

Abstract
Focal adhesion kinase (FAK) and vascular endothelial growth factor receptor 3 (VEGFR3) are tyrosine kinases, which function as key modulators of survival and metastasis signals in cancer cells. Previously, we reported that small molecule chlorpyramine hydrochloride (C4) specifically targets the interaction between FAK and VEGFR3 and exhibits anti-tumor efficacy. In this study, we designed and synthesized a series of 1 (C4) analogs on the basis of structure activity relationship and molecular modeling. The resulting new compounds were evaluated for their binding to the FAT domain of FAK and anti-cancer activity. Amongst all tested analogs, compound 29 augmented anti-proliferative activity in multiple cancer cell lines with stronger binding to the FAT domain of FAK and disrupted the FAK–VEGFR3 interaction. In conclusion, we hope that this work will contribute to further studies of more potent and selective FAK–VEGFR3 protein–protein interaction inhibitors.
Keywords: Focal adhesion kinase, Vascular endothelial growth factor receptor-3, Protein–protein interaction, Focal adhesion targeting domain, FAK scaffold inhibitor
1. Introduction
Focal adhesion kinase (FAK) is a non-receptor tyrosine kinase that is localized to focal adhesions, and forms points of contacts between cells and the extracellular matrix. The structural components of FAK encompass a huge variety of binding partners, phosphorylation sites and signaling crosstalk with multiple extracellular and intracellular proteins [1-3]. The FAK-FERM domain interacts with several proteins such as p53, EGFR, c-Met, RIP, p85 subunit of PI3K, and β-integrins, whereas the FAK-FAT domain binds to paxillin, talin, and Grb2 [4-6]. Thus, FAK serves as a signaling hub and scaffolding protein to direct numerous signaling pathways to achieve specific cellular outcomes [7]. FAK is indispensable to cancer cells, where it functions to promote the overall aggressiveness of tumor growth by controlling cellular processes such as, adhesion, migration, proliferation and survival [8-10]. Studies with dominant negative FAK mutants, antisense oligonucleotides and siRNA’s for FAK, and germline deletions in FAK, exemplify the role of FAK in cancer cell progression and metastasis [11-15]. Massive presence of FAK in almost all solid tumor types [16], coupled with its involvement with deregulated proteins in cancer [17], offers a perfect setting for FAK specific therapeutics. Existing FAK kinase inhibitors such as PF-562–271, PF-04554878 and GSK2256098 have been shown to directly inhibit FAK kinase activity and tumor growth [18]. However, it is important to note that the aforementioned drugs being kinase inhibitors compete for the ATP binding site, thus raising the possibility for these drugs to exhibit non-specific kinase inhibition and related toxicities [19].
VEGFR3 is part of the VEGF receptor tyrosine kinase family that is involved in the regulation of normal and pathological endothelial cell specific responses, from embryogenesis to adulthood. Ligands VEGF-C and VEGF-D bind to VEGFR3, which is essential for the growth and maintenance of the lymphatic vasculature [20,21]. However, VEGFR3 [22,23] and its ligands VEGF-C [24,25] and VEGF-D [26,27] have been shown to promote lymphangiogenesis and metastatic spread in various tumor models. Several inhibitors of lymphangiogenesis are being developed such as, neutralizing monoclonal antibodies to VEGF-C and VEGF-D, peptidomimetics based on VEGF-C and VEGF-D, and kinase inhibitors of VEGFR3 [28]. Our lab’s approach has been to understand the implications of FAK specific protein–protein interactions and develop inhibitors that preferentially target FAK-protein complexes. We have shown that VEGFR3 binds to the C-terminal FAT domain of FAK and the interaction between FAK and VEGFR3 is crucial for tumor cell viability [29]. Despite the numerous challenges encountered while developing inhibitors that target protein–protein interactions [30], in our previous efforts, we discovered small molecule 1 (chlorpyramine hydrochloride) (Fig. 1) which successfully binds and disrupts the FAK–VEGFR3 interaction [31]. Further testing with 1 showed reduction in tumor burden and neovascularization in mouse models of melanoma, breast cancer, pancreatic cancer [32], and glioblastoma.
Fig. 1.

Structure of parent compound 1 and commercially available compounds structurally similar to 1.
Although compound 1 is an attractive candidate, it exerts anti-tumor and anti-angiogenic effects at high micromolar concentrations. In order to gain a detailed understanding of structure activity relationships and improve the anti-tumor potency and physicochemical properties of 1, we altered the structure of 1 to address the importance of certain chemical motifs. The newly synthesized analogs were screened in vitro using several cancer cell lines and 3D predicted binding modes of all analogs with the FAT domain of FAK were generated through molecular modeling. We also ranked the analogs on the basis of scoring functions. Biological studies with the selected analogs were performed to investigate their anti-proliferative activity, binding and disruption of the FAK–VEGFR3 complex, and mechanism of cell death. Collectively, our findings show that analog 29, which displayed maximum potency and specificity amongst all tested analogs, is a novel compound which warrants further investigation in the drug development pipeline for FAK–VEGFR3 specific inhibitors.
2. Results and discussion
2.1. Structure activity relationship (SAR) studies
We previously verified the potential anti-cancer activities of commercially available compounds such as 2, 3, 4, 5, (Fig. 1) and 14 (Table 1) which are chemically similar to parent drug 1 and found that none of these drugs showed any improvement in activity over 1. This prompted us to perform SAR studies on 1. Parent drug 1 (Fig. 2) was an excellent starting point for exploring rational drug design and optimization, as the core template of 1 was amenable to rapid structural modifications. To investigate the impact of various substituents of 1 on biological efficacy, a series of novel derivatives were obtained by replacing the pyridine moiety with other aromatic systems (A), or by introducing other N-alkyl or aryl substituents instead of the N-p-chlorobenzyl group (B) and finally altering the N, N-dimethyl ethyl chain (C) (Fig. 2). For our study, a series of compounds were synthesized and are listed in Table 1. We determined their biological efficacy and performed molecular modeling analysis using the known crystal structure of the FAT domain of FAK. The grid scores of the analogs from the molecular docking study are shown in Table 1.
Table 1.
A list of compounds related to 1 investigated for SAR studies.
| Compd. number | Changes at “A” | Changes at “B” | Changes at “C” | Grid score |
|---|---|---|---|---|
| 1 |
|
|
|
−30.4 |
| 2 |
|
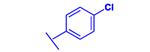
|
|
−29.08 |
| 3 |
|
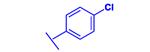
|
|
−29.91 |
| 4 |
|
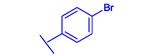
|
|
−31.13 |
| 5 |
|
|
|
−33.29 |
| 7 |
|
|
|
−24.27 |
| 8 |
|
|
|
−29.72 |
| 9 |
|
|
|
−25.35 |
| 10 |
|
|
|
−33.69 |
| 11 |
|
|
|
−38.64 |
| 12 |
|
|
|
−43.76 |
| 13 |
|
|
|
−43.29 |
| 14 |
|
|
|
−29.36 |
| 15 |
|
|
|
−34.77 |
| 16 |
|
|
|
−32.75 |
| 17 |
|
|
|
−35.04 |
| 18 |
|
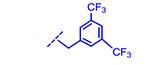
|
|
−33.35 |
| 19 |
|
|
|
−32.10 |
| 20 |
|
|
|
−24.32 |
| 22 |
|
|
|
Not calcd. |
| 23 |
|
|
|
−33.49 |
| 24 |
|
|
|
−40.21 |
| 25 |
|
|
|
−31.81 |
| 27 |
|
|
|
−29.25 |
| 28 |
|
|
|
−34.11 |
| 29 |
|
|
|
−34.41 |
Fig. 2.

Structure of parent compound 1 containing three functionalities A, B, and C.
2.2. Chemistry
For the synthesis of the desired analogs, a three step parallel synthesis protocol was followed, and the synthetic details are illustrated in Schemes 1–3. For investigating the importance of the N-p-chlorobenzyl group, a series of N-substituted analogs were synthesized as depicted in Scheme 1. The key precursor was prepared by reacting chloropyridine 6 with N, N-dimethyl ethyl-enediamine in a sealed tube at 140° C, which gave the desired key intermediate 7. The intermediate amine 7 was functionalized by reacting with various alkyl/aryl halides in the presence of lithium amide on refluxing temperature, and this gave the corresponding pyridine derivatives 9–19 in good yields. Particularly, the benzyl derivatives 14–18 were isolated in higher yields than the corresponding N-alkyl analogs 9–13. The analog of compound 1 with an extended carbon unit 19 was prepared by reacting with N, N-dimethyl ethyl group with an increased carbon unit (Scheme 1). The pyridine moiety in 1 was replaced with benzene and quinoline on reacting the chlorobenzene 21 and 2-chloroquinoline 26 with N, N-dimethylethylamine and thus, compounds 22 and 27 (Schemes 2 and 3) were isolated in excellent yields. By using the intermediates 22 and 27, the corresponding benzyl 23–25 and quinoline analogs 28 and 29 were synthesized (Table 1) in modest yields and the reaction conditions were not optimized.
Scheme 1.

N-alkyl, aryl, and substituted aryl analogs of 1.
Scheme 3.
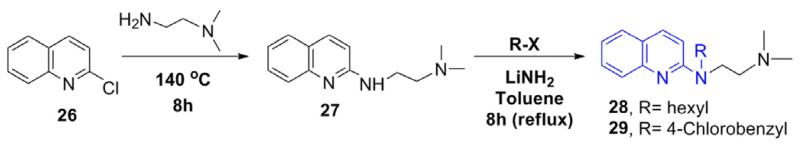
Synthesis of quinoline analogs of 1.
Scheme 2.

Synthesis of phenyl analogs of 1.
2.3. Comparative in vitro efficacy of 1 analogs
First, we analyzed the basal expression levels of target proteins, FAK and VEGFR3 in A375 and C8161 (melanoma), MDA-MB-231 (breast cancer), MiaPaCa-2-luc and Panc-1-luc (pancreatic cancer), and U87 (glioblastoma) cell lines (Fig. 3a). Robust FAK expression with varying expression levels of VEGFR3 was seen in all tested cell lines. Growth inhibitory activities of 1 analogs were determined using the MTS assay (Fig. 3b). The activities of all newly synthesized analogs were compared to parent drug 1. Anti-proliferative activity of the analogs was significantly affected by the type and position of chemical modifications. Replacement of the pyridine ring (region ‘A’) with a quinoline moiety, 29 or a benzene ring, 25, showed improved potency in multiple cancer cell lines. Replacement of the chloro group in the 4-chlorobenzyl moiety (region ‘B’) with bromo, 15, iodo, 16, thio, 17, and trifluoromethyl, 18, groups did not show improvement in activity over 1. Removal of the 4-chlorobenzyl moiety, analog 7 abolished activity. Additionally, analogs 9, 10, 11, 12, and 13 share a common theme where they all bear varying lengths of the alkyl chain. Of these, 11, 12, and 13 showed excellent potency in all cell lines used for screening. We found that an increase in length of the carbon chain directly correlated with an increase in cytotoxicity and log P values (Table S1) of these compounds, with 9 showing no activity and 13 being the most potent in this series. Removal of the N, N-dimethyl ethyl group (region ‘C’), 20 and introduction of 1 carbon in the N-linker, 19 did not improve activity. Lastly, dual alterations were made to the pyridine ring (region ‘A’) and the 4-chlorobenzyl group (region ‘B’). Analogs 23 and 24 both have benzene rings, but varying length of alkyl chains. 24, having the 12 carbon chain dramatically increased cytotoxicity in some cancer cell lines whereas 23, with the 6 carbon chain failed to show improved activity in all tested cell lines. Overall, the trend of increased alkyl chain length enhances cytotoxicity was seen with analogs 23 and 24 and analogs 9, 10, 11, 12, and 13. Also, analog 24 had the highest log P of 4.8 which may not favor an optimal drug like character [33] (Table S1). Next, we observed that removal of the 4-chlorobenzyl group with the presence of a quinoline ring, 27, did not enhance potency. Analog 28, with a 6 carbon chain and quinoline ring showed no improvement in anti-proliferative activity. Lastly, when the 4-chlorobenzyl group (region ‘B’) was removed and one extra carbon was introduced to the N, N-dimethyl ethyl group (region ‘C’), analog 8 did not show enhanced activity.
Fig. 3.

In vitro screening of 1 and its analogs. (A) Basal expression levels of FAK and VEGFR3 protein were analyzed in the indicated cancer cell lines. GAPDH was used as a loading control. (B) Anti-proliferative effects of 1 analogs in the indicated cell lines that were treated with 50 μM of the each analog for 72 h. Error bars represent ±SD. (C) Immunoprecipitation with FAK antibody in MCF7-VEGFR3 cells after treatment with the selected analogs for 24 h followed by immunoblotting with total VEGFR3 and total FAK antibodies.
On the basis of in vitro screening results, it appears that retaining the hetero-aromatic moiety in region ‘A’ plays an important role in biological efficacy. Replacing the chloro group from the p-chlorobenzyl functionality in region ‘B’ of 1 with bromo-, 15, or iodo-, 16, reduced efficacy probably due to their bulkier nature. Diminished activity of 3, 5-bis-(trifluoromethyl) benzyl analog, 18, again suggests the importance of the chlorobenzyl group in 1. Interestingly, replacing the chlorobenzyl group with long alkyl side chains, analogs 11, 12, 13 and 24 due to their flexible nature might show improved activity due to increased hydrophobic contacts with the FAT protein and could also contribute to an increase in toxicity due to non-specific protein binding. Any modification made to region ‘C’ failed to improve activity, which suggests that retaining this group in parent compound 1 is important. Based on these results, we further investigated the effects of analogs 25 and 29 on FAK–VEGFR3 disruption in MCF7-VEGFR3 cells [34], which overexpress both FAK and VEGFR3, and performed immunoprecipitation with FAK antibody (Fig. 3c). 1 was used as a positive control based on previous reports [31]. Analog 7 was included as a negative control, since it showed no anti-proliferative activity in all cell lines used for screening (Fig. 3b) and also had the lowest grid score from modeling analysis (Table 1). Changes in VEGFR3 protein levels co-precipitated with FAK were analyzed after addition of the selected analogs for 24 h. Decrease in VEGFR3 protein levels was seen following treatment with 10 μM of 29, confirming that it successfully inhibits the FAK–VEGFR3 interaction. We also analyzed the effect of analog 29 on kinase specificity (Table 2). A panel of 10 kinases was tested for inhibition caused by analog 29 at 1 μM. Select kinases were included in this assay as they are closely related to FAK or VEGFR3 proteins or are involved in FAK downstream signaling cascades. We found that 29 did not significantly inhibit (<40%) any of the tested kinases. This data demonstrates that analog 29 does not cause FAK–VEGFR3 disruption by targeting the kinase activity of both these proteins and proteins involved in the FAK signaling pathway. Analog 29 retained the target specific properties of parent compound 1 and hence, in the rest of the study we have focused our efforts on evaluating the biological efficacy of analog 29.
Table 2.
Kinase inhibitory profile of analog 29.
| Kinase tested | % Inhibitiona |
|---|---|
| AKT1 (PKB α) | 7 |
| EGFR (ErbB1) | 4 |
| FLT1 (VEGFR1) | 1 |
| FLT4 (VEGFR3) | 4 |
| IGF1R | 11 |
| KDR (VEGFR2) | 14 |
| PDGFRA (PDGFR α) | 8 |
| PTK2 (FAK) | 13 |
| PTK2B (FAK2) | 0 |
| SRC | 3 |
Percent inhibition was calculated according to the Z’-LYTE® Data Analysis protocol (Life Technologies).
2.4. Molecular modeling and binding analysis
In silico docking was performed on all analogs and the grid scores were computed (Table 1). The compound with the highest predicted affinity for the FAT domain was analog 12 (−43.7 kcal/mol), followed by 13 (−43.3 kcal/mol), 24 (−40.2 kcal/mol), and 11 (−38.6 kcal/mol). The high grid scores of these compounds are most likely due to the hydrophobic interactions of the long alkyl chains with the groove of the FAT protein. While analog 29 had a modest score (−34.4 kcal/mol), it was chosen for additional computational analysis due to its anti-proliferative activity in MiaPaCa-2-luc, Panc-1-luc, and U87 cell lines (Fig. 3b) and importantly, its target specificity for the FAK–VEGFR3 interaction (Fig. 3c). Also, analog 29 had a slightly favorable grid score than the parent compound 1 (−30.4 kcal/mol). Based on the surface representation of the FAT protein, there appears to be a pocket on the FAT protein which could favor drug binding. This pocket is comprised of hydrophobic residues Tyr 925, Val 928, Val 932, and Leu 1035 (Fig. 4a). The predicted docking pose of 29 suggests that it occupies this pocket on the FAT protein. Analog 29 sits in a charged cavity surrounded by residues Ser 910, Thr 929, Lys 1032, and Asp 1036 (Fig. 4b). A cation-π interaction between the quinoline ring of 29 and Lys 1032 along with a potential π stacking between the 4-chlorobenzyl group of 29 and the aromatic side chain of Tyr 925 are the key interactions observed. The N, N-dimethyl ethyl group might have a secondary effect of orienting the two other groups to this hydrophobic pocket. It is worthwhile exploring the importance of this functionality to promote hydrogen bonding with the nearby residues. The proximity of analog 29 to Tyr 925 is of utmost importance and could be the main reason for its enhanced activity, since Tyr 925 is a key residue that has been shown to be involved in the activation of the Grb2-Ras-MAPK pathway which accelerates tumor progression and angiogenesis [35,36]. To further investigate the importance of the 4-chlorobenzyl group, we analyzed the predicted binding modes of analogs 27 and 28 to the FAT protein since both these analogs are structurally similar to analog 29 (Table 1). Analogs 27 (Fig. S21) and 28 (Fig. S22) occupy a binding region slightly above from that of analog 29. Analog 27 does not appear to make any key interactions with its surrounding residues, Ile 936, Ser 939, His 1025, and Ala 1028. This is also reflected by its relatively low grid score (−29.25). Although, the quinoline ring of analog 28 participates in a hydrogen bond formation with Lys 1032 and the alkyl chain sits in the hydrophobic groove (Val 932, Ile 936, and Ala 1028) of FAT, this analog showed no improvement in anti-proliferative activity. In summary, the docking analysis strongly suggests that retaining a heteroaromatic moiety and the 4-chlorobenzyl group are critical for activity and that Tyr 925 and the residues which form the druggable pocket on FAT (Fig. 4a) could play an important role in the binding and specificity of 1 analogs. To further validate the specificity of 29 for the FAT domain of FAK, we evaluated its binding efficacy compared to parent compound 1 by using the Octet RED Assay based on the principle of bio-layer interferometry [37]. Binding constants obtained from this assay revealed that the Kd (M) of 29 (Kd (M) = 1.8 × 10−8) was significantly greater than 1 (Kd (M) = 5.7 × 10 −7) and analog 7 which was included as a negative control did not show any binding to FAT protein.
Fig. 4.

Predicted binding mode of analog 29. (A) Surface representation of the FAT domain of FAK (gray) showing docking of compound 29 (magenta stick). Potentially druggable pocket on FAT is indicated in orange. (B) Ribbon representation of the FAT domain (gray) showing the binding pose of analog 29 (magenta stick) and residues within 5 Å of 29 (green stick). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
2.5. Cellular activity of analog 29
From the in vitro screening, we found that analog 29 showed maximum anti-proliferative effects in pancreatic cancer cell lines. Hence, we conducted further analyses of this analog using a pancreatic cancer model system. We first analyzed the expression levels of FAK and VEGFR3 in a panel of six pancreatic cancer cell lines, SU8686, AsPc, Panc 213, Panc 10.5, Panc-1-luc and MiaPaCa-2-luc (Fig. 5a). The MCF7-VEGFR3 cell line was included as a positive control cell line. High levels of FAK and varying levels of VEGFR3 were found in all these cell lines, with MCF7-VEGFR3 showing very high levels of both FAK and VEGFR3. Next, we determined the IC50 of 29 in all these cell lines and found that Panc-1-luc and MiaPaCa-2-luc cell lines were particularly sensitive to drug treatment (Fig. 5b). MCF7-VEGFR3 cells showed maximum susceptibility to cytotoxic effects of 29. We then assessed the effects of 29 on colony formation using MiaPaCa-2-luc and MCF-VEGFR3 cells (Fig. 5c). 29 inhibited colony formation in both cell lines at 10 μM compared to compound 1 which achieved the same effect at a concentration >10 μM. Analog 7 was also tested for colony formation as a negative control.
Fig. 5.

In vitro cytotoxicity profile of analog 29 in pancreatic cancer cell lines. (A) Analysis of FAK and VEGFR3 protein expression in six pancreatic cancer cell lines and MCF7-VEGFR3 cell line. GAPDH was used as a loading control. (B) IC50 values of 29 in the indicated cell lines. Error bars represent ±SD. (C) MiaPaCa-2-luc or MCF7-VEGFR3 cells were treated with 29 at the indicated concentrations. After 14 days, colonies were stained with 0.25% of 1, 9-dimethyl-methylene blue in 50% methanol for 1 h. Representative images from three independent experiments have been shown. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
We further evaluated the ability of 29 to disrupt the FAK–VEGFR3 interaction in MiaPaCa-2-luc cells. 1 was included as a positive control. Immunoprecipitation of FAK was carried out and levels of FAK pan-phosphorylated tyrosine and co-precipitated VEGFR3 were evaluated after drug treatment for 24 h (Fig. 6a). Levels of VEGFR3 and phosphorylated tyrosine residues associated with FAK protein decreased in a dose dependent manner following drug exposure. Similarly, reverse immunoprecipitation using VEGFR3 antibody, confirmed a dose dependent increase in FAK–VEGFR3 disruption along with reduction in VEGFR3 pan-phosphorylated tyrosine levels after 24 h treatment of 1 and 29 (Fig. 6b). Densitometry analysis of the protein levels obtained from the immunoprecipitation results confirmed a dose dependent decrease after treatment with 1 and 29 in both directions, using FAK (Fig. 6a) and VEGFR3 antibodies (Fig. 6b). Together, these results confirm that 29 enhanced target selectivity and specifically inhibited the FAK–VEGFR3 interaction in a dose dependent manner.
Fig. 6.

Dose dependent disruption of FAK–VEGFR3 binding by analog 29 in MiaPaCa-2-luc cells. (A) Immunoprecipitation with FAK antibody or (B) VEGFR3 antibody after 24 h treatment with 1 and 29 followed by immunoblotting with pan phosphorylated tyrosine, total FAK and total VEGFR3 antibodies. Isotype and no antibody lanes indicate negative controls.
2.6. Mechanism of cell death of analog 29
Given that 29 exert cytotoxic effects in multiple cancer cell lines, we investigated the involvement of caspase-3/7 in the cell death mechanism induced in MiaPaCa-2-luc cells. We used two dyes, TMRM, a cell permeable red-orange fluorescent dye which is used to measure the membrane potential of mitochondria and CellEvent™, a cell permeable bright green fluorescent dye which exhibits fluorescence only in the presence of caspase-3/7 activation. Staurosporine, a well-studied activator of apoptosis [38] was included as a positive control in this experiment. Increasing doses of 29 led to increased apoptosis, which was marked by a loss of TMRM signal with a concomitant increase in the CellEvent™ signal (Fig. 7a). Apoptotic cells with bright green (in the web version) staining were further counted for each condition and data were quantified (Fig. 7b). We found a statistically significant increase in apoptosis at 70 μM of 29 compared to control cells. We further tested the dose dependent apoptotic effects of 29 on caspase-3 and PARP cleavage in MiaPaCa-2-luc and MCF-VEGFR3 cell lines after 24 h drug treatment (Fig. 7c). In both cell lines, an increase in caspase-3 and PARP cleavage product was seen with increasing doses of 29. In summary, our results confirm that analog 29 induces dose dependent apoptotic cell death involving caspase-3/7 and PARP activation. The apoptotic cell death caused by analog 29 is consistent with the effects seen with compound 1 which support our previous conclusions that the FAK–VEGFR3 interaction exerts a pro-survival effect on cancer cells [29,31].
Fig. 7.

Analog 29 induced cell death via activation of the apoptotic pathway. (A) MiaPaCa-2-luc cells were treated with analog 29 at the indicated concentrations. After 24 h cells were stained with CellEvent™, a caspase-3, 7 dye and TMRM, a mitochondrial dye. Representative images are shown from three independent experiments. (B) Apoptotic cells were counted from multiple microscopic fields for each condition. Error bars represent ±SEM. *p < 0.05 (C) Addition of analog 29 to MiaPaCa-2-luc and MCF-VEGFR3 cells at the indicated concentrations for 24 h. Antibodies for caspase-3 and PARP were used to detect apoptosis. GAPDH serves as a loading control.
3. Conclusions
In this report, we investigated the importance of specific chemical motifs inherent in 1 with an aim to develop small molecules with improved anti-cancer activity, enhanced potency, and target specificity for the FAK–VEGFR3 interaction. Our SAR approach was to first divide molecule 1 into three parts in order to understand the importance of the chemical groups for anti-cancer activity. Next, we designed and synthesized novel analogs of parent compound 1 and subsequently tested them using an anti-proliferative screening assay along with molecular modeling with an aim to filter out potential FAK–VEGFR3 inhibitors for further studies. Our results highlight the importance of the heteroaromatic moiety and p-chlorobenzyl group being critical for activity. Analog 29 caused an increase in cytotoxicity in multiple cancer cell lines compared to parent drug 1. We have shown that analog 29 shows target specificity for the FAK–VEGFR3 interaction site using both, a cell based assay and a label free binding assay. Importantly, analog 29 showed greater selectivity for MCF7-VEGFR3 cells that overexpress both target proteins, FAK and VEGFR3, as compared to MiaPaCa-2-luc cells that have moderate levels of both proteins.
Compound 1 and the selected analog 29 are unconventional anti-cancer drugs, as they are not kinase inhibitors but in fact disrupt protein–protein interactions, in this case between FAK and VEGFR3. A few examples of well characterized protein–protein interaction inhibitors are SP4206 that disrupts the interaction between interleukin 2 (IL-2) and the α-chain of the IL-2 receptor (IL-2Rα) [39], BH3-mimetic ABT-737 [40] which possesses high-affinity binding to the hydrophobic BH3-binding site of recombinant Bcl-XL, ABT-737, and Nutlins that were found to disrupt MDM2–p53 complexes [41]. By inhibiting the interplay between FAK and VEGFR3, we have previously shown that 1 downregulated major FAK and VEGFR3 tyrosine phosphorylation sites that in turn affect key protein signaling networks associated with cancer cell survival, angiogenesis, and metastasis [32,34]. Here, we have discovered that 29 successfully disrupted the FAK–VEGFR3 interaction and induced apoptotic cancer cell death, thus we anticipate 29 to mimic the overall anti-cancer effects of 1, albeit at much lower concentrations. Analog 29 shows much promise as a specific FAK–VEGFR3 inhibitor and could be used to treat a variety of solid cancers. Further development and optimization of 29 is currently underway.
4. Experimental section
4.1. Chemical synthesis
All chemicals were of reagent grade and used as such. All reagents were purchased from Aldrich chemical company and were used as received. Solvents were dried using standard methods unless stated otherwise. Reactions were carried out under nitrogen atmosphere and were monitored by pre-coated (0.20 cm) silica TLC plastic sheet (20 cm × 20 cm) strips (POLYGRAM SIL N-HR). 1H NMR spectra were recorded on Varian 400 spectrometers at 303 K in CDCl3 or CD3OD. Proton chemical shifts (δ) are reported in parts per million (ppm) relative to CDCl3 (7.26 ppm), CD3OD (3.34 ppm) or TMS (0.00 ppm). Coupling constants (J) are reported in Hertz (Hz) and s, d, t, q, p, m and br refer to singlet, doublet, triplet, quartet, pentet, multiplet and broad respectively. Mass spectral data (Electro Spray Ionization, ESI by fusion) were obtained from Biopolymer Facility, Roswell Park Cancer Institute, Buffalo, NY, HRMS data were obtained from the Mass Spectrometry Facility, Michigan State University, East Lansing, MI.
4.2. N, N-dimethyl-N’-(pyridine-2-yl)ethane-1,2-diamine (7)
In a dry 25 mL sealed tube, 2-chloropyridine 6 (10 eq, 1.13 g) and N, N-dimethyl ethylenediamine (11 eq, 1.20 g) were taken and heated the entire mixture to 140 °C over a period of 12 h with constant stirring. Then, the reaction mixture was cooled down to room temperature and the resulting crude was purified on a silica gel column by eluting (5% MeOH-DCM) as a solvent mixture to afford the desired derivative 7 (0.676 g) in 41% yield. 1H NMR 400 MHz (CDCl3) δ: 8.08 (dd, J = 4.8 Hz, J = 0.8 Hz, 1H); 7.38 (dt, J = 7.2 Hz, J = 2 Hz, 1H); 6.54 (dt, J = 5.6 Hz, J = 0.8 Hz, 1H); 6.40 (d, J = 8.4 Hz, 1H); 5.04 (brS, NH, 1H); 3.36 (q, J = 5.6 Hz, 2H); 2.55 (t, J = 6.0 Hz 2H); 2.27 (s, 6H); 13C NMR (CDCl3) δ: 158.7, 147.9, 137.1, 112.5, 107.4, 58.0, 45.1, 39.2; MASS (ESI): m/z 166 ([MH]+, 100%); HRMS: C9H15N3 [M]+ Calcd 165.1266, Obsd. 165.1275.
4.3. N, N-dimethyl-N’-(pyridine-2-yl)propane-1,2-diamine (8)
Compound 8 was synthesized by following the procedure for compound 7. Yield is 48%. 1H NMR 400 MHz (CDCl3) δ: 8.03 (dd, J = 4.8 Hz, J = 0.8 Hz, 1H); 7.38 (td, J = 7.2 Hz, J = 2 Hz, 1H); 6.48–6.54 (m, 2H); 5.64 (brS, NH, 1H); 3.50 (t, J = 6.4 Hz, 2H); 2.89 (t, J = 6.4 Hz 2H); 2.64 (s, 6H); 2.05 (quin, J = 6.0 Hz, 2H); 13C NMR (CDCl3) δ: 147.0, 137.5, 112.7, 109.0, 56.1, 43.3, 38.5, 30.9, 24.7; MASS (ESI): m/z 180 ([MH]+, 100%); HRMS: C10H17N3 [M]+ Calcd 179.1422, Obsd. 179.1488.
4.4. N, N-dimethyl-N’-(phenyl)ethane-1,2-diamine (22)
Compound 22 was synthesized by following the procedure for compound 7. Yield is 40%. 1H NMR 400 MHz (CD3OD) δ: 7.07–7.11 (m, 2H); 6.59–6.64 (m, 3H); 6.48–3.21 (t, J = 6.8 Hz, 2H); 2.57 (t, J = 7.2 Hz, 2H); 2.29 (s, 6H); 13C NMR (CD3OD) δ: 148.5, 129.1, 117.1, 112.8, 58.0, 45.1, 41.1; MASS (ESI): m/z 165 ([MH]+, 100%); HRMS: C10H16N2 [M]+ Calcd 164.1313, Obsd. 164.1374.
4.5. N, N-dimethyl-N’-(quinolin-2-yl)ethane-1,2-diamine (27)
Compound 27 was obtained by following the procedure for compound 7. Yield is 41%. 1H NMR 400 MHz (CD3OD) δ: 7.82 (d, J = 8.8 Hz, 1H); 7.58 (t, J = 8.4 Hz, 2H), 7.47 (t, J 8.0 Hz, 1H); 7.16 (t, J = 8.0 Hz, 1H); 6.72 (d, J = 8.8 Hz, 1H); 3.60 (t, J = 6.8 Hz, 2H); 2.64 (t, J = 6.8 Hz 2H); 2.34 (s, 6H); 13C NMR (CD3OD) δ: 156.9, 148.0, 136.9, 129.4, 127.4, 126.0, 123.3, 121.8, 112.1, 58.0, 45.1, 38.7; MASS (ESI): m/z 216 ([MH]+, 100%); HRMS: C13H17N3 [M]+ Calcd 215.3000, Obsd. 215.3226.
4.6. General procedure for the synthesis of 1 analogs
The compound of type 7 (1eq) and dry LiNH2 (1.2 eq) were taken in a dry RB flask and was added dry CH3CN (5 mL) as a solvent. After stirring for 5 min under Ar atm, the alkyl/benzyl halide (1.2 eq) was added drop by drop via syringe. The stirring was continued for 8 h at reflux temperature. The TLC indicated the formation of the new product spot. The reaction mixture was filtered and the filtrate was evaporated under reduced pressure. The crude obtained was purified on a silica gel column. Elution of 3% MeOH-DCM solvent mixture delivered the expected alkylated derivatives in 40–68% yield.
4.6.1. N-[propyl]-N-[2-(dimethylamino)ethyl]pyridin-2-amine (9)
Compound 9 was obtained by following the general procedure mentioned above; Yield 51%. 1H NMR 400 MHz (CDCl3) δ: 8.01 (d, J = 4.4 Hz, 1H), 7.35 (t, J = 7.6 Hz, 1H); 6.70 (d, J = 8.4 Hz, 1H); 6.57 (t, J = 6.0 Hz, 1H); 3.98 (m, 2H); 3.82 (m, 2H); 3.38–3.60 (m, 2H), 3.39 (s, 6H); 1.80 (m, 2H), 0.98 (t, J = 7.2 Hz, 3H); 13C NMR (CDCl3) δ: 157.5, 147.2, 137.3, 113.5, 110.1, 66.8, 62.7, 52.3, 36.0, 16.4, 10.5; MASS (ESI): m/z 208 ([MH]+, 100%); HRMS: C12H21N3 [M]+ Calcd 207.1735, Obsd. 207.1797.
4.6.2. N-[hexyl]-N-[2-(dimethylamino)ethyl]pyridin-2-amine (10)
Compound 10 was obtained by following the general procedure mentioned above; Yield 54%. 1H NMR 400 MHz (CD3OD) δ: 7.96 (d, J = 4.0 Hz, 1H), 7.31 (t, J = 6.8 Hz, 1H); 6.68 (d, J = 7.2 Hz, 1H); 6.52 (t, J = 6.8 Hz, 1H); 3.94 (m, 2H); 3.76 (m, 2H); 3.55 (m, 2H); 3.37 (s, 6H); 1.67 (m, 2H); 1.21–1.30 (m, 6H), 0.81 (t, J = 5.6 Hz, 3H); 13C NMR (CD3OD) δ: 157.6, 148.0, 137.0, 111.2, 105.6, 56.5, 49.2, 46.3, 45.3, 31.7, 27.4, 26.7, 22.6, 14.0; MASS (ESI): m/z 250 ([MH]+, 100%); HRMS: C15H27N3 [M]+ Calcd 249.2205, Obsd. 249.2211.
4.6.3. N-[octyl]-N-[2-(dimethylamino)ethyl]pyridin-2-amine (11)
Compound 11 was obtained by following the general procedure mentioned above; Yield 51%. 1H NMR 400 MHz (CDCl3) δ: 8.08 (dd, J = 5.4 Hz, J = 1.2 Hz, 1H), 7.52 (td, J = 8.6 Hz, J = 2.2 Hz, 1H); 6.59 (d, J = 8.6 Hz, 1H); 6.56 (t, J = 6.6 Hz, 1H); 4.04 (t, J = 6.0 Hz, 2H); 3.82 (t, J = 6.0 Hz, 2H); 3.56–3.62 (m, 2H); 3.46 (s, 6H); 1.60 (m, 2H); 1.26–1.42 (m, 10H), 0.87 (t, J = 7.2 Hz, 3H); 13C NMR (CD3OD) δ: 157.1, 147.4, 137.4, 112.2, 106.2, 50.5, 48.7, 41.7, 31.5, 29.1, 29.0, 28.8, 26.6, 25.9, 22.2, 12.9; MASS (ESI): m/z 278 ([MH]+, 100%); HRMS: C17H31N3 [M]+ Calcd 277.2518, Obsd. 277.2529.
4.6.4. N-[decyl]-N-[2-(dimethylamino)ethyl]pyridin-2-amine (12)
Compound 12 was obtained by following the general procedure mentioned above; Yield 53%. 1H NMR 400 MHz (CD3OD) δ: 8.09 (dd, J = 5.4 Hz, J = 1.2 Hz, 1H), 7.54 (td, J = 8.8 Hz, J = 2.0 Hz, 1H); 6.69 (d, J = 8.8 Hz, 1H); 6.61 (t, J = 6.4 Hz, 1H); 3.78 (t, J = 6.0 Hz, 2H); 3.42 (t, J = 7.6 Hz, 2H); 3.08 (t, J = 6.0 Hz, 2H); 2.75 (s, 6H); 1.62 (m, 2H); 1.26–1.42 (m, 14H), 0.89 (t, J = 7.2 Hz, 3H); 13C NMR (CD3OD) δ: 157.7, 147.0, 137.4, 111.2, 106.2, 50.0, 48.8, 45.6, 43.8, 31.6, 29.3, 29.2, 29.0, 26.9, 26.5, 22.2, 12.9; MASS (ESI): m/z 306 ([MH]+, 100%); HRMS: C19H35N3 [M]+ Calcd 305.2831, Obsd. 305.2612.
4.6.5. N-[dodecyl]-N-[2-(dimethylamino)ethyl]pyridin-2-amine (13)
Compound 13 was obtained by following the general procedure mentioned above; Yield 42%. 1H NMR 400 MHz (CD3OD) δ: 8.02 (d, J = 4.4 Hz, 1H), 7.43 (td, J = 8.0 Hz, J = 1.6 Hz, 1H); 6.61 (t, J = 6.0 Hz, 1H); 6.55 (d, J = 8.4 Hz, 1H); 3.81 (t, J = 6.4 Hz, 2H); 3.52 (t, J = 6.4 Hz, 2H); 3.36–3.42 (m, 2H); 3.16 (s, 6H); 1.78 (m, 2H); 1.21–1.34 (m, 18H), 0.89 (t, J = 6.4 Hz, 3H); 13C NMR (CD3OD) δ: 157.8, 146.7, 138.2, 112.5, 107.1, 58.3, 49.5, 48.2, 44.8, 42.7, 31.6, 29.33, 29.31, 29.2, 29.1, 29.0, 26.57, 26.51, 22.3, 13.0; MASS (ESI): m/z 334 ([MH]+, 100%); HRMS: C21H39N3 [M]+ Calcd 333.3144, Obsd. 333.3210.
4.6.6. N-[(4-bromophenyl)methyl]-N-[2-(dimethylamino)ethyl] pyridin-2-amine (15)
Compound 15 was obtained by following the general procedure mentioned above; Yield 64%. 1H NMR 400 MHz (CD3OD) δ: 8.02 (d, J = 4.4 Hz, 1H); 7.68 (d, J = 8.4 Hz, 2H); 7.51 (d, J = 8.4 Hz, 2H), 7.42 (td, J = 8.0 Hz, J = 1.6 Hz, 1H); 6.62 (t, J = 6.8 Hz, 1H); 6.56 (d, J = 8.4 Hz, 1H); 4.60 (s, 2H); 3.90 (t, J = 6.8 Hz, 2H); 3.53 (t, J = 6.4 Hz, 2H); 3.12 (s, 6H); 13C NMR (CD3OD) δ: 157.9, 146.9, 137.3, 134.5, 132.1, 126.5, 125.1, 113.0, 109.3, 67.2, 62.9, 49.4, 35.0; MASS (ESI): m/z 335 ([MH]+, 100%); HRMS: C16H20BrN3 [M]+ Calcd 333.0841, Obsd. 333.0698.
4.6.7. N-[(4-iodophenyl)methyl]-N-[2-(dimethylamino)ethyl] pyridin-2-amine (16)
Compound 16 was obtained by following the general procedure mentioned above; Yield 61%. 1H NMR 400 MHz (CD3OD) δ: 8.01 (dd, J = 5.2 Hz, J = 01.2 Hz, 1H); 7.87 (d, J = 8.0 Hz, 2H); 7.42 (td, J = 9.6 Hz, J = 2.0 Hz, 1H); 7.35 (d, J = 8.4 Hz, 2H), 6.60 (td, J = 7.2 Hz, J = 0.8 Hz, 1H); 6.56 (d, J = 8.4 Hz, 1H), 4.58 (s, 2H); 3.90 (t, J = 6.8 Hz, 2H); 3.53 (t, J = 6.4 Hz, 2H); 3.11 (s, 6H); 13C NMR (CD3OD) δ: 157.9, 146.9, 138.2, 137.3, 134.5, 126.9, 113.0, 109.3, 96.9, 67.4, 62.9, 49.5, 35.0; MASS (ESI): m/z 382 ([MH]+, 100%); HRMS: C16H20IN3 [M]+ Calcd 381.0702, Obsd. 381.0672.
4.6.8. N-[(4-methylthiophenyl)methyl]-N-[2-(dimethylamino) ethyl]pyridin-2-amine (17)
Compound 17 was obtained by following the general procedure mentioned above; Yield 64%. 1H NMR 400 MHz (CDCl3) δ: 8.01 (dd, J = 5.2 Hz, J = 1.2 Hz, 1H); 7.52 (d, J = 8.4 Hz, 2H); 7.33 (td, J = 8.8 Hz, J = 2.0 Hz, 1H); 7.21 (d, J = 8.4 Hz, 2H), 6.69 (d, J = 8.4 Hz, 1H); 6.54 (t, J = 6.8 Hz, 1H); 4.91 (s, 2H); 4.05–4.09 (m, 2H); 3.84 (t, J = 5.2 Hz, 2H); 3.29 (s, 6H); 2.45 (s, 3H); 13C NMR (CDCl3) δ: 157.6, 147.0, 142.9, 137.3, 133.4, 126.0, 122.8, 113.3, 110.3, 67.6, 63.7, 50.5, 36.3, 14.9; MASS (ESI): m/z 302 ([MH]+, 100%); HRMS: C17H23N3S [M]+ Calcd 301.1613, Obsd. 301.1596.
4.6.9. N-[(3,5-bis-trifluoromethyl-phenyl)methyl]-N-[2-(dimethylamino)ethyl]pyridin-2-amine (18)
Compound 18 was obtained by following the general procedure mentioned above; Yield 63%. 1H NMR 400 MHz (CD3OD) δ: 8.25 (s, 2H), 8.19 (s, 1H), 8.02 (d, J = 4.4 Hz, 1H); 7.43 (td, J = 8.0 Hz, J = 2.0 Hz, 1H); 6.57–6.63 (m, 2H), 4.84 (s, 2H); 3.94 (t, J = 6.4 Hz, 2H); 3.63 (t, J = 6.4 Hz, 2H); 3.18 (s, 6H); 13C NMR (CD3OD) δ: 157.9, 146.9, 137.3, 133.3, 130.5, 124.4, 124.3, 121.6, 113.1, 109.4, 66.1, 63.6, 49.5, 35.1; MASS (ESI): m/z 392 ([MH]+, 100%); HRMS: C18H19F6IN3 [M]+ Calcd 391.1483, Obsd. 391.1446.
4.6.10. N-[(4-chlorophenyl)methyl]-N-[2-(dimethylamino)propyl] pyridin-2-amine (19)
Compound 19 was obtained by following the general procedure mentioned above; Yield 68%. 1H NMR 400 MHz (CD3OD) δ: 7.96 (d, J = 4.4 Hz, 1H); 7.45–7.53 (m, 5H); 6.58–6.32 (m, 2H); 4.54 (s, 2H); 3.44 (t, J = 6.4 Hz, 2H); 3.30–3.45 (m, 2H); 3.05 (s, 6H); 2.13–2.20 (m, 2H); 13C NMR (CD3OD) δ: 158.0, 145.3, 138.0, 136.7, 134.2, 129.0, 126.0, 112.2, 109.5, 66.5, 62.1, 49.2, 37.7, 22.6; MASS (ESI): m/z 304 ([MH]+, 100%); HRMS: C17H22ClN3 [M]+ Calcd 303.1502, Obsd. 303.1598.
4.6.11. N-[hexyl]-N-[2-(dimethylamino)ethyl]phenylamine (23)
Compound 23 was obtained by following the general procedure mentioned above; Yield 44%. 1H NMR 400 MHz (CDCl3) δ: 7.20–7.23 (m, 2H), 6.34–7.19 (m, 3H); 3.42–3.46 (m, 2H); 3.28 (t, J = 7.6 Hz, 1H); 2.48–2.50 (m, 2H); 2.31 (s, 6H); 1.57–1.61 (m, 2H); 1.31–1.36 (m, 6H); 0.92 (t, J = 5.6 Hz, 3H); 13C NMR (CDCl3) δ: 147.8, 129.2, 115.5, 111.7, 56.3, 51.3, 49.2, 45.8, 31.7, 27.2, 26.7, 22.6, 14.0; MASS (ESI): m/z 249 ([MH]+, 100%); HRMS: C16H28N2 [M]+ Calcd 248.2252, Obsd. 248.2266.
4.6.12. N-[dodecyl]-N-[2-(dimethylamino)ethyl]phenylamine (24)
Compound 24 was obtained by following the general procedure mentioned above; Yield 42%. 1H NMR 400 MHz (CDCl3) δ: 7.12–7.16 (m, 2H), 6.67 (d, J = 7.6 Hz, 2H); 6.58 (t, J = 7.6 Hz, 1H); 3.43 (t, J = 8.0 Hz, 2H); 3.27 (t, J = 8.2 Hz, 2H); 2.48 (t, J = 8.4 Hz, 2H); 2.29 (s, 6H); 1.56 (p, J = 7.2 Hz, 2H); 1.28–1.32 (m, 18H); 0.89 (t, J = 6.8 Hz, 3H); 13C NMR (CDCl3) δ: 147.9, 129.2, 115.4, 111.6, 56.4, 51.2, 49.4, 45.9, 31.9, 29.66, 29.6, 29.53, 29.50, 29.3, 27.5, 27.1, 22.6, 14.1; MASS (ESI): m/z 333 ([MH]+, 100%); HRMS: C22H40N2 [M]+ Calcd 332.3191, Obsd. 332.3114.
4.6.13. N-[(4-chlorophenyl)methyl]-N-[2-(dimethylamino)ethyl] phenylamine (25)
Compound 25 was obtained by following the general procedure mentioned above; Yield 52%. 1H NMR 400 MHz (CDCl3) δ: 7.23 (dd, J = 22.8 Hz, J = 8.4 Hz, 4H); 7.13 (t, J = 7.6 Hz, 2H); 6.69 (d, J = 8.0 Hz, 2H); 6.63 (t, J = 7.2 Hz, 1H); 4.53 (s, 2H); 3.55 (t, J = 7.6 Hz, 2H); 2.55 (t, J = 7.6 Hz, 2H); 2.28 (s, 6H). 13C NMR (CDCl3) δ: 147.9, 129.2, 115.4, 111.6, 56.4, 51.2, 49.4, 45.9, 31.9, 29.66, 29.6, 29.53, 29.50, 29.3, 27.5, 27.1, 22.6, 14.1; MASS (ESI): m/z 289 ([MH]+, 100%); HRMS: C17H21ClN2 [M]+ Calcd 288.1393, Obsd. 288.1412.
4.6.14. N-[hexyl]-N-[2-(dimethylamino)ethyl]quinolin-2-amine (28)
Compound 28 was obtained by following the general procedure mentioned above; Yield 49%. 1H NMR 400 MHz (CD3OD) δ: 7.91 (d, J = 8.8 Hz, 1H), 7.63 (d, J = 8.8 Hz, 2H), 7.51 (t, J = 8.0 Hz, 1H), 7.23 (t, J = 7.6 Hz, 1H); 6.76 (d, J = 9.2 Hz, 1H); 3.98 (t, J = 6.0 Hz, 2H); 3.64 (t, J = 6.4 Hz, 2H); 3.43–3.47 (m, 2H); 3.21 (s, 6H); 1.83 (m, 2H); 1.26–1.36 (m, 6H), 0.87 (t, J = 6.4 Hz, 3H); 13C NMR (CD3OD) δ: 156.4, 147.2, 137.3, 129.1, 127.3, 125.3, 123.5, 122.1, 112.4, 64.3, 61.6, 50.8, 34.9, 30.9, 25.6, 22.2, 22.0, 12.8; MASS (ESI): m/z 300 ([MH]+, 100%); HRMS: C19H29N3 [M]+ Calcd 299.2361, Obsd. 299.2380.
4.6.15. N-[(4-chlorophenyl)methyl]-N-[2-(dimethylamino)ethyl] quinolin-2-amine (29)
Compound 29 was obtained by following the general procedure mentioned above; Yield 58%. 1H NMR 400 MHz (CD3OD) δ: 7.89 (d, J = 8.8 Hz, 1H), 7.61–7.64 (m, 4H), 7.48–7.54 (m, 3H); 7.23 (t, J = 6.8 Hz, 1H); 6.80 (d, J = 8.8 Hz, 1H); 4.69 (s, 2H), 4.10 (t, J = 6.8 Hz, 1H); 3.67 (t, J = 6.8 Hz, 1H); 3.19 (s, 6H); 13C NMR (CD3OD) δ: 156.4, 147.4, 137.2, 136.8, 134.4, 129.1, 129.0, 127.3, 126.1, 125.4, 123.6, 122.1, 112.5, 67.1, 62.7, 49.5, 34.8; MASS (ESI): m/z 340 ([MH]+, 100%); HRMS: C20H22ClN3 [M]+ Calcd 339.1502, Obsd. 339.1494.
4.7. N-(4-chlorobenzyl)pyridin-2-amine (20)
Compound 20 was obtained by following the known procedure; Yield 40%. 1H NMR 400 MHz (CDCl3) δ: 8.10 (m, 1H); 7.40 (dt, J = 7.2 Hz, J = 2 Hz, 1H); 7.29 (s, 4H); 6.60 (dt, J = 5.6 Hz, J = 1.2 Hz, 1H); 4.48 (d, J = 6.0 Hz, 1H); 4.86 (brS, NH, 1H). MASS (ESI): m/z 219 ([MH]+). (Known compound).
4.8. Cell culture
MiaPaCa-2-luc and Panc-1-luc cell lines were transfected with retroviral expression vector pMSCV-LucSh (provided by Dr. Andrew M. Davidoff, St. Jude Children’s Research Hospital), which contains a luciferase and zeocin resistance fusion gene that was used to create cell lines stably expressing luciferase. The C8161 human melanoma line was kindly provided by Dr. Bernard Weissman, University of North Carolina, Chapel Hill, North Carolina. MDA-MB-231, U87, A375, SU8686, AsPc, Panc 213 and Panc 10.5 were purchased from American Type Culture Collection (ATCC, Rockville, MD, USA). The MCF7-VEGFR3 cell line has been previously developed and characterized [34]. All cell lines were maintained in DMEM supplemented with 10% fetal bovine serum and incubated at 37 °C in 5% CO2.
4.9. Antibodies and reagents
FAK mouse monoclonal antibody (clone 4.47), VEGFR3 clone 9D9, phosphorylated tyrosine 4G10 (Millipore, MA). VEGFR3 and phosphorylated VEGFR3 rabbit polyclonal antibodies (Santa Cruz Biotechnology Inc, Santa Cruz, CA). Caspase-3 and PARP rabbit polyclonal antibodies (Cell Signaling Technology, Danvers, MA). GAPDH (Invitrogen, Grand Island, NY). Chlorpyramine hydrochloride (Sigma). Staurosporine (EMD Millipore Corporation, Billerica, MA), CellEvent™ Caspase-3/7 Green Detection Reagent and Tetramethylrhodamine, Methyl Ester, Perchlorate (TMRM) (Life Technologies, Grand Island, NY).
4.10. Cell viability assay
Cancer cell lines were seeded at a density of 5000 cells/well in 96 well plates and allowed to attach overnight. Drugs were added at the indicated concentrations and plates were incubated at 37 °C for 72 h. 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)- 2-(4-sulfophenyl)-2H-tetrazolium compound from Promega (Madison, WI) was added to each well and plates were incubated at 37 °C for approximately 1 h. Absorbance was read on a microplate reader (BioTek) at 570 nm. The absorbance values were used to calculate the viability and data was normalized to control values. Each experiment was repeated three times independently with 3–5 replicate wells for each drug concentration or DMSO/untreated controls.
4.11. Biolayer interoferometry
The binding studies were performed by Lampire Biological Laboratories, PA. The human FAT domain protein was biotinylated and the interaction between the small molecules and the FAT domain protein was determined by biolayer interferometry using an Octet Red 96 instrument (FortéBio Inc.). Super streptavidin biosensor tips (FortéBio, Inc) were pre-wetted with buffer (FortéBio) to establish a baseline before protein immobilization. Association and dissociation measurements were carried out over 210 s. Two concentrations of the various small molecules were used. Kinetic parameters (Kon, Koff, and Kd) measurements were calculated using the FortéBio data analysis software.
4.12. Western blotting
Indicated cancer cell lines were seeded at a density such that they were ~80% confluent after 24 h. Drugs were added and cells were incubated at 37 °C for 24 h. Cells were washed twice with ice cold PBS followed by cell lysis using 1% NP40 (Nonidet P-40) lysis buffer containing PhosSTOP and Complete inhibitor cocktail tablets (Roche). After centrifugation of the cell lysate, protein concentration of the supernatant fraction was determined using Pierce BCA Protein Assay Reagent A and B (Thermo Scientific). Briefly, protein extracts (30 μg) were separated on 7.5% or 4–20% mini-PROTEAN TGX gels (Bio-Rad). Proteins were transferred to a PVDF (polyvinylidene fluoride) membrane. Non-specific interactions were blocked using 5% BSA in 0.1% Tween 20/PBS solution for 1 h at room temperature. Specific primary antibodies were added to the membrane and allowed to incubate overnight at 4 °C. Peroxidase conjugated secondary antibodies were then exposed to the membrane for 1 h at room temperature, followed by enhanced chemiluminescence detection (ECL, GE Healthcare) using BioBlot BXR film for western blot (LPS, Inc) developed by Kodak X-OMAT Processor (SourceOne Healthcare Technologies).
4.13. Immunoprecipitation
Protein A/Plus Protein G Sepharose beads (Calbiochem) were added to the cell lysate suspension (500 μg or 800 μg protein) and the volume was made up to 500 μl using ice cold immunoprecipitation buffer (NP40 containing protease and phosphatase inhibitors) to pre-clear by constant rotation for 1 h at 4 °C. After centrifugation at 3000 rpm at 4 °C, the supernatant was collected and appropriate antibodies, FAK 4.47, VEGFR3 clone 9D9 or control IgG were added to facilitate protein-antibody binding. Next, after rotating the lysate containing antibodies for 2 h at 4 °C, beads were added and the resultant lysate mixture was placed on a rotator overnight at 4 °C. Beads were washed three times with ice cold immunoprecipitation buffer before boiling the samples with Laemmli Sample Buffer (Bio-Rad) for 5 min at 95 °C. Samples were loaded onto a 7.5% Mini-PROTEAN TGX gels (Bio-Rad) followed by the western blotting procedure described above.
4.14. Molecular modeling
Derivatives of 1 were built using ChemDraw 11.0 Suite (CambridgeSoft), and saved in MOL2 format. Structures were prepped for molecular docking using the “Dock Prep” function in the UCSF Chimera software [42]. As part of the docking preparations, partial charges were calculated using the AMBER module ANTECHAMBER using the Gasteiger method. All ligand structures were energy minimized following partial charge calculations. In silico scoring of the 1 analogs was performed with UCSF DOCK 6.6 (dock.compio.ucsf.edu). The target of the molecular docking was the FAT domain of FAK (PDB ID: 1K05) [43]. The docking parameters included testing up to 10,000 orientations of each derivative while allowing for ligand flexibility, followed by energy minimization of the docked pose. The scoring function used was the DOCK 6.6 grid scoring method, which consists of the non-bonded terms of the AMBER molecular mechanics force field. The final grid score was computed as the sum of van der Waals, electrostatic, and ligand internal energy terms for each derivative. Images depicting the predicted binding poses of various analogs to the FAT domain of FAK (surface and ribbon representation) were created using PyMOL v.1.3.
4.15. Colony formation
700 cells/well were seeded in 6 well plates, immediately followed by the addition of the compounds at specified concentrations. Plates were then incubated at 37 °C for 10–14 days or until colonies appeared. Cells were rinsed with PBS, 1% methylene blue staining solution was added to each well and plates were allowed to sit at room temperature for 1 h. The staining solution was aspirated, cells were washed with phosphate buffered saline (PBS) and distilled water and the plates were allowed to dry overnight. Plates with stained colonies were scanned using HP Scanjet G3110. Each experiment was done in duplicates and was repeated three times independently.
4.16. Kinase specificity
The Z’-LYTE SelectScreen® Single Point biochemical assay was performed by Life Technologies. Percent inhibition was calculated as per the Z’-LYTE® Data Analysis protocol (Life Technologies). Concentration of analog 29 used in the reaction mixture was 1 μM and ATP was 100 μM. Inhibition data represent mean kinase inhibition from two independent runs.
4.17. Detection of apoptosis
MiaPaCa-2-luc or MCF7-VEGFR3 cells were plated at a density of 10,000 cells/well and allowed to adhere overnight at 37 °C. Drugs were added and 24 h later, cells were rinsed with ice cold PBS, followed by addition of 5 μM CellEvent caspase-3/7 dye and 2.5 μM mitochondrial specific TMRM dye in PBS for 30 min at 37 °C. Plates were protected from light during incubation and subsequent steps. Cells were washed with ice cold PBS and images were captured on camera using a Zeiss Axio Observer A1 microscope. Images from multiple fields were taken across each sample. Apoptotic cells, whose nuclei were bright green, were counted from 6 to 8 fields for each condition. Percent apoptosis was calculated and data were normalized to controls. Representative images are shown from three independent experiments.
4.18. Statistical analysis
Comparisons between groups were made using a Students t test. Data were considered significant when p < 0.05.
Supplementary Material
Acknowledgments
This work was supported by the National Cancer Institute grant RO1-CA65910 (W.G.C), Photolitec Grant (R.K.P), and Roswell Park Cancer Institute. This work utilized core resources supported by the NCI Cancer Center Support Grant CA016156 (Trump, DL).
Abbreviations
- FAK
focal adhesion kinase
- VEGFR
vascular endothelial growth factor receptor
- FERM
4.1 protein–ezrin–radixin–moesin
- FAT
focal adhesion targeting
- EGFR
endothelial growth factor receptor
- c-MET
MNNG HOS Transforming gene
- PI3K
phosphatidylinositide 3-kinase
- RIP
receptor interacting protein kinase
- Grb2
Growth factor receptor bound protein 2
- ATP
adenosine 5′-triphosphate
- BH3
Bcl-2 homology domain
- Bcl-XL
B-cell lymphoma extra-large
- MDM2
mouse double minute 2 homolog
- TMRM
tetramethylrhodamine, methyl ester
Footnotes
Disclosure of potential conflicts of interest
William Cance and Elena Kurenova are stockholders and founders of CureFAKtor Pharmaceuticals, LLC. University of Florida and Roswell Park Cancer Institute have patents pending and awarded that are based on their work developing FAK inhibitors.
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.ejmech.2014.04.041.
References
- [1].Parsons JT. Focal adhesion kinase: the first ten years. Journal of Cell Science. 2003;116:1409–1416. doi: 10.1242/jcs.00373. [DOI] [PubMed] [Google Scholar]
- [2].Schaller MD, Borgman CA, Cobb BS, Vines RR, Reynolds AB, Parsons JT. pp125fak a structurally distinctive protein-tyrosine kinase associated with focal adhesions. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:5192–5196. doi: 10.1073/pnas.89.11.5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Zhao J, Guan J-L. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Reviews. 2009;28:35–49. doi: 10.1007/s10555-008-9165-4. [DOI] [PubMed] [Google Scholar]
- [4].Frame MC, Patel H, Serrels B, Lietha D, Eck MJ. The FERM domain: organizing the structure and function of FAK. Nature Reviews. Molecular Cell Biology. 2010;11:802–814. doi: 10.1038/nrm2996. [DOI] [PubMed] [Google Scholar]
- [5].Shen Y, Schaller MD. Focal adhesion targeting: the critical determinant of FAK regulation and substrate phosphorylation. Molecular Biology of the Cell. 1999;10:2507–2518. doi: 10.1091/mbc.10.8.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kurenova E, Xu L-H, Yang X, Baldwin AS, Jr., Craven RJ, Hanks SK, Liu Z.-g., Cance WG. Focal adhesion kinase suppresses apoptosis by binding to the death domain of receptor-interacting protein. Molecular and Cellular Biology. 2004;24:4361–4371. doi: 10.1128/MCB.24.10.4361-4371.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Cance WG, Kurenova E, Marlowe T, Golubovskaya V. Disrupting the scaffold to improve focal adhesion kinase-targeted cancer therapeutics. Science Signalling. 2013;6:pe10. doi: 10.1126/scisignal.2004021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].McLean GW, Carragher NO, Avizienyte E, Evans J, Brunton VG, Frame MC. The role of focal-adhesion kinase in cancer – a new therapeutic opportunity. Nature Reviews. Cancer. 2005;5:505–515. doi: 10.1038/nrc1647. [DOI] [PubMed] [Google Scholar]
- [9].Lark AL, Livasy CA, Dressler L, Moore DT, Millikan RC, Geradts J, Iacocca M, Cowan D, Little D, Craven RJ, Cance W. High focal adhesion kinase expression in invasive breast carcinomas is associated with an aggressive phenotype. Modern Pathology: An Official Journal of the United States and Canadian Academy of Pathology, Inc. 2005;18:1289–1294. doi: 10.1038/modpathol.3800424. [DOI] [PubMed] [Google Scholar]
- [10].Owens LV, Xu L, Craven RJ, Dent GA, Weiner TM, Kornberg L, Liu ET, Cance WG. Overexpression of the focal adhesion kinase (p125FAK) in invasive human tumors. Cancer Research. 1995;55:2752–2755. [PubMed] [Google Scholar]
- [11].Schlaepfer DD, Mitra SK, Ilic D. Control of motile and invasive cell phenotypes by focal adhesion kinase. Biochimica et Biophysica Acta. 2004;1692:77–102. doi: 10.1016/j.bbamcr.2004.04.008. [DOI] [PubMed] [Google Scholar]
- [12].van Nimwegen MJ, Verkoeijen S, van Buren L, Burg D, van de Water B. Requirement for focal adhesion kinase in the early phase of mammary adenocarcinoma lung metastasis formation. Cancer Research. 2005;65:4698–4706. doi: 10.1158/0008-5472.CAN-04-4126. [DOI] [PubMed] [Google Scholar]
- [13].Duxbury MS, Ito H, Benoit E, Zinner MJ, Ashley SW, Whang EE. RNA interference targeting focal adhesion kinase enhances pancreatic adenocarcinoma gemcitabine chemosensitivity. Biochemical and Biophysical Research Communications. 2003;311:786–792. doi: 10.1016/j.bbrc.2003.10.060. [DOI] [PubMed] [Google Scholar]
- [14].McLean GW, Komiyama NH, Serrels B, Asano H, Reynolds L, Conti F, Hodivala-Dilke K, Metzger D, Chambon P, Grant SG, Frame MC. Specific deletion of focal adhesion kinase suppresses tumor formation and blocks malignant progression. Genes & Development. 2004;18:2998–3003. doi: 10.1101/gad.316304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Smith CS, Golubovskaya VM, Peck E, Xu LH, Monia BP, Yang X, Cance WG. Effect of focal adhesion kinase (FAK) downregulation with FAK antisense oligonucleotides and 5-fluorouracil on the viability of melanoma cell lines. Melanoma Research. 2005;15:357–362. doi: 10.1097/00008390-200510000-00003. [DOI] [PubMed] [Google Scholar]
- [16].Golubovskaya VM, Kweh FA, Cance WG. Focal adhesion kinase and cancer. Histology and Histopathology. 2009;24:503–510. doi: 10.14670/HH-24.503. [DOI] [PubMed] [Google Scholar]
- [17].Siesser PMF, Hanks SK. The signaling and biological implications of FAK overexpression, %R 10.1158/1078-0432.CCR-06-0456. Cancer, Clinical Cancer Research: An Official Journal of the American Association for Cancer Research. 2006;12:3233–3237. doi: 10.1158/1078-0432.CCR-06-0456. [DOI] [PubMed] [Google Scholar]
- [18].Schultze A, Fiedler W. Clinical importance and potential use of small molecule inhibitors of focal adhesion kinase. Anti-Cancer Agents in Medicinal Chemistry. 2011;11:593–599. doi: 10.2174/187152011796817727. [DOI] [PubMed] [Google Scholar]
- [19].Hartmann JT, Haap M, Kopp HG, Lipp HP. Tyrosine kinase inhibitors – a review on pharmacology, metabolism and side effects. Current Drug Metabolism. 2009;10:470–481. doi: 10.2174/138920009788897975. [DOI] [PubMed] [Google Scholar]
- [20].Oliver G. Lymphatic vasculature development. Nature Reviews. Immunology. 2004;4:35–45. doi: 10.1038/nri1258. [DOI] [PubMed] [Google Scholar]
- [21].Saharinen P, Tammela T, Karkkainen MJ, Alitalo K. Lymphatic vasculature: development, molecular regulation and role in tumor metastasis and inflammation. Trends in Immunology. 2004;25:387–395. doi: 10.1016/j.it.2004.05.003. [DOI] [PubMed] [Google Scholar]
- [22].Smith NR, Baker D, James NH, Ratcliffe K, Jenkins M, Ashton SE, Sproat G, Swann R, Gray N, Ryan A, Jürgensmeier JM, Womack C. Vascular endothelial growth factor receptors VEGFR-2 and VEGFR-3 are localized primarily to the vasculature in human primary solid cancers. Clinical Cancer Research. 2010;16:3548–3561. doi: 10.1158/1078-0432.CCR-09-2797. [DOI] [PubMed] [Google Scholar]
- [23].He Y, Rajantie I, Pajusola K, Jeltsch M, Holopainen T, Yla-Herttuala S, Harding T, Jooss K, Takahashi T, Alitalo K. Vascular endothelial cell growth factor receptor 3-mediated activation of lymphatic endothelium is crucial for tumor cell entry and spread via lymphatic vessels. Cancer Research. 2005;65:4739–4746. doi: 10.1158/0008-5472.CAN-04-4576. [DOI] [PubMed] [Google Scholar]
- [24].Karpanen T, Egeblad M, Karkkainen MJ, Kubo H, Yla-Herttuala S, Jaattela M, Alitalo K. Vascular endothelial growth factor C promotes tumor lymphangiogenesis and intralymphatic tumor growth. Cancer Research. 2001;61:1786–1790. [PubMed] [Google Scholar]
- [25].Mandriota SJ, Jussila L, Jeltsch M, Compagni A, Baetens D, Prevo R, Banerji S, Huarte J, Montesano R, Jackson DG, Orci L, Alitalo K, Christofori G, Pepper MS. Vascular endothelial growth factor-C-mediated lymphangiogenesis promotes tumour metastasis. The EMBO Journal. 2001;20:672–682. doi: 10.1093/emboj/20.4.672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Von Marschall Z, Scholz A, Stacker SA, Achen MG, Jackson DG, Alves F, Schirner M, Haberey M, Thierauch KH, Wiedenmann B, Rosewicz S. Vascular endothelial growth factor-D induces lymphangiogenesis and lymphatic metastasis in models of ductal pancreatic cancer. International Journal of Oncology. 2005;27:669–679. [PubMed] [Google Scholar]
- [27].Stacker SA, Caesar C, Baldwin ME, Thornton GE, Williams RA, Prevo R, Jackson DG, Nishikawa S, Kubo H, Achen MG. VEGF-D promotes the metastatic spread of tumor cells via the lymphatics. Nature Medicine. 2001;7:186–191. doi: 10.1038/84635. [DOI] [PubMed] [Google Scholar]
- [28].Achen MG, Mann GB, Stacker SA. Targeting lymphangiogenesis to prevent tumour metastasis. British Journal of Cancer. 2006;94:1355–1360. doi: 10.1038/sj.bjc.6603120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Garces CA, Kurenova EV, Golubovskaya VM, Cance WG. Vascular endothelial growth factor receptor-3 and focal adhesion kinase bind and suppress apoptosis in breast cancer cells. Cancer Research. 2006;66:1446–1454. doi: 10.1158/0008-5472.CAN-05-1661. [DOI] [PubMed] [Google Scholar]
- [30].Arkin MR, Wells JA. Small-molecule inhibitors of protein-protein interactions: progressing towards the dream, Nature Reviews. Drug Discovery. 2004;3:301–317. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- [31].Kurenova EV, Hunt DL, He D, Magis AT, Ostrov DA, Cance WG. Small molecule chloropyramine hydrochloride (C4) targets the binding site of focal adhesion kinase and vascular endothelial growth factor receptor 3 and suppresses breast cancer growth in vivo. Journal of Medicinal Chemistry. 2009;52:4716–4724. doi: 10.1021/jm900159g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kurenova E, Liao J, He DH, Hunt D, Yemma M, Bshara W, Seshadri M, Cance WG. The FAK scaffold inhibitor C4 disrupts FAK-VEGFR-3 signaling and inhibits pancreatic cancer growth. Oncotarget. 2013;4:1632–1646. doi: 10.18632/oncotarget.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hann MM, Oprea TI. Pursuing the leadlikeness concept in pharmaceutical research. Current Opinion in Chemical Biology. 2004;8:255–263. doi: 10.1016/j.cbpa.2004.04.003. [DOI] [PubMed] [Google Scholar]
- [34].Kurenova EV, Hunt DL, He D, Fu AD, Massoll NA, Golubovskaya VM, Garces CA, Cance WG. Vascular endothelial growth factor receptor-3 promotes breast cancer cell proliferation, motility and survival in vitro and tumor formation in vivo. Cell Cycle. 2009;8 doi: 10.4161/cc.8.14.9101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Mitra SK, Mikolon D, Molina JE, Hsia DA, Hanson DA, Chi A, Lim ST, Bernard-Trifilo JA, Ilic D, Stupack DG, Cheresh DA, Schlaepfer DD. Intrinsic FAK activity and Y925 phosphorylation facilitate an angiogenic switch in tumors. Oncogene. 2006;25:5969–5984. doi: 10.1038/sj.onc.1209588. [DOI] [PubMed] [Google Scholar]
- [36].Deramaudt TB, Dujardin D, Hamadi A, Noulet F, Kolli K, De Mey J, Takeda K, Ronde P. FAK phosphorylation at Tyr925 regulates crosstalk between focal adhesion turnover and cell protrusion. Molecular Biology of the Cell. 2011;22(7):964–975. doi: 10.1091/mbc.E10-08-0725. mbc.E10-08-0725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Abdiche Y, Malashock D, Pinkerton A, Pons J. Determining kinetics and affinities of protein interactions using a parallel real-time label-free biosensor. the Octet, Analytical Biochemistry. 2008;377:209–217. doi: 10.1016/j.ab.2008.03.035. [DOI] [PubMed] [Google Scholar]
- [38].Belmokhtar CA, Hillion J, Segal-Bendirdjian E. Staurosporine induces apoptosis through both caspase-dependent and caspase-independent mechanisms. Oncogene. 2001;20:3354–3362. doi: 10.1038/sj.onc.1204436. [DOI] [PubMed] [Google Scholar]
- [39].Wilson CG, Arkin MR. Small-molecule inhibitors of IL-2/IL-2R: lessons learned and applied. Current Topics in Microbiology and Immunology. 2011;348:25–59. doi: 10.1007/82_2010_93. [DOI] [PubMed] [Google Scholar]
- [40].Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O’Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW, Rosenberg SH. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- [41].Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- [42].Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimeraea visualization system for exploratory research and analysis. Journal of Computational Chemistry. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- [43].Arold ST, Hoellerer MK, Noble ME. The structural basis of localization and signaling by the focal adhesion targeting domain. Structure. 2002;10:319–327. doi: 10.1016/s0969-2126(02)00717-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.