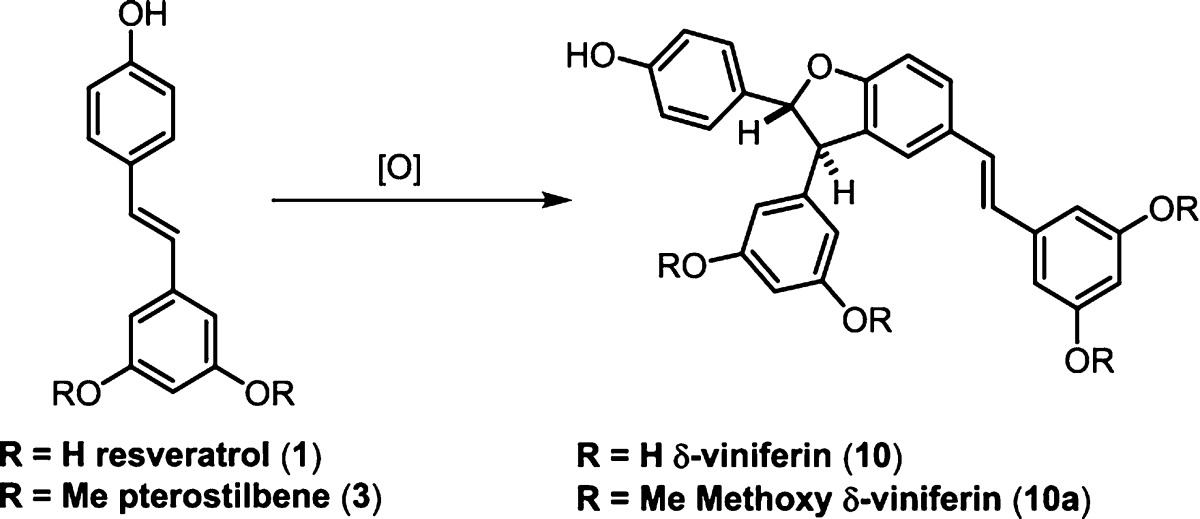
The 3–8′ dimers (Scheme 10) are one of the least structurally diverse group of resveratrol natural products. The most abundant compound in this class, δ-viniferin (10),103 also commonly called the “resveratrol-trans-dehydrodimer”, is found in many families of resveratrol producing plants. δ-Viniferin was inadvertently synthesized by Langcake and Pryce during their seminal isolation studies on grape phytoalexin compounds when they attempted to produce ε-viniferin (7) from resveratrol using horseradish peroxidase (HRP) and H2O2.36 A subsequent report by Jeandet103 was the first to identify δ-viniferin as a major constituent of resveratrol oxidation by fungal laccases. These findings, along with a report by Cichewicz and co-workers,38 advanced the notion that the resveratrol dimers are not exclusively synthesized endogenously by the plant, but can be generated extracellularly by other organisms. The 3–8′ dimers formed in this manner are typically isolated as racemates, since laccases/peroxidases, which differ by the metal oxidant in their active sites (Cu104 vs Fe105), are fairly promiscuous enzymes capable of oxidizing a wide range of substrates. When resveratrol is oxidized by inorganic oxidants, δ-viniferin is typically the major product, indicating an inherent regioisomeric preference during an uncontrolled dimerization reaction. The biomimetic oxidation of resveratrol has since been studied by numerous groups following these seminal contributions.106−111 Because this compound predominately forms during the uncontrolled oxidation of resveratrol, δ-viniferin is often referred to as a “non-natural” resveratrol dimer. Despite this classification, 10 has been isolated as an optically active constituent of Rheum maximowiczii (Polygonaceae) called maximol A along with its alkene isomer 65 (Scheme 10).112 Interestingly, the optical rotation of maximol A nearly matches the [α]D of 99.5% enantiopure δ-viniferin obtained by chiral separation of a racemic sample.113
Scheme 10. 3–8′ Dimers are Commonly the Major Isomers Formed on Exposure of Resveratrol to Oxidants.

5. Biosynthesis of Higher-Order Resveratrol Oligomers
The chemical defense against pathogenic invasion is a continuous evolutionary arms race between the invasive species and the host plant. In this scenario, the survival of the plant relies partly on its ability to rapidly generate novel defense compounds in response to pathogenesis. The regioisomeric dimerization modes described in the preceding sections constitute the foundation for the biosynthesis of a diverse collection of higher-order resveratrol oligomers. Indeed, biosynthesis of the higher-order natural products proceeds in a similar fashion to the resveratrol dimers, and several structural motifs found in the resveratrol dimers are conserved in the more complex oligomeric compounds. The resveratrol dimers are almost universally generated by an oxidative radical coupling. In contrast, the higher-order oligomers can be the product of either an oxidative coupling to resveratrol/ε-viniferin or an intermolecular Friedel–Crafts reaction (Scheme 11). One stringent structural requirement for the oxidative oligomerization is the presence of the 4-hydroxy stilbene moiety found in both resveratrol and 7. At a glance, the biosynthesis of the resveratrol oligomers would appear to be the product of an iterative oligomerization sequence, wherein a resveratrol dimer such as pallidol (18) would undergo successive homologation reactions, in a fashion similar to polyketide natural products (Scheme 11).114 Instead, it would appear that the majority of oligomerization reactions are convergent processes, wherein tetramers are the product of a dimerization of dimers, pentamers a crossed dimerization of a trimer and a dimer, and so forth.
Scheme 11. Biosynthesis of Higher-Order Oligomers Is Convergent in Nature and Employs Both Radical and Polar Mechanisms.

As is the case with other resveratrol based natural products, several stereoisomers of the same carbon skeleton have been isolated. This has led to speculation on whether the oligomerization occurs in a random or controlled fashion. However, there is little known about the biogenic pathways responsible for the formation of these natural products. In the following sections, we will depict the dimerization and cyclization reactions that are likely involved in the biosynthesis of these oligomers. The molecules chosen are representative of as many of the carbon skeletons found within this class of natural products.
5.1. Biosynthesis of 8–10′ Trimers
The 8–10′ trimers have been isolated from most plant families that biosynthesize resveratrol oligomers such as Vitaceae, Dipterocarpaceae, Cyperaceae, etc. In 2012, Pan et al. established the biosynthetic relationship between ε-viniferin (7), gnetin H (68),53 and miyabenol C (69)115 via a HRP-mediated cross trimerization of 7 and resveratrol (1) to afford 68 and 69 in 3.3% and 3.4% yields, respectively (Scheme 12).52 The gnetin H isolated from that reaction was subjected to a Brønsted-acid-mediated isomerization, yielding ampelopsin G (70) (4.5%), amurensin G (71) (8.4%), wilsonol B (72) (2.7%), and vitisin E (73) (34%), suggesting a common biosynthetic origin. These studies were also able to confirm the absolute configuration for compounds 68–73.
Scheme 12. Pan’s Biosynthetic Reactions on the Formation of the 10–8′ Trimers.
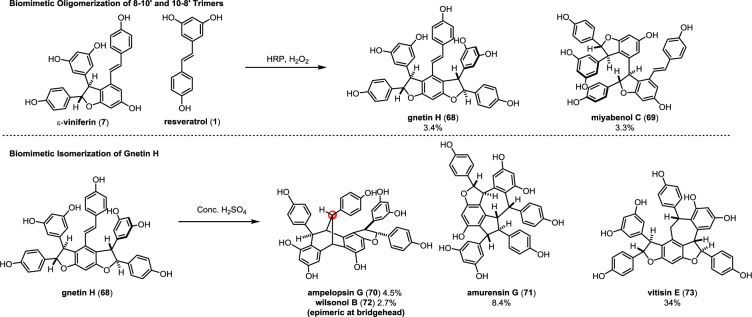
The 8–10′ trimers have two distinct modes of oligomerization: (1) the 10 position of ε-viniferin (7) can cross couple with the 8′ position of resveratrol, to afford the gnetin H (68) regioisomer (Scheme 12, path A) or (2) the 8 position ε-viniferin can cross couple with the 10′ position of resveratrol to give the miyabenol C (69) scaffold (path G).
As the apparent biosynthetic precursor for many of the resveratrol oligomers, gnetin H (68) can conceivably be interconverted into other oligomers through a series of acid-mediated Friedel–Crafts reactions. Protonation/fragmentation of the 11b dihydrobenzofuran (Scheme 13 for numbering scheme) reconstitutes the highly reactive quinone methide 74 (Scheme 13, path B). In a fashion analogous to the biosynthesis of ampelopsins D/F (25/16), a vinylogous Friedel–Crafts (5-exo-trig) cyclization of 74 would generate para-quinone methide 75 through the formation of the C8b–C7a bond (path D). This intermediate is presumably the precursor for two isomeric natural product scaffolds. A 7-exo-trig cyclization (C10c–C7b) yields 5,7-fused indane skeleton found in the natural products vaticanol A (76) (Scheme 13, path E),116 amurensin G (71),95 and suffruticosol B (77),117 among others (see Scheme 14). This ring system is prevalent among resveratrol trimers found in several families of oligostilbene producing plants.
Scheme 13. Divergent Biosynthesis of Regioisomeric 10–8′ and 8–10′ Trimers.

Scheme 14. Oxidized 8–10′ Trimers from Dipterocarpaceae.

Alternatively, intermediate 75 can be nucleophilically trapped by a transannular (6-exo-trig) Friedel–Crafts cyclization (C10a–C7b, path F), generating the [3.2.1]bicyclooctane cores of ampelopsin G (70),69 and wilsonol B (72).52 Finally, the direct protonation of the gnetin H (68) stilbene could promote cyclization to the natural product vitisin E (73),118 via intermediate 78.
Although the corresponding oxidation/cyclization reactions have not been reported for miyabenol C (69), its relationship between other natural products can be deduced in a similar manner as described above. Acidification of 69 and fragmentation of its dihydrobenzofuran would regenerate para-quinone methide 79 (Scheme 12, path H), which can undergo an intramolecular vinylogous Friedel–Crafts (5-exo-trig) cyclization to form the C8a–C7b bond (path K). The resultant quinone methide 80 can undergo two transannular cyclization reactions: an intramolecular 6-exo-trig cyclization (C14b–C7a, path L) yields the carbon skeleton of carasiphenol B (81)119 while an alternative 9-exo-trig cyclization (C10c–C7a, path M) yields the natural product suffruticosol C (82).117 Finally, the direct oxidation of miyabenol C (69) can initiate an intramolecular formal [3 + 2] cycloaddition to form a 9-membered ring, yielding the cyclic trimer α-viniferin (6) (path I).
5.2. Biosynthesis of Oxidized 8–10′/10–8′ Trimers
Several oxidized resveratrol trimers have been isolated from plants in the Dipterocarpaceae family. In 2005, Ito and co-workers isolated a series of resveratrol trimers including the cotylelophenols A (83), B (84), and C (85),120 which are oxidized derivatives of pauciflorol A (86)75 (Scheme 14). This series of natural products provides an exquisite snapshot of the biogenic relationship between these compounds that is likely relevant to the previously discussed oxidized dibenzocycloheptane dimers in section 4.2. Cotylelophenol C (85) is formed from pauciflorol A (86) in a net 4 electron oxidation, and is in the same oxidation level as cotylelophenol B (84). These natural products are likely interconverted via a Brønsted-acid-mediated rearrangement. Cotylelophenol B is the logical biosynthetic precursor to cotylelophenol A (83) via a stereoselective 1,2 aryl shift, mirroring the relationship between vaticahainol B (46) and A (47) (Scheme 6). In 2014, Ge and Tan reported the isolation and structural elucidation of dipterocarpol C (87),121 a remarkable oxidized derivative of vaticanol E122 that is presumably formed from 84 upon hydration of the quinone methide and subsequent oxidative cleavage of the vicinal diol. The presence of the hydrolytically labile phenol ester is the first direct evidence for the oxidative cleavage of the dihydrobenzofuran moiety in the resveratrol oligomers. The analogous structure for the related natural products hemsleyanol E (49)/diptoindonesin D (50) has not yet been reported (Scheme 6).
A similar set of oxidations have been observed for the cyclic 8–10′ trimers, wherein dearomatization of (+)-α-viniferin (6)123 yields grandiphenol C (88)124 which can interconvert to caraphenol A (89)43 by a dehydration reaction. An oxidation/rearrangement of benzofuran 89 affords grandiphenol D (90),124 while oxidative cleavage converts 89 into hopeachinol B (91).125
Taken together, these natural products provide a compelling unified mechanism for the biogenesis of the oxidized Dipterocarpaceous resveratrol oligomers. The biosynthetic machinery responsible for the oxidation of 86, 6, and the dimers 34 and 35 (section 4.2, Scheme 6) appears to be highly conserved, given the structural homology of several of the isolated natural products. The isolation of 87 provides a clear mechanistic rationale for the biogenesis of the resveratrol dimers hemsleyanol E (49) and diptoindonesin D (50). Very little is known about the enzymes that are responsible for this oxidation chemistry which so far has only been identified in Dipterocarpaceae. The resveratrol dimer viniferifuran (amurensin H) (51) (Section 4.2, Scheme 6) and caraphenol A (89) (Scheme 14) have been isolated from Vitaceae and Fabaceae, but it is not known at this time if these are synthesized in a similar fashion.
5.3. Biosynthesis of 8–10′ Tetramers
In contrast to the regioisomeric crossed 8–10′/10–8′ trimerization modes of 7 with resveratrol (Scheme 12), the 8–10′/10–8′ homocoupling modes of 7 are degenerate and therefore form para-quinone methide 92 as a single regioisomer (Scheme 15, path A). As a result, there is a narrower product distribution available from the divergent reactivity of hypothetical intermediate 92. A vinylogous Friedel–Crafts (5-exo-trig) cyclization forms the C8b–C7c bond of quinone methide 93 (path A). This intermediate, by analogy to ampelopsins F/G (16/70) (Schemes 4 and 12, respectively), can undergo a transannular 6-exo-trig cyclization resulting in the tetramer vaticanol C (94) (C14c–C7b, path B) or a 7-exo-trig affording the vaticanol B (95)116 skeleton (C10a–C7b, path C). An intramolecular oxa-conjugate addition of 92 yields the natural product flexuosol A (97) (path D);126 however, this natural product maps onto the meso stereoisomer of gnetin H (68), ampelopsin E.76 Finally, a 7-exo cyclization of quinone methide 92 (C10d–C7c, path E) yields the hypothetical intermediate 96, which upon oxidation can undergo a formal dearomative [3 + 2] annulation, forming the bicyclo-[3.2.1]-octen-dione core of kobophenol B (13) (see section 3.4 for isolation).
Scheme 15. Proposed Biosynthesis of the 8–10′ Tetramers.

The miyabenol oligomers are an unusual set of regioisomeric 8–10′ oligomers that possess dihydrobenzofuran motifs that alternate in absolute configuration (Scheme 15, path B). The biosynthesis of miyabenol A (98)115,127 is somewhat ambiguous and can possibly operate through iterative homologations of resveratrol, analogous to its trimeric counterpart miyabenol C (69) (section 5.1). The direct dimerization of two homochiral molecules (−)-ε-viniferin can also account for the alternating configurations found in the dihydrobenzofurans of 98. Miyabenol A (98) can undergo an additional oxidative formal [3 + 2], yielding the cyclic tetramer miyabenol B (99), which shares structural homology to the trimer α-viniferin (6). Impressively, Kawabata and co-workers were able to successfully interconvert miyabenol A (98) to miyabenol B (99) using vanadium oxytrichloride in 27% yield, representing one of the earliest applications of this strategy to validate the biogenic relationship between related resveratrol oligomers.127
5.4. Biosynthesis of 8–8′ Trimers and Tetramers
5.4.1. Biosynthesis of 8–8′ Trimers
Although the 8–8′ trimers are biosynthesized through a crossed coupling of 7 and 1, there are fewer possible cyclization and dimerization reaction pathways as compared to their 8–10′ counterparts, since there is only one intermediate generated during the radical coupling. Linear bis-para-quinone methide 100 has two electrophilic sites, allowing it to undergo dissymmetric cyclization reactions (Scheme 16). The existence of this intermediate is strongly supported by the isolation of the tetraarylfuran dimer, nepalensinol C (101), which is the product of the double addition of water to 100 (path A).128 Intermediate 100 can undergo two regioisomeric 5-exo-trig cyclization reactions, that are product determining. Upon cyclization to form the C10a–C7b bond of 102 (path B), a subsequent Friedel–Crafts (8-exo-trig) cyclization produces dibenzocyclooctane natural products distichol (103),129 or its stereoisomers canaliculatol,130 and stenophyllol B (path C).131 Alternatively, formation of the C14b–C7a bond (path D) produces the highly versatile intermediate 104. A second 5-exo-trig arylation of 104 (C10a–C7b, path E) affords the pallidol (18) derivative carasiphenol C (66),132 whereas the intramolecular 7-exo-trig cyclization of 104 forms ampelopsin C (105)82 (path F), a member of a larger group of stereoisomeric 5,7-fused indane trimers. Finally, the nucleophilic trapping of 104 with water (path G) yields nepalensinol A (106)128 and is indirect evidence for the existence of 104 itself.
Scheme 16. Proposed Biosynthesis of the 8–8′ Trimers (A) and Tetramers (B).

5.4.2. Biosynthesis of 8–8′ Tetramers
Like the 8–8′ trimers, the 8–8′ tetramers are widely distributed between the various families of oligomer producing plants. They are the products of the oxidative dimerization of two molecules of ε-viniferin (7), with 107 as the presumed biosynthetic intermediate for a diverse series of resveratrol tetramers (Scheme 16, path B). This bis-para-quinone methide can undergo either symmetric cyclization modes, delivering products such as hopeaphenol (9, path F),34 vaticaffinol (11, path A),60 and ampelopsin H (67, path B),76 or dissymmetric cyclizations to form viniferol A (109, path C)133 and hemsleyanol C (108, path D).93 It is reasonable to infer that these cyclizations occur through the same mechanisms as the trimers and dimers. The tetramer shoreaketone (14) (see section 3.5) features a highly functionalized spirocyclic cyclopentane that derives from quinone methide 110 through a dearomative Friedel–Crafts cyclization between carbons C1c and C7b. An oxa-conjugate addition onto the presumed spirocyclohexadienone (not pictured) forms the O11d–C2c bond to generate the spirocyclohexene found in shoreaketone. Ito and co-workers were able to demonstrate a Brønsted-acid-mediated rearrangement of shoreaketone (14) to (+)-isohopeaphenol methyl ether (15a) in quantitative yield (see section 3.5), establishing its absolute configuration and its identity as an 8–8′ tetramer.65
5.4.3. Empirical Support for Proposed Biosyntheses of 8–8′ Trimers and Tetramers
In order to investigate the biosynthesis of the resveratrol oligomers, Niwa51 and Pan52 have studied the oxidative cross-coupling of (+)-ε-viniferin (7) and resveratrol using horseradish peroxidase (HRP) and H2O2 (Scheme 17, path A). Although these reactions were nonselective, they were successful in procuring isolable quantities of davidiol A (111)134 from the reaction in 1–3% overall yield. Niwa and co-workers were also able to cross-dimerize 7 with pterostilbene (3) to get 111a, a methylated analogue of davidiol A in 2.7% yield. The Pan group was also able to isolate a very small quantity (<1%) of the natural product wilsonol A (112),52 a diastereomer of carasiphenol C (66) that they had isolated from V. wilsonae, which was not observed previously. Since the absolute configuration of (+)-ε-viniferin (7) was known, both groups were able to deduce the relative and absolute configurations of davidiol A (111) and wilsonol A (112) using these biomimetic reactions.
Winterhalter and co-workers corroborated these findings during their investigations on the large scale HRP/H2O2 oxidation reaction of resveratrol and ε-viniferin.109 In this complex mixture of products, they isolated and characterized two non-natural 3–8′ trimers named resviniferins A (113) and B (114) (Scheme 17, path A). Although they were unable to assign the relative configurations of these compounds, 113 has the same gross structure as the natural product diptoindonesin B (115) and is likely (bio)synthesized in similar fashion.135
The groups of Niwa136 and Pan52 have also performed biomimetic peroxidase mediated homocoupling of (+)-ε-viniferin (7) to study the biogenesis of the resveratrol tetramers. This oxidation unselectively produces an array of resveratrol oligomers. From this mixture, they were able to isolate the 8–8′ tetramers (+)-hopeaphenol (9, 9%) and (−)-isohopeaphenol (15, 1%), both of which matched the spectral data of the authentic samples (Scheme 17, path B). However, in a fashion analogous to the conversion of resveratrol to δ-viniferin (10), ε-viniferin (7) can also dimerize in a 3–8′ mode to generate tetramers such as vitisin B (116, 6%) and C (117, 3%). They also found that vitisin B (116)137 could be quantitatively converted to vitisin A (118)138 by acidic cyclization in MeOH. Vitisin A (118) was further reacted in the same conditions to produce vitisin D (119) in 37% yield. Since the absolute configuration of 7 was known (isolated from Vitis coignetiae), Niwa and co-workers were able to infer the absolute configurations of (+)-vitisin A (118), (+)-vitisin B (116), (−)-vitisin C (117),139 (+)-vitisin D (119),118 (+)-hopeaphenol (9), (−)-isohopeaphenol (15), and (−)-viniferal139 by analogy.52,136
5.5. Biosynthesis of 3–8′ Trimers and Tetramers
The 3–8′ oligomers are a smaller class of resveratrol natural products possessing the same trans-dihydrobenzofuran found in δ-viniferin (10) that were initially isolated from Vitaceous plants. It is unknown whether or not these natural products are biosynthesized endogenously, like δ-viniferin (10), or if they are the result of uncontrolled oxidation. Unlike 10, the 3–8′ oligomers are typically isolated as optically active compounds since they derive from chiral starting materials. The trimer parthenocissin B (120), for instance, appears to be the product of oxidative cross coupling of parthenocissin A (61)101 and resveratrol (1), while the tetramer vitisin B (116) is the 3–8′ dimer of two molecules of ε-viniferin (7) (Scheme 18). Further complicating the biogenic origins of these natural products is the fact that, for several 3–8′ oligomers, both dihydrobenzofuran diastereoisomers can be found in the plant extract, suggesting that these radical couplings are the result of nonspecific oxidation. Unlike the 8–8′ or the 8–10′ trimers and tetramers, which are primarily the result of ε-viniferin or resveratrol oligomerization, a surprising variety of substrates appear to be competent coupling partners for the 3–8′ oligomerization (Scheme 18). These include indane stilbenes quadrangularin A (17)/parthenocissin A (61) as plausible precursors for laetevirenols A–F,102 linear dimeric stilbene amurensin A (59) for wenchowenol (121),140 or ε-viniferin (7) for vitisins A (118), B (116), and C (117) (vide infra).137−139
Scheme 18. Proposed Biogenesis of the 3–8′ Trimers and Tetramers.

5.5.1. Unified Biosynthesis of the Laetevirenols
In 2008, the Pan group isolated an unusual series of dihydrobenzofuran-containing indane natural products called the laetevirenols102 which are parthenocissin A/quadrangularin A (61/17) derivatives that contain an unusual 3–8′ dihydrobenzofuran motif. Through a series of biomimetic oxidation reactions, Pan and co-workers were able to establish the biogenic relationship between quadrangularin A (17), the parthenocissins, and the laetevirenols (Scheme 19). Irradiation of parthenocissin A (61) with UV light effected its conversion into (−)-quadrangularin A (17) in 30% yield. A smaller fraction of the starting material was converted into the unusual dihydroacephenanthrylene natural product laetevirenol A (62) through a UV-light mediated oxidative 6π-electrocyclization in 10% yield. An HRP-mediated, intermolecular oxidative coupling of (−)-quadrangularin A (17) and resveratrol provided a 1:1 mixture of laetevirenol D (122) and laetevirenol C (123), supporting the hypothesis that quadrangularin A (17) is a precursor for these natural products. A similar set of experiments demonstrated an analogous biogenic relationship between (−)-parthenocissin A (61), (−)-parthenocissin B (120), and laetevirenol E (124), which were also isolated as a 1:1 mixture of diastereomers.
Scheme 19. Pan’s Biogenic Studies of the Parthenocissins and Laetevirenols.

Although these oxidative coupling reactions do not occur with any diastereoselectivity, it is important to mention that parthenocissin B (120) was isolated in 5-fold greater abundance than 122 and 124 and 500-fold greater abundance than 123 from the plant material. Irradiation of parthenocissin B (120) with UVA afforded laetevirenol D (122) in 24% yield (confirming their structural relationship as alkene isomers), and phenanthrene derivative laetevirenol B (125) in 21% yield. Analogous isomerization of laetevirenol E (124) to laetevirenol C (123) was evident by HPLC. The absolute configurations of these compounds was established by comparison of its CD spectra with the spectra of (−)-ampelopsin D (25), whose absolute configuration is known.50
The intermolecular oxidative cross coupling of quadrangularin A (17), a trisubstituted stilbene, and ε-viniferin (7) was unprecendented; previous biomimetic dimerization reactions were homo/heterocouplings of resveratrol (1) or ε-viniferin (7), which are disubstituted stilbenes. The next year, the same group disclosed the isolation of two related resveratrol tetramers, laetevirenols F/G (126/127). Due to the large distance between the 7c/8c protons and the indane 7a/8a protons, the relative configuration of each of these natural products is currently unknown. They proposed a tentative structure for 126 and 127, which they inferred by comparison of their HPLC behavior to that of the structurally related trimers 120 and 124, though they stress that this is speculative and the true configuration of the 7c/8c stereogenic centers may be reversed. The authors postulate that the biogenesis of these tetramers is the result of an iterative oxidative homologation of resveratrol onto 120, since it is the most abundant trimer extracted from the plant, though it is also possible that 126/127 are the result of a more convergent heterocoupling of parthenocissin A (61) and δ-viniferin (10).
5.6. Oligomerization via Polar Mechanisms
The impressive chemical diversity of the resveratrol natural products is the result of the multiple regioisomeric radical coupling and Friedel–Crafts cyclization pathways available upon the oxidation of resveratrol. For a majority of the resveratrol dimers, trimers, and tetramers, oligomerization occurs through an intermolecular radical coupling, followed by an intramolecular Friedel–Crafts cyclization. Typically, an intramolecular cyclization or tautomerization of an intermediate para-quinone methide can outcompete most intermolecular processes. However, intermolecular functionalization can in fact occur. Several resveratrol dimers such as parthenostilbenin A/B (54) and quadrangularin B/C (55) (Scheme 7) are the products of solvolysis of electrophilic intermediates. It is perhaps unsurprising to find that the electron rich arenes of the resveratrol natural products themselves can also react with the quinone methides generated during the dimerization of resveratrol. For instance natural products cajyphenol A (128) and B (129), isolated from Cayratia japonica (Vitaceae) are regioisomeric tetramers of the cross coupling of quadrangularin A (17) and its penultimate biosynthetic intermediate 52 (Scheme 20). A more complex natural product pauciflorol D (130) (Dipterocarpaceae),78 is a resveratrol heptamer that arises from the trapping of the 8–8′ trimeric quinone methide 102 (precursor for canaliculatol and distichol (103), Scheme 16, part A)129,130 by the 8–10′ tetramer vaticaphenol A (131)141 (Scheme 20). This intermolecular oligomerization mode is common to many resveratrol oligomers including pentamers,142 hexamers,143 heptamers,144 and octamers.145 Thus, the biogenesis of the higher-order oligomers can be reduced to the same intermediates invoked for the biosynthesis of the dimers, trimers, and tetramers. Nonetheless, it remains unclear whether the plant exerts any form of stereo- or regiospecificity, or if the observed products of the oligomerization process are just reflective of the inherent reactivity of the random coupling of bulky reaction partners. Additionally, it is unknown whether the active electrophiles (e.g., 52 and 104) are intercepted immediately upon formation or if they are generated from the corresponding solvent adducts (e.g., 53 or 106). This reversible ionization may also be operative in the previously described biosynthetic routes to the resveratrol dimers, trimers, and tetramers, and has been implemented in de novo synthetic approaches to these natural products (see section 7).146−148
Scheme 20. Oligomerization by Intermolecular Trapping of Quinone Methide Intermediates.

5.6.1. Biosynthesis of Stemonoporol, Copalliferol A and B
Stemonoporol (132)149 and copalliferols A (133)150 and B (134)151 are among the first trimeric resveratrol natural products identified, isolated from several Dipterocarpaceous plants by Sotheeswaran and co-workers in the early 1980s. These highly unusual resveratrol trimers feature a benzocycloheptane core (as opposed to the dibenzocycloheptane core found in the 8–10′ dimers, section 4.2) and were the first resveratrol oligomers discovered that were not derived from ε-viniferin (7). The core cycloheptane structure of stemonoporol (132) and the copalliferols A and B (133/134) is suggestive of a unique trimerization mechanism. The 8–8′ dimerization of resveratrol forms the reactive bis-para-quinone methide 22 (possibly formed reversibly from amurensin A (59)). Interception of 22 by resveratrol (1) through an intermolecular, vinylogous Friedel–Crafts reaction forging C8a–C7b bond (Scheme 21), followed by tautomerization or hydrolysis, would afford 135. A subsequent intramolecular 7-exo-trig cyclization of trimeric intermediate 135 (bond C10c–C7a) yields benzocycloheptane 136. The oxidation of the B1 phenol, followed by intramolecular Friedel–Crafts cyclization of the A2 resorcinol, provides the intermediate spirocyclohexadienone 137, which, upon a phenol–dienone rearrangement152 and ionization of the carbon 7c, yields the divergent para-quinone methide 138. An intramolecular 5-exo-trig cyclization (path A) forges the final ring of copalliferol A (133). Tautomerization of 138, on the other hand, yields stemonoporol (132). The biogenic relationship between these two natural products was established by Sotheeswaran and co-workers, who converted 132 to 133 via a cyclization reaction using formic acid.153 Copalliferol B 134 is presumably generated from a series of related reactions.
Scheme 21. Biogenic Hypothesis for the Formation of Stemonoporol and Copalliferols A and B.

5.7. Potential Role of Dirigent Proteins in Resveratrol Oligomer Biosynthesis
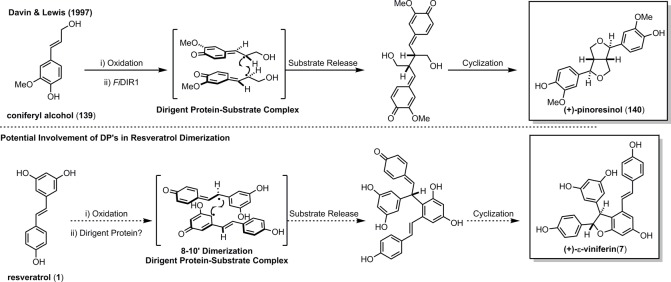
Most of what is currently known about the biosynthesis of the resveratrol natural products has been inferred through isolation studies and biomimetic reactions. Due to its structure, the oxidation of (1) is facile, and can be mediated by a number of constituents found in plants including endogenous peroxidases, exogenous fungal laccases, and even reactive oxygen species generated by UV irradiation.38,103,154 A long-standing question, which has yet to be answered, is what are the factors that dictate product determination? In the absence of any stereocontrolling element, resveratrol displays little regioisomeric or stereochemical preference during its dimerization or oligomerization. However, a majority of the resveratrol natural products are isolated as optically active compounds, implying that the radical coupling is mediated by some chiral entity. In 1997, Davin and Lewis isolated an unusual protein, FiDIR1, which could mediate the regiospecific and enantioselective dimerization of coniferyl alcohol (139) into the dimeric lignan (+)-pinoresinol (140) (Scheme 22).155 These “dirigent” proteins (Latin: dirigere, to align or guide) did not possess any oxidase activity in their own right, but rather, they had the remarkable ability to capture and dimerize the phenoxyl radical of conferyl alcohol (139) faster than the background rate of self-dimerization/oligomerization.156 They also found that FiDIR1 could enantioselectively dimerize the phenoxyl radicals of 139 generated from different peroxidases, laccases, and even inorganic oxidants such as ceric ammonium nitrate (CAN). In the absence of the dirigent protein, 139 dimerizes as a complex mixture of racemic regioisomers in a fashion similar to resveratrol. Like the resveratrol oligomers, several regioisomeric and antipodal lignans have been isolated.157 The structural similarity of the resveratrol oligomers and lignans, their homologous biosynthesis, and identities as phytoalexins, has led to speculation that the resveratrol dimerization is controlled by dirigent proteins;42,154,157,158 however, there have not been any studies that have successfully identified the cellular components responsible for the stereoselective biosynthesis of the resveratrol oligomers. Thus, this tantalizing hypothesis remains unsubstantiated. The only certainty is that there is much work to be done to fully elucidate the biosynthesis of this fascinating class of molecules.
Scheme 22. Potential Role of Dirigent Proteins in the Stereoselective Coupling of Resveratrol.
6. Biomimetic Syntheses
The challenge of biomimetic synthesis has captivated the synthetic community for over a century. Conceptually developed by Robinson following his historic synthesis of tropinone in 1917,159,160 the strategy of imitating a biogenic reaction sequence has been used to validate biosynthetic hypotheses, and in many instances is recognized as the most efficient means to construct complex natural products. The resveratrol natural products are particularly well-suited for biomimetic synthesis because of the convergent nature of their oligomerization, which can be used to quickly introduce molecular complexity. The challenge in the synthesis of resveratrol based natural products is principally an issue of chemoselectivity, since the products of dimerization have similar electrochemical potentials as resveratrol itself and are prone to overoxidation. As described in the preceding sections, the arene rings of resveratrol can undergo a number of cyclization reactions upon oligomerization, which have proven difficult to control. These inherent properties of resveratrol as well as a lack of appropriate chemoselective reaction methodologies have hindered progress in this area.
In the following sections we will describe several biomimetic approaches to the resveratrol natural products. This task is somewhat complicated by the biogenic studies of natural product isolation groups who sought to establish their absolute configuration through semisynthesis. These studies are often cited to highlight the disadvantages of biomimetic approaches to the resveratrol oligomers. However, it is important to acknowledge that the primary motivations for these investigations were elucidating the structure and providing empirical support for the biogenesis of these compounds, rather than their total synthesis. In this review, we will attempt to differentiate these investigations to properly contextualize their findings and to accurately present the successes and unsolved problems in the biomimetic synthesis of these natural products.
6.1. Biomimetic Synthesis of δ-Viniferin
The first synthesis of a resveratrol dimer, δ-viniferin (10), was described by Langcake and Pryce in 1977, during their isolation and structure elucidation of α-(6) and ε-(7) viniferin.36 Drawing on the biosynthesis of a structurally related lignan natural product licarin A,35 they proposed that an oxidative oligomerization of resveratrol could conceivably generate ε-viniferin (7). By subjecting resveratrol to the horseradish peroxidase (HRP)/H2O2 oxidation conditions developed by Wallis and co-workers,161 Langcake and Pryce isolated δ-viniferin (10) in 40% yield. These reaction conditions have since become a popular tool for structure elucidation of the resveratrol natural products and have been highly influential in subsequent biomimetic syntheses.
Since this seminal contribution, several groups have successfully dimerized resveratrol into δ-viniferin (10) using a variety of oxidation strategies, including enzymatic,107,148,162 organic,106,163 inorganic,108,164,165 and photochemical oxidations.111 The selectivity and yield for direct dimerization of resveratrol to δ-viniferin (10) have improved dramatically since the initial experiment by Langcake and Pryce. Table 1 summarizes the dimerization reactions that have been developed for the selective synthesis of 10. The chemical oxidation by Sako (Table 1, entry 8), using AgOAc, was the first high-yielding biomimetic nonezymatic oxidation reaction and could be performed on a preparative scale.165 The high regioselectivity of this dimerization reaction was also observed with ε-viniferin (7), where they obtained 40% yield of vitisin B (116) and 32% of an unidentified tetrameric didehydrodimer for an impressive 72% combined yield (Scheme 23). Two years later, Niwa (Table 1, entries 9 and 11) disclosed two separate conditions that were chemoselective and were slightly higher yielding using MnO2 or FeCl3 as stoichiometric oxidants. Recently, a catalytic method using graphitic nitride to dimerize resveratrol via an aerobic photochemical oxidation was reported by Wang and co-workers, which provided δ-viniferin (10) in high yield and with good regioselectivity (Table 1, entry 7).
Table 1. Biomimetic Syntheses of δ-Viniferin.
| entry | reaction conditions | yield | ref |
|---|---|---|---|
| 1a | CuII SO4, MeCN, rt | 16% | (164) |
| 2a | DPPH, MeOH | 18% | (106) |
| 3a | various peroxidases, acetone/EtOH | 13–21% | (108) |
| 4a | laccase, n-BuOH | 31% | (107) |
| 5b | AgOAc, DCM | 36% | (168) |
| 6a | galvinoxyl radical, ethanol, rt | 41% | (163) |
| 7a | graphitic carbon nitride (hv, 410 nm), lutidine, air, MeCN, rt | 85% | (111) |
| 8a | AgOAc, MeOH, 50 °C | 86% | (165) |
| 9a | MnO2, DCM, rt | 91% | (108) |
| 10a | HRP/H2O2, H2O/acetone, pH 8 | 93% | (148) |
| 11a | FeCl3, acetone, rt | 97% | (108) |
R = H resveratrol (1).
R = Me pterostilbene (3).
Scheme 23. Sako’s Biomimetic Synthesis of Vitisin B.

6.2. Biomimetic Synthesis of ε-Viniferin
Although the 3–8′ dimerization of resveratrol can be obtained in high yield by a variety of methods, that regioisomer is rare among the resveratrol oligomers, thus limiting the utility for a general synthesis of other members of the natural product family. The selective synthesis of the 8–10′ dimer ε-viniferin (7), on the other hand, would be much more impactful from a synthetic standpoint, since it is the biogenic precursor for a majority of the resveratrol natural products. The principal challenge in the biomimetic synthesis of 7 is that its oxidation potential is nearly the same as resveratrol (Eox = +1.15 V vs +1.14 V vs SCE, respectively165). This limitation does not exist for the synthesis of δ-viniferin (10), where dimerization alkylates the phenol of resveratrol which dramatically attenuates its reactivity. As a consequence there are very few reaction conditions that can successfully mediate the conversion of resveratrol (1) to 7.
In 1998, Pezet isolated a laccase-like stilbene oxidase from the plant pathogen B. cinerea that had the remarkable ability to dimerize resveratrol to ε-viniferin (7) in 97% yield (Table 2, entry 1).154 Resveratrol analogues, such as 4,4′-dihydroxystilbene or pterostilbene (3) (11,13-dimethoxyresveratrol), were also competent substrates that could be rapidly oxidized by the enzyme. In corroboration with other studies, the isolated stilbene oxidase was only active on substrates containing a free phenol; alkylated substrates such as 4-methoxystilbene were unreactive. Although the optical rotation of 7 generated in the reaction was not measured, these results suggest that some fraction of ε-viniferin (7) could in principle be an exogenously synthesized natural product. It is unclear how the stilbene oxidase controls the regioselectivity of this dimerization, since other peroxidases and laccases tend to produce δ-viniferin (10) preferentially. This oxidative dimerization was conducted on a microgram scale, and it has yet to be implemented in preparative synthesis.
Table 2. Biomimetic Syntheses of ε-Viniferin.

| entry | reaction conditions | yield | ref |
|---|---|---|---|
| 1 | B. cinerea laccase-like stilbene oxidase | 97% | (154) |
| 2 | FeCl3, MeOH/H2O, rt | 30% | (166) |
| 3 | K3Fe(CN)6, K2CO3, MeOH/H2O, rt | 22%a | (108) |
| 4 | Tl(NO3)3, MeOH, –50 °C | 30% | (108) |
Obtained as a 1:1:0.7 mixture of 7:10:18.
A scalable and selective biomimetic syntheses of ε-viniferin (7) did not appear until 2004, when Yao and co-workers described the dimerization of resveratrol to 7 with FeCl3 in MeOH in 30% yield and with 40% recovery of resveratrol (Table 2, entry 2).166 This reaction was performed on 0.5 g scale and has since been implemented for the synthesis of other resveratrol dimers.167 They were able to derivatize 7 to phenanthroline 141 upon photooxidation of 7 in 20% yield (Scheme 24).
Scheme 24. Oxidative Photocyclization of 7.

One year later, Niwa and co-workers also reported a series of reaction conditions that could selectively generate ε-viniferin (7).108 If potassium hexacyanoferrate (K3Fe(CN)6) was used as the stoichiometric oxidant, 22% of 7 could be isolated as a mixture of resveratrol dimers with 40% recovered resveratrol (Table 2, entry 3). Much higher product selectivity could be achieved using Tl(NO3)3 as the oxidant. This reaction was found to be highly temperature sensitive, yielding 30% 7 as the sole reaction product (56% recovered starting material) at −50 °C, but gave complete decomposition at −30 °C (Table 2, entry 4). Collectively, these methodologies highlight the importance of oxidant identity in the regioselective synthesis of resveratrol dimers. It is unclear how these oxidants avoid overoxidation while maintaining high levels of product selectivity. These remarkable findings are promising leads toward a reagent-controlled, chemoselective synthesis of the resveratrol oligomers.
6.3. Biomimetic Synthesis of Other Resveratrol Dimers
Despite the previous successes in the synthesis of the dihydrobenzofuran dimers, strategies to selectively synthesize more complex dimeric architectures, such as quadrangularin A (17), pallidol (18), or ampelopsin D/F (25/16), remained a significant synthetic challenge. In their studies on the biogenic origins of the resveratrol dimers, Niwa and co-workers disclosed several oxidation conditions which could yield various nonbenzofuran containing natural products.108 Using the same conditions described in section 6.2, they found that K3Fe(CN)6 could oxidize resveratrol, yielding a mixture of 7, 10, and up to 16% of pallidol (18) (Table 3, entry 1). Oxidizing resveratrol with HRP in acetone afforded a mixture of 10 (13% yield) and 18 (10% yield) (Table 3, entry 2), while soybean peroxidase in ethanol produced a mixture of δ-viniferin (10) (12% yield), pallidol (18) (10% yield), quadrangularin B/C (55) (9% yield), and leachinol F/G (53) (5% yield) (Table 3, entry 3), with a 24% combined yield of 8–8′ dimers. Although the chemoselectivity of these biomimetic oxidations was modest at best, this study was the first to demonstrate the feasibility of performing a reagent controlled dimerization of resveratrol, developing distinct conditions to synthesize 3–8′, 8–10′, and 8–8′ dimers.
Table 3. Biomimetic Dimerization of Resveratrol.

| entry | reaction conditions | 10 | 16 | 53/55 | 18 | 57 | ref |
|---|---|---|---|---|---|---|---|
| 1 | K3Fe(CN)6, K2CO3, MeOH/H2O, rt | 23% | — | — | 16% | — | (108) |
| 2 | horseradish peroxidase, aq acetone | 13% | — | — | 10% | — | (108) |
| 3 | soybean peroxidase, aq EtOH | 12% | — | 14%b | 10% | — | (108) |
| 4a | FeCl3·6H2O, CH2Cl2 | — | 7% | — | 10% | — | (168) |
| 5a | FeCl3·6H2O, CH2Cl2/MeOH (7:3 v/v) | — | — | — | — | 29% | (168) |
| 6a | Pt anode/cathode in MeCN/0.2 M LiClO4 (+1.00 V vs Ag/AgNO3) | — | 8% | — | 14% | — | (170) |
| 7 | horseradish peroxidase, acetone/pH 8.0 buffer | 93%c | — | — | — | — | (148) |
| (89%)d | |||||||
| 8 | horseradish peroxidase, acetone/pH 6.0 buffer | — | — | 55%c,e | — | — | (148) |
| (24%)d,e | |||||||
| 9 | horseradish peroxidase, acetone/pH 5.0 buffer | — | — | — | 20%c | — | (148) |
| (19%)d | |||||||
| 10 | horseradish peroxidase, acetone/pH 4.0 buffer | 28%c | — | — | — | — | (148) |
| (26%)d,f | |||||||
| 11 | NaNO2/pH 3.0 buffer, MeOH | 2% | — | — | — | 1% | (171) |
R = Me (pterostilbene, 3).
2:1 mixture of 55/53.
HPLC yield.
Isolated yield.
Product was 53.
cis-δ-viniferin is major pdt.
A subsequent study by Velu and co-workers sought to understand the effects of solvent polarity and oxidant choice on chemoselectivity during dimerization of resveratrol analogues.168 Using FeCl3 as their single electron oxidant, they found that they could influence product distribution by switching between “hard” and “soft” solvents or oxidants.169 When 3 was oxidized with FeCl3 in CH2Cl2, they observed 10% and 7% yield of methylated pallidol (18a) and ampelopsin F (16a), respectively (Table 3, entry 4). In a mixture of CH2Cl2 and methanol, 3 dimerized primarily to give the tricuspidatol derivative 57a in 29% yield (Table 3, entry 5). These results compare favorably to the enzymatic oxidation of resveratrol by B. cinerea laccase enzymes which yielded the isomeric tetraaryl furan restrytisols A/B (56/63) (section 4.3).38 The authors also observed that solvent could influence the product distribution of several non-natural resveratrol analogues, independent of the oxidant identity, albeit in relatively low isolated yields. Kam and co-workers isolated a similar yield of permethylated 16 and 18 in the electrochemical oxidation of permethylated 1 (Table 3, entry 6), corroborating those findings.170 In both of these approaches, the modest yields were the result of stilbene overoxidation into an intractable mixture of polymeric material.
In 2012, Pan and co-workers examined the effects of reaction pH on the product distribution of a peroxidase mediated dimerization of resveratrol.148 They performed peroxidase-mediated dimerizations of 1 at pH 4.0, 5.0, 6.0, and 8.0 (Table 3, entries 7–10). By adjusting the reaction basicity to pH 8.0, they isolated 89% yield of 10 as the major regioisomer. In mildly acidic conditions at pH 6.0, they observed a dramatic change in product distribution, with a 55% HPLC yield of a 1:1 mixture of leachinol F/G (53), isolating 24% yield on preparative scale. At pH 5.0 they detected an enrichment in pallidol (18) formation (19% isolated), while at pH 4.0, they isolated primarily cis-10 in 26% yield (28% HPLC yield). At more acidic conditions (pH of 3.0), they observed no conversion, due to the increased oxidation potential of resveratrol under those conditions. A prior report by d’Ischia and co-workers utilizing NaNO2 as an oxidant at the same pH identified 10 and 57 in minute quantities (<2%), corroborating the poor reactivity at low pH (Table 3, entry 11).171 These results indicate the pH sensitivity of the dimerization reaction, enabling the selective enrichment of one regioisomer over the other. This simplified purification, which was performed by HPLC due to the polarity of the compound mixtures. Furthermore, they performed the dimerization on a preparative scale for the purposes of synthesis and biological evaluation (see section 8.4). They could interconvert leachianols F/G (53) (as a 1:1 mixture), into parthenocissin A (61) using BF3·OEt2 in 90% yield (Scheme 25). Subsequent irradiation of 61 with >306 nm light cleanly isomerized it to the corresponding (E)-isomer, quadrangularin A (17) in 77% yield. This report clearly demonstrated the pH-sensitive nature of the dimerization of resveratrol and its impact on product distribution. The scalability of the procedure is also notable; this method was capable of producing 61 and 17 in 50 and 30 mg quantities, respectively.
Scheme 25. Interconversion of Leachinol F/G to Parthenocissin A and Quadrangularin A.

6.3.1. Hou and Li Synthesis of Quadrangularin A via an Arene Protecting Group Strategy
Despite the fact that the resveratrol oligomers have been known since the 1960s, very little effort was initially invested toward their total synthesis. Studies such as those discussed above on the biomimetic dimerization of resveratrol (1) or pterostilbene (3), highlighted significant challenges in achieving high chemoselectivity, regioselectivity, and useful yields. The important innovations during this time period were the identification of the impact of oxidant, solvent choice, and pH on product selectivity. However, given the complexity of these reactions, it was impossible to predict which natural products would be generated under chosen conditions, and research in this area was largely driven by empirical observation.
In 2006, Hou and co-workers disclosed a target-oriented biomimetic synthesis of quadrangularin A (17), which was the first synthesis of a fully elaborated indane containing resveratrol dimer.172 The key innovation of this approach was the identification of the 2,6-di-t-butyl motif as a blocking group to prevent unwanted 3–8′ coupling that is typical for the uncontrolled oxidation of resveratrol, a method pioneered by Müller173 and Wallis174 in their investigations on lignan biosynthesis.
Stilbene 145 was synthesized from 3,5-dihydroxybenzoic acid in six steps. Although 143 and 144 were synthesized de novo, the benzyl bromide of 142 and benzaldehyde 144 are now commercially available. Treatment of tert-butyl stilbene 145 with AlCl3 and dimethylaniline enabled the selective dealkylation of its benzyl ethers generating 2,6-di-tert-butyl resveratrol 146 in 78% yield. This compound was then oxidized with HRP and H2O2 in an acetone/water mixture over 2 days, affording the cyclized para-quinone methide 147 in 35% yield on 0.5 g scale. Curiously, this reaction also produces trimeric intermediates 149 and 150 in 11% and 14% yield, which are the products of trapping of the monomer-based resorcinol onto product 147 (Scheme 26, section B). Compound 147 is a direct analogue of the 8–8′ dimerization intermediate 52 outlined in section 4.3 and provides support for the presence of quinone-methide intermediates in the biosynthesis of quadrangularin A (17). Tautomerization of 147 with basic alumina in benzene provides protected quadrangularin A (148) in 64% yield, along with 31% recovered starting material. Finally, a Lewis-acid-mediated retro-Friedel–Crafts reaction dealkylates 148 with concomitant extrusion of isobutylene,175,176 affording quadrangularin A (17) in 85% yield. Overall, this reaction sequence provides quadrangularin A (17) in 10 total steps in 17% overall yield and is an excellent demonstration of the step-efficiency that biomimetic synthesis can offer. In a later communication, Li and co-workers attempted a direct deprotection of mixture of 147, 148, and 150, which lead to an interesting skeletal rearrangement to compounds 151 and 152, non-natural analogues of ampelopsin F (16).177
Scheme 26. Hou Synthesis of Quadrangularin A.

6.3.2. Li’s Approach to Quadrangularin A, Pallidol, and Ampelopsin F by a Regioselective Oxidative Coupling
Expanding upon the work of the Hou group, Li and co-workers reported a series of oxidations of stilbene 146 using inorganic oxidants in several solvent systems.178 In this communication, the Li group reports reaction conversions and percent recovered starting material, but in several cases, these values add up to more than 100%, making yield estimation difficult. They found that when 146 was oxidized by 1 equiv of either Ag2CO3 or MnO2 in CH2Cl2, they observed 59% and 54% reaction conversion to quinone methide 147 with 64% and 76% recovered starting material, respectively (Table 4). Using FeCl3·H2O as an oxidant in 2:1 benzene/acetone, they observed 55% reaction conversion to protected pallidol (153) with 45% recovery of starting material. Finally, opposite regioselectivity could be achieved if FeCl3·6H2O was used as the oxidant in CH2Cl2, delivering protected ampelopsin F (154) in 45% reaction conversion with 25% recovered starting material. These results corroborate the findings described by Niwa108 and Velu,168 demonstrating that it is possible to influence product distribution by changing solvent and/or stoichiometric oxidant. Deprotection of 153 and 154 with AlCl3 in toluene/MeNO2 proceeded uneventfully for pallidol (18) and ampelopsin F (16) in 85% and 76% isolated yields, respectively. The synthesis of ampelopsin F (16) is the first example of a selective, biomimetic synthesis of an 8–10′ dimer.
Table 4. Regioselective Dimerizations of 146.

| entry | reaction conditions | 147 | 153 | 154 |
|---|---|---|---|---|
| 1 | Ag2CO3, CH2Cl2 | 59%a | — | — |
| 2 | MnO2, CH2Cl2 | 54%a | — | — |
| 3 | FeCl3·6H2O, benzene/acetone 2:1 | — | 55%a | — |
| 4 | FeCl3·6H2O, CH2Cl2 | — | — | 45%a |
Reported as % conversion.
6.3.3. Li’s Synthesis of Protected Tetraaryl Furan Dimers
In 2014, Li and co-workers reported the synthesis protected analogues of tricuspidatol A (155a/b) and restrytisol B (156a/b) (Scheme 27).179 Upon acylation of the resorcinol of 146 with Ac2O or Boc2O and oxidation with FeCl3·6H2O they observed the formation of 155a/b and its nonsymmetrical isomer 156a/b. The acylated stilbene 146a gave higher combined yield of the tetraaryl furan dimers 155a and 156a (48% yield as a 1:1 mixture), whereas the Boc-protected derivative 146b afforded a slightly lower yield (32% combined as a 1:1.3 mixture) of 155a/155b. Interestingly, deprotection of the mixture of 155a and 156a in neat CF3SO2H did not provide tricuspidatol (57) or restrytisol B (63) but rather pallidol (18) in 80% yield.
Scheme 27. Li’s Synthesis of Protected Restrytisol Dimers and Interconversion to Pallidol.

6.3.4. Stephenson’s Synthesis of Quadrangularin A and Pallidol from Bioinspired Quinone-Methides
Recently, Stephenson and co-workers disclosed a scalable biomimetic synthesis of quadrangularin A (17) and pallidol (18).180 Drawing on design principles delineated by the Hou and Li groups, they developed a highly efficient dimerization of the tert-butylated resveratrol derivative 146. Their initial approach, using the strongly oxidizing photocatalyst, Ir(tpy)(ttpy)(PF6)3,181 was competent for the selective aerobic oxidation of 157 to the cyclized para-quinone methide 158 over a period of 2 days in 38% yield (Table 5, entry 1).
Table 5. Optimization of Aerobic Resveratrol Dimerization.
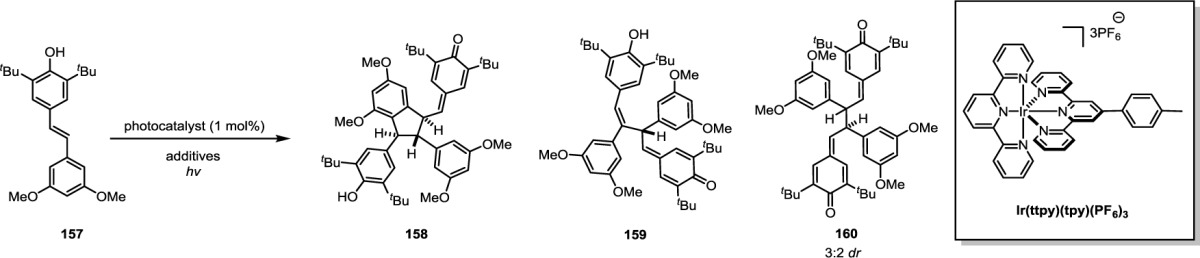
| entry | photocatalyst | solvent | oxidant | additive | time | yield | product |
|---|---|---|---|---|---|---|---|
| 1 | Ir(ttpy)(tpy)(PF6)3 | acetone | O2 | — | 2 d | 38% | 158 |
| 2 | Ir(ttpy)(tpy)(PF6)3 | acetone/MeOH | O2 | NaOMe | 5 h | 47% | 159 |
| 3a | — | acetone/MeOH | O2 | NaOMe | 2 h | 55% | 159 |
| 4a | — | CCl4 | O2 | KOtBu | 1 h | 60–80% | 160 |
| 5b | — | THF | FeCp2PF6 | KHMDS | 30 min | 99% | 160 |
Reaction was shielded from light.
Reaction performed on decagram scale.
Owing to the pH-dependence of the oxidation potential of resveratrol and phenols,182 they sought to improve the efficiency of the reaction by performing it under alkaline conditions. In the presence of excess NaOMe and 1% photocatalyst under aerobic conditions, full consumption of 157 was observed in 5 h to give a slightly improved 47% yield (Table 5, entry 2). However, instead of quinone methide 158, they isolated the linear dimer 159. Control experiments revealed that the photocatalyst was not needed and that the reaction was proceeding via aerobic oxidation (Table 5, entry 3). Further optimization revealed that when the aerobic oxidation of 157 was performed in CCl4 and KOtBu, quinone methide 160 was the major product (Table 5 entry 4), indicating that product selectivity was both pH- and solvent-dependent. Importantly, dimers 159 and 160 are analogues of the two other biosynthetic intermediates of the 8–8′ dimers and are precursors to pallidol (18) and amurensin A (59) (see section 4.3). A modification of the reaction conditions using 1.05 equiv of FeCp2PF6 as a stoichiometric oxidant and KHMDS as the base improved scalability and increased the yield to 95–99% on a decagram scale.
Starting from commercially available benzyl bromide 161, stilbene 145 was prepared in two steps in 81% overall yield. The ferrocenium-mediated dimerization of 145 in THF yielded a 4:3 mixture of meso/dl diastereomers of 162 in quantitative yield. The relative configuration of the vicinal stereocenters in 162 had important consequences on product formation (see section 4.3). Namely, upon exposure to BF3·OEt2, 162 was transformed to a mixture of protected pallidol (164, 43%) and quinone methide 163 (45%). Hydrogenation followed by an AlCl3 mediated de-tert-butylation reaction afforded pallidol (18) in 27% overall yield from 161.
The synthesis of quadrangularin A (17) proceeded in a similar fashion, starting from the oxidative dimerization of 145. It was discovered that bis-para-quinone methide 145 could be interconverted to the mono-para-quinone methide quantitatively by tautomerization under strongly basic conditions. By adding another equivalent of KHMDS to the original dimerization reaction, they could synthesize 165 in one step with no loss of yield. Quadrangularin A (17) could be accessed from 165 by one-, two-, or three-step sequences, through the intermediacy of 166 and 148, in 51%, 88%, and 65% yield, respectively (Scheme 28). Here, Lewis acids served to promote cyclization, global debenzylation, and removal of the tert-butyl groups, while Me5-benzene sequestered the benzyl cations generated during the dealkylation step.183,184 Quadrangularin A (17), therefore, could be accessed in 4 steps/41%, 5 steps/64%, or 6 steps/47% overall yield from commercially available 161. The scalability of this route enabled the rigorous evaluation of the antioxidant properties of these natural products and their congeners (see section 8.4).
Scheme 28. Total Synthesis of Quadrangularin A and Pallidol by Stephenson and Coworkers.

7. de Novo Synthetic Approaches
The strategies outlined in the previous section were incredibly important to advance the understanding of resveratrol reactivity under conditions designed to emulate the chemical environment in which its oligomerization is achieved within plant cells. However, until recently,180 these strategies have failed to meet material requirements for systematic physical and biochemical evaluation. Given their impressive biological activities, elusive mechanisms of action, and fascinating molecular architectures, the resveratrol oligomers have been the target of synthetic chemists for years. In 2007,185 Snyder and co-workers published a powerful de novo strategy to resveratrol dimers that has evolved and flourished in the interim, resulting in a series of pioneering and inspirational syntheses of complex molecules within this class. This work and the contributions of other research groups to this area are presented below.
7.1. Synthesis of Resveratrol Dimers Using Cationic Cyclizations and Skeletal Rearrangements
7.1.1. Snyder’s Synthesis of Indane-Containing Resveratrol Dimers From a Common Building Block
While there is tremendous chemical diversity within the resveratrol class of oligomeric natural products, they are characterized by highly conserved carbogenic cores (see section 4−5). Of the various structural subtypes, Snyder and co-workers elected to target indane-derived resveratrol natural products ampelopsin D (25) and quadrangularin A (17), realizing that a modular approach to these compounds would potentially lay the foundation for the synthesis of higher-order oligomers possessing these core structures. Specifically, the indane-containing resveratrol oligomers can be divided into two regioisomeric classes depending on the relative positioning of the phenol (A1) and resorcinol (A2) components of the resveratrol monomer whose ethylene bridge comprises two of the three carbons in the indane ring (Scheme 29, inset). This observation enabled the retrosynthetic design of triaryl starting materials 167 and 173 (Scheme 29) which, by careful temperature control, could be channeled through chemoselective acid-promoted cyclizations to generate regioisomeric trans-configured 2,3-diaryl indanyl cations 168 and 174.185
Scheme 29. Synthesis of Indane-Based Resveratrol Dimers from a Common Building Block.

When the cyclization was promoted by TFA, the intermediate cation was trapped by the conjugate base (trifluoroacetate) in solution. Quenching of the trifluoroacetate adduct with adventitious water in the presence of base resulted in formation of the corresponding benzyl alcohol 169, which was subsequently oxidized to the protected indanone 170. Pauciflorol F (28) was revealed by a global demethylation using BBr3.
Alternatively, by employing a Brønsted acid whose conjugate base is less nucleophilic (TsOH), the intermediate indanyl cation could be captured by a benzyl mercaptan, yielding 171, which served as a nucleophilic proxy to append a para-methoxy styryl group at this position through an ensuing Meyer’s modified Ramberg–Bäcklund reaction.186,187 This gave a 5:1 mixture of E/Z olefin isomers that were separated chromatographically. The (E)-isomer (172), a protected derivative of the natural product ampelopsin D (25), was demethylated using BBr3 to give a 5:1 mixture of the desired natural product (25) and its internal alkene isomer, isoampelopsin D (see Scheme 5, A). Isolation of these materials required peracetylation prior to chromatographic separation and subsequent KCN promoted hydrolysis. A logical redesign of their starting alcohol (173) enabled access to the regioisomeric materials isopauciflorol F (176) and quadrangularin A (17) through an identical reaction sequence. Notably, a 5-step method for the interconversion of pauciflorol F (28) to ampelopsin D (25) (and isopauciflorol F (176) to quadrangularin A (17)) has since been developed (see section 7.1.3).188
7.1.2. Snyder’s Interconversion of Indane Dimers to [3.3.0] and [3.2.1] Bicyclooctanes
In an extension of these seminal studies, Snyder sought to exploit the inherent polarization of the methoxy styrene moiety present in the natural products ampelopsin D (25) and quadrangularin A (17) to effect their interconversion to the remaining two dimeric structural subtypes: the bicyclo[3.2.1]octanes and the bicyclo[3.3.0]octanes.189 Each of these transformations was to be accomplished via electrophilic activation of these olefins to generate cationic quinone methide intermediates. Intramolecular trapping by a proximal resorcinol would provide the desired carbogenic cores. While this would ideally be accomplished using a Brønsted acid, the authors had already observed that both 172 and 178 are susceptible to olefin isomerization under acidic conditions. Therefore, electrophilic bromination reagents were chosen as alternative promoters, which smoothly transformed 172 and 178 to the trihalo natural product precursors 180 and 182 through the intermediacy of quinone methide cations 179 and 181, respectively (Scheme 30). Sequential hydrodehalogenation and demethylation provided the natural products ampelopsin F (16) and pallidol (18).
Scheme 30. Electrophile-Promoted Interconversion of Indane Dimers to [3.2.1] and [3.3.0] Bicicyclooctanes.
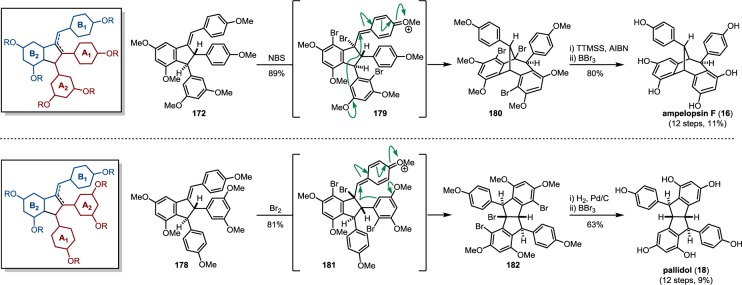
By quenching the electrophilic cyclization at various time intervals, the researchers were able to elucidate the order of halogenation events leading to each of the bicyclooctane cores. Namely, bromination of the electron-rich resorcinol rings takes place prior to olefin activation, with halogenation of the B2-ring occurring first for both regioisomers. This can be rationalized by the additional stabilization of the Wheland intermediate by two alkyl substituents for the B2-ring as compared to one for the A2-ring. The high yields obtained for the electrophilic cyclizations warrant further discussion. Theoretically, halonium formation could take place from either face of the olefin, yet only one of the resultant configurations enables the subsequent bicyclooctane formation. To account for this, the authors propose that bromination of the A2-rings provides enough steric encumbrance to bias olefin activation to occur from the opposite face. Rehybridization (sp2–sp3) of the bromine-bearing aliphatic carbon induces molecular reorganization, orienting the dearomatized B1-ring para-quinone methide such that it can be captured by the vicinal A2-ring (in pallidol synthesis) or the transannular A2-ring (in ampelopsin F synthesis).
To explore whether olefin geometry had any influence over these events, they subjected the (Z) olefin isomer of 178 to the bromination conditions and obtained either 178 or 182, depending on whether the reaction was quenched at −78 °C or warmed to room temperature, respectively. This result suggests that another mechanism may be operative, whereby bromonium formation is not facially selective but occurring reversibly via exchange with free olefin in solution.190,191 This mechanism is likely active when bromination takes place so as to position the quinone methide anti to the nucleophilic resorcinol, whereas the mechanism proceeding through the syn configuration is rendered irreversible by the Friedel–Crafts cyclization event.
7.1.3. Synthesis and Structural Revision of Caraphenols B and C by Snyder and Brill
As described in section 4−5, nearly all of the indane-based resveratrol dimers possess either a trans,trans or cis,trans relative configuration about the vicinal indane stereocenters. Two exceptions to this were caraphenols B (27) and C (60) (Scheme 31), which were originally proposed to have an all-cis arrangement of the indane substituents.43 Synthesis of each of the proposed structures revealed that these natural products had been misassigned. As most of the indane dimers possess a trans,trans relative configuration, Snyder and Brill completed the synthesis of these frameworks with this arrangement and confirmed their structures by comparison to the spectroscopic data for the natural compounds.188 Relying on their previous syntheses of pauciflorol F (28) and isopauciflorol F (176), a reaction sequence was developed for functionalization of the indanone. These aryl ketones have been historically challenging to functionalize, and several attempts to use these indanones as intermediates in the synthesis of resveratrol oligomers have failed (Scheme 31).
Scheme 31. Synthesis and Structural Reassignment of Caraphenols B and C.

Treatment of permethylated indanones 170 and 175 with Tebbe’s reagent192 provided the corresponding exocyclic methylene, which was subjected to anti-Markovnikov hydration and oxidation. Notably, the hydroboration proceeds from the opposite face of the neighboring D ring, thus yielding the cis,trans diastereomer of 183 and 185. Grignard addition to the aldehydes and oxidation of the resultant benzyl alcohols afforded 184 and 186. The α-stereocenter was not configurationally stable under the acidic conditions required for demethylation, and some of 184 epimerized to the corresponding trans,trans diastereomer in the course of this reaction. The epimers were isolated by HPLC, and the trans,trans compound was identified as caraphenol B (27). For 186, epimerization of the α-center was performed prior to demethylation under alkaline conditions to yield the thermodynamically favored trans,trans isomer. However, over the course of the demethylation with BBr3, some of the material epimerized back to the cis,trans isomer. Again, the trans,trans configured product was identified as caraphenol C (60) by comparison with spectra for the natural sample.
7.1.4. Nicolaou and Chen’s Synthesis of Hopeahainol A and Hopeanol
Just three years after their initial isolation, Nicolaou and Chen disclosed an impressive synthesis of the biosynthetically related resveratrol dimers, hopeahainol A (40) and hopeanol (31).92 These unusual structures are each thought to derive from skeletal rearrangement of resveratrol dimers, presumably via an oxidative rearrangement of malibatol A (44) (see section 4.2). As these structures can conceivably be interconverted under thermodynamic control, Nicolaou, Chen, and co-workers targeted the synthesis of hopeahainol A (40), whose cycloheptane core would be assembled through a 7-exo cyclization onto a cationic para-quinone methide intermediate (Scheme 32).
Scheme 32. Total Synthesis of Hopeanol and Hopeahainol A by Nicolaou and Chen.

In the forward sense, benzylic alcohol 187 was esterified with phenylglyoxalic acid 188 to give keto ester 189. Exposure of 189 to 4-methoxyphenyl magnesium bromide resulted in selective addition to the aryl ketone, followed by desilylation gave 190 as a 1:1 mixture of diastereomers. While 190 was formed as a diastereomeric mixture, this was inconsequential for subsequent synthetic operations, as the stereochemical information at these centers was ultimately destroyed.
As previously demonstrated by the Snyder group in the context of resveratrol oligomer synthesis, treatment of benzylic alcohols such as 190 with Brønsted acids promotes rapid ionization to generate reactive carbocations. Indeed, exposure of 190 to TsOH facilitates para-OMe-assisted ionization to generate a highly electrophilic para-quinone methide cation which engages the pendant resorcinol ring in Friedel–Crafts alkylation, yielding lactone 191. Treatment of the lactone with KOtBu followed by acidification with saturated aq NH4Cl resulted in rearrangement to γ-lactone 192. Geometric considerations suggest that this mechanism proceeds via an E2-elimination of the lactone, followed by condensation of the free phenol onto the acid. Acetylation of the remaining phenol (193), followed by epoxidation of the stilbene gave 194 as a 1:1 mixture of epoxide stereoisomers. Exposure of this material to Lewis acid (SnCl4) promoted regioselective epoxide opening to generate the cationic quinone methide 195, which was trapped by the adjacent resorcinol ring through a 7-exo Friedel–Crafts alkylation to give 196 as a 2:1 mixture of diastereomers. Comparison of 196 (a protected adduct of vaticahainol A 47) with the natural products reveals that it is only two oxidation states away from the protected form of hopeahainol A (40). This redox adjustment was made in a single operation using an excess of IBX,193,194 giving the α-keto quinone methide compound 197. Exchange of the acetyl group for a methyl group afforded permethyl hopeahainol A (199), whose 1H NMR matched those reported for this derivative.86 Treatment with BBr3 effected global demethylation to give the desired hopeahainol A (40, 4.6 mg). Methanolysis of the strained γ-lactone under alkaline conditions produced the ring-opened intermediate 200, which adopts a conformation that permits spontaneous rearrangement to hopeanol (31, 0.8 mg). This impressive transformation occurs in the reverse direction of that which had been proposed; in fact, attempted conversions of hopeanol (31) to hopeahainol A (40) were unsuccessful under a variety of conditions, demonstrating the utility of synthesis in elucidating plausible biosynthetic pathways. In their follow up report,195 these researchers were able to synthesize the natural products in enantiomerically pure form by employing an Itsuno–Corey196,197 reduction to establish the absolute configuration of starting bibenzyl 187. Through their studies, Nicolaou and Chen were able to confirm the reported acetylcholinesterase inhibitory activity of hopeahainol A (40) but not the reported cytotoxic activity of hopeanol (31).
7.1.5. Snyder’s Syntheses of Heimiol A, Hopeahainol D, Hopeahainol A, and Hopeanol
Encouraged by their success with electrophilic cascade cyclizations of triaryl building blocks, Snyder and co-workers targeted the synthesis of dibenzocycloheptanes, which are present in a number of resveratrol based oligomers.185 Here, the preferred cyclization mode of the stilbene precursor was modified by utilizing benzophenone 201, an oxidized variant of the previously employed doubly benzylic alcohols 167/173. Upon electrophilic activation of the stilbene with Br2, the desired cycloheptanone was formed via the intermediacy of a cationic para-quinone methide (Scheme 33). However, upon exposure to AgOAc in the presence of AcOH, this material underwent an unexpected aryl migration (presumably via dearomatized phenonium 204), ultimately providing the non-natural compounds 206 and 207, which are methylated congeners of the natural products hemsleyanol E (49) and diptoindonesin D (50), respectively (section 4.2, Scheme 6).
Scheme 33. Preliminary Efforts Towards Dibenzocycloheptanones by Snyder and Coworkers.

On the basis of these preliminary studies, Snyder and co-workers sought to exploit the instability of the benzylic halide substrates of type 202 for the synthesis of strained bicyclic natural products heimiol A (37) and hopeahainol D (38).198 Through redesign of the parent triaryl building block 201, these researchers were able to access the core structure of these molecules using a halolactonization/Friedel–Crafts cascade (Scheme 34). Namely, homologation of triaryl ketone 201 through a three-step sequence entailing Johnson–Corey–Chaykovsky epoxidation,199,200 Meinwald rearrangement,201 and Pinnick–Lindgren oxidation202,203 yielded the corresponding acid 208. Here, the pendant carboxylic acid serves as a nucleophilic trap for the labile benzylic halide formed upon electrophilic activation of the stilbene. Whether δ-lactone or cycloheptanone formation takes place first is unclear, but rapid construction of the bicyclo[3.2.2]nonane core structure 209 present in epimeric natural products heimiol A (37) and hopeahainol D (38) was achieved. A number of electrophilic halide sources were screened, with the I+ source IDSI [(Et2SI)2Cl·SbCl6]204 being the only one to provide the desired product 209 in useful yields. Unfortunately the yield of this transformation is not reported, but it can be inferred that it proceeds in 40–50% conversion based on previously reported yields for the global demethylation of other resveratrol dimers.185,189
Scheme 34. Synthesis of Heimiol A and Hopeahainol D by Snyder et al.
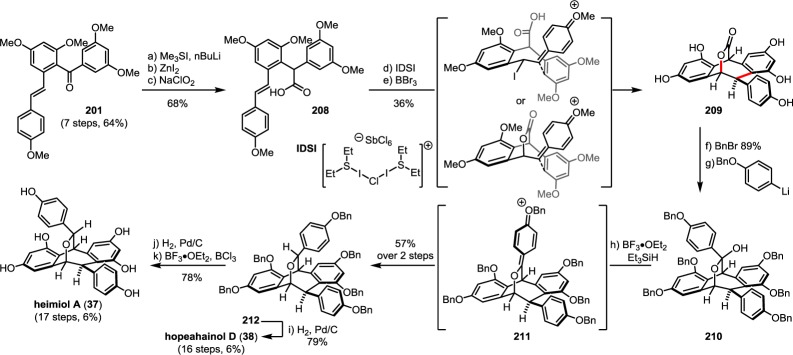
At this point, the methyl groups were exchanged for benzyl ethers for fear that the harsh conditions required for demethylation may result in destruction of a late-stage intermediate. Benzyl protection at the outset was not feasible because it interfered with the homologation sequence (201 → 208). Nucleophilic addition of 4-benzyloxyphenyl lithium to the perbenzylated lactone 209 gave a single diastereomer of lactol 210, which the authors rationalized on the basis of steric encumbrance from the phenol ring of perbenzylated 209. A subsequent Kishi reduction205served to rectify the oxidation state of 211 to that of the natural products and resulted in epimerization of the stereogenic center formed in the previous step, presumably via an intermediate para-quinone methide cation. Hydrogenolysis of 212 gave the natural product hopeahainol D (38, 3.3 mg) whereas epimerization could again be achieved under Lewis acidic conditions, thus yielding the natural product heimiol A (37, 0.5 mg).
On the basis of their ability to form cycloheptanones using electrophilic cyclization chemistry, Snyder and co-workers were able to design a route to the natural products hopeanol (31) and hopeahainol A (40) (Scheme 35).206 It was found that the cyclization of triaryl precursor 201 could be accomplished using Brønsted acid rather than electrophilic halides. This was an important development as it enabled the key cyclization without introducing atoms to the molecular framework that are not part of the natural products. Notably, a shorter route (4 steps, 56%) to 201 was now available through direct, regioselective bromination of permethyl resveratrol. Vinylogous protonation of 201 using para-TsOH generated an intermediate cationic para-quinone methide that was captured through a 7-exo cyclization by the pendant resorcinol ring to give 213. Leveraging the homologation chemistry employed in their synthesis of heimiol A (37) and hopeahainol D (38) (vide supra), the researchers were able to convert the cycloheptanone 213 into the diol 214. Here, the use of a Brønsted acid (AcOH) to promote the epoxide opening resulted in trapping by the conjugate base (acetate) rather than Meinwald rearrangement. Oxidation of the primary alcohol to aldehyde 215 followed by addition of 4-methoxyphenyl magnesium bromide gave diol 216a/b as a 1.3:1 mixture of epimers in favor of 216b. Next, a number of Brønsted acids were screened in an effort to promote a Pinacol rearrangement207,208 of the diol. They found that the 1 equiv of VAPOL phosphoric acid 217(209) could efficiently promote the Pinacol rearrangement of 216b with high diastereoselectivity (>18:1 dr). Varying yields and epimeric ratios at the newly formed quaternary center of 218 were obtained depending on the acid used, with enantiopure phosphoric acids giving the best results. While the carbon framework of 218 was consistent with that of the natural products, the oxidation state needed to be increased by four. Three of these oxidations could be accomplished in a single step by treatment of 218 with Jones reagent, giving 27% of spirocyclohexadienone 220 through the intermediacy of phenonium 219. Surprisingly, the aldehyde was not oxidized under these conditions. Compound 220 could be converted to the protected hopeanol (221) through Pinnick–Lindgren oxidation and esterification, but its deprotection was not possible. A number of synthetic precursors also failed to deprotect using standard BBr3 conditions, resulting in decomposition and/or rearrangement of the material. Ultimately, the researchers found that acid 222, obtained by Pinnick–Lindgren oxidation of 218, could be dealkylated with BBr3, with concomitant formation of the γ-lactone yielding dibenzocycloheptane 223. This compound was then immediately protected with benzyl bromide. The three remaining oxidations were accomplished in one pot using ceric ammonium nitrate (CAN). This gave a perbenzyl derivative which could be dealkylated using BCl3 to reveal the natural product hopeahainol A (40, 60 mg) in good yield. Methanolysis using the conditions reported by Nicolaou and Chen gave hopeanol (31, 11 mg).
Scheme 35. Synthesis of Hopeahainol A and Hopeanol by Snyder et al.

7.1.6. Chen’s Synthesis of Malibatol A and Shoreaphenol (Hopeafuran)
In 2010, Chen and co-workers disclosed a synthesis of malibatol A (44) and shoreaphenol (45), two benzofuran-containing resveratrol dimers (Scheme 36).210 Chen’s strategy to these natural products was conceptually similar to that of Kim and Choi (see section 7.2.3),219 consisting of benzofuran construction followed by electrophile-promoted 7-membered ring formation. From benzophenone 224, reminiscent of those used by Snyder and co-workers in their synthesis of resveratrol dimers,185 benzofuran synthesis was achieved by deprotonation of the benzyloxy ether using lithium tetramethylpiperidide (LiTMP), attack of the resultant alkyl lithium onto the ketone, and acid-promoted dehydration. Epoxidation followed by a one-pot cyclization/demethylation sequence gave malibatol A (44). Further oxidation afforded shoreaphenol (hopeafuran) (45).
Scheme 36. Chen’s Synthesis of Malibatol A and Shoreaphenol.

7.1.7. Synthesis of Laetevirenol A by Heo et al
In 2012, Heo and co-workers disclosed a synthesis of laetevirenol A (62) (Scheme 37),211 a member of a dimeric subclass of resveratrol oligiomers containing phenanthrene ring systems that are likely generated biosynthetically through an oxidative 6π-electrocyclization from parthenocissin A (61), as demonstrated by Pan et al. (see section 5.5.1).102 Retrosynthetically, the strategy was to construct the phenanthrene first through a one-pot Suzuki–Miyaura coupling/aldol condensation cascade, with late-stage formation of the indane through an intramolecular Friedel–Crafts cyclization onto triaryl-substituted olefin 232. Phenylacetonitrile 226, prepared in two steps from dimethoxy benzyl bromide, was subjected to cross coupling with aldehyde 227 under microwave irradiation in the presence of base, providing the desired phenanthrene 228 in good yield. Hydrolysis of the nitrile and peptide coupling afforded the corresponding Weinreb amide, which was efficiently converted to triaryl ketone 230 upon treatment with the phenyl Grignard reagent 229. The final arene was introduced through another Grignard addition, and dehydration to the required olefin 232 was achieved using Brønsted acid. Toluene sulfonic acid proved to be best-suited for this transformation, affording a 5:1 ratio of E/Z alkene isomers. In fact, prolonged exposure to TsOH with heating also promoted the desired intramolecular Friedel–Crafts reaction to 235, albeit in modest yield. Alternatively, exposure of 232 to FeCl3 promoted oxidative cyclization to acephenanthrylene 233, as was found by Sarpong (section 7.2.2). This was converted into cis-permethyl laetevirenol A (234) via catalytic hydrogenation.216 Demethylation of 235 proceeded quantitatively to give laetevirenol A (62).
Scheme 37. Synthesis of Laetevirenol A by Heo et al.

7.2. Synthesis of Resveratrol Dimers Using Transition-Metal Catalysis
7.2.1. She and Pan’s Synthesis of Pauciflorol F by a Pd-Mediated Intramolecular Heck Cyclization
In 2006, She, Pan, and co-workers disclosed the first de novo approach to resveratrol dimers, featuring a Pd-catalyzed intramolecular 5-endo Mizoroki–Heck cyclization212,213 onto an allylic alcohol (Scheme 38).214 While the key substrate 236 could be synthesized in just 5 steps using well-established methods, two of these steps were redox manipulations of the benzylic carbon. Nonetheless, reacting 236 with Pd(OAc)2/PPh3 resulted in its conversion to indenone 237, presumably via a tandem Mizoroki–Heck cyclization/Saegusa–Ito oxidation215 sequence. The researchers were able to reduce the α,β-unsaturated enone to the corresponding cis-2,3-dihydroindanone (not shown) and epimerize the α-stereogenic center under alkaline conditions to give 170. Finally, global demethylation using BBr3 revealed the natural product pauciflorol F (28).
Scheme 38. Synthesis of Pauciflorol F by She and Pan.

7.2.2. Sarpong’s Synthesis of Pauciflorol F and related Carbogenic Cores by a Pd-Mediated Intermolecular Heck Cyclization Cascade
Two years after the seminal publication by Snyder,185 Sarpong and co-workers disclosed an alternative approach to the carbogenic cores of resveratrol dimers.216 Snyder had demonstrated that bromoresveratrol derivative 238 could be efficiently lithiated and reacted with benzaldehydes to form the corresponding alcohols. Sarpong and co-workers envisioned that metalation of this substrate with a redox active catalyst such as palladium would, with appropriately designed substrates, lead to the construction of the indane and indanone cores of several resveratrol dimers. In particular, coupling of 238 with resveratrol-derived tolane 239 enabled rapid assembly of benzofulvene dimers representing the carbon skeletons of pallidol and related congeners (Scheme 39). Here, oxidative insertion of Pd0 into the C–Br bond produces a π-acidic PdII–aryl species that coordinates the electron-rich tolane. Polarization of the alkyne by the para-disposed methoxy group facilitates regioselective migratory insertion to the PdII–aryl bond, in turn generating a nucleophilic vinyl palladium species 240. Upon coordination of the neighboring olefin, this vinyl–Pd undergoes sequential insertion and β-hydride elimination to yield 241, an unsaturated form of the natural product parthenocissin A (61). Treatment of 241 with FeCl3·6H2O resulted in the production of 246, likely through a series of single electron oxidation/deprotonation events (242 → 245).
Scheme 39. Mizoroki–Heck/Friedel–Crafts Sequence To Access Oxidized Forms of Resveratrol Dimers.

As previously described, She and co-workers demonstrated that indenone dimer 237 could be converted to the natural product pauciflorol F (28) (Scheme 38).214 On the basis of the high level of regiocontrol observed in their synthesis of benzofulvene dimers,216 Sarpong and co-workers envisioned that a similar Heck coupling strategy using a different aryl-halide electrophile could provide a highly convergent route to 237, and therefore an efficient synthesis of pauciflorol F (28).217 Namely, the use of a brominated benzaldehyde precursor 247, rather than the previously employed stilbene derivative, would provide the corresponding indenone 237, through a Larock-type annulation (Scheme 40).218 In practice, the cascade cyclization proceeded in good yield (81%), although a 1:1 mixture of regioisomers 237/249 with respect to the tolane-derived arenes was obtained.
Scheme 40. Sarpong’s Synthesis of Pauciflorol F Using a Larock Annulation.

Reduction of 237 to the corresponding cis-2,3-diaryl indane was accomplished by hydrogenation over Pd/C, as was performed by She et al.214 An interesting observation was made by Sarpong and co-workers during their subsequent attempts to epimerize cis-170 to its trans isomer, leading them to bring into question the structural assignments made by Snyder and She in their respective publications.189,214 Instead of the reported trans-indenone 170, the authors isolated the α-ketol product 250. By comparison of the spectral data for 250 to those reported for 170, it appeared as though each of these groups had also obtained 250 in similar transformations. Undesired formation of 250 could be avoided by performing the reduction and epimerization in one pot, suggesting that O2 was promoting the enol oxidation. However, Sarpong and co-workers again ran into difficulty when they attempted to deprotect 170 using the conditions employed by She and Snyder, isolating just 35% (conditions not shown) of pauciflorol F (28) as compared to the reported 86% and 60% yields.185,214 The use of α-ketol 250 in these experiments would require both reduction (via disproportionation of 13) and global dealkylation to complete the synthesis of 28. To gain insight into the feasibility of this transformation, Sarpong and co-workers subjected 250 to the reported conditions for the deprotection and obtained just 10% of pauciflorol F (28), suggesting that disproportionation may be possible but is inefficient under these conditions. On the other hand, performing the deprotection of 170 in the presence of added reductant (Et3SiH) improved the yield of the natural product pauciflorol F (28) substantially.
7.2.3. Kim’s Syntheses of Benzofuran-Containing Resveratrol Dimers Using C–H Activation
In 2009, Kim and Choi reported a general strategy to access the privileged 7,5-ring system ubiquitous in resveratrol-derived oligostilbenoids,219 relying on the well-established acid-promoted cyclodehydration of α-aryloxyketones to form benzofuran 252 from 251 (Scheme 41).220 Bi(OTf)3 was found to be optimal from the Lewis and Brønsted acids that were screened. Arylation at C-2 of the benzofuran was accomplished through a Pd-mediated Heck cross coupling that is initiated via direct C–H activation.221 Here, C-2 palladation likely takes place through either an electrophilic aromatic substitution (SEAr) or concerted metalation-deprotonation (CMD)222 process to form bisaryl-PdII complex 253. Reductive elimination gives a dimer of benzofuran 252 and generates the required Pd0-complex for oxidative addition to 4-bromoanisole. The authors consistently isolated a mixture of desired 254 and the dimer of 252, suggesting that the metalation/reductive elimination pathway is competitive with oxidative addition to the Ar–X bond. This is perhaps unsurprising given that an electron-rich aryl halide was used, 20 mol % of the PdII precatalyst was employed, and the reaction was run open to air.
Scheme 41. Synthesis of Benzofuran-based Resveratrol Dimers Using C–H Activation.

Reduction of ester 254 provided the aldehyde 255, which was the branching point for the synthesis of each of the three natural product analogues. Olefination by a standard Horner–Wadsworth–Emmons protocol yielded viniferifuran analogue 256. Epoxidation of 255 by a Johnson–Corey–Chaykovsky protocol afforded racemic 257. Exposure of 257 to acidic conditions at cryogenic temperatures gave malibatol A analogue 258 and its epimer in a product distribution that was influenced by the identity of the acid catalyst. Subsequent oxidation yielded shoreaphenol analogue 259.
In a subsequent report, Kim and co-workers achieved the synthesis of diptoindonesin G (263) using intermediates 251 and 254 (Scheme 42).223 From 254, a tandem Friedel–Crafts acylation/global demethylation was possible using the highly acidic BBr3. Alternatively, treatment of 251 with the less acidic BCl3 promoted both cyclodehydration and Friedel–Crafts acylation, but only led to dealkylation of the phenol ortho to the so-formed diaryl ketone. Arylation of the benzofuran was again accomplished by direct C–H activation, this time using modified Fagnou conditions.224 In combination with the use of bulky phosphine ligands, these conditions dramatically improved the yield of the cross-coupling as compared to the previously and eliminated formation of significant amounts of homodimeric products.
Scheme 42. Synthesis of Diptoindonesin G using C–H Activation.

7.2.4. Yang and Heo’s Syntheses of Pauciflorol F by a Pd-Mediated α-Arylation
In 2011, Yang and co-workers reported an approach to pauciflorol F (28) employing an α-arylation reaction of 3-aryl-indanone 265 (Scheme 43).225 Here, 265 was prepared through a Nazarov cyclization226 of the chalcone precursor 264, a strategy that has since been utilized by other groups for the synthesis of this natural product.211,227,228 While the synthesis was racemic, the arylation was diastereoselective, providing exclusively the trans-2,3-diaryl indanone derivative (170). Under the optimized cross-coupling conditions, an excess of KHMDS was used, resulting in the formation indenone 237 as a byproduct through product enolization and Saegusa–Ito oxidation. Notably, the product distribution was influenced by the precatalyst, ligand, and base employed. Heo and co-workers were able to largely eliminate this problem by the use of fewer equivalents of a weaker base (NaOtBu).211 Additionally, these researchers were able to achieve the first asymmetric synthesis of (+)-pauciflorol F (28) by subjecting She’s indenone intermediate 237 (vide supra)214 to an enantioselective baker’s yeast-promoted conjugate reduction. Flynn and co-workers subsequently achieved the asymmetric synthesis of both enantiomers of pauciflorol F using a chiral auxiliary-directed Nazarov cyclization,228 and found that Heo and co-workers had misassigned the configuration of the natural product generated through their synthesis.
Scheme 43. Synthesis of Pauciflorol F Using a Nazarov/C–H Arylation Sequence.

7.2.5. Studer’s Synthesis of Indane Dimers by a Pd-Catalyzed Decarboxylative Arylation and Oxidative Heck Coupling
As discussed previously, the two types of indane dimers formed in nature fundamentally differ only in the substitution pattern of the phenol and resorcinol rings about the C–2 and C–3 positions of the indane ring. Studer and co-workers therefore envisaged that a synthetic route that introduced these aryl groups from a common intermediate would be an attractive and flexible strategy for oligomer synthesis.146 Retrosynthetically, these researchers arrived at intermediate 266 as a suitable candidate for this purpose (Scheme 44). Previously, Studer et al. had demonstrated that doubly allylic quaternary carboxylates such as 266 can be decarboxylated and regio- and stereospecifically coupled with a variety of aryl iodides.229,230 Application of this method to 266 gave the desired arylated indene products 267 and 268 with complete regioselectivity in moderate yields (65% and 57%, respectively, Scheme 44).
Scheme 44. Synthesis of Resveratrol Dimers using a Decarboxylative Arylation/Oxidative Mizoroki–Heck Sequence.

To complete the syntheses of the gross structures of regioisomers quadrangularin A (17) and ampelopsin D (25), these researchers again employed their own cross-coupling methodology: a nitroxide-mediated oxidative Heck arylation.231 As with the previous step in the synthesis, this reaction was more effective for the quadrangularin A core 178 than for 172, the core of ampelopsin D. Of note, TEMPO derivative 269 had to be used for the ampelopsin D substrate in order to suppress formation of an unidentified byproduct. The coupling reactions were completely regioselective for each of the boronic acids used, adding exclusively to the less substituted carbon of the indene. Additionally, the subsequent β-hydride eliminations were completely regio- and stereoselective, providing exclusively the exocyclic alkenes as the (E)-isomers. The authors rationalize the (E)-selectivity on the basis of allylic strain minimization. Finally, C–C bond formation took place from the opposite face of the indene than the C–3 aryl group, thereby giving products 172 and 178 with the requisite trans relative configuration. Each of these compounds were deprotected using BBr3 to reveal the natural products quadrangularin A (17) and ampelopsin D (25). Quadrangularin A precursor 178 was converted to the natural product pallidol (18) by a known two-step procedure.227
7.2.6. Shaw’s Asymmetric Synthesis of δ-Viniferin by C–H Insertion
Recently, Shaw and co-workers disclosed a general strategy to dihydrobenzofuran synthesis via the intramolecular C–H insertion of diaryl rhodium-carbenoids to methylenes of para-methoxybenzyl protected phenols.232 This is a highly impactful development as 2,3-diaryl-dihydrobenzofurans have traditionally been difficult to synthesize in an efficient manner. The reported method utilizes easy to prepare starting materials, and occurs with high levels of diastereo- and enantiocontrol. Interestingly, the insertion preferentially yields the cis-diastereomer. However, it has been shown that cis-dihydrofurans bearing an anisole or phenol at the 2-position can be epimerized under acidic233 or basic234 conditions. Shaw and co-workers exploited this for the asymmetric synthesis of δ-viniferin (10) (Scheme 45).
Scheme 45. Asymmetric Synthesis of δ-Viniferin Using a Rh-Catalyzed C–H Insertion.

The requisite diaryl hydrazone 271 was easily synthesized by the condensation of hydrazine on to benzophenone 270. Oxidation of the hydrazone to the corresponding diazo compound by MnO2 followed by exposure of the crude material to Rh2(S-PTAD)4 effected the smooth conversion to cis-dihydrobenzofuran 272 with high diastereo- and enantioselectivity. A one-pot demethylation/epimerization was accomplished by treatment with BCl3/TBAI, and the phenols were reprotected to give peracetyl compound 273. A Mizoroki–Heck olefination with styrene 274 followed by global deprotection provided (+)-(E)-δ-viniferin (10),112 representing the first asymmetric synthesis of a dihydrobenzofuran-containing resveratrol dimer.
7.3. Synthesis of Higher-Order Resveratrol Oligomers: Reagent-Controlled Bromination of Resveratrol Dimers as a General Strategy for Trimer and Tetramer Synthesis
The stereo- and regiochemical outcome of the bromonium-promoted cascade cyclizations developed by Snyder et al. (section 7.1.1) for the synthesis of bicyclic dimers has significant consequences for the synthesis of higher-order resveratrol oligomers. Although the oxidations involved in the biosynthesis of resveratrol oligomers proceed via single electron transfers, these polar oxidations nonetheless provide insight into the substrate contribution to the regiochemistry observed during oligomer biogenesis.235,236 While the requirement for hydrodehalogenation negatively impacts the step- and atom-economy for the synthesis of dimeric natural products, the complete regioselectivity observed for these transformations has enabled the synthesis of a number of higher-order oligomers. Recall that the carbocyclic scaffolds present at the cores of these dimeric natural products are conserved across the entire series of natural products belonging to this class. Therefore, these seemingly extraneous aryl bromides actually provide functional handles with which to target the construction of 2,3-diaryldihydrobenzofurans for the synthesis of trimeric and tetramer resveratrol-derived oligomeric natural products (Scheme 46, path A). Through an impressive series of syntheses, Snyder and co-workers have successfully capitalized on this opportunity.237 These endeavors are described in the sections below.
Scheme 46. Paradigm for the Synthesis of Higher-Order Oligomers through Iterative (A) Homologation and (B) Dihydrobenzofuran Synthesis.
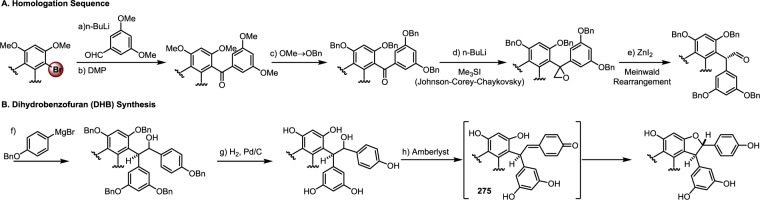
7.3.1. Synthesis of Higher-Order Oligomers Containing Bicyclooctane Cores
Treatment of the tribromo-pallidol derivative 182 with 2 equiv of n-butyllithium resulted in selective metal–halogen exchange of the aryl bromides (Scheme 47). The product monobromide (not shown) was then exposed to 1 equiv of NBS resulting in selective bromination of the less hindered resorcinol ring to give 276. Another lithium halogen exchange, this time using tert-butyllithium (3 equiv), resulted in formation of the corresponding dianion, which was selectively reacted with 3,5 dimethoxy benzaldehyde at the aryl lithium (not shown). This is somewhat surprising given the greater reactivity expected of a tertiary aliphatic anion (pKa ∼53) versus that of an aryl-lithium species (pKa ∼43), and suggests that perhaps sterics are responsible for the observed selectivity. The product was then homologated to aldehyde 277 using conditions employed in the synthesis of dimeric products heimiol (37) and hopeahainol D (38) (see Scheme 46, path A).198 The intermediate aldehyde 277 was obtained as a 5.5:1 mixture of chromatographically separable diastereomers. During the course of the homologation sequence, the methoxy protecting groups were exchanged for benzyl ethers. In spite of the harsh conditions required for demethylation (35–45 equiv BBr3, sealed tube, 70 °C, 3–4 days), the deprotected compound was obtained in 85–95% yield.
Scheme 47. Synthesis of Higher-Order Oligomers Containing Bicyclo[3.3.0]octane Cores.

Construction of the critical dihydrobenzofuran moiety was achieved through a three-step sequence (Scheme 46, path B) consisting of Grignard addition of 4-benzyloxyphenyl magnesium bromide to the aldehyde 277, global hydrogenolysis, and acidic resin-promoted C–O bond formation, presumably via the intermediacy of benzyl alcohol derived cationic para-quinone methide intermediates such as 275. The benzylic alcohols survive global hydrogenolysis and were carried on as a mixture of diastereomers (dr ∼10:1) through the dihydrobenzofuran synthesis to give carasiphenol C (66, 25 mg). The trans-product of 66 is thermodynamically favored, and is likely enriched under the acidic conditions employed for the cyclization. Isolation of the natural product was accomplished by reverse-phase HPLC.
For the synthesis of resveratrol tetramer ampelopsin H (67) (Scheme 47), debrominated pallidol derivative 278 was brominated regioselectively on each of the resorcinol rings to give C2-symmetrical 279, which was subjected to two-directional238,239 homologation to yield dialdehyde 280 in good yield (Scheme 46, A). Aldehyde 280 was obtained as a diastereomeric mixture (dr ∼1.8:1), from which recrystallization of the major (C2-symmetric, shown) diastereomer was possible. Epimerization of the minor isomer could also be achieved, but required strong base (KHMDS), which the authors propose is due to the resistance of the aldehyde to enolization caused by its proximity to the electron rich aromatic rings. From here, the identical dihydrobenzofuran synthesis (Scheme 46, path B) afforded the desired natural product ampelopsin H (67, 6.7 mg), again as an approximately 10:1 mixture of the desired natural product and what is presumably a dihydrobenzofuran stereoisomer.
With a [3.3.0] trimer and tetramer completed, these researchers turned their attention to the [3.2.1] ampelopsin F derivative 281 as a starting point for extension of this chemistry to higher-order oligomers containing this core.237 Unlike permethyl pallidol (278), regioselective monobromination of permethyl ampelopsin F (281) was only possible through a combination of reagent and stoichiometry control. Specifically, treatment of 281 with 1 equiv of NBS resulted in selective bromination of the C14a on the resorcinol A2 yielding 282. The use of an in-house developed reagent, bromodiethylsulfonium bromopentachloroantimonate(V) (BDSB),240 selectively yielded 283 (Scheme 48). Other brominating reagents exclusively yielded 282 or 284. Finally, addition of 2 equiv of NBS smoothly transformed 281 to the dibromide 284. Application of the same alkylation/homologation/protecting group exchange/dihydrobenzofuran formation sequence to these intermediates provided the natural products, carasiphenol B (81, 2.4 mg), ampelopsin G (70, 2.0 mg), and vaticanol C (94, 1.2 mg). Impressively, the team demonstrated that this strategy was scalable and could isolate 55 mg of >90% pure carasiphenol C (66) from one deprotection/cyclization sequence.
Scheme 48. Synthesis of Higher-Order Oligomers Containing Bicyclo[3.2.1]octane Cores.

7.3.2. Synthesis of Vaticanol A, an Indane-Derived Resveratrol Trimer
In 2014, Snyder and co-workers completed the first total synthesis of a resveratrol oligomer containing a 7,5-fused indane ring system, vaticanol A (76) (Scheme 49).147 Starting from permethyl pauciflorol F (170) the dibromoampelopsin D derivative 289 was prepared through a 6-step sequence previously developed for the synthesis of caraphenols B (27) and C (60).188 Note that the styrenyl phenol is orthogonally protected with respect to the remaining arenes in the molecule; this was performed in an attempt to achieve selective quinone methide formation later in the synthetic sequence. Formation of the dianion of 289 through lithium-halogen exchange with subsequent addition of 3,5-dimethoxybenzaldehyde gave the monoalkylated product 290 without formation of its regioisomer or the doubly alkylated product. Oxidation gave biaryl ketone 291, which contains 5 of the 6 aromatic rings found in the natural product. Hydrogenolysis of 291 cleaves the benzyl ether and stereoselectively reduces the olefin, yielding 292 in 16% yield from 170. A variety of oxidants were screened in an effort to selectively dehydrogenate the exposed phenol to its corresponding quinone methide. Ultimately, it was found that prolonged exposure to DDQ at room temperature oxidized the B1 phenol of 292, initiating 7-exo-trig cyclization (C10c–C7b). The subsequent global methylation gave 293 as 1:1 mixture of diastereomers at C7b. A selective demethylation of the C13 methoxy group afforded dibenzocycloheptanone 294 in 15% yield over the 3 steps.
Scheme 49. Total Synthesis of Vaticanol A Through in Situ Generation and Reactions of para-Quinone Methides.
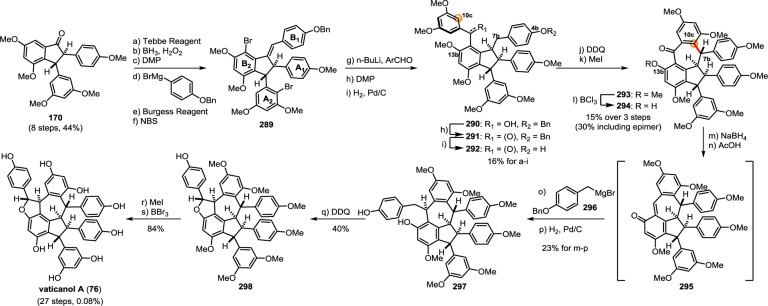
At this stage, 5 of the 6 aryl rings of the natural product were built into the framework. To complete the synthesis, a redox manipulation, addition of the final arene, construction of the dihydrobenzofuran, and global deprotection were required. The team planned to introduce the aryl ring through nucleophilic addition to an in-situ-generated ortho-quinone methide, which required adjustment of the oxidation state of 294. Reduction with NaBH4 provided the corresponding alcohol in 91% as a mixture of epimers, which, upon treatment with acetic acid, ionized to the ortho-quinone methide 295. In situ exposure of this material to an excess of Grignard reagent 296 and hydrogenolysis of the benzyl ether afforded 297 in 25% over the three steps, along with 25% of the undesired product epimer. Oxidative dihydrobenzofuran closure was achieved by treatment of 297 with DDQ, providing the thermodynamically favored trans diastereomer of 298. Attempts to directly deprotect 298 with BBr3 were unsuccessful, necessitating methylation of the free phenol prior to exposure to BBr3. This yielded a sample containing vaticanol A (76, 0.9 mg), whose spectral data matched those reported by Tanaka and co-workers. This remarkable achievement was possible through careful design of protecting group strategy which enabled selective in situ generation and cyclizations of para-quinone methides and is the state of the art in the synthesis of the resveratrol natural products.
7.3.3. Synthesis of 9-Membered Ring Containing Resveratrol Trimer, Caraphenol A
Recently, Snyder and Wright disclosed a synthesis of caraphenol A (89), a resveratrol trimer belonging to a subclass of resveratrol oligomers containing strained 9-membered rings (Scheme 50).241 On the basis of a series of model studies, these researchers demonstrated the viability of Friedel–Crafts chemistry for 9-membered ring formation under conformationally controlled conditions. As such, the synthesis of caraphenol A (89) commenced with 299, which was prepared in 3 steps from 3,5-dibenzyloxy benzyl alcohol. Here, the researchers used the previously developed homologation/dihydrobenzofuran synthesis sequence (Scheme 46, paths A + B). The phenols were reprotected as benzyl ethers, and the silyl protecting group cleaved yielding benzofuran 301 in 54% yield from 299. The resulting primary benzyl alcohol 301 was oxidized to the corresponding carboxylic acid through sequential Dess–Martin/Pinnick–Lindgren oxidations, and the product acid was esterified with 302 to give tetraaryl dihydrobenzofuran 303 in 81% overall yield. In a clever synthetic operation, this compound was subjected to an anionic Fries rearrangement242,243 and in situ silyl ether formation to provide 304 in 80% yield, which possesses the correct connectivity of the aryl rings that will ultimately comprise the 9-membered ring. Another three-step homologation of the benzophenone, identical to that performed at the beginning of the synthesis, afforded benzyl alcohol 305. Oxidation with DMP provided the corresponding ketone 306 in 82% overall yield from 304, which, upon exposure to HCl, underwent cyclodehydration. Cleavage of the silyl ether afforded 308/307 as a 2.8:1 mixture of atropisomers. 307 could be readily interconverted to the required atropisomer 308 by thermal equilibration. Functional group interconversion of benzyl alcohol 308 to allyl alcohol 309 was achieved in two steps by a Dess–Martin oxidation followed by the addition of vinyl lithium to the corresponding ketone as 1:1 mixture of isomers. The doubly activated allylic/benzylic alcohol was ionized with MsOH, triggering a 9-exo-trig Friedel–Crafts reaction to provide the carbocycle 310. Oxidative cleavage of the alkene 310 provided aldehyde 311 in 46% yield from 306. Addition of the aryl Grignard reagent to 311 yielded the penultimate intermediate 312, which was subjected to the usual conditions for debenzylation/dihydrobenzofuran formation (Scheme 46). The natural product, caraphenol A (89), was thus obtained in 23 steps from commercially available materials in an impressive 7.8% overall yield. Astonishingly, Wright and Snyder report isolating 317 mg in the final reaction, preparing as much as 600 mg of 89 thus far!
Scheme 50. Total Synthesis of Caraphenol A by Snyder and Wright.

8. Beyond the Plant: Therapeutic Potential of Resveratrol Oligomers
Since the popularization of resveratrol at the end of the 20th century, an enormous number of studies have been undertaken in an effort to define its pharmacological profile. In spite of these efforts, resveratrol’s mode of action appears to be fairly indiscriminate when one considers the number of intracellular processes it has been claimed to influence. This “broad spectrum” biological activity is likely a reflection of the intrinsic reactivity of the trihydroxylated stilbene as a redox-active molecule. Mounting evidence suggests that resveratrol and its oligomers exert their effects via interference with signal transduction cascades and epigenetic pathways rather than direct inhibition of enzymes designated for specific purposes. Nonetheless, implementation of resveratrol in human therapeutic applications is forestalled by its poor bioavailability and an incomplete understanding of its pharmacodynamics. In light of this, interest in the pharmacological potential of resveratrol oligomers has recently increased, and preliminary explorations into their bioactivity are promising. Indeed, the probability of selective modification of targets within a chiral cellular environment is rapidly enhanced as a relatively small 2-dimensional molecule (resveratrol) is transformed into a complex 3-dimensional structure. Here, we present frontiers in this area of science with an emphasis on the mechanisms underlying observed biological activities.
8.1. Resveratrol Oligomers as Anticancer Agents
The biochemical processes accompanying carcinogenesis are loosely categorized into three major phases: tumor initiation, promotion, and progression.244 In 1997, Pezzuto and co-workers reported that resveratrol was a potential cancer chemopreventive agent, exhibiting anti-initiation, antipromotion, and antiprogression activities.12 In this study, these effects manifested themselves as antioxidant and antimutagenic behavior, inhibition of cyclooxygenase (COX-1, ED50 = 15 μM) and hydroperoxidase (ED50 = 3.7 μM), and induction of promyelocytic leukemia cell differentiation (the phenomenon by which cancerous cells appear and behave like normal cells of the tissue in which they exist), respectively. Readers interested in the potential of resveratrol as an anticancer agent are directed to a recent review.20 Here, we will focus on the resveratrol oligomers, which unsurprisingly have also attracted significant attention in this regard. However, most of these biological studies have required the laborious extraction and purification of the oligomers from plant matter.245 Because the resveratrol-producing stilbene synthase is not constitutively expressed, these compounds are typically isolated as a minute fraction of the plant’s weight. Obviously, this approach is limiting with respect to the diversity of assays that can be performed and the purity with which the subject compounds can be obtained. Nonetheless, the oligomers have demonstrated a broad range of interesting biological activities, and hold promise as a source of natural products-inspired drug development.
As is typical of most explorations into the biological activities of natural product classes, the resveratrol oligomers have primarily been examined for their anticancer properties (Table 6). This topic has recently been reviewed,246 and therefore we will attempt to provide a concise overview of these studies with context as to the significance of the selected examples. The anticarcinogenic effects of resveratrol oligomers have principally been ascribed to cytotoxicity and induction of apoptosis, although antimutagenic and antiproliferative activities have also been observed. Unfortunately, many investigations in this area did not extend beyond initial toxicity studies of the oligomers against one or multiple cancer cell lines, which provide no information about the mechanisms of action. Several groups have since taken a more rigorous approach to these studies, and as a result we are beginning to understand the specific actions of these compounds within cells to which their observed biological activities can be attributed.
Table 6. Cytotoxicities of Resveratrol Oligomers Against Various Cancer Cell Lines.
| cell lines; refs | most active compd | (EC50, μM)a |
|---|---|---|
| Cervical (Adeno)carcinoma | ||
| HeLa; (87, 247−251) | pauciflorol B | 2.8 |
| HSG; (252) | sophorastilbene A | 108.0 |
| KB; (66, 87, 141, 251, 253, 145) | peracetyl ampelopsin A | 0.8 |
| Ovarian (Adeno)carcinoma | ||
| 1A9; (253) | peracetyl ampelopsin A | 1.0 |
| SK-OV-3; (254) | cis-vitisin A | 0.17 |
| Breast (Adeno)carcinoma | ||
| MCF-7; (247−250, 253, 255−258) | peracetyl ampelopsin A | 2.8 |
| MDA-MB-231; (250, 257, 259) | hopeachinol E | 14.3 |
| BJMC-3879; (286) | vaticanol C | 8.0 |
| BC-1; (141) | (−)-vatdiospyroidol | 4.2 |
| Leukemia | ||
| P-388; (260, 289) | (−)-hopeaphenol | 5.2 |
| EHEB; (261) | N/A | N/A |
| WSU-CLL and ESKOL; (261) | ε-viniferin | 6.7 |
| Jurkat; (262) | miyabenol C | 29.4 |
| HL-60; (247, 252, 258, 289, 263−267) | vaticanol C | 3.0 |
| K-562; (262, 265) | miyabenol C | 18.7 |
| Myeloma | ||
| U-266; (262) | miyabenol C | 12.1 |
| RPMI-8226; (262) | miyabenol C | 20.8 |
| Lymphoma | ||
| U-937; (262, 265, 268) | heyneanol A | 6.6 |
| Colon (Adeno)carcinoma | ||
| HCT-116; (257, 259, 269, 270) | α-viniferin | 6.6 |
| HT-29; (248, 249, 257, 269) | α-viniferin | 32.6 |
| Caco-2; (269) | α-viniferin | 16.1 |
| Col-2; (141) | (−)-vatdiospyroidol | 2.1 |
| HCT-8; (253) | peracetyl ampelopsin | 5.7 |
| SW-480; (265, 270, 285, 271) | A vaticanol C | 3.2 |
| DLD-1; (265, 285) | vaticanol C | N/A |
| COLO-201; (265, 285) | vaticanol C | N/A |
| Lung (Adeno)carcinoma | ||
| A-549; (250, 253, 254, 258, 272, 273) | cis-vitisin A | 0.93 |
| NCI-H446; (269, 272, 273) | α-viniferin | 16.1 |
| NCI-H460; (272) | kobophenol A | 200 |
| NCI-H1299; (250) | vaticanol A | 19.7 |
| SPC-A-1; (272) | ||
| Liver Carcinoma | ||
| Hep-G2; (248, 249, 258, 259, 274) | hopeachinol J | 12.4 |
| SMMC-7721; (259) | hopeachinol F | 10.4 |
| Clear Cell Renal Carcinoma | ||
| Caki-1; (253) | peracetyl ampelopsin A | 5.5 |
| Prostate (Adeno)carcinoma | ||
| LNCaP; (265) | vaticanol C | N/A |
| PC-3; (265) | α-viniferin | N/A |
| Neuroblastoma | ||
| SH-SY5Y; (265, 275) | vaticanol C | N/A |
| C6; (249) | gnetin H | 18.7 |
| Sarcoma | ||
| S-180; (276) | extract-Vat. indica | 29.5 |
| SaOS-2; (253) | peracetyl ε-viniferin | 11.3 |
| Oral Squamous Carcinoma | ||
| HSC-2; (252) | sophorastilbene A | 27.0 |
| HSC-3; (252) | (+)-α-viniferin | 60.0 |
| Malignant Melanoma | ||
| SK-MEL-2; (253, 254) | vitisin B | 4.9 |
All values reported as (μg/mL) have been converted to EC50 (μM) for the sake of comparison.
8.2. Potential Modes of Action: Case Studies on the Cellular Effects of Two Resveratrol Oligomers
8.2.1. α-Viniferin: A Resveratrol Cyclotrimer with Diverse Biological Activity
The structure of α-viniferin (6) was first proposed by Langcake and Pryce in 1977.37,58 This oligomer has now been identified from a number of plants in the Fabaceae and Cyperaceae families that have traditionally been employed in East Asian folk medicine. It has consistently outperformed other compounds within this class in assays of cytotoxicity, anti-inflammatory activity, and enzyme inhibition. As such, it represents one of the most well-studied resveratrol oligomers in the context of biological activity. Here, we will present the molecular mechanisms by which this compound exerts its biological effects, which could very well be representative of the mode of action for the larger class of natural products. Much of the knowledge that we have on this subject has come from the dedicated efforts of Kim and co-workers, who have thoroughly investigated the cellular effects of α-viniferin (6) from both a phenotypic and genotypic perspective.277−279
Initial work revealed that 6 could inhibit the cyclooxygenase activity of prostaglandin H2 synthase (IC50 = 7.0 μM), a pharmacological target for the treatment of fever, inflammation, and heart disease. To better understand this behavior, these researchers explored the effects of 6 on the activity and expression of cyclooxygenase (COX) isozymes and inducible nitric oxide synthase (iNOS). α-Viniferin showed a dose-dependent inhibitory effect on COX-2 activity but only weakly inhibited COX-1. COX-1 is expressed constitutively in most tissues and is essential to functions other than inflammatory response, while COX-2 is up-regulated in response to inflammatory mediators such as lipopolysaccharides (LPS) or cytokines. Additionally, 6 inhibited the transcription of COX-2 genes in LPS-activated murine (mouse) macrophages. Likewise, 6 inhibited the synthesis of the iNOS transcript. Transcription of the iNOS gene, like that of COX-2, is performed as a response mechanism. The nitrite content in the culture medium of these macrophages could also be used as an indicator of nitric oxide (NO) production when measured against a control. Nitric oxide (NO) plays an important role in cellular signaling, and iNOS has been implicated in autoimmune and inflammatory processes. A significant reduction in NO production was found when 6 was administered with LPS simultaneously (IC50 = 2.7 μM), but this effect was not observed when the macrophages were treated 12 h after stimulation, indicating a preventive (down-regulation) but not a therapeutic (inhibitory) role for 6. The mRNA initiation site of the murine iNOS gene is preceded by a TATA box, and the promoter region is composed of several conserved binding sequences for recruitment of transcription factors such as NF-κB, STAT, C/EBP, CREB, and OCT.280 In their initial study, Kim and co-workers found that 6 did not inhibit NF-κB transactivation in LPS-stimulated murine macrophages, in contrast to activity that had previously been reported for monomeric resveratrol (1).281
In a follow-up study, Kim and co-workers were able to elucidate several ways by which α-viniferin influences signal transduction pathways and ultimately autoimmune and inflammatory response in murine macrophages.279 Interferon (IFN)-γ is a pleiotropic cytokine involved in both innate immune surveillance and adaptive immunity against pathogenic infection. By participation in an array of cell signaling pathways, this homodimeric protein, produced chiefly in helper T (Th)-1 cells and natural killer (NK) cells, influences cell growth and differentiation, potentiates responses to mitogens, and modulates gene expression and redox homeostasis. Its two transmembrane receptor subunits, termed α and β, do not associate under normal cellular conditions but are assembled upon binding of INF-γ.282 The intracellular domains of these subunits bear tyrosine kinases known as “Janus”-kinases (JAK)-1 and -2, which upon assembly can phosphorylate the intracellular domain of the complete (α2β2) INF-γ receptor complex. Phosphorylation of each of the α-subunits activates the complex for recognition by two molecules of a transcription factor known as “the signal transducer and activator of transcription-1” (STAT-1), which upon binding are phosphorylated at tyrosine (Tyr701) residues by the associated JAK kinases. The two phosphorylated STAT-1 molecules form a homodimer upon dissociation from the complex, and are each further phosphorylated at serine (Ser727) residues by a p38 mitogen-activated protein (MAP)-kinase.282 The resultant activated STAT-1 translocates to the nucleus, where it specifically binds to the IFN-γ activated consensus sequence (GAS)-motif on the promoter regions of various genes including those of inducible nitric oxide synthase (iNOS), CXC chemokines (chemoattractants), and IFN regulatory factor (IRF)-1.279 The latter is a transcription factor that serves as a feedback loop to enforce transcriptional control of IFN-γ-regulated secondary genes. Dysregulation of this complex signal transduction pathway has been implicated in immunosuppression, evasion of immune detection by malignant neoplasms, and tumor angiogenesis. Kim and co-workers were able to establish that α-viniferin (6) suppresses IFN-γ-induced STAT-1 activation in murine macrophages by inhibiting Ser727 phosphorylation by the p38 MAP kinase. Specifically, 6 was found to inhibit STAT-1 phosphorylation by the MAP kinase 3 (MAPK-3), also known as extracellular signal-regulated kinase (ERK)-1. As a consequence of this inhibition, differential attenuation of the mRNA levels of INF-γ/STAT-1-inducible inflammatory mediators iNOS (and [NO] levels), CXC chemokines, and IRF-1 was achieved in a dose-dependent manner.
Recently, the anti-inflammatory mechanism of α-viniferin (6) in BV2 murine microglial cells was explored.283 In this study, previously reported activities such as decreased production of NO and prostaglandins were confirmed, and new mechanisms by which 6 influences lipopolysaccharide (LPS)-induced expression of proinflammatory mediators were elucidated. Namely, it was found that 6 downregulates iNOS and COX-2 expression via inhibition of the activity of the transcription factors NF-κβ. These proteins are involved in an enormous number of cellular processes, including immune/inflammatory responses, growth and development, and apoptosis.284 Analogously to STAT-1, phosphorylation is a prerequisite for NF-κβ activation, homo/heterodimerization, and translocation. However, unlike STAT-1, NF-κβ itself is not phosphorylated. Rather, cytoplasmic NF-κβ is typically sequestered by the one of the aptly named “inhibitor of kappa B” (IκB) proteins, of which there are three (α, β, and γ). Independently, these proteins each mask the conserved N-terminal nuclear localization sequence (NLS) of NF-κβ. In response to various inflammatory nuclei, the α-, β-, and γ-subunits aggregate to form the IκB kinase (IKK) complex, resulting in phosphorylation of the α-subunit and release of NF-κβ for translocation to the nucleus.284 The equilibrium controlling this phosphorylation event is influenced by a number of kinases, including phosphoinositide 3-kinase (PI3K); MAP-kinases; and protein kinases A (PKA), B (known as Akt), and C (PKC).283 Dysregulation of NF-κβ-mediated events has been implicated in the pathogenesis of cancer, arthritis, inflammation, neurodegenerative diseases, and heart disease. Kim and co-workers demonstrated that 6 was able to downregulate iNOS and COX-2 expression by interfering with PI3K/Akt-mediated release of NF-κβ from the IKK complex. Furthermore, these researchers showed that heme oxygenase-1 (HO-1) expression was upregulated by 6, which in turn inhibits NO and prostaglandin production. HO-1 catalyzes the degradation of heme to biliverdin, carbon monoxide, and iron, and is involved in cellular redox homeostasis. As HO-1 is not constitutively expressed, its expression is induced by a variety of oxidative stimuli and is regulated by nuclear transcription factor erythroid 2-related factor 2 (Nrf2). In this study, α-Viniferin significantly increased the DNA-binding activity of Nrf2 in a dose-dependent manner and increased the expression of Nrf2 in the nuclear compartment.
8.2.2. Vaticanol C: A Cytotoxic Resveratrol Tetramer
In 2002, Ito and co-workers isolated a resveratrol tetramer that they named vaticanol C (94) from the Dipterocarpaceous plant, Vatica rassak.271,285 In this initial report, it was found that 94 induced apoptosis in three colon cancer cell lines, with observable morphological changes such as nuclear condensation and fragmentation, as well as DNA ladder formation. In a series of follow-up publications, these groups were able to elucidate several of the mechanisms contributing to the induction of apoptosis in a variety of cancer cell lines by 94. A family of cysteine-dependent aspartate-directed proteases (Caspases) are known to play essential roles in the initiation (initiator caspases) and execution (effector caspases) of programmed cell death. Western blot analyses revealed that 94 activated the effector caspases-3, but not initiator caspase-8, although it was later found that 94 does activate caspase-8 in mammary carcinoma cells BJMC 3879.286 It is known that the effector phase of apoptosis is largely dictated by upstream events at the mitochondria, where mitochondrial membrane permeabilization, loss of potential, and release of cytochrome c are triggered via the action of the permeability transition pore complex (PTPC). As such, these researchers examined the influence of vaticanol C (94) on these mitochondrial processes. It was shown that the amount of released cytochrome c following treatment with 94 increased time-dependently, and that the mitochondrial membrane potential was markedly decreased after 12 h. This is interesting as ε-viniferin (7) and hopeaphenol (9) have each demonstrated the ability to prevent loss of mitochondrial membrane potential, while vitisin A (118) showed similar activity to vaticanol C (94).287,288 Additionally, the initiator caspase-9, which is activated through binding with the apoptotic protease activating factor-(APAF)-1/cytochrome c complex, was indeed found to be activated concurrently with the release of cytochrome c in the cells treated with 94. These events can be prevented by the antiapoptotic regulatory protein Bcl-2, and it was found that growth suppression by 94 was significantly reduced in Bcl-2 overexpressant clones. Notably, this preventive effect was not observed for the anticancer agent taxol. Collectively, these results seem to suggest that 94 may be affecting the mitochondrial membrane proteins that constitute the PTPC, although no direct evidence for this was presented.
In a subsequent study on the apoptotic effects of vaticanol C (94) in the HL-60 human leukemia cell line, Ohguchi and co-workers discovered an additional signal transduction pathway controlling cell survival that is affected by this molecule.289 The Ras-Raf-MEK-ERK pathway290 and the PI3K/Akt pathway291 are additional kinase-mediated cascades that couple signals cell surface receptors to transcription factors and that play important roles in regulating apoptotic cellular events. In this study, it was found that levels of phosphorylated MEK, ERK, and Akt were significantly reduced in cells treated with 94, but only at fairly high concentrations (10 μM). These effects were coupled with a significant loss of Bcl-2-associated death promoter (Bad) phosphorylation. Bad is a pro-apoptotic member of the Bcl-2 protein family, which upon phosphorylation by Akt releases the pro-survival proteins Bcl-xL and Bcl-2 that it normally sequesters through complexation. The phosphorylated Bad then binds to the 14-3-3 protein, while Bcl-xL and Bcl-2 prevent cytochrome c release from the mitochondrial membrane by inhibiting pore formation. Thus, it was suggested that vaticanol C (94) may induce apoptosis via a mechanism involving prevention of Bad-phosphorylation and therefore inhibition of pro-survival signaling pathways.
Recently, Inoue and co-workers discovered that vaticanol C (94) activates peroxisome proliferator-activated receptor (PPAR)-α and β/δ in vitro and in vivo.292 PPARs are a family of nuclear receptor proteins that function as ligand-dependent transcription factors and that play various roles in lipid and carbohydrate metabolism as well as cell differentiation and development. Each of the PPARs forms heterodimers with the 9-cis-retinoic acid receptor (RXR) prior to binding consensus response elements in the promoter regions of the genes whose expression they regulate.293 In this study, it was found that 94 activates PPARα and β/δ in cell-based reporter assays, which was confirmed by induced expression of PPAR-responsive genes in wild-type, but not PPARα-knockout mice. Thus, 94 has potential in prevention of lifestyle-related diseases, as PPARα is currently targeted by marketed drugs for hyperlipidemia.
8.3. Modulation of Enzyme Activity by Resveratrol Natural Products
In addition to the cytotoxicity, anti-inflammatory, and other properties described above, a large number of studies have been performed to determine the ability of various resveratrol oligomers to inhibit specific enzymes. Selected examples of these biological activities are outlined in Table 7.
Table 7. Selected Studies of Enzyme Inhibition by Resveratrol Oligomers.
| target enzymes and pathological implications | active compds; refs | IC50 (μM) |
|---|---|---|
| Cytochrome P 450 (CYP) | ||
| heme-containing superfamily of oxidase enzymes responsible for the metabolism of both endogenous and exogenous substrates; these are the main enzymes involved in drug metabolism | α-viniferin; (294) | 0.9 |
| suffruticosol B; (263) | N/A | |
| ε-viniferin; (295) | N/A | |
| Cyclooxygenase (COX)-1 and -2 | ||
| key enzymes responsible for the conversion of arachidonic acid to prostaglandins; implicated in pain, fever, inflammation and tumorigenesis. | δ-viniferin-glucopyranoside; 296 | 3.7 |
| α-viniferin; (277, 278, 283) | 4.9 | |
| Inducible Nitric Oxide Synthase (iNOS) | ||
| transcription of iNOS gene is controlled by a variety of inflammatory mediators such as lipopolyssacharides (LPS) or cytokines; nitric oxide (NO) plays an important role in cellular signaling and iNOS has been implicated in autoimmune and inflammatory processes | vitisin A; (297, 298) | 3.9 |
| vitisin B; (297) | 4.7 | |
| ε-viniferin; (261, 299) | N/A | |
| α-viniferin; (283, 279) | N/A | |
| Protein Kinase C (PKC) | ||
| phosphorylating enzymes with key roles in signal transduction such as regulating transcription, membrane structure, immune response, and cell growth; overexpression implicated in cancer, diabetes, cardiovascular disease, and Alzheimer’s disease, among others | α-viniferin; (300, 301) | 62.5 |
| kobophenol A; (300) | 52.0 | |
| miyabenol C; (273, 300) | 27.5 | |
| Sphingosine Kinase 1 (SK1) | ||
| lipid kinase that catalyzes the formation of sphingosine-1-phosphate, an important signaling molecule involved in vascular and immune regulation; SK1 has been implicated in cancer progression. | ampelopsin A; (256) | N/A |
| balanocarpol; (256) | N/A | |
| Acetylcholine Esterase (AChE) | ||
| catalyzes the hydrolysis of the neurotransmitter acetylcholine to acetate and choline at near-diffusion rates, a critical reaction for synaptic transmission; inhibition has been targeted for treatment of neurodegenerative diseases such as Alzheimer’s | α-viniferin; (302, 303) | 2.0 |
| vitisin A; (304) | 1.0 | |
| heyneanol A; (304) | 1.7 | |
| others; 86, 91, 121, 195, 277, 304 | ||
| HIV-1 Integrase/MOS-1 Transposase | ||
| polynucleotidyl transferases catalyze the excision, transfer, and reintegration of DNA fragments, an essential process for genome evolution; retroviruses such as HIV rely on integrases for the incorporation of viral DNA into the host genome | hopeaphenol; (305) | 7.0 |
| pallidol; (305) | 9.0 | |
| Xanthine Oxidase (XO) | ||
| oxidase enzyme containing a molybdopterin-ligated molybdenum oxo- that catalyzes the oxidation of hypoxanthine to xanthine and further to uric acid; XO also produces ROS and has been targeted for chemotherapeutic applications in cardiovascular diseases | vaticanol A; (91) | 23.3 |
| Topoisomerase II (Topo II) | ||
| modifies DNA by breaking and religating each of the strands of the phosphodiester backbone, resulting in strain-releasing relaxation of supercoiled DNA and catenation/decatenation of circular DNA, both important processes for cell reproduction (cancer)306 | hemsleyanol C; (316) | 1.0 |
| α-viniferin-glucopyranoside; 316 | 4.0 | |
| nepalensinol B; (128) | 0.022 | |
| nepalensinol F; (307) | 5.5 | |
| Multidrug Resistance-Associated Protein 1 (MRP1) | ||
| functions as a transporter of various endogenous substrates such as folates, glutathione disulfide (GSSH), glutathionated-, glucoronidated- or sulfated-steroids, leukotrienes, prostaglandins and bile salts, as well as exogenous xenobiotics including a wide range of therapeutic agents308 | (+)-α-viniferin; (309) | 0.8 |
| sophorastilbene A; (309) | 3.1 |
8.4. Resveratrol and its Oligomers as Antioxidants
Oxidative damage not only to membrane lipids but also to nucleic acids and enzymes can have deleterious effects on various processes that are fundamental to cell function and survival. Due to the polyphenolic structure of resveratrol and its oligomers, the biological activities of these compounds are often ascribed to their radical-trapping antioxidant activity.8 However, these conclusions are lacking adequate experimental support, as investigators often assay only the position of their thermodynamic equilibrium with oxidants in solution, and not whether the chemistry is kinetically competitive under biologically relevant conditions.11,310 As a result, it has been inaccurately asserted by many that certain resveratrol oligomers are or are not “antioxidants” on the basis of their ability to scavenge 2,2-diphenyl-1-picrylhydrazyl (DPPH) or other radicals in solution phase assays. These experiments often ignore not only the rates at which the analytes are acting as “radical-trapping antioxidants”, but also the fact that antioxidant behavior is not limited to radical scavenging ability.321 Many compounds can influence the redox environment within the cell through other mechanisms, such as induction of the expression of endogenous detoxifying and antioxidant defense enzymes and proteins, including glutathione, glutathione peroxidase, and heme oxygenase.
Despite the hype surrounding the antioxidant properties of resveratrol, it was not until 2003 that someone performed a kinetic study on its radical-trapping ability. Valgimigli and co-workers determined the rate of peroxyl radical-trapping by resveratrol in homogeneous chlorobenzene solution to be 2.0 × 105 M–1 s–1,311 which is approximately 16 times lower than that of α-tocopherol, (α-TOH, kinh = 3.2 × 106 M–1 s–1), nature’s ubiquitous radical-trapping antioxidant.312 Recently, Pratt and co-workers systematically evaluated the radical trapping efficacies of resveratrol dimers quadrangularin A (17) and pallidol (18) along with their 2,6-di-tert-butylated analogues in homogeneous organic solution, lipid bilayers, and cell culture.180 Similar to resveratrol, the dimeric natural products and their analogues did not react with peroxyl radicals in homogeneous solution at a rate that was competitive with α-TOH. However, in lipid bilayers tBu2-resveratrol 146 and tBu2-quadrangularin A 148 were each found to be highly effective; in fact, these compounds were superior to α-TOH itself (10- to 16-fold for (146) and 4-fold for (148), depending on whether the oxidation was mediated by lipophilic or aqueous peroxyl radicals). Finally, the effective concentrations for inhibition of lipid peroxidation in cultured human erythroblasts were determined. Again, the natural products were far less effective than their tert-butylated analogues, and 146 was shown to have an EC50 one-third that of α-TOH. Not only are the natural products expected to be less soluble than the tert-butylated analogues in the lipid milieu, but the reactivity of the phenolic O–H can also be reduced due to its H-bonding interaction with phosphatidylcholine moieties and/or water at the lipid/aqueous interface, an interaction that is diminished by the bulky tert-butyl moieties. Now that systematic studies of these types of compounds have extended past homogeneous solution into lipid bilayers and cell culture, they challenge the popular belief that the biological activities of the natural products derive from their ability to trap radicals. Curiously, although tBu2-pallidol 153 was inefficient at preventing lipid peroxidation in bilayers, it was surprisingly potent (EC50 = 0.39 μM) in cell culture, suggesting that it likely operates through a mechanism other than direct radical trapping. To account for this, Pratt and co-workers postulated that 153 may act as a modulator of the Kelch-like ECH-associated protein 1 (Keap1)-nuclear factor erythroid 2-related factor 2 (Nrf2)-antioxidant response element (ARE) pathway.313,314
The Keap1-Nrf2-ARE pathway represents one of the central mechanisms by which cells combat oxidative stress and xenobiotic-induced damage. Under normal conditions, Keap1 binds to and represses the transcription factor Nrf2 in the cytoplasm by promoting its ubiquitination by the appended Cullin (CuI)-3-based ubiquitin E3 ligase and subsequent proteasomal degradation.313,314 Oxidants and electrophiles present during oxidative stress cause modification of cysteine residues in the Keap1 backbone, inducing conformational changes that lead to the release of Nrf2. This basic leucine zipper (bZIP) transcription factor then translocates to the nucleus where it heterodimerizes with small Maf proteins (also bZIP transcription factors) and activates genes containing antioxidant response elements (ARE) in their promoter regions, including those for glutathione and other antioxidant proteins, drug metabolizing enzymes and transporters, and other transcription factors. It is known that cancer cells hijack this signal transduction pathway to support their malignant growth, and dysregulation has also been associated with the pathogenesis of multiple sclerosis (MS) and diabetic nephropathy.313,314
Recently, Pan and co-workers evaluated the ability of three resveratrol dimers, quadrangularin A (17), pallidol (18), and parthenocissin A (61), to activate Nrf2 in vitro.315 The breast cancer cell line, MCF-7, was transfected with an ARE-luciferase reporter plasmid containing 10 copies of the ARE (5′-GTGACAAAGCA-3′), and the cells were exposed to resveratrol and the three dimers (7.5–30 μM in 0.1% v/v DMSO as vehicle). Resveratrol induced ARE-luciferase activity dose-dependently, with a 2.5-fold increase at 7.5 μM. Parthenocissin A (61) and quadrangularin A (17) were each found to have no effect at all doses, while pallidol (18) showed a 3-fold induction at the highest dose (30 μM). This finding lends credence to the supposition by Pratt and co-workers that the tert-butylated pallidol analogue 153 may be acting on this signal transduction pathway in human erythroblasts, especially when considering the improved cell permeability that the bulky substituents are expected to impart. Further studies are needed to determine whether other oligomers and their analogues exhibit more potent Nrf2 activation, and if certain structural features such as a C2-axis of symmetry correlate with activity.316
9. Concluding Remarks
The broad spectrum biological activities of resveratrol and its oligomers, as evidenced by the range of cell lines against which they were found to be cytotoxic and the multitude of enzymes which they were demonstrated to either inhibit or downregulate, speaks to the pharmacological potential of this class of natural products. At the same time, such diverse biological activity is a bit disconcerting, as it implies that these molecules are perhaps nonspecific with respect to their pharmacodynamics. In spite of this, the moderate potency (low micromolar) observed in the majority of the in vitro assays delineated above and the historically low bioavailability of chemopreventive and chemotherapeutic polyphenols render them relatively nontoxic.317,318 Taken together, this data raises concerns about whether the concentrations required for efficacy are attainable in vivo. While the aglycones of polyphenols can be absorbed fairly efficiently in the small intestine, many of the naturally occurring glycoconjugates are not, although the aglycones typically undergo rapid metabolic glucuronidation and sulfation in the small intestine, liver, and kidneys. However, in order for the hydrophilic metabolites to be excreted, cellular efflux transporters must operate in concert with these phase II bioconjugation pathways, a process which could have profound effects on the bioavailability and apparent half-life of the polyphenols.319
While the resveratrol oligomers possess only micromolar potencies in the in vitro assays in which they have been examined, this should not justify their uncritical dismissal as candidate scaffolds for drug development. Indeed, the “nanomolar rule” (the selective development of compounds with nanomolar in vitro potency) has recently been challenged,320 and it is imperative that we not allow advances in predictive technologies to engender a myopic approach to drug discovery. To better understand the behavior of these molecules in biological systems and enable the development of more potent analogs, systematic structure–activity relationship (SAR) studies must be performed. Limited commentary on the structural features correlated to bioactivity exists in the literature, and typically consists of retrospective conjecture based on observed IC50 values for a given set of isolated natural products. It would be beneficial for researchers to perform these studies on analogues that have been rationally designed, as this would enable a more generalized understanding of which physical properties can be exploited in the development of more potent congeners. To date, progress in this area has been hampered by existing synthetic technologies, which have largely been incapable of providing materials in sufficient quantities for their systematic evaluation, although several strategies have recently been reported that hold promise in this regard (see sections 6 and 7). While resveratrol oligomers are a relatively new target in the area of complex molecule synthesis, they offer unique challenges and therefore opportunities for valuable contributions to synthetic chemistry and an improved biological understanding of one of the most widely distributed groups of natural products in the plant kingdom.
Acknowledgments
The authors gratefully acknowledge Dr. James Douglas, Dr. Elizabeth Swift, and Mr. Joel Beatty for their useful comments and suggestions during the preparation of this paper.
Biographies

Mitchell H. Keylor received a B.S. in Biochemistry from Stonehill College in 2011 and an M.A. in Chemistry from Boston University in 2013. He is currently a Ph.D. candidate at the University of Michigan under the direction of Professor Corey Stephenson, where he is pursuing the biomimetic total synthesis of resveratrol-derived oligomeric natural products.

Bryan S. Matsuura received his B.A. from Boston University in 2009 and started his Ph.D. studies in the same year. He is currently a Ph.D. candidate under the supervision of Professor Corey R. J. Stephenson, studying the biomimetic synthesis of the resveratrol oligomers and the degradation of biomass derived polymers using visible-light photoredox catalysis.

Corey R. J. Stephenson completed his Ph.D. at the University of Pittsburgh under Professor Peter Wipf followed by postdoctoral research with Professor Erick M. Carreira at ETH Zürich. In 2007, he joined the Department of Chemistry at Boston University as an Assistant Professor, and in 2013, he was promoted to Associate Professor and moved to the University of Michigan. His research interests are broadly focused on complex molecule synthesis, biomass degradation, and catalysis.
Author Contributions
† M.H.K. and B.S.M. contributed equally.
Financial support for this research from the NIH-NIGMS (GM096129) and the University of Michigan is gratefully acknowledged.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Kinsella K. G. Changes in Life Expectancy 1900–1990. Am. J. Clin. Nutr. 1992, 55, 1196S–1202S. [DOI] [PubMed] [Google Scholar]
- Soleas G. J.; Diamandis E. P.; Goldberg D. M. Resveratrol: A Molecule Whose Time Has Come? And Gone?. Clin. Biochem. 1997, 30, 91–113. [DOI] [PubMed] [Google Scholar]
- Rimm E. B.; Klatsky A.; Grobbee D.; Stampfer M. J. Review Of Moderate Alcohol Consumption And Reduced Risk Of Coronary Heart Disease: Is The Effect Due To Beer, Wine, Or Spirits?. BMJ [Br. Med. J.] 1996, 312, 731–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siemann E. H.; Creasy L. L. Concentration of the Phytoalexin Resveratrol in Wine. Am. J. Enol. Vitic. 1992, 43, 49–52. [Google Scholar]
- Nonomura S.; Kanagawa H.; Makimoto A. Chemical Constituents of Polygonaceous Plants. I Studies on the Components of Ko-jô-kon (Polygonum cuspidatum SIEB. et ZUCC.). Yakugaku Zasshi 1963, 83, 988–990. [PubMed] [Google Scholar]
- National Research Council Committee on Diet and Health. Diet and Health: Implications for Reducing Chronic Disease Risk; The National Academies Press: Washington, DC, 1989. [PubMed] [Google Scholar]
- Renaud S.; de Lorgeril M. Wine, Alcohol, Platelets, and the French Paradox for Coronary Heart Disease. Lancet 1992, 339, 1523–1526. [DOI] [PubMed] [Google Scholar]
- Frankel E. N.; German J. B.; Kinsella J. E.; Parks E.; Kanner J. Inhibition of Oxidation of Human Low-Density Lipoprotein by Phenolic Substances in Red Wine. Lancet 1993, 341, 454–457. [DOI] [PubMed] [Google Scholar]
- Law M.; Wald N. Why Heart Disease Mortality is Low in France: the Time Lag Explanation. BMJ [Br. Med. J.] 1999, 318, 1471–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs F. D.; Chambless L. E. Is the Cardioprotective Effect of Alcohol Real?. Alcohol 2007, 41, 399–402. [DOI] [PubMed] [Google Scholar]
- He S.; Yan X. From Resveratrol to Its Derivatives: New Sources of Natural Antioxidant. Curr. Med. Chem. 2013, 20, 1005–1017. [PubMed] [Google Scholar]
- Jang M.; Cai L.; Udeani G. O.; Slowing K. V.; Thomas C. F.; Beecher C. W. W.; Fong H. H. S.; Farnsworth N. R.; Kinghorn A. D.; Mehta R. G.; Moon R. C.; Pezzuto J. M. Cancer Chemopreventive Activity of Resveratrol, a Natural Product Derived from Grapes. Science 1997, 275, 218–220. [DOI] [PubMed] [Google Scholar]
- Banks A. S.; Kon N.; Knight C.; Matsumoto M.; Gutiérrez-Juárez R.; Rossetti L.; Gu W.; Accili D. SirT1 Gain of Function Increases Energy Efficiency and Prevents Diabetes in Mice. Cell Metab. 2008, 8, 333–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne J. C.; Lambert P. D.; Schenk S.; Carney D. P.; Smith J. J.; Gagne D. J.; Jin L.; Boss O.; Perni R. B.; Vu C. B.; Bemis J. E.; Xie R.; Disch J. S.; Ng P. Y.; Nunes J. J.; Lynch A. V.; Yang H.; Galonek H.; Israelian K.; Choy W.; Iffland A.; Lavu S.; Medvedik O.; Sinclair D. A.; Olefsky J. M.; Jirousek M. R.; Elliott P. J.; Westphal C. H. Small Molecule Activators of SIRT1 as Therapeutics for the Treatment of Type 2 Diabetes. Nature 2007, 450, 712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankel E. N.; Waterhouse A. L.; Kinsella J. E. Inhibition of Human LDL Oxidation by Resveratrol. Lancet 1993, 341, 1103–1104. [DOI] [PubMed] [Google Scholar]
- Baur J. A.; Pearson K. J.; Price N. L.; Jamieson H. A.; Lerin C.; Kalra A.; Prabhu V. V.; Allard J. S.; Lopez-Lluch G.; Lewis K.; Pistell P. J.; Poosala S.; Becker K. G.; Boss O.; Gwinn D.; Wang M.; Ramaswamy S.; Fishbein K. W.; Spencer R. G.; Lakatta E. G.; Le Couteur D.; Shaw R. J.; Navas P.; Puigserver P.; Ingram D. K.; de Cabo R.; Sinclair D. A. Resveratrol Improves Health and Survival of Mice on a High-Calorie Diet. Nature 2006, 444, 337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baur J. A.; Sinclair D. A. Therapeutic Potential of Resveratrol: The in Vivo Evidence. Nat. Rev. Drug Discovery 2006, 5, 493–506. [DOI] [PubMed] [Google Scholar]
- Aggarwal B. B.; Bhardwaj A.; Aggarwal R. S.; Seeram N. P.; Shishodia S.; Takada Y. Role of Resveratrol in Prevention and Therapy of Cancer: Preclinical and Clinical Studies. Anticancer Res. 2004, 24, 2783–2840. [PubMed] [Google Scholar]
- Frémont L. Biological Effects of Resveratrol. Life Sci. 2000, 66, 663–673. [DOI] [PubMed] [Google Scholar]
- Harikumar K. B.; Aggarwal B. B. Resveratrol: A Multitargeted Agent for Age-Associated Chronic Diseases. Cell Cycle 2008, 7, 1020–1035. [DOI] [PubMed] [Google Scholar]
- Vang O.; Ahmad N.; Baile C. A.; Baur J. A.; Brown K.; Csiszar A.; Das D. K.; Delmas D.; Gottfried C.; Lin H.-Y.; Ma Q.-Y.; Mukhopadhyay P.; Nalini N.; Pezzuto J. M.; Richard T.; Shukla Y.; Surh Y.-J.; Szekeres T.; Szkudelski T.; Walle T.; Wu J. M. What Is New for an Old Molecule? Systematic Review and Recommendations on the Use of Resveratrol. PLoS One 2011, 6, e19881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manach C.; Scalbert A.; Morand C.; Rémésy C.; Jiménez L. Polyphenols: Food Sources and Bioavailability. Am. J. Clin. Nutr. 2004, 79, 727–747. [DOI] [PubMed] [Google Scholar]
- Rice-Evans C. A.; Miller N. J.; Paganga G. Structure-Antioxidant Activity Relationships of Flavonoids and Phenolic Acids. Free Radical Biol. Med. 1996, 20, 933–956. [DOI] [PubMed] [Google Scholar]
- Vogt T. Phenylpropanoid Biosynthesis. Mol. Plant 2010, 3, 2–20. [DOI] [PubMed] [Google Scholar]
- Schöppner A.; Kindl H. Purification and Properties of a Stilbene Synthase from Induced Cell Suspension Cultures of Peanut. J. Biol. Chem. 1984, 259, 6806–6811. [PubMed] [Google Scholar]
- Vannozzi A.; Dry I. B.; Fasoli M.; Zenoni S.; Lucchin M. Genome-Wide Analysis of the Grapevine Stilbene Synthase Multigenic Family: Genomic Organization and Expression Profiles upon Biotic and Abiotic Stresses. BMC Plant Biol. 2012, 12, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorham J.; J. Coughlan S. Inhibition of Photosynthesis by Stilbenoids. Phytochemistry 1980, 19, 2059–2064. [Google Scholar]
- Lopez-Lluch G.; Santa Cruz-Calvo S.; Navas P. Resveratrol in Cancer: Cellular and Mitochondrial Consequences of Proton Transport Inhibition. Curr. Pharm. Des. 2012, 18, 1338–1344. [DOI] [PubMed] [Google Scholar]
- Flamini R.; Mattivi F.; Rosso M. D.; Arapitsas P.; Bavaresco L. Advanced Knowledge of Three Important Classes of Grape Phenolics: Anthocyanins, Stilbenes and Flavonols. Int. J. Mol. Sci. 2013, 14, 19651–19669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regev-Shoshani G.; Shoseyov O.; Bilkis I.; Kerem Z. Glycosylation of Resveratrol Protects It from Enzymic Oxidation. Biochem. J. 2003, 374, 157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langcake P.; Pryce R. J. The Production of Resveratrol by Vitis Vinifera and Other Members of the Vitaceae as a Response to Infection or Injury. Physiol. Plant Pathol. 1976, 9, 77–86. [Google Scholar]
- Langcake P.; McCarthy W. V. The Relationship of Resveratrol Production to Infection of Grapevine Leaves by Botrytis Cinerea. Vitis 1979, 18, 244–253. [Google Scholar]
- Langcake P. Disease Resistance of Vitis Spp. and the Production of the Stress Metabolites Resveratrol, ε-Viniferin, α-Viniferin and Pterostilbene. Physiol. Plant Pathol. 1981, 18, 213–226. [Google Scholar]
- Coggon P.; King T. J.; Wallwork S. C. The Structure of Hopeaphenol. Chem. Commun. (London) 1966, 439–440. [Google Scholar]
- Aiba C. J.; Corrêa R. G. C.; Gottlieb O. R. Natural Occurrence of Erdtman’s Dehydrodiisoeugenol. Phytochemistry 1973, 12, 1163–1164. [Google Scholar]
- Langcake P.; Pryce R. J. Oxidative Dimerisation of 4-Hydroxystilbenes in Vitro: Production of a Grapevine Phytoalexin Mimic. J. Chem. Soc., Chem. Commun. 1977, 208–210. [Google Scholar]
- Langcake P.; Pryce R. J. A New Class of Phytoalexins from Grapevines. Experientia 1977, 33, 151–152. [DOI] [PubMed] [Google Scholar]
- Cichewicz R. H.; Kouzi S. A.; Hamann M. T. Dimerization of Resveratrol by the Grapevine Pathogen Botrytis Cinerea. J. Nat. Prod. 2000, 63, 29–33. [DOI] [PubMed] [Google Scholar]
- Coggon P.; McPhail A. T.; Wallwork S. C. Structure of Hopeaphenol: X-Ray Analysis of the Benzene Solvate of Dibromodeca-O-Methylhopeaphenol. J. Chem. Soc. B 1970, 884–897. [Google Scholar]
- Coggon P.; Janes N. F.; King F. E.; King T. J.; Molyneux R. J.; Morgan J. W. W.; Sellars K. 61. Hopeaphenol, an Extractive of the Heartwood of Hopea Odorata and Balanocarpus Heimii. J. Chem. Soc. 1965, 406–409. [Google Scholar]
- Xiao K.; Zhang H.-J.; Xuan L.-J.; Zhang J.; Xu Y.-M.; Bai D.-L. In Studies in Natural Products Chemistry; Bioactive Natural Products (Part N); Atta-ur-Rahman, Ed.; Elsevier: New York, 2008; Vol. 34, pp 453–646. [Google Scholar]
- Cichewicz R. H.; Kouzi S. A. In Studies in Natural Products Chemistry (Part G); Atta-ur-Rahman, Ed.; Bioactive Natural Products; Elsevier, 2002; Vol. 26, pp 507–579. [Google Scholar]
- Luo H.-F.; Zhang L.-P.; Hu C.-Q. Five Novel Oligostilbenes from the Roots of Caragana Sinica. Tetrahedron 2001, 57, 4849–4854. [Google Scholar]
- Ito T.; Abe N.; Oyama M.; Iinuma M. Oligostilbenoids from Dipterocarpaceaeous Plants: A New Resveratrol Tetramer from Vateria Indica and the Revised Structure of Isohopeaphenol. Helv. Chim. Acta 2008, 91, 1989–1998. [Google Scholar]
- Sotheeswaran S.; Champika Diyasena M. N.; Leslie Gunatilaka A. A.; Bokel M.; Kraus W. Further Evidence for the Structure of Vaticaffinol and a Revision of Its Stereochemistry. Phytochemistry 1987, 26, 1505–1507. [Google Scholar]
- Moscowitz A.; Mislow K.; Glass M. A. W.; Djerassi C. Optical Rotatory Dispersion Associated with Dissymmetric Non-Conjugated Chromophores. An Extension of the Octant Rule1–3. J. Am. Chem. Soc. 1962, 84, 1945–1955. [Google Scholar]
- Snatzke G. Semiempirical Rules in Circular Dichroism of Natural Products. Pure Appl. Chem. 1979, 51, 769–785. [Google Scholar]
- Berova N.; Ellestad G.; Nakanishi K.; Harada N. In Bioactive Compounds from Natural Sources, 2nd ed.; CRC Press: Boca Raton, FL, 2011; pp 133–166. [Google Scholar]
- Kurihara H.; Kawabata J.; Ichdcawa S.; Mizutani J. (−)-εViniferin and Related Oligo-Stilhenes from Carex Pumila Thunb. (Cyperaceae). Agric. Biol. Chem. 1990, 54, 1097–1099. [Google Scholar]
- Takaya Y.; Yan K.-X.; Terashima K.; Ito J.; Niwa M. Chemical Determination of the Absolute Structures of Resveratrol Dimers, Ampelopsins A, B, D and F. Tetrahedron 2002, 58, 7259–7265. [Google Scholar]
- Niwa M.; He Y.-H.; Takaya Y.; Terashima K. Determination of Absolute Structure of (+)-Davidiol A. Heterocycles 2006, 68, 93–100. [Google Scholar]
- Jiang L.; He S.; Sun C.; Pan Y. Selective 1O2 Quenchers, Oligostilbenes, from Vitis Wilsonae: Structural Identification and Biogenetic Relationship. Phytochemistry 2012, 77, 294–303. [DOI] [PubMed] [Google Scholar]
- Lins A. P.; Yoshida M.; Gottlieb O. R.; Gottlieb H. E.; Kubitzki K. Gnetins in Welwitschia. Bull. Soc. Chim. Belg. 1986, 95, 737–748. [Google Scholar]
- Gottlieb O. R.; Mourão J. C.; Yoshida M.; Mascarenhas Y. P.; Rodrigues M.; Rosenstein R. D.; Tomita K. Absolute Configuration of the Benzofuranoid Neolignans. Phytochemistry 1977, 16, 1003–1006. [Google Scholar]
- Takaoka M. Of the phenolic substances of white hellebore (Veratrum grandiflorum Loes. Fil.). J. Fac. Sci. Hokkaido Univ. 1940, 3, 1–16. [Google Scholar]
- Kawabata J.; Fukushi E.; Hara M.; Mizutani J. Detection of Connectivity between Equivalent Carbons in a C2Molecule Using Isotopomeric Asymmetry: Identification of Hopeaphenol in Carex Pumila. Magn. Reson. Chem. 1992, 30, 6–10. [Google Scholar]
- Ingham J. L. 3,5,4′-Trihydroxystilbene as a Phytoalexin from Groundnuts (Arachis Hypogaea). Phytochemistry 1976, 15, 1791–1793. [Google Scholar]
- Pryce R. J.; Langcake P. α-Viniferin: An Antifungal Resveratrol Trimer from Grapevines. Phytochemistry 1977, 16, 1452–1454. [Google Scholar]
- Kitanaka S.; Ikezawa T.; Yasukawa K.; Yamanouchi S.; Takida M.; Sung H. K.; Kim I. H. (+)-α-Viniferin, an Anti-Inflammatory Compound from Caragana Chamlagu Root. Chem. Pharm. Bull. 1990, 38, 432–435. [DOI] [PubMed] [Google Scholar]
- Sultanbawa M. U. S.; Surendrakumar S.; Wazeer M. I. M.; Bladon P. Novel Resveratrol Tetramer, Vaticaffinol, from Vatica Affinis Thw. (dipterocarpaceae). J. Chem. Soc., Chem. Commun. 1981, 1204–1206. [Google Scholar]
- Lins A. P.; Ribeiro M. N. D. S.; Gottlieb O. R.; Gottlieb H. E. Gnetins: Resveratrol Oligomers From Gnetum Species. J. Nat. Prod. 1982, 45, 754–761. [Google Scholar]
- Huang Y.-L.; Tsai W.-J.; Shen C.-C.; Chen C.-C. Resveratrol Derivatives from the Roots of Vitis Thunbergii. J. Nat. Prod. 2005, 68, 217–220. [DOI] [PubMed] [Google Scholar]
- Kawabata J.; Mishima M.; Kurihara H.; Mizutani J.; Kobophenol B. A Tetrastilbene from Carex Pumila. Phytochemistry 1991, 30, 645–647. [Google Scholar]
- Ito T.; Furusawa M.; Iliya I.; Tanaka T.; Nakaya K.; Sawa R.; Kubota Y.; Takahashi Y.; Riswan S.; Iinuma M. Rotational Isomerism of a Resveratrol Tetramer, Shoreaketone, in Shorea Uliginosa. Tetrahedron Lett. 2005, 46, 3111–3114. [Google Scholar]
- Ito T.; Oyama M.; Sajiki H.; Sawa R.; Takahashi Y.; Iinuma M. Absolute Structure of Shoreaketone: A Rotational Isomeric Resveratrol Tetramer in Dipterocarpaceaeous Plants. Tetrahedron 2012, 68, 2950–2960. [Google Scholar]
- Ito T.; Tanaka T.; Nakaya K.; Iinuma M.; Takahashi Y.; Naganawa H.; Ohyama M.; Nakanishi Y.; Bastow K. F.; Lee K.-H. A Novel Bridged Stilbenoid Trimer and Four Highly Condensed Stilbenoid Oligomers in Vatica Rassak. Tetrahedron 2001, 57, 7309–7321. [Google Scholar]
- Ito T.; Tanaka T.; Iinuma M.; Nakaya K.; Takahashi Y.; Sawa R.; Naganawa H.; Chelladurai V. Two New Oligostilbenes with Dihydrobenzofuran from the Stem Bark of Vateria Indica. Tetrahedron 2003, 59, 1255–1264. [Google Scholar]
- Rivière C.; Pawlus A. D.; Mérillon J.-M. Natural Stilbenoids: Distribution in the Plant Kingdom and Chemotaxonomic Interest in Vitaceae. Nat. Prod. Rep. 2012, 29, 1317–1333. [DOI] [PubMed] [Google Scholar]
- Oshima Y.; Ueno Y.; Hisamichi K.; Takeshita M. Ampelopsins F and G, Novel Bridged Plant Oligostilbenes from Ampelopsis Brevipedunculata Var. Hancei Roots (vitaceae). Tetrahedron 1993, 49, 5801–5804. [Google Scholar]
- Ducrot P.-H.; Kollmann A.; Bala A. E.; Majira A.; Kerhoas L.; Delorme R.; Einhorn J.; Cyphostemmins A.-B. Two New Antifungal Oligostilbenes from Cyphostemma Crotalarioides (Vitaceae). Tetrahedron Lett. 1998, 39, 9655–9658. [Google Scholar]
- Adesanya S. A.; Nia R.; Martin M.-T.; Boukamcha N.; Montagnac A.; Païs M. Stilbene Derivatives from Cissus Quadrangularis. J. Nat. Prod. 1999, 62, 1694–1695. [Google Scholar]
- Khan M. A.; Nabi S. G.; Prakash S.; Zaman A. Pallidol, a Resveratrol Dimer from Cissus Pallida. Phytochemistry 1986, 25, 1945–1948. [Google Scholar]
- Huang K.-S.; Lin M.; Cheng G.-F. Anti-Inflammatory Tetramers of Resveratrol from the Roots of Vitis Amurensis and the Conformations of the Seven-Membered Ring in Some Oligostilbenes. Phytochemistry 2001, 58, 357–362. [DOI] [PubMed] [Google Scholar]
- Ito T.; Tanaka T.; Iinuma M.; Iliya I.; Nakaya K.; Ali Z.; Takahashi Y.; Sawa R.; Shirataki Y.; Murata J.; Darnaedi D. New Resveratrol Oligomers in the Stem Bark of Vatica Pauciflora. Tetrahedron 2003, 59, 5347–5363. [Google Scholar]
- Finefield J. M.; Sherman D. H.; Kreitman M.; Williams R. M. Enantiomeric Natural Products: Occurrence and Biogenesis. Angew. Chem., Int. Ed. 2012, 51, 4802–4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima Y.; Ueno Y. Ampelopsins D, E, H and Cis-Ampelopsin E, Oligostilbenes from Ampelopsis Brevipedunculata Var. Hancei Roots. Phytochemistry 1993, 33, 179–182. [Google Scholar]
- Fujii F.; He Y.-H.; Terashima K.; Takaya Y.; Niwa M. Three New Stilbeneoligomers from the Roots of Vitis vinifera ‘Kyohou’. Heterocycles 2005, 65, 2461–2469. [Google Scholar]
- Ito T.; Tanaka T.; Iinuma M.; Nakaya K.; Takahashi Y.; Sawa R.; Murata J.; Darnaedi D. Three New Resveratrol Oligomers from the Stem Bark of Vatica Pauciflora. J. Nat. Prod. 2004, 67, 932–937. [DOI] [PubMed] [Google Scholar]
- Ge H. M.; Xu C.; Wang X. T.; Huang B.; Tan R. X. Hopeanol: A Potent Cytotoxin with A Novel Skeleton from Hopea Exalata. Eur. J. Org. Chem. 2006, 2006, 5551–5554. [Google Scholar]
- Diyasena M. N. C.; Sotheeswaran S.; Surendrakumar S.; Balasubramanian S.; Bokel M.; Kraus W. Balanocarpol, a New Polyphenol from Balanocarpus Zeylanicus(trimen) and Hopea jucunda(Thw.)(Dipterocarpaceae). J. Chem. Soc., Perkin Trans. 1 1985, 1807–1809. [Google Scholar]
- Tanaka T.; Ito T.; Ido Y.; Son T.-K.; Nakaya K.; Iinuma M.; Ohyama M.; Chelladurai V. Stilbenoids in the Stem Bark of Hopea Parviflora. Phytochemistry 2000, 53, 1015–1019. [DOI] [PubMed] [Google Scholar]
- Oshima Y.; Ueno Y.; Hikino H.; Ling-Ling Y.; Kun-Ying Y. Ampelopsins A, B and C, New Oligostilbenes of Ampelopsis Brevipedunculata VAR Hancei. Tetrahedron 1990, 46, 5121–5126. [Google Scholar]
- Muhammad N.; Din L. B.; Sahidin I.; Hashim S. F.; Ibrahim N.; Zakaria Z.; Yaacob W. A. Acuminatol and Other Antioxidative Resveratrol Oligomers from the Stem Bark of Shorea Acuminata. Molecules 2012, 17, 9043–9055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber J.-F. F.; Wahab I. A.; Marzuki A.; Thomas N. F.; Kadir A. A.; Hadi A. H. A.; Awang K.; Latiff A. A.; Richomme P.; Delaunay J.; Heimiol A. a New Dimeric Stilbenoid from Neobalanocarpus Heimii. Tetrahedron Lett. 2001, 42, 4895–4897. [Google Scholar]
- Ge H.-M.; Yang W.-H.; Zhang J.; Tan R.-X. Antioxidant Oligostilbenoids from the Stem Wood of Hopea Hainanensis. J. Agric. Food Chem. 2009, 57, 5756–5761. [DOI] [PubMed] [Google Scholar]
- Ge H. M.; Zhu C. H.; Shi D. H.; Zhang L. D.; Xie D. Q.; Yang J.; Ng S. W.; Tan R. X. Hopeahainol A: An Acetylcholinesterase Inhibitor from Hopea Hainanensis. Chem.—Eur. J. 2008, 14, 376–381. [DOI] [PubMed] [Google Scholar]
- Liu J. Y.; Ye Y. H.; Wang L.; Shi D. H.; Tan R. X. New Resveratrol Oligomers from the Stem Bark of Hopea Hainanensis. Helv. Chim. Acta 2005, 88, 2910–2917. [Google Scholar]
- Dai J.-R.; Hallock Y. F.; Cardellina J. H.; Boyd M. R. HIV-Inhibitory and Cytotoxic Oligostilbenes from the Leaves of Hopea malibato. J. Nat. Prod. 1998, 61, 351–353. [DOI] [PubMed] [Google Scholar]
- Patra A.; Dey A. K.; Kundu A. B.; Saraswathy A.; Purushothaman K. K. Shoreaphenol, a Polyphenol from Shorea Robusta. Phytochemistry 1992, 31, 2561–2562. [Google Scholar]
- Tanaka T.; Ito T.; Ido Y.; Nakaya K.; Iinuma M.; Chelladurai V. Hopeafuran and a C-Glucosyl Resveratrol Isolated from Stem Wood of Hopea Utilis. Chem. Pharm. Bull. 2001, 49, 785–787. [DOI] [PubMed] [Google Scholar]
- Qin Y. H.; Zhang J.; Cui J. T.; Guo Z. K.; Jiang N.; Tan R. X.; Ge H. M. Oligostilbenes from Vatica Mangachapoi with Xanthine Oxidase and Acetylcholinesterase Inhibitory Activities. RSC Adv. 2011, 1, 135–141. [Google Scholar]
- Nicolaou K. C.; Wu T. R.; Kang Q.; Chen D. Y.-K. Total Synthesis of Hopeahainol A and Hopeanol. Angew. Chem., Int. Ed. 2009, 48, 3440–3443. [DOI] [PubMed] [Google Scholar]
- Tanaka T.; Ito T.; Nakaya K.; Iinuma M.; Takahashi Y.; Naganawa H.; Riswan S. Six New Heterocyclic Stilbene Oligomers from Stem Bark of Shorea Hemsleyana. Heterocycles 2001, 55, 729–740. [Google Scholar]
- Sahidin H. E.; Juliawaty L. D.; Syah Y. M.; Din L. B.; Ghisalberti E. L.; Latip J.; Said I. M.; Achmad S. A. Cytotoxic Properties of Oligostilbenoids from the Tree Barks of Hopea Dryobalanoides. Z. Naturforsch. 2005, 60, 723–727. [DOI] [PubMed] [Google Scholar]
- Huang K. A New Oligostilbene from the Roots of Vitis Amurensis. Chin. Chem. Lett. 1999, 10, 775–776. [Google Scholar]
- Gordon H. L.; Freeman S.; Hudlicky T. Stability Relationships in Bicyclic Ketones. Synlett 2005, 2911–2914. [Google Scholar]
- Ohyama M.; Tanaka T.; Iinuma M. Five Resveratrol Oligomers from Roots of Sophora Leachiana. Phytochemistry 1995, 38, 733–740. [Google Scholar]
- Kim H. J.; Saleem M.; Seo S. H.; Jin C.; Lee Y. S. Two New Antioxidant Stilbene Dimers, Parthenostilbenins A and B from Parthenocissus tricuspidata. Planta Med. 2005, 71, 973–976. [DOI] [PubMed] [Google Scholar]
- Lins A. P.; Felicio J. D.; Braggio M. M.; Roque L. C. A Resveratrol Dimer from Parthenocissus Tricuspidata. Phytochemistry 1991, 30, 3144–3146. [Google Scholar]
- Huang K.-S.; Lin M. Oligostilbenes from the Roots of Vitis Amurensis. J. Asian Nat. Prod. Res. 1999, 2, 21–28. [DOI] [PubMed] [Google Scholar]
- Tanaka T.; Iinuma M.; Murata H. Stilbene Derivatives in the Stem of Parthenocissus Quinquefolia. Phytochemistry 1998, 48, 1045–1049. [Google Scholar]
- He S.; Wu B.; Pan Y.; Jiang L. Stilbene Oligomers from Parthenocissus Laetevirens: Isolation, Biomimetic Synthesis, Absolute Configuration, and Implication of Antioxidative Defense System in the Plant. J. Org. Chem. 2008, 73, 5233–5241. [DOI] [PubMed] [Google Scholar]
- Breuil A.-C.; Adrian M.; Pirio N.; Meunier P.; Bessis R.; Jeandet P. Metabolism of Stilbene Phytoalexins by Botrytis Cinerea: 1. Characterization of a Resveratrol Dehydrodimer. Tetrahedron Lett. 1998, 39, 537–540. [Google Scholar]
- Solomon E. I.; Sundaram U. M.; Machonkin T. E. Multicopper Oxidases and Oxygenases. Chem. Rev. 1996, 96, 2563–2606. [DOI] [PubMed] [Google Scholar]
- Isaac I. S.; Dawson J. H. Haem Iron-Containing Peroxidases. Essays Biochem. 1999, 34, 51–69. [DOI] [PubMed] [Google Scholar]
- Wang M.; Jin Y.; Ho C.-T. Evaluation of Resveratrol Derivatives as Potential Antioxidants and Identification of a Reaction Product of Resveratrol and 2,2-Diphenyl-1-Picryhydrazyl Radical. J. Agric. Food Chem. 1999, 47, 3974–3977. [DOI] [PubMed] [Google Scholar]
- Nicotra S.; Cramarossa M. R.; Mucci A.; Pagnoni U. M.; Riva S.; Forti L. Biotransformation of Resveratrol: Synthesis of Trans-Dehydrodimers Catalyzed by Laccases from Myceliophtora Thermophyla and from Trametes Pubescens. Tetrahedron 2004, 60, 595–600. [Google Scholar]
- Takaya Y.; Terashima K.; Ito J.; He Y.-H.; Tateoka M.; Yamaguchi N.; Niwa M. Biomimic Transformation of Resveratrol. Tetrahedron 2005, 61, 10285–10290. [Google Scholar]
- Wilkens A.; Paulsen J.; Wray V.; Winterhalter P. Structures of Two Novel Trimeric Stilbenes Obtained by Horseradish Peroxidase Catalyzed Biotransformation of Trans-Resveratrol and (−)-E-Viniferin. J. Agric. Food Chem. 2010, 58, 6754–6761. [DOI] [PubMed] [Google Scholar]
- Tamboli V. F.; Re N.; Coletti C.; Defant A.; Mancini I.; Tosi P. A Joint Experimental and Theoretical Investigation on the Oxidative Coupling of Resveratrol Induced by Copper and Iron Ions. Int. J. Mass Spectrom. 2012, 319–320, 55–63. [Google Scholar]
- Song T.; Zhou B.; Peng G.-W.; Zhang Q.-B.; Wu L.-Z.; Liu Q.; Wang Y. Aerobic Oxidative Coupling of Resveratrol and Its Analogues by Visible Light Using Mesoporous Graphitic Carbon Nitride (mpg-C3N4) as a Bioinspired Catalyst. Chem.—Eur. J. 2014, 20, 678–682. [DOI] [PubMed] [Google Scholar]
- Shikishima Y.; Takaishi Y.; Honda G.; Ito M.; Takeda Y.; Kodzhimatov O. K.; Ashurmetov O. Phenylbutanoids and Stilbene Derivatives of Rheum Maximowiczii. Phytochemistry 2001, 56, 377–381. [DOI] [PubMed] [Google Scholar]
- Han C.; Xu J.; Wang X.; Xu X.; Luo J.; Kong L. Enantioseparation of Racemic Trans-δ-Viniferin Using High Speed Counter-Current Chromatography Based on Induced Circular Dichroism Technology. J. Chromatogr. A 2014, 1324, 164–170. [DOI] [PubMed] [Google Scholar]
- Staunton J.; Weissman K. J. Polyketide Biosynthesis: A Millennium Review. Nat. Prod. Rep. 2001, 18, 380–416. [DOI] [PubMed] [Google Scholar]
- Suzuki K.; Shimizu T.; Kawabata J.; Mizutani J. New 3, 5, 4′-Trihydroxystilbene (Resveratrol) Oligomers from Carex Fedia Nees Var. miyabei (Franchet) T. Koyama (Cyperaceae). Agric. Biol. Chem. 1987, 51, 1003–1008. [Google Scholar]
- Tanaka T.; Ito T.; Nakaya K.; Iinuma M.; Riswan S. Oligostilbenoids in Stem Bark of Vatica Rassak. Phytochemistry 2000, 54, 63–69. [DOI] [PubMed] [Google Scholar]
- Sarker S. D.; Whiting P.; Dinan L.; Šik V.; Rees H. H. Identification and Ecdysteroid Antagonist Activity of Three Resveratrol Trimers (Suffruticosols A, B and C) from Paeonia Suffruticosa. Tetrahedron 1999, 55, 513–524. [Google Scholar]
- Ohshima Y.; Shinoda K.; Takaya Y.; Ohta T.; Niwa M.; Hisamichi K.; Takeshita M. Vitisins D and E, Novel Oligostilbenes from Vitis Coignetilae Stem Barks. Heterocycles 1997, 46, 169–172. [Google Scholar]
- Wang S.-G.; Ma D.-Y.; Hu C.-Q. Two New Oligostilbenes from Caragana Sinica. J. Asian Nat. Prod. Res. 2004, 6, 241–248. [DOI] [PubMed] [Google Scholar]
- Ito T.; Ali Z.; Furusawa M.; Iliya I.; Tanaka T.; Nakaya K.; Murata J.; Darnaedi D.; Oyama M.; Iinuma M. Two Novel Trimeric Resveratrol Derivatives from Cotylelobium Lanceolatum. Chem. Biodiversity 2005, 2, 1200–1216. [DOI] [PubMed] [Google Scholar]
- Chen C.; Jiang R.; Wang G.; Jiao R.; Tancharoen C.; Sudto K.; Vajarothai S.; Hannongbua S.; Ge H.; Tan R. Oligostilbenoids with Acetylcholinesterase Inhibitory Activity from Dipterocarpus Alatus. Planta Med. 2014, 80, 1641–1646. [DOI] [PubMed] [Google Scholar]
- Ito T.; Tanaka T.; Ido Y.; Nakaya K.; Iinuma M.; Takahashi Y.; Naganawa H.; Riswan S. Five New Oligostilbenes with One or Two Dihydrofurans from the Stem Bark of Vatica Rassak. Heterocycles 2001, 55, 557–567. [Google Scholar]
- Ito T.; Tanaka T.; Ido Y.; Nakaya K.; Iinuma M.; Riswan S. Stilbenoids Isolated from Stem Bark of Shorea Hemsleyana. Chem. Pharm. Bull. 2000, 48, 1001–1005. [DOI] [PubMed] [Google Scholar]
- Ito T.; Abe N.; Oyama M.; Tanaka T.; Murata J.; Darnaedi D.; Iinuma M. Two Novel Resveratrol Trimers from Dipterocarpus Grandiflorus. Helv. Chim. Acta 2009, 92, 1203–1216. [Google Scholar]
- Ge H. M.; Yang W. H.; Shen Y.; Jiang N.; Guo Z. K.; Luo Q.; Xu Q.; Ma J.; Tan R. X. Immunosuppressive Resveratrol Aneuploids from Hopea Chinensis. Chem.—Eur. J. 2010, 16, 6338–6345. [DOI] [PubMed] [Google Scholar]
- Li W.; Li B.; Chen Y.; Flexuosol A. a New Tetrastilbene from Vitis Flexuosa. J. Nat. Prod. 1998, 61, 646–647. [DOI] [PubMed] [Google Scholar]
- Kawabata J.; Mishima M.; Kurihara H.; Mizutani J. Stereochemistry of Two Tetrastilbenes from Carex Species. Phytochemistry 1995, 40, 1507–1510. [Google Scholar]
- Yamada M.; Hayashi K.; Hayashi H.; Ikeda S.; Hoshino T.; Tsutsui K.; Tsutsui K.; Iinuma M.; Nozaki H. Stilbenoids of Kobresia Nepalensis (Cyperaceae) Exhibiting DNA Topoisomerase II Inhibition. Phytochemistry 2006, 67, 307–313. [DOI] [PubMed] [Google Scholar]
- Sultanbawa M. U. S.; Surendrakumar S.; Bladon P. Distichol an Antibacterial Polyphenol from Shorea Disticha. Phytochemistry 1987, 26, 799–801. [Google Scholar]
- Bokel M.; Diyasena M. N. C.; Gunatilaka A. A. L.; Kraus W.; Sotheeswaran S. Canaliculatol, an Antifungal Resveratrol Trimer from Stemonoporous Canaliculatus. Phytochemistry 1988, 27, 377–380. [Google Scholar]
- Ohyama M.; Tanaka T.; Iinuma M.; Burandt C. L. Phenolic Compounds Isolated from the Roots of Sophora Stenophylla. Chem. Pharm. Bull. 1998, 46, 663–668. [Google Scholar]
- Wang S.; Ma D.; Hu C. Three New Compounds from the Aerial Parts of Caragana Sinica. Helv. Chim. Acta 2005, 88, 2315–2321. [Google Scholar]
- Yan K.-X.; Terashima K.; Takaya Y.; Niwa M. A Novel Oligostilbene Named (+)-Viniferol A from the Stem of Vitis Vinifera “Kyohou”. Tetrahedron 2001, 57, 2711–2715. [Google Scholar]
- Tanaka T.; Ito T.; Iinuma M.; Ohyama M.; Ichise M.; Tateishi Y. Stilbene Oligomers in Roots of Sophora Davidii. Phytochemistry 2000, 53, 1009–1014. [DOI] [PubMed] [Google Scholar]
- Syah Y. M.; Aminah N. S.; Hakim E. H.; Aimi N.; Kitajima M.; Takayama H.; Achmad S. A. Two Oligostilbenes, Cis- and Trans-Diptoindonesin B, from Dryobalanops Oblongifolia. Phytochemistry 2003, 63, 913–917. [DOI] [PubMed] [Google Scholar]
- Takaya Y.; Yan K.-X.; Terashima K.; He Y.-H.; Niwa M. Biogenetic Reactions on Stilbenetetramers from Vitaceaeous Plants. Tetrahedron 2002, 58, 9265–9271. [Google Scholar]
- Oshima Y.; Kamijou A.; Ohizumi Y.; Niwa M.; Ito J.; Hisamichi K.; Takeshita M. Novel Oligostilbenes from Vitis Coignetiae. Tetrahedron 1995, 51, 11979–11986. [Google Scholar]
- Oshima Y.; Kamijou A.; Moritani H.; Namao K.; Ohizumi Y. Vitisin A and Cis-Vitisin A, Strongly Hepatotoxic Plant Oligostilbenes from Vitis Coignetiae (Vitaceae). J. Org. Chem. 1993, 58, 850–853. [Google Scholar]
- Ito J.; Niwa M. Absolute Structures of New Hydroxystilbenoids, Vitisin C and Viniferal, from Vitis Vinifera “Kyohou”. Tetrahedron 1996, 52, 9991–9998. [Google Scholar]
- Gu B.; Xu Y.; He S. A New Resveratrol Trimer from the Roots and Stems of Vitis Wenchowensis. Molecules 2013, 18, 7486–7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo E.-K.; Chai H.; Constant H. L.; Santisuk T.; Reutrakul V.; Beecher C. W. W.; Farnsworth N. R.; Cordell G. A.; Pezzuto J. M.; Kinghorn A. D. Resveratrol Tetramers from Vatica Diospyroides. J. Org. Chem. 1999, 64, 6976–6983. [Google Scholar]
- Abe N.; Ito T.; Oyama M.; Sawa R.; Takahashi Y.; Chelladurai V.; Iinuma M. Occurrence of C-Glucoside of Resveratrol Oligomers in Hopea Parviflora. Chem. Pharm. Bull. 2011, 59, 239–248. [DOI] [PubMed] [Google Scholar]
- Abe N.; Ito T.; Oyama M.; Sawa R.; Takahashi Y.; Iinuma M. Resveratrol Derivatives from Vatica Albiramis. Chem. Pharm. Bull. 2011, 59, 452–457. [DOI] [PubMed] [Google Scholar]
- Ito T.; Tanaka T.; Iinuma M.; Nakaya K.; Takahashi Y.; Sawa R.; Murata J.; Darnaedi D. Three New Resveratrol Oligomers from the Stem Bark of Vatica Pauciflora. J. Nat. Prod. 2004, 67, 932–937. [DOI] [PubMed] [Google Scholar]
- Ito T.; Tanaka T.; Nakaya K.; Iinuma M.; Takahashi Y.; Naganawa H.; Ohyama M.; Nakanishi Y.; Bastow K. F.; Lee K.-H. A New Resveratrol Octamer, Vateriaphenol A. Vateria Indica. Tetrahedron Lett. 2001, 42, 5909–5912. [Google Scholar]
- Klotter F.; Studer A. Total Synthesis of Resveratrol-Based Natural Products Using a Palladium-Catalyzed Decarboxylative Arylation and an Oxidative Heck Reaction. Angew. Chem., Int. Ed. 2014, 53, 2473–2476. [DOI] [PubMed] [Google Scholar]
- Jepsen T. H.; Thomas S. B.; Lin Y.; Stathakis C. I.; de Miguel I.; Snyder S. A. Harnessing Quinone Methides: Total Synthesis of (±)-Vaticanol A. Angew. Chem. 2014, 126, 6865–6869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.; Lu J.; Xu X.; Hu R.; Pan Y. pH-Switched HRP-Catalyzed Dimerization of Resveratrol: A Selective Biomimetic Synthesis. Green Chem. 2012, 14, 3281–3284. [Google Scholar]
- Samaraweera U.; Sotheeswaran S.; S. Sultanbawa M. U. Polyphenols from Stemonoporus Species. Phytochemistry 1982, 21, 2585–2587. [Google Scholar]
- Sultanbawa M. U. S.; Surendrakumar S.; Bladon P. New Antibacterial Polyphenol, Copalliferol A, from Vateria Copallifera(retz.) Alston (dipterocarpaceae). J. Chem. Soc., Chem. Commun. 1980, 619–620. [Google Scholar]
- Gunawardena Y. A. G. P.; Sotheeswaran S.; Sultanbawa M. U. S.; Surendrakumar S.; Bladon P. Another Antibacterial Polyphenol, Copalliferol B, from Vateria Copallifera (dipterocarpaceae). Phytochemistry 1986, 25, 1498–1500. [Google Scholar]
- Wilds A. L.; Djerassi C. The Dienone-Phenol Rearrangement Applied to Chrysene Derivatives. The Synthesis of 3-Hydroxy-1-Methylchrysene and Related Compounds1. J. Am. Chem. Soc. 1946, 68, 1715–1719. [DOI] [PubMed] [Google Scholar]
- Sotheeswaran S.; Sultanbawa M. U. S.; Surendrakumar S.; Bladon P. Polyphenols from Dipterocarp Species. Copalliferol A and Stemonoporol. J. Chem. Soc., Perkin Trans. 1 1983, 699–702. [Google Scholar]
- Pezet R. Purification and Characterization of a 32-kDa Laccase-like Stilbene Oxidase Produced by Botrytis Cinerea Pers.:Fr. FEMS Microbiol. Lett. 1998, 167, 203–208. [Google Scholar]
- Davin L. B.; Wang H.-B.; Crowell A. L.; Bedgar D. L.; Martin D. M.; Sarkanen S.; Lewis N. G. Stereoselective Bimolecular Phenoxy Radical Coupling by an Auxiliary (Dirigent) Protein Without an Active Center. Science 1997, 275, 362–367. [DOI] [PubMed] [Google Scholar]
- Kim K.-W.; Smith C. A.; Daily M. D.; Cort J. R.; Davin L. B.; Lewis N. G. Trimeric Structure of (+)-Pinoresinol-Forming Dirigent Protein at 1.95 Å Resolution with Three Isolated Active Sites. J. Biol. Chem. 2015, 290, 1308–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickel B.; Constantin M.-A.; Pfannstiel J.; Conrad J.; Beifuss U.; Schaller A. An Enantiocomplementary Dirigent Protein for the Enantioselective Laccase-Catalyzed Oxidative Coupling of Phenols. Angew. Chem., Int. Ed. 2010, 49, 202–204. [DOI] [PubMed] [Google Scholar]
- Pezet R.; Gindro K.; Viret O.; Spring J.-L. Glycosylation and Oxidative Dimerization of Resveratrol Are Respectively Associated to Sensitivity and Resistance of Grapevine Cultivars to Downy Mildew. Physiol. Mol. Plant Pathol. 2004, 65, 297–303. [Google Scholar]
- Robinson R. LXIII.—A Synthesis of Tropinone. J. Chem. Soc., Trans. 1917, 111, 762–768. [Google Scholar]
- Robinson R. LXXV.—A Theory of the Mechanism of the Phytochemical Synthesis of Certain Alkaloids. J. Chem. Soc., Trans. 1917, 111, 876–899. [Google Scholar]
- Sarkanen K. V.; Wallis A. F. A. Oxidative Dimerizations of (E)- and (Z)-Isoeugenol (2-Methoxy-4-pro-Penylphenol) and (E)- and (Z)-2,6-Dimethoxy-4-Propenylphenol. J. Chem. Soc., Perkin Trans. 1 1973, 1869–1878. [Google Scholar]
- Ponzoni C.; Beneventi E.; Cramarossa M. R.; Raimondi S.; Trevisi G.; Pagnoni U. M.; Riva S.; Forti L. Laccase-Catalyzed Dimerization of Hydroxystilbenes. Adv. Synth. Catal. 2007, 349, 1497–1506. [Google Scholar]
- Shang Y.-J.; Qian Y.-P.; Liu X.-D.; Dai F.; Shang X.-L.; Jia W.-Q.; Liu Q.; Fang J.-G.; Zhou B. Radical-Scavenging Activity and Mechanism of Resveratrol-Oriented Analogues: Influence of the Solvent, Radical, and Substitution. J. Org. Chem. 2009, 74, 5025–5031. [DOI] [PubMed] [Google Scholar]
- Fan G.-J.; Liu X.-D.; Qian Y.-P.; Shang Y.-J.; Li X.-Z.; Dai F.; Fang J.-G.; Jin X.-L.; Zhou B. 4,4′-Dihydroxy-Trans-Stilbene, a Resveratrol Analogue, Exhibited Enhanced Antioxidant Activity and Cytotoxicity. Bioorg. Med. Chem. 2009, 17, 2360–2365. [DOI] [PubMed] [Google Scholar]
- Sako M.; Hosokawa H.; Ito T.; Iinuma M. Regioselective Oxidative Coupling of 4-Hydroxystilbenes: Synthesis of Resveratrol and ε-Viniferin (E)-Dehydrodimers. J. Org. Chem. 2004, 69, 2598–2600. [DOI] [PubMed] [Google Scholar]
- Yao C.-S.; Lin M.; Wang Y.-H. Synthesis of the Active Stilbenoids by Photooxidation Reaction of Trans-E-Viniferin. Chin. J. Chem. 2004, 22, 1350–1355. [Google Scholar]
- Wang G.-W.; Wang H.-L.; Capretto D. A.; Han Q.; Hu R.-B.; Yang S.-D. Tf2O-Catalyzed Friedel–Crafts Alkylation to Synthesize Dibenzo[a,d]cycloheptene Cores and Application in the Total Synthesis of Diptoindonesin D, Pauciflorial F, and (±)-Ampelopsin B. Tetrahedron 2012, 68, 5216–5222. [Google Scholar]
- Velu S. S.; Buniyamin I.; Ching L. K.; Feroz F.; Noorbatcha I.; Gee L. C.; Awang K.; Wahab I. A.; Weber J.-F. F. Regio- and Stereoselective Biomimetic Synthesis of Oligostilbenoid Dimers from Resveratrol Analogues: Influence of the Solvent, Oxidant, and Substitution. Chem.—Eur. J. 2008, 14, 11376–11384. [DOI] [PubMed] [Google Scholar]
- Pearson R. G. Hard and Soft Acids and Bases. J. Am. Chem. Soc. 1963, 85, 3533–3539. [Google Scholar]
- Hong F.-J.; Low Y.-Y.; Chong K.-W.; Thomas N. F.; Kam T.-S. Biomimetic Oxidative Dimerization of Anodically Generated Stilbene Radical Cations: Effect of Aromatic Substitution on Product Distribution and Reaction Pathways. J. Org. Chem. 2014, 79, 4528–4543. [DOI] [PubMed] [Google Scholar]
- Panzella L.; De Lucia M.; Amalfitano C.; Pezzella A.; Evidente A.; Napolitano A.; d’ Ischia M. Acid-Promoted Reaction of the Stilbene Antioxidant Resveratrol with Nitrite Ions: Mild Phenolic Oxidation at the 4′-Hydroxystiryl Sector Triggering Nitration, Dimerization, and Aldehyde-Forming Routes. J. Org. Chem. 2006, 71, 4246–4254. [DOI] [PubMed] [Google Scholar]
- Li W.; Li H.; Li Y.; Hou Z. Total Synthesis of (±)-Quadrangularin A. Angew. Chem., Int. Ed. 2006, 45, 7609–7611. [DOI] [PubMed] [Google Scholar]
- Müller E.; Mayer R.; Spanagel H.-D.; Scheffler K. Über Sauerstoffradikale, XIX. Dehydrierung Hydroxyphenyl-Substituierter Äthylene. Justus Liebigs Ann. Chem. 1961, 645, 53–65. [Google Scholar]
- Sarkanen K. V.; Wallis A. F. A. Oxidative Dimerizations of (E)- and (Z)-Isoeugenol (2-Methoxy-4-pro-Penylphenol) and (E)- and (Z)-2,6-Dimethoxy-4-Propenylphenol. J. Chem. Soc., Perkin Trans. 1 1973, 1878–1881. [Google Scholar]
- Tashiro M.; Itoh T.; Yoshiya H.; Fukata G. A Preparative Route To Alkoxyphenols Using t-Butyl Function as a Positional Protective Group. Org. Prep. Proced. Int. 1984, 16, 155. [Google Scholar]
- Saleh S. A.; Tashtoush H. I. De-Tert-Butylation of Substituted Arenes. Tetrahedron 1998, 54, 14157–14177. [Google Scholar]
- Li W.; Li H.; Wang A.; Luo Y.; Zang P.. Unexpected Formation of Two Novel Resveratrol Dimers. J. Chem. Res. 2010, 34. [Google Scholar]
- Li W.; Li H.; Luo Y.; Yang Y.; Wang N. Biosynthesis of Resveratrol Dimers by Regioselective Oxidative Coupling Reaction. Synlett 2010, 4, 1247–1250. [Google Scholar]
- Yang Y.; Liu Q.; Chen P.; Li W. FeCl3·6H2O Oxidation of Protected Resveratrol for the Synthesis of Tetraarylfuran-Type Oligostilbenes. Tetrahedron Lett. 2014, 55, 4455–4457. [Google Scholar]
- Matsuura B. S.; Keylor M. H.; Li B.; Lin Y.; Allison S.; Pratt D. A.; Stephenson C. R. J. A Scalable Biomimetic Synthesis of Resveratrol Dimers and Systematic Evaluation of Their Antioxidant Activities. Angew. Chem., Int. Ed. 2015, 54, 3754–3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin J.-P.; Dixon I. M.; Sauvage J.-P.; Williams J. A. G.; Barigelletti F.; Flamigni L. Synthesis and Photophysical Properties of Iridium(III) Bisterpyridine and Its Homologues: A Family of Complexes with a Long-Lived Excited State. J. Am. Chem. Soc. 1999, 121, 5009–5016. [Google Scholar]
- Corduneanu O.; Janeiro P.; Brett A. M. O. On the Electrochemical Oxidation of Resveratrol. Electroanalysis 2006, 18, 757–762. [Google Scholar]
- Yoshino H.; Tsuchiya Y.; Saito I.; Tsujii M. Promoting Effect of Pentamethylbenzene on the Deprotection of O-Benzyltyrosine and Nε-Benzyloxycarbonyllysine with Trifluoroacetic Acid. Chem. Pharm. Bull. 1987, 35, 3438–3441. [Google Scholar]
- Okano K.; Okuyama K.; Fukuyama T.; Tokuyama H. Mild Debenzylation of Aryl Benzyl Ether with BCl 3 in the Presence of Pentamethylbenzene as a Non-Lewis-Basic Cation Scavenger. Synlett 2008, 1977–1980. [Google Scholar]
- Snyder S. A.; Zografos A. L.; Lin Y. Total Synthesis of Resveratrol-Based Natural Products: A Chemoselective Solution. Angew. Chem., Int. Ed. 2007, 46, 8186–8191. [DOI] [PubMed] [Google Scholar]
- Taylor R. J. K. Recent Developments in Ramberg -Bäcklund and Episulfone Chemistry. Chem. Commun. 1999, 3, 217–227. [Google Scholar]
- Meyers C. Y.; Malte A. M.; Matthews W. S. Ionic Reactions of Carbon Tetrachloride. Survey of Reactions with Ketones, Alcohols, and Sulfones. J. Am. Chem. Soc. 1969, 91, 7510–7512. [Google Scholar]
- Snyder S. A.; Brill Z. G. Structural Revision and Total Synthesis of Caraphenol B and C. Org. Lett. 2011, 13, 5524–5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder S. A.; Breazzano S. P.; Ross A. G.; Lin Y.; Zografos A. L. Total Synthesis of Diverse Carbogenic Complexity within the Resveratrol Class from a Common Building Block. J. Am. Chem. Soc. 2009, 131, 1753–1765. [DOI] [PubMed] [Google Scholar]
- Brown R. S. Investigation of the Early Steps in Electrophilic Bromination through the Study of the Reaction with Sterically Encumbered Olefins. Acc. Chem. Res. 1997, 30, 131–137. [Google Scholar]
- Rodebaugh R.; Fraser-Reid B. Evidence for Cyclic Bromonium Ion Transfer in Electrophilic Bromination of Alkenes: Reaction of Ω-Alkenyl Glycosides with Aqueous N-Bromosuccinimide. Tetrahedron 1996, 52, 7663–7678. [Google Scholar]
- Tebbe F. N.; Parshall G. W.; Reddy G. S. Olefin Homologation with Titanium Methylene Compounds. J. Am. Chem. Soc. 1978, 100, 3611–3613. [Google Scholar]
- Hartmann C.; Meyer V. Ueber Jodobenzoësäure. Ber. Dtsch. Chem. Ges. 1893, 26, 1727–1732. [Google Scholar]
- Frigerio M.; Santagostino M. A Mild Oxidizing Reagent for Alcohols and 1,2-Diols: O-Iodoxybenzoic Acid (IBX) in DMSO. Tetrahedron Lett. 1994, 35, 8019–8022. [Google Scholar]
- Nicolaou K. C.; Kang Q.; Wu T. R.; Lim C. S.; Chen D. Y.-K. Total Synthesis and Biological Evaluation of the Resveratrol-Derived Polyphenol Natural Products Hopeanol and Hopeahainol A. J. Am. Chem. Soc. 2010, 132, 7540–7548. [DOI] [PubMed] [Google Scholar]
- Hirao A.; Itsuno S.; Nakahama S.; Yamazaki N. Asymmetric Reduction of Aromatic Ketones with Chiral Alkoxy-Amineborane Complexes. J. Chem. Soc., Chem. Commun. 1981, 315–317. [Google Scholar]
- Corey E. J.; Bakshi R. K.; Shibata S.; Chen C. P.; Singh V. K. A Stable and Easily Prepared Catalyst for the Enantioselective Reduction of Ketones. Applications to Multistep Syntheses. J. Am. Chem. Soc. 1987, 109, 7925–7926. [Google Scholar]
- Snyder S. A.; Wright N. E.; Pflueger J. J.; Breazzano S. P. Total Syntheses of Heimiol A, Hopeahainol D, and Constrained Analogues. Angew. Chem., Int. Ed. 2011, 50, 8629–8633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson A. W.; LaCount R. B. The Chemistry of Ylids. VI. Dimethylsulfonium Fluorenylide—A Synthesis of Epoxides1. J. Am. Chem. Soc. 1961, 83, 417–423. [Google Scholar]
- Corey E. J.; Chaykovsky M. Dimethyloxosulfonium Methylide ((CH3)2SOCH2) and Dimethylsulfonium Methylide ((CH3)2SCH2). Formation and Application to Organic Synthesis. J. Am. Chem. Soc. 1965, 87, 1353–1364. [Google Scholar]
- Meinwald J.; Labana S. S.; Chadha M. S. Peracid Reactions. III.1 The Oxidation of Bicyclo [2.2.1]heptadiene2. J. Am. Chem. Soc. 1963, 85, 582–585. [Google Scholar]
- Lindgren B. O.; Nilsson T.; Husebye S.; Mikalsen Ø.; Leander K.; Swahn C.-G. Preparation of Carboxylic Acids from Aldehydes (Including Hydroxylated Benzaldehydes) by Oxidation with Chlorite. Acta Chem. Scand. 1973, 27, 888–890. [Google Scholar]
- Bal B. S.; Childers W. E. Jr.; Pinnick H. W. Oxidation of A,β-Un Saturated Aldehydes. Tetrahedron 1981, 37, 2091–2096. [Google Scholar]
- Snyder S. A.; Treitler D. S.; Brucks A. P. Simple Reagents for Direct Halonium-Induced Polyene Cyclizations. J. Am. Chem. Soc. 2010, 132, 14303–14314. [DOI] [PubMed] [Google Scholar]
- Lewis M. D.; Cha J. K.; Kishi Y. Highly Stereoselective Approaches to.alpha.- and.beta.-C-Glycopyranosides. J. Am. Chem. Soc. 1982, 104, 4976–4978. [Google Scholar]
- Snyder S. A.; Thomas S. B.; Mayer A. C.; Breazzano S. P. Total Syntheses of Hopeanol and Hopeahainol A Empowered by a Chiral Brønsted Acid Induced Pinacol Rearrangement. Angew. Chem., Int. Ed. 2012, 51, 4080–4084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fittig R. 41. Ueber Einige Derivate Des Acetons. Justus Liebigs Ann. Chem. 1860, 114, 54–63. [Google Scholar]
- Butlerow A. Ueber Trimethylessigsäure. Justus Liebigs Ann. Chem. 1873, 170, 151–162. [Google Scholar]
- Liang T.; Zhang Z.; Antilla J. C. Chiral Brønsted Acid Catalyzed Pinacol Rearrangement. Angew. Chem., Int. Ed. 2010, 49, 9734–9736. [DOI] [PubMed] [Google Scholar]
- Chen D. Y.-K.; Kang Q.; Wu T. R. Modular Synthesis of Polyphenolic Benzofurans, and Application in the Total Synthesis of Malibatol A and Shoreaphenol. Molecules 2010, 15, 5909–5927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B. H.; Choi Y. L.; Shin S.; Heo J.-N. Stereoselective Palladium-Catalyzed A-Arylation of 3-Aryl-1-Indanones: An Asymmetric Synthesis of (+)-Pauciflorol F. J. Org. Chem. 2011, 76, 6611–6618. [DOI] [PubMed] [Google Scholar]
- Mizoroki T.; Mori K.; Ozaki A. Arylation of Olefin with Aryl Iodide Catalyzed by Palladium. Bull. Chem. Soc. Jpn. 1971, 44, 581. [Google Scholar]
- Heck R. F.; Nolley J. P. Palladium-Catalyzed Vinylic Hydrogen Substitution Reactions with Aryl, Benzyl, and Styryl Halides. J. Org. Chem. 1972, 37, 2320–2322. [Google Scholar]
- Bo C.; Lu J.-P.; Xie X.-G.; She X.-G.; Pan X.-F. Total Synthesis of (±)-Pauciflorol F. Chin. J. Org. Chem. 2006, 26, 1300–1302. [Google Scholar]
- Ito Y.; Hirao T.; Saegusa T. Synthesis of.alpha.,.beta.-Unsaturated Carbonyl Compounds by palladium(II)-Catalyzed Dehydrosilylation of Silyl Enol Ethers. J. Org. Chem. 1978, 43, 1011–1013. [Google Scholar]
- Jeffrey J. L.; Sarpong R. An Approach to the Synthesis of Dimeric Resveratrol Natural Products via a Palladium-Catalyzed Domino Reaction. Tetrahedron Lett. 2009, 50, 1969–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffrey J. L.; Sarpong R. Concise Synthesis of Pauciflorol F Using a Larock Annulation. Org. Lett. 2009, 11, 5450–5453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larock R. C.; Doty M. J.; Cacchi S. Synthesis of Indenones via Palladium-Catalyzed Annulation of Internal Alkynes. J. Org. Chem. 1993, 58, 4579–4583. [Google Scholar]
- Kim I.; Choi J. A Versatile Approach to Oligostilbenoid Natural Products – Synthesis of Permethylated Analogues of Viniferifuran, Malibatol A, and Shoreaphenol. Org. Biomol. Chem. 2009, 7, 2788–2795. [DOI] [PubMed] [Google Scholar]
- Abu-Hashem A. A.; Hussein H. A. R.; Aly A. S.; Gouda M. A. Synthesis of Benzofuran Derivatives via Different Methods. Synth. Commun. 2014, 44, 2285–2312. [Google Scholar]
- Ohta A.; Akita Y.; Ohkuwa T.; Chiba M.; Fukunaga R.; Miyafuji A.; Nakata T.; Tani N.; Aoyagi Y. Palladium-Catalyze Arylation of Furan, Thiophene, Benzo [b]furan and Benzo[b]thiophene. Heterocycles 1990, 31, 1951–1958. [Google Scholar]
- Kapdi A. R. Organometallic Aspects of Transition-Metal Catalysed Regioselective C–H Bond Functionalisation of Arenes and Heteroarenes. Dalton Trans. 2014, 43, 3021–3034. [DOI] [PubMed] [Google Scholar]
- Kim K.; Kim I. Total Synthesis of Diptoindonesin G via a Highly Efficient Domino Cyclodehydration/Intramolecular Friedel–Crafts Acylation/Regioselective Demethylation Sequence. Org. Lett. 2010, 12, 5314–5317. [DOI] [PubMed] [Google Scholar]
- Liégault B.; Lapointe D.; Caron L.; Vlassova A.; Fagnou K. Establishment of Broadly Applicable Reaction Conditions for the Palladium-Catalyzed Direct Arylation of Heteroatom-Containing Aromatic Compounds. J. Org. Chem. 2009, 74, 1826–1834. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Philips D.; Pan S. A Concise Synthesis of Paucifloral F and Related Indanone Analogues via Palladium-Catalyzed α-Arylation. J. Org. Chem. 2011, 76, 1902–1905. [DOI] [PubMed] [Google Scholar]
- Nazarov I. N.; Zaretskaya I. I. Izv. Akad. Nauk. SSSR, Ser. Khim 1941, 211–224. [Google Scholar]
- Zhong C.; Zhu J.; Chang J.; Sun X. Concise Total Syntheses of (±)isopaucifloral F, (±)quadrangularin A, and (±)pallidol. Tetrahedron Lett. 2011, 52, 2815–2817. [Google Scholar]
- Kerr D. J.; Miletic M.; Manchala N.; White J. M.; Flynn B. L. Asymmetric Synthesis of (+)- and (−)-Pauciflorol F: Confirmation of Absolute Stereochemistry. Org. Lett. 2013, 15, 4118–4121. [DOI] [PubMed] [Google Scholar]
- Chou C.-M.; Chatterjee I.; Studer A. Stereospecific Palladium-Catalyzed Decarboxylative C(sp3)–C(sp2) Coupling of 2,5-Cyclohexadiene-1-Carboxylic Acid Derivatives with Aryl Iodides. Angew. Chem., Int. Ed. 2011, 50, 8614–8617. [DOI] [PubMed] [Google Scholar]
- Koch E.; Studer A. Regioselective Threefold Aromatic Substitution of Benzoic Acid Derivatives by Dearomatization, Regioselective Functionalization, and Rearomatization. Angew. Chem., Int. Ed. 2013, 52, 4933–4936. [DOI] [PubMed] [Google Scholar]
- He Z.; Kirchberg S.; Fröhlich R.; Studer A. Oxidative Heck Arylation for the Stereoselective Synthesis of Tetrasubstituted Olefins Using Nitroxides as Oxidants. Angew. Chem., Int. Ed. 2012, 51, 3699–3702. [DOI] [PubMed] [Google Scholar]
- Soldi C.; Lamb K. N.; Squitieri R. A.; González-López M.; Di Maso M. J.; Shaw J. T. Enantioselective Intramolecular C–H Insertion Reactions of Donor–Donor Metal Carbenoids. J. Am. Chem. Soc. 2014, 136, 15142–15145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurosawa W.; Kan T.; Fukuyama T. An Efficient Synthesis of OpticallyActive trans −2-Aryl-2,3-Dihydrobenzofuran-3-carboxylicAcid Esters via C-H Insertion Reaction. Synlett 2003, 1028–1030. [Google Scholar]
- Natori Y.; Tsutsui H.; Sato N.; Nakamura S.; Nambu H.; Shiro M.; Hashimoto S. Asymmetric Synthesis of Neolignans (−)-Epi-Conocarpan and (+)-Conocarpan via Rh(II)-Catalyzed C–H Insertion Process and Revision of the Absolute Configuration of (−)-Epi-Conocarpan. J. Org. Chem. 2009, 74, 4418–4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis E. M.; Croteau R.. Cyclization Enzymes in the Biosynthesis of Monoterpenes, Sesquiterpenes, and Diterpenes. In Biosynthesis; Leeper D. F. J., Vederas P. D. J. C., Eds.; Topics in Current Chemistry; Springer: Berlin, 2000; pp 53–95. [Google Scholar]
- Maimone T. J.; Baran P. S. Modern Synthetic Efforts toward Biologically Active Terpenes. Nat. Chem. Biol. 2007, 3, 396–407. [DOI] [PubMed] [Google Scholar]
- Snyder S. A.; Gollner A.; Chiriac M. I. Regioselective Reactions for Programmable Resveratrol Oligomer Synthesis. Nature 2011, 474, 461–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poss C. S.; Schreiber S. L. Two-Directional Chain Synthesis and Terminus Differentiation. Acc. Chem. Res. 1994, 27, 9–17. [Google Scholar]
- Magnuson S. R. Two-Directional Synthesis and Its Use in Natural Product Synthesis. Tetrahedron 1995, 51, 2167–2213. [Google Scholar]
- Snyder S. A.; Treitler D. S. Et2SBr SbCl5Br: An Effective Reagent for Direct Bromonium-Induced Polyene Cyclizations. Angew. Chem., Int. Ed. 2009, 48, 7899–7903. [DOI] [PubMed] [Google Scholar]
- Wright N. E.; Snyder S. A. 9-Membered Carbocycle Formation: Development of Distinct Friedel–Crafts Cyclizations and Application to a Scalable Total Synthesis of (±)-Caraphenol A. Angew. Chem. 2014, 126, 3477–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fries K.; Finck G. Über Homologe Des Cumaranons Und Ihre Abkömmlinge. Ber. Dtsch. Chem. Ges. 1908, 41, 4271–4284. [Google Scholar]
- Fries K.; Pfaffendorf W. Über Ein Kondensationsprodukt Des Cumaranons Und Seine Umwandlung in Oxindirubin. Ber. Dtsch. Chem. Ges. 1910, 43, 212–219. [Google Scholar]
- Wattenberg L. W. Prevention—therapy—basic Science and the Resolution of the Cancer Problem: Presidential Address. Cancer Res. 1993, 53, 5890–5896. [PubMed] [Google Scholar]
- Davis R. A.; Beattie K. D.; Xu M.; Yang X.; Yin S.; Holla H.; Healy P. C.; Sykes M.; Shelper T.; Avery V. M.; Elofsson M.; Sundin C.; Quinn R. J. Solving the Supply of Resveratrol Tetramers from Papua New Guinean Rainforest Anisoptera Species That Inhibit Bacterial Type III Secretion Systems. J. Nat. Prod. 2014, 77, 2633–2640. [DOI] [PubMed] [Google Scholar]
- Xue Y.-Q.; Di J.-M.; Luo Y.; Cheng K.-J.; Wei X.; Shi Z. Resveratrol Oligomers for the Prevention and Treatment of Cancers. Oxid. Med. Cell. Longevity 2014, 2014, e765832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johari A. T. T. Antioxidant, Antimicrobial and Cytotoxic Activities of Resveratrol Oligomers of Shorea Macroptera Dyer. Aust. J. Basic Appl. Sci. 2012, 6, 431–436. [Google Scholar]
- Kim H.-J.; Lee W.-J.; Park Y.-H.; Cho S.-H.; Choi S.-W. Differential Effects of Resveratrol and Its Oligomers Isolated from Seeds of Paeonia Lactiflora (Peony) on Proliferation of MCF - 7 and ROS 17/2.8 Cells. J. Food Sci. Nutr. 2003, 8, 356–364. [Google Scholar]
- Kim H.; Chang E.; Bae S.; Shim S.; Park H.; Rhee C.; Park J.; Choi S. Cytotoxic and Antimutagenic Stilbenes from Seeds of Paeonia Lactiflora. Arch. Pharmacal Res. 2002, 25, 293–299. [DOI] [PubMed] [Google Scholar]
- Qiao H.; Chen X.; Xu L.; Wang J.; Zhao G.; Hou Y.; Ge H. M.; Tan R.-X.; Li E. Antitumor Effects of Naturally Occurring Oligomeric Resveratrol Derivatives. FASEB J. 2013, 27, 4561–4571. [DOI] [PubMed] [Google Scholar]
- Patcharamun W.; Sichaem J.; Siripong P.; Khumkratok S.; Jong-aramruang J.; Tip-pyang S. A New Dimeric Resveratrol from the Roots of Shorea Roxburghii. Fitoterapia 2011, 82, 489–492. [DOI] [PubMed] [Google Scholar]
- Chowdhury S. A.; Kishino K.; Satoh R.; Hashimoto K.; Kikuchi H.; Nishikawa H.; Shirataki Y.; Sakagami H. Tumor-Specificity and Apoptosis-Inducing Activity of Stilbenes and Flavonoids. Anticancer Res. 2005, 25, 2055–2063. [PubMed] [Google Scholar]
- Ohyama M.; Tanaka T.; Ito T.; Iinuma M.; Bastow K. F.; Lee K.-H. Antitumor Agents 200. Cytotoxicity of Naturally Occurring Resveratrol Oligomers and Their Acetate Derivatives. Bioorg. Med. Chem. Lett. 1999, 9, 3057–3060. [DOI] [PubMed] [Google Scholar]
- Choi C.-W.; Choi Y.-H.; Cha M.-R.; Yoo D.-S.; Kim Y.-S.; Yon G.-H.; Choi S.-U.; Kim Y.-H.; Ryu S.-Y. A New Glycoside of Resveratrol Dimer from Stem Bark of Vitis Vinifera. Bull. Korean Chem. Soc. 2010, 31, 3448–3450. [Google Scholar]
- Xuan L.; Shi J.; Yao C.; Bai J.; Qu F.; Zhang J.; Hou Q. Vam3, a Resveratrol Dimer, Inhibits Cigarette Smoke-Induced Cell Apoptosis in Lungs by Improving Mitochondrial Function. Acta Pharmacol. Sin. 2014, 35, 779–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim K. G.; Gray A. I.; Pyne S.; Pyne N. J. Resveratrol Dimers Are Novel Sphingosine Kinase 1 Inhibitors and Affect Sphingosine Kinase 1 Expression and Cancer Cell Growth and Survival. Br. J. Pharmacol. 2012, 166, 1605–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L.; Henry G. E.; Seeram N. P. Identification and Bioactivities of Resveratrol Oligomers and Flavonoids from Carex Folliculata Seeds. J. Agric. Food Chem. 2009, 57, 7282–7287. [DOI] [PubMed] [Google Scholar]
- Wibowo A.; Ahmat N.; Hamzah A. S.; Sufian A. S.; Ismail N. H.; Ahmad R.; Jaafar F. M.; Takayama H.; Malaysianol A. a New Trimer Resveratrol Oligomer from the Stem Bark of Dryobalanops Aromatica. Fitoterapia 2011, 82, 676–681. [DOI] [PubMed] [Google Scholar]
- Cheng Y.-Q.; Jiang R.; Huang W.; Wei W.; Chen C.-J.; Tan R.-X.; Ge H.-M.; Hopeachinols E–K Novel Oligostilbenoids from the Stem Bark of Hopea Chinensis. RSC Adv. 2014, 4, 28901–28907. [Google Scholar]
- Muhtadi; Hakim E. H.; Juliawaty L. D.; Syah Y. M.; Achmad S. A.; Latip J.; Ghisalberti E. L. Cytotoxic Resveratrol Oligomers from the Tree Bark of Dipterocarpus Hasseltii. Fitoterapia 2006, 77, 550–555. [DOI] [PubMed] [Google Scholar]
- Quiney C.; Dauzonne D.; Kern C.; Fourneron J.-D.; Izard J.-C.; Mohammad R. M.; Kolb J.-P.; Billard C. Flavones and Polyphenols Inhibit the NO Pathway during Apoptosis of Leukemia B-Cells. Leuk. Res. 2004, 28, 851–861. [DOI] [PubMed] [Google Scholar]
- Barjot C.; Tournaire M.; Castagnino C.; Vigor C.; Vercauteren J.; Rossi J.-F. Evaluation of Antitumor Effects of Two Vine Stalk Oligomers of Resveratrol on a Panel of Lymphoid and Myeloid Cell Lines: Comparison with Resveratrol. Life Sci. 2007, 81, 1565–1574. [DOI] [PubMed] [Google Scholar]
- Kang J. H.; Park Y. H.; Choi S. W.; Yang E. K.; Lee W. J. Resveratrol Derivatives Potently Induce Apoptosis in Human Promyelocytic Leukemia Cells. Exp. Mol. Med. 2003, 35, 467–474. [DOI] [PubMed] [Google Scholar]
- Ito T.; Tanaka T.; Ali Z.; Akao Y.; Nozawa Y.; Takahashi Y.; Sawa R.; Nakaya K.; Murata J.; Darnaedi D.; Iinuma M. A New Resveratrol Hexamer from Upuna Borneensis. Heterocycles 2004, 63, 129–136. [Google Scholar]
- Ito T.; Akao Y.; Yi H.; Ohguchi K.; Matsumoto K.; Tanaka T.; Iinuma M.; Nozawa Y. Antitumor Effect of Resveratrol Oligomers against Human Cancer Cell Lines and the Molecular Mechanism of Apoptosis Induced by Vaticanol C. Carcinogenesis 2003, 24, 1489–1497. [DOI] [PubMed] [Google Scholar]
- Ito T.; Iliya I.; Tanaka T.; Nakaya K.; Akao Y.; Nozawa Y.; Murata J.; Darnaedi D.; Iinuma M. Stilbenoids from Leaves of Upuna Borneensis. Heterocycles 2005, 65, 173–179. [Google Scholar]
- Iliya I.; Akao Y.; Matsumoto K.; Nakagawa Y.; Zulfiqar A.; Ito T.; Oyama M.; Murata H.; Tanaka T.; Nozawa Y.; Iinuma M. Growth Inhibition of Stilbenoids in Welwitschiaceae and Gnetaceae through Induction of Apoptosis in Human Leukemia HL60 Cells. Biol. Pharm. Bull. 2006, 29, 1490–1492. [DOI] [PubMed] [Google Scholar]
- Lee E.-O.; Kwon B.-M.; Song G.-Y.; Chae C.-H.; Kim H.-M.; Shim I.-S.; Ahn K.-S.; Kim S.-H. Heyneanol A Induces Apoptosis via Cytochrome c Release and Caspase Activation in Human Leukemic U937 Cells. Life Sci. 2004, 74, 2313–2326. [DOI] [PubMed] [Google Scholar]
- González-Sarrías A.; Gromek S.; Niesen D.; Seeram N. P.; Henry G. E. Resveratrol Oligomers Isolated from Carex Species Inhibit Growth of Human Colon Tumorigenic Cells Mediated by Cell Cycle Arrest. J. Agric. Food Chem. 2011, 59, 8632–8638. [DOI] [PubMed] [Google Scholar]
- Colin D.; Gimazane A.; Lizard G.; Izard J.-C.; Solary E.; Latruffe N.; Delmas D. Effects of Resveratrol Analogs on Cell Cycle Progression, Cell Cycle Associated Proteins and 5-Fluoro-Uracil Sensitivity in Human Derived Colon Cancer Cells. Int. J. Cancer 2009, 124, 2780–2788. [DOI] [PubMed] [Google Scholar]
- Marel A.-K.; Lizard G.; Izard J.-C.; Latruffe N.; Delmas D. Inhibitory Effects of Trans-Resveratrol Analogs Molecules on the Proliferation and the Cell Cycle Progression of Human Colon Tumoral Cells. Mol. Nutr. Food Res. 2008, 52, 538–548. [DOI] [PubMed] [Google Scholar]
- Zhi S.; Guang X. Growth Inhibitory Effect of Stilbenoids on Lung Cancer Cell Lines. Shanghai Yike Daxue Xuebao 1998, 25, 327–330. [Google Scholar]
- Zhi S.; Chunyan T.; Yuxiong W.; Hong H.; Guang X. Effects of Miyabenol C on Protein Kinase C in Two Lung Carcinoma Cell Lines. Shanghai Yike Daxue Xuebao 1999, 26, 395–399. [Google Scholar]
- Colin D.; Lancon A.; Delmas D.; Lizard G.; Abrossinow J.; Kahn E.; Jannin B.; Latruffe N. Antiproliferative Activities of Resveratrol and Related Compounds in Human Hepatocyte Derived HepG2 Cells Are Associated with Biochemical Cell Disturbance Revealed by Fluorescence Analyses. Biochimie 2008, 90, 1674–1684. [DOI] [PubMed] [Google Scholar]
- Jang M.; Cho E. J.; Piao X.-L. Protective Effects of Resveratrol Oligomers from Vitis Amurensis against Sodium Nitroprusside-Induced Neurotoxicity in Human Neuroblastoma SH-SY5Y Cells. Arch. Pharmacal Res. 2014, 1–7. [DOI] [PubMed] [Google Scholar]
- Mishima S.; Matsumoto K.; Futamura Y.; Araki Y.; Ito T.; Tanaka T.; Iinuma M.; Nozawa Y.; Akao Y. Antitumor Effect of Stilbenoids from Vateria Indica against Allografted Sarcoma S-180 in Animal Model. J. Exp. Ther. Oncol. 2003, 3, 283–288. [DOI] [PubMed] [Google Scholar]
- Lee S.-H.; Shin N.-H.; Kang S.-H.; Park J.; Chung S.; Min K.; Kim Y. α-Viniferin: A Prostaglandin H2 Synthase Inhibitor from Root of Carex Humilis. Planta Med. 1998, 64, 204–207. [DOI] [PubMed] [Google Scholar]
- Chung E. Y.; Kim B. H.; Lee M. K.; Yun Y.-P.; Lee S. H.; Min K. R.; Kim Y. Anti-Inflammatory Effect of the Oligomeric Stilbene Alpha-Viniferin and Its Mode of the Action through Inhibition of Cyclooxygenase-2 and Inducible Nitric Oxide Synthase. Planta Med. 2003, 69, 710–714. [DOI] [PubMed] [Google Scholar]
- Chung E. Y.; Roh E.; Kwak J.-A.; Lee H.-S.; Lee S. H.; Lee C.-K.; Han S.-B.; Kim Y. α-Viniferin Suppresses the Signal Transducer and Activation of Transcription-1 (STAT-1)–Inducible Inflammatory Genes in Interferon-γ–Stimulated Macrophages. J. Pharmacol. Sci. 2010, 112, 405–414. [DOI] [PubMed] [Google Scholar]
- Xie Q. W.; Whisnant R.; Nathan C. Promoter of the Mouse Gene Encoding Calcium-Independent Nitric Oxide Synthase Confers Inducibility by Interferon Gamma and Bacterial Lipopolysaccharide. J. Exp. Med. 1993, 177, 1779–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai S. H.; Lin-Shiau S. Y.; Lin J. K. Suppression of Nitric Oxide Synthase and the down-Regulation of the Activation of NFκB in Macrophages by Resveratrol. Br. J. Pharmacol. 1999, 126, 673–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach E. A.; Aguet M.; Schreiber R. D. The IFN-γ Receptor: A Paradigm for Cytokine Receptor Signaling. Annu. Rev. Immunol. 1997, 15, 563–591. [DOI] [PubMed] [Google Scholar]
- Dilshara M. G.; Lee K.-T.; Kim H. J.; Lee H.-J.; Choi Y. H.; Lee C.-M.; Kim L. K.; Kim G.-Y. Anti-Inflammatory Mechanism of α-Viniferin Regulates Lipopolysaccharide-Induced Release of Pro-inflammatory Mediators in BV2Microglial Cells. Cell. Immunol. 2014, 290, 21–29. [DOI] [PubMed] [Google Scholar]
- Perkins N. D. Integrating Cell-Signalling Pathways with NF-κB and IKK Function. Nat. Rev. Mol. Cell Biol. 2007, 8, 49–62. [DOI] [PubMed] [Google Scholar]
- Ito T.; Akao Y.; Tanaka T.; Iinuma M.; Nozawa Y.; Vaticanol C. a Novel Resveratrol Tetramer, Inhibits Cell Growth through Induction of Apoptosis in Colon Cancer Cell Lines. Biol. Pharm. Bull. 2002, 25, 147–148. [DOI] [PubMed] [Google Scholar]
- Shibata M.-A.; Akao Y.; Shibata E.; Nozawa Y.; Ito T.; Mishima S.; Morimoto J.; Otsuki Y.; Vaticanol C. a Novel Resveratrol Tetramer, Reduces Lymph Node and Lung Metastases of Mouse Mammary Carcinoma Carrying p53 Mutation. Cancer Chemother. Pharmacol. 2007, 60, 681–691. [DOI] [PubMed] [Google Scholar]
- Fu J.; Jin J.; Cichewicz R. H.; Hageman S. A.; Ellis T. K.; Xiang L.; Peng Q.; Jiang M.; Arbez N.; Hotaling K.; Ross C. A.; Duan W. Trans-(−)-ε-Viniferin Increases Mitochondrial Sirtuin 3 (SIRT3), Activates AMP-Activated Protein Kinase (AMPK), and Protects Cells in Models of Huntington Disease. J. Biol. Chem. 2012, 287, 24460–24472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seya K.; Kanemaru K.; Sugimoto C.; Suzuki M.; Takeo T.; Motomura S.; Kitahara H.; Niwa M.; Oshima Y.; Furukawa K.-I. Opposite Effects of Two Resveratrol (trans-3,5,4′-Trihydroxystilbene) Tetramers, Vitisin A and Hopeaphenol, on Apoptosis of Myocytes Isolated from Adult Rat Heart. J. Pharmacol. Exp. Ther. 2009, 328, 90–98. [DOI] [PubMed] [Google Scholar]
- Ohguchi K.; Akao Y.; Matsumoto K.; Tanaka T.; Ito T.; Iinuma M.; Nozawa Y. Vaticanol C-Induced Cell Death Is Associated with Inhibition of Pro-Survival Signaling in HL60 Human Leukemia Cell Line. Biosci. Biotechnol. Biochem. 2005, 69, 353–356. [DOI] [PubMed] [Google Scholar]
- Chang F.; Steelman L. S.; Lee J. T.; Shelton J. G.; Navolanic P. M.; Blalock W. L.; Franklin R. A.; McCubrey J. A. Signal Transduction Mediated by the Ras/Raf/MEK/ERK Pathway from Cytokine Receptors to Transcription Factors: Potential Targeting for Therapeutic Intervention. Leukemia 2003, 17, 1263–1293. [DOI] [PubMed] [Google Scholar]
- Courtney K. D.; Corcoran R. B.; Engelman J. A. The PI3K Pathway as Drug Target in Human Cancer. J. Clin. Oncol. 2010, 28, 1075–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto T.; Nakata R.; Tamura E.; Kosuge Y.; Kariya A.; Katsukawa M.; Mishima S.; Ito T.; Iinuma M.; Akao Y.; Nozawa Y.; Arai Y.; Namura S.; Inoue H.; Vaticanol C. a Resveratrol Tetramer, Activates PPARα and PPARβ/δ in Vitro and in Vivo. Nutr. Metab. 2010, 7, 46–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juge-Aubry C.; Pernin A.; Favez T.; Burger A. G.; Wahli W.; Meier C. A.; Desvergne B. DNA Binding Properties of Peroxisome Proliferator-Activated Receptor Subtypes on Various Natural Peroxisome Proliferator Response Elements: Importance of the 5′-Flanking Region. J. Biol. Chem. 1997, 272, 25252–25259. [DOI] [PubMed] [Google Scholar]
- Sim J.; Jang H. W.; Song M.; Kim J. H.; Lee S. H.; Lee S. Potent Inhibitory Effect of Alpha-Viniferin on Human Cytochrome P450. Food Chem. Toxicol. 2014, 69, 276–280. [DOI] [PubMed] [Google Scholar]
- Piver B.; Berthou F.; Dreano Y.; Lucas D. Differential Inhibition of Human Cytochrome P450 Enzymes by ε-Viniferin, the Dimer of Resveratrol: Comparison with Resveratrol and Polyphenols from Alcoholized Beverages. Life Sci. 2003, 73, 1199–1213. [DOI] [PubMed] [Google Scholar]
- Waffo-Teguo P.; Lee D.; Cuendet M.; Mérillon J.-M.; Pezzuto J. M.; Kinghorn A. D. Two New Stilbene Dimer Glucosides from Grape (Vitis Vinifera) Cell Cultures. J. Nat. Prod. 2001, 64, 136–138. [DOI] [PubMed] [Google Scholar]
- Sung M. J.; Davaatseren M.; Kim W.; Park S. K.; Kim S.-H.; Hur H. J.; Kim M. S.; Kim Y.-S.; Dae Young K. Vitisin A Suppresses LPS-Induced NO Production by Inhibiting ERK, p38, and NF-κB Activation in RAW 264.7 Cells. Int. Immunopharmacol. 2009, 9, 319–323. [DOI] [PubMed] [Google Scholar]
- Nassra M.; Krisa S.; Papastamoulis Y.; Kapche G.; Bisson J.; André C.; Konsman J.-P.; Schmitter J.-M.; Mérillon J.-M.; Waffo-Téguo P. Inhibitory Activity of Plant Stilbenoids against Nitric Oxide Production by Lipopolysaccharide-Activated Microglia. Planta Med. 2013, 79, 966–970. [DOI] [PubMed] [Google Scholar]
- Billard C.; Izard J.-C.; Roman V.; Kern C.; Mathiot C.; Mentz F.; Kolb J.-P. Comparative Antiproliferative and Apoptotic Effects of Resveratrol, ε-Viniferin and Vine-shots Derived Polyphenols (Vineatrols) on Chronic B Lymphocytic Leukemia Cells and Normal Human Lymphocytes. Leuk. Lymphoma 2002, 43, 1991–2002. [DOI] [PubMed] [Google Scholar]
- Xu G.; Zhang L. P.; Chen L. F.; Hu C. Q. Inhibition of Protein Kinase C by Stilbenoids. Yaoxue Xuebao 1994, 29, 818–822. [PubMed] [Google Scholar]
- Kulanthaivel P.; Janzen W. P.; Ballas L. M.; Jiang J. B.; Hu C.-Q.; Darges J. W.; Seldin J. C.; Cofield D. J.; Adams L. M. Naturally Occurring Protein Kinase C Inhibitors; II. Isolation of Oligomeric Stilbenes from Caragana Sinica. Planta Med. 1995, 61, 41–44. [DOI] [PubMed] [Google Scholar]
- Sung S. H.; Kang S. Y.; Lee K. Y.; Park M. J.; Kim J. H.; Park J. H.; Kim Y. C.; Kim J.; Kim Y. C. (+)-α-Viniferin, a Stilbene Trimer from Caragana Chamlague, Inhibits Acetylcholinesterase. Biol. Pharm. Bull. 2002, 25, 125–127. [DOI] [PubMed] [Google Scholar]
- Yan T.; Wang T.; Wei W.; Jiang N.; Qin Y.; Tan R.; Ge H. Polyphenolic Acetylcholinesterase Inhibitors from Hopea Chinensis. Planta Med. 2012, 78, 1015–1019. [DOI] [PubMed] [Google Scholar]
- Jang M. H.; Piao X. L.; Kim J. M.; Kwon S. W.; Park J. H. Inhibition of Cholinesterase and Amyloid-β Aggregation by Resveratrol Oligomers from Vitis Amurensis. Phytother. Res. 2008, 22, 544–549. [DOI] [PubMed] [Google Scholar]
- Pflieger A.; Waffo Teguo P.; Papastamoulis Y.; Chaignepain S.; Subra F.; Munir S.; Delelis O.; Lesbats P.; Calmels C.; Andreola M.-L.; Merillon J.-M.; Auge-Gouillou C.; Parissi V. Natural Stilbenoids Isolated from Grapevine Exhibiting Inhibitory Effects against HIV-1 Integrase and Eukaryote MOS1 Transposase In Vitro Activities. PLoS One 2013, 8, e81184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodish H.; Berk A.; Zipursky S. L.; et al. The Role of Topoisomerases in DNA Replication. Molecular Cell Biology, 4th ed.; W. H. Freeman: New York, 2000; Section 12.3. Available from http://www.ncbi.nlm.nih.gov/books/NBK21703/. [Google Scholar]
- Yamada M.; Hayashi K.; Hayashi H.; Tsuji R.; Kakumoto K.; Ikeda S.; Hoshino T.; Tsutsui K.; Tsutsui K.; Ito T.; Iinuma M.; Nozaki H. Nepalensinols D–G. New Resveratrol Oligomers from Kobresia Nepalensis (Cyperaceae) as Potent Inhibitors of DNA Topoisomerase II. Chem. Pharm. Bull. 2006, 54, 354–358. [DOI] [PubMed] [Google Scholar]
- Munoz M.; Henderson M.; Haber M.; Norris M. Role of the MRP1/ABCC1Multidrug Transporter Protein in Cancer. IUBMB Life 2007, 59, 752–757. [DOI] [PubMed] [Google Scholar]
- Bobrowska-Hägerstrand M.; Lillås M.; Mrówczyñska L.; Wróbel A.; Shirataki Y.; Motohashi N.; Hägerstrand H. Resveratrol Oligomers Are Potent MRP1 Transport Inhibitors. Anticancer Res. 2006, 26, 2081–2084. [PubMed] [Google Scholar]
- Ingold K. U.; Pratt D. A. Advances in Radical-Trapping Antioxidant Chemistry in the 21st Century: A Kinetics and Mechanisms Perspective. Chem. Rev. 2014, 114, 9022–9046. [DOI] [PubMed] [Google Scholar]
- Amorati R.; Ferroni F.; Pedulli G. F.; Valgimigli L. Modeling the Co-Antioxidant Behavior of Monofunctional Phenols. Applications to Some Relevant Compounds. J. Org. Chem. 2003, 68, 9654–9658. [DOI] [PubMed] [Google Scholar]
- Burton G. W.; Ingold K. U. Vitamin E: Application of the Principles of Physical Organic Chemistry to the Exploration of Its Structure and Function. Acc. Chem. Res. 1986, 19, 194–201. [Google Scholar]
- Suzuki T.; Motohashi H.; Yamamoto M. Toward Clinical Application of the Keap1–Nrf2 Pathway. Trends Pharmacol. Sci. 2013, 34, 340–346. [DOI] [PubMed] [Google Scholar]
- Magesh S.; Chen Y.; Hu L. Small Molecule Modulators of Keap1-Nrf2-ARE Pathway as Potential Preventive and Therapeutic Agents. Med. Res. Rev. 2012, 32, 687–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.; Xu X.; Tao Z.; Wang X. J.; Pan Y. Resveratrol Dimers, Nutritional Components in Grape Wine, Are Selective ROS Scavengers and Weak Nrf2 Activators. Food Chem. 2015, 173, 218–223. [DOI] [PubMed] [Google Scholar]
- Yamada M.; Hayashi K.; Ikeda S.; Tsutsui K.; Tsutsui K.; Ito T.; Iinuma M.; Nozaki H. Inhibitory Activity of Plant Stilbene Oligomers against DNA Topoisomerase II. Biol. Pharm. Bull. 2006, 29, 1504–1507. [DOI] [PubMed] [Google Scholar]
- Willenberg I.; Michael M.; Wonik J.; Bartel L. C.; Empl M. T.; Schebb N. H. Investigation of the Absorption of Resveratrol Oligomers in the Caco-2 Cellular Model of Intestinal Absorption. Food Chem. 2015, 167, 245–250. [DOI] [PubMed] [Google Scholar]
- Walle T. Bioavailability of Resveratrol. Ann. N. Y. Acad. Sci. 2011, 1215, 9–15. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Hu M. Natural Polyphenol Disposition via Coupled Metabolic Pathways. Expert Opin. Drug Metab. Toxicol. 2007, 3, 389–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong C. C.; Cheng K.-W.; Rigas B. Preclinical Predictors of Anticancer Drug Efficacy: Critical Assessment with Emphasis on Whether Nanomolar Potency Should Be Required of Candidate Agents. J. Pharmacol. Exp. Ther. 2012, 341, 572–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Li B; Pratt D. A. Methods for Determining the Efficacy Of Radical-Trapping Antioxidants. Free Radic. Biol. Med. 2015, 10.1016/j.freeradbiomed.2015.01.020. [DOI] [PubMed] [Google Scholar]; b Amorati R.; Valgimigli L. Advantages and Limitations of Common Testing Methods for Antioxidants. Free Radic. Res. 2015, 10.3109/10715762.2014.996146. [DOI] [PubMed] [Google Scholar]