

Abstract
Islet antigen (IA)-2, IA-2β, and glutamate decarboxylase (GAD65) are major autoantigens in type 1 diabetes (T1D). Autoantibodies to these autoantigens appear years before disease onset and are widely used as predictive markers. Little is known, however, about what regulates the expression of these autoantigens. The present experiments were initiated to test the hypothesis that microRNAs (miRNAs) can target and affect the levels of these autoantigens. Bioinformatics was used to identify miRNAs predicted to target the mRNAs coding IA-2, IA-2β, and GAD65. RNA interference for the miRNA processing enzyme Dicer1 and individual miRNA mimics and inhibitors were used to confirm the effect in mouse islets and MIN6 cells. We show that the imprinted 14q32 miRNA cluster contains 56 miRNAs, 32 of which are predicted to target the mRNAs of T1D autoantigens and 12 of which are glucose-sensitive. Using miRNA mimics and inhibitors, we confirmed that at least 7 of these miRNAs modulate the mRNA levels of the T1D autoantigens. Dicer1 knockdown significantly reduced the mRNA levels of all 3 autoantigens, further confirming the importance of miRNAs in this regulation. We conclude that miRNAs are involved in regulating the expression of the major T1D autoantigens.—Abuhatzira, L., Xu, H., Tahhan, G., Boulougoura, A., Schäffer, A. A., Notkins, A. L. Multiple microRNAs within the 14q32 cluster target the mRNAs of major type 1 diabetes autoantigens IA-2, IA-2β, and GAD65.
Keywords: autoimmune diabetes, miRNA, miR-342, glucose-sensitive
Islet antigen (IA)-2, IA-2β, and glutamic acid decarboxylase (GAD)-65 are major autoantigens in type 1 diabetes (T1D). Autoantibodies against these autoantigens are found in 70–80% or more of patients with newly diagnosed T1D and appear years before the onset of clinical disease (1). These antibodies have become important diagnostic and predictive biomarkers. However, little is known about the regulation of these autoantigens.
IA-2 and IA-2β, encoded by protein tyrosine phosphatase receptor, type N (PTPRN) and PTPRN2, respectively, are integral membrane proteins of dense core vesicles (DCVs) and are highly expressed in neuroendocrine cells throughout the body (2). These 2 proteins are members of the transmembrane protein tyrosine phosphatase (PTP) family but are enzymatically inactive with known substrates because of a critical amino acid substitution in the PTP domain (3). Recent studies have shown that IA-2β has low phosphatidylinositol phosphatase activity (4). Knockout of Ptprn, Ptprn2, or both in mice leads to glucose intolerance and impaired insulin secretion as a result of a decrease in the half-life and number of DCVs in β cells in the pancreas (2, 5, 6), whereas overexpression of IA-2 in the mouse insulinoma (MIN6) cell line leads to an increase in insulin secretion and an increase in half-life and number of DCVs (2). In addition to its effect on insulin, overexpression of IA-2 in the MIN6 cell line promotes a preapoptotic state which, upon exposure to a high concentration of glucose, results in G2/M arrest and apoptosis (3).
GAD65, encoded by GAD2, is the major enzyme necessary for the production of the neurotransmitter GABA and is highly abundant in brain and pancreatic β cells (7). GABA is stored in synaptic-like vesicles (8) and may have a local paracrine effect on the regulation of glucagon secretion (9). In addition, it has been reported that GABA can play an inhibitory role in inflammation and autoimmunity in mice and can regulate the survival and replication of human β cells (10). In normal isolated islets, exposure to glucose for 6–96 h can specifically increase the transcription of Gad2 (coding GAD65) (11, 12). Likewise, glucose stimulation of MIN6 cells and mouse islets results in increased transcription of both Ptprn and Ptprn2 (coding IA-2 and IA-2β, respectively) (13). Thus, GAD65, IA-2, and IA-2β are all responsive to high levels of glucose and are linked to both secretory pathways and β-cell apoptosis/survival. Moreover, a whole-genome expression analysis comparing pancreatic islets from controls and deceased patients who had T1D found that the expression level of different T1D autoantigens—insulin, GAD65 (GAD2), IA-2 (PTPRN), IA-2β (PTPRN2), islet-specific glucose-6-phosphatase (IGRP; G6PC2), and ZnT8 (SLC30A8)—were markedly reduced in the T1D islets (14). These findings support the possibility that in addition to serving as disease markers, these proteins are aberrantly regulated in patients with T1D and, as such, play an active role in disease pathogenesis.
MicroRNAs (miRNAs) are endogenous, small, noncoding RNA sequences (21–23 nucleotides) that function as posttranscriptional regulators by binding the 3′UTR of their target mRNAs, leading to translational repression, transcript degradation, or both (15). The miRNA expression profile of β cells is altered in both type 1 and type 2 diabetes, suggesting that miRNAs are involved in the disease process (16). Indeed, miRNA inactivation in β cells of adult mice results in a diabetic phenotype (17) and several miRNAs (e.g., miR-375, -24, and -7) are crucial for normal pancreatic development and function (18). miRNAs also are known to play an important role in the development of the immune system and in innate and adaptive immune responses (19, 20). Increasing evidence also links miRNA deregulation to the pathogenesis of autoimmune disorders and to humoral responses against autoantigens (21, 22).
It is estimated that >40% of miRNAs are located near at least 1 other miRNA, and some of them are organized in larger clusters (23). Clustered miRNAs correlate highly in expression across 24 different human organs (24), where they may target the same gene or a group of functionally related genes in the same pathway (15, 25).
The present experiments were initiated to look for miRNAs that regulate the expression of the major T1D autoantigens IA-2, IA-2β, and GAD65, with emphasis on miRNA clusters that contain multiple miRNAs that are predicted to target one or more of these autoantigens.
MATERIALS AND METHODS
Bioinformatics identification of miRNAs, potential targets, and miRNA clustering
miRNAs predicted to target PTPRN, PTPRN2, and GAD2 were identified by using the Targetscan (release 5.2; Whitehead Institute for Biomedical Research, Cambridge, MA, USA) and miRWalk (Ruprecht-Karls-Universität Heidelberg, Mannheim, Germany) algorithms, combined with a literature query for pancreatic expression, glucose sensitivity, and T1D association. miRBase (Griffith-Junes Lab, University of Manchester, Manchester, United Kingdom) and GenBank (National Center for Biotechnology Information, Bethesda, MD, USA) were used to determine the genomic location and mature sequences of each of these miRNAs.
miRNA clusters were identified with MetaMirClust (http://fgfr.ibms.sinica.edu.tw/MetaMirClust/MirClustStat.php/, Center for Sustainability Science, Academia Sinica, Taiwan) with a threshold of 500 kb.
MIN6 cell line
Cells were cultured at 37°C in 95% air and 5% CO2 in DMEM supplemented with 15% heat-inactivated fetal bovine serum, 2 mM l-glutamine, and 0.1 mg/ml penicillin/streptomycin. Glucose concentration in MIN6 growth medium was 25 mM, unless otherwise stated.
Mouse islets
Experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals [National Institutes of Health (NIH), Bethesda, MD, USA] and have been approved by the Animal Subjects Review Board of the National Institute of Dental and Craniofacial Research/NIH. Islets from 3- to 6-mo-old C57BL/6 female mice were isolated, with slight modification of the published method (26), according to the manufacturer’s protocol for collagenase P (Roche, Indianapolis, IN, USA). The mice were euthanized, and collagenase P solution (1 mg/ml) was injected into the bile duct to inflate the pancreas, followed by removal and incubation at 37°C for digestion until the pancreas formed a milky solution with only a few clumps. After digestion, islets were purified in density gradient Histopaque1077, 1083, and 1119 (Sigma-Aldrich, St. Louis, MO, USA). Islets were manually selected, washed in Krebs-Ringer 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer, and cultured overnight in RPMI 1640 full medium (supplemented with 15% heat-inactivated fetal bovine serum, 100 U/ml penicillin, and 0.1 mg/ml streptomycin) containing 5 mM glucose.
Glucose-stimulated gene expression assay in MIN6 cells and mouse islets
Gene expression assays were performed as described elsewhere (13). MIN6 cells were washed with PBS ×1 and transferred to low-glucose DMEM (5 mM glucose) for 72 h, to adjust to normal glucose conditions (5 mM), because their regular growth medium contains high levels of glucose (25 mM). Cells were washed and incubated for 1 h in buffer A [5 mM KCl, 120 mM NaCl, 24 mM NaHCO3, 1 mM MgCl2, 2 mM CaCl2, 1 mg/ml ovalbumin, and 15 mM HEPES (pH 7.4)], supplemented with 3.3 mM glucose before stimulation with various concentrations of glucose (16.7 and 25 mM compared with 3.3 or 5 mM) for 4–72 h.
Mouse islets were plated in 24-well plates (30–40 islets per well), allowed 24 h recovery after isolation, washed twice in buffer A, and transferred to RPMI 1640 full medium containing 3.3 mM glucose overnight to stabilize basal gene expression. The islets were washed twice with buffer A without glucose and stimulated for 72 h in RPMI 1640 full medium, with either 3.3 or 16.7 mM glucose.
Total RNA, miRNA, and quantitative real-time PCR
Total RNA and miRNA were extracted with the miRNeasy Mini kit (Qiagen, Germantown, MD, USA), according to the manufacturer’s protocol. RNA (200–600 ng) was used for cDNA synthesis by the miScript II reverse transcription kit with HiFlex buffer (Qiagen) for simultaneous conversion of all RNA and miRNA species into cDNA. cDNA then was subjected to quantitative (q)real-time PCR analysis. Mature miRNA expression analysis was performed in duplicate or triplicate with the miScript SYBR Green PCR Kit (Qiagen), using forward primers that overlapped the specific mature miRNA sequences (primers available upon request); matched with Universal Primer (Qiagen); and normalized to the expression level of the U6 small nuclear RNA. Gene expression analysis was performed in duplicate or triplicate with mRNA-specific primers and SYBR Green PCR Master Mix (Life Technologies-Applied Biosystems, Grand Island, NY, USA) and normalized to β-actin or Dhfr as controls. Samples were run on the 7500 Real-Time PCR System (Life Technologies-Applied Biosystems) platform in the following conditions: initial activation step, 15 min at 95°C, followed by 40 cycles of denaturation for 15 s at 94°C, annealing for 30 s at 55°C, and extension for 30 s at 72°C. Relative expression was calculated based on the 2−ΔΔCt method, using the following formula: ΔΔCt = [ΔCt control gene (normal sample) − ΔCt control gene (treated sample)] – [ΔCt target gene (normal sample) − ΔCt target gene (treated sample)].
siRNA-mediated knockdown of Dicer1
For targeted knockdown of Dicer1 expression, MIN6 cells were transfected separately with 4 different Flexitube short interfering RNAs (siRNAs) (Qiagen) directed against the mouse Dicer1 sequence: target site1, ATCGATCATATGTCCAGTCTA; target site 2, AACGGATCTTACAGCAATTAA; target site 3, CACCATATCCATCGAGCTGAA; and target site 6: CAGGAGGAGGTACTTAGGAAA. As controls, we used a mock transfection and AllStars negative control siRNA (Qiagen), which has no homology with any known mammalian genes. Cells were transfected in replicate in 24-well plates with 50 nM siRNA in the presence of HiPerfect Transfection Reagent (Qiagen) and harvested for RNA 48 h after transfection. qReal-time PCR analysis for Dicer expression levels was performed according to the conditions described above, with Dicer1 QuantiTect Primer Assay (cat. QT00114702; Qiagen), and calculated relative to the expression level of Dhfr.
Western blot analysis
Whole-cell extracts from MIN6 cells were prepared in Laemmli sample buffer (Bio-Rad, Hercules, CA, USA). Equal amounts were run on Criterion precast gels (Bio-Rad), blotted to PVDF membrane (Millipore, Billerica, MA, USA), and treated with anti IA-2β (PTPRN2 antibody C18297; Assay Biotech, Sunnyvale, CA, USA) or anti-GAD65 (Pierce PA5-21297; by ThermoScientific, Rockford, IL, USA). The antibody α-tubulin (T5168, Sigma-Aldrich) was used as the loading control. Images were quantified with Image J software.
miRNA mimic and inhibitor transfections
MIN6 cells and fresh mouse pancreatic islets were transfected in replicate experiments with miRNA mimics, inhibitors, or controls (Qiagen, see list below), with HiPerfect Transfection Reagent (Qiagen), used according to the manufacturer’s instructions, and processed for expression and Western blot analyses 72 h after transfection. Because many of these miRNAs are induced by high levels of glucose, for the transfections with the different miRNA inhibitors and appropriate controls, MIN6 cells were grown in normal (5 mM) glucose conditions for 72 h before transfection and throughout the 72 h incubation time after transfection.
Mouse pancreatic islets were transfected as intact islets without disrupting their structure [adapted from Lakey et al. (27) with modifications]. Freshly isolated islets were plated in 24-well plates and allowed to recover for 24 h. Each well containing 40–50 islets was transfected separately (HiPerfect transfection reagent; Qiagen) with the different miRNA mimics, inhibitors, or negative controls and subjected to RNA and miRNA extraction (miRNeasy kit) and qReal-time PCR, 72 h after transfection.
The following miRNA inhibitors, mimics, and controls (Qiagen) were used for the transfection experiments: negative controls: Miscript inhibitors negative control and AllStars negative control siRNA. miRNA inhibitors: anti-mmu-miR-541-5p (cat. MIN0003170); anti-mmu-miR-665-3p (cat. MIN0003733); anti-mmu-miR-495-3p (cat. MIN0003456); anti-mmu-miR-370-3p (cat. MIN0001095); anti-mmu-miR-487b-3p (cat. MIN0003184); anti-mmu-miR-654-5p (cat. MIN0004897); anti-mmu-miR-758-3p (cat. MIN0003889); anti-mmu-miR-409-3p (cat. MIN0001090); anti-mmu-miR-377-3p (cat. MIN0000741); anti-mmu-miR-485-5p (cat. MIN0003128); anti-mmu-miR-337-3p (cat. MIN0000578); and anti-mmu-miR-342-3p (cat. MIN0000590). miRNA mimics: syn-mmu-miR-370-3p (cat. MSY0001095); syn-mmu-miR-665-3p (cat. MSY0003733); syn-mmu-miR-541-5p (cat. MSY0003170); syn-mmu-miR-495-3p (cat. MSY0003456); syn-mmu-miR-337-3p (cat. MSY0000578); and syn-mmu-miR-342-3p (cat. MSY0000590).
RNA immunoprecipitation PCR with anti-argonaute 2
miRNA:mRNA immunoprecipitation was performed with the Magna RNA immunoprecipitation PCR (RIP) kit (Millipore) according to the manufacturer’s protocol. MIN6 cells were transfected with either the negative control or miR-342-3p mimic. At 72 h after transfection, the cells were washed with ice-cold PBS ×1 and removed by scraping. The cell pellets were lysed in 200 µl complete RIP lysis buffer. A portion (10%) of the cell lysate was taken before precipitation, as the input fraction. Whole-cell lysate (100 µl) was incubated with 900 µl RIP buffer containing magnetic beads conjugated with 5 µg rat monoclonal anti-argonaute (Ago)2 antibody clone.11A9 (SAB4200085; Sigma-Aldrich) or negative control IgG and rotated overnight at 4°C (bound fraction). Samples were washed 6 times with RIP wash buffer and incubated with proteinase K buffer at 55°C for 30 min with shaking for protein digestion. Total RNA was extracted by the phenol:chloroform method. Pellets were precipitated in 10 µl of water, and equal amounts (40 ng) of RNA were used for reverse transcription and qReal-time PCR with mRNA-specific primers. Results were calculated as relative enrichment of bound over input fraction, using the equation: 2(input – bound) target gene/2(input – bound) control gene. Gapdh and β-actin served as controls.
Statistical analysis
One-way ANOVA, followed by the Bonferroni correction, was performed for multiple-group comparisons. The 2-tailed Student’s t test was used for comparison between 2 groups. Statistical analysis was performed with Prism 6 software (GraphPad, San Diego, CA, USA). P < 0.05 indicated statistical significance. To evaluate the density of miRNAs on 14q32, predicted to target PTPRN, PTPRN2, and GAD2, we used a χ2 test and wrote a program in C to simulate random placement of miRNAs in clusters.
RESULTS
The major T1D autoantigens and miRNA clusters
Using the TargetScan and miRWalk algorithms, we identified 419 miRNA precursors that are predicted to target the mRNA of PTPRN (coding IA-2), PTPRN2 (coding IA-2β), and/or GAD2 (coding GAD65). To our surprise, we found that 32 of these miRNAs are located on the same genomic region in human chromosome band 14q32, within the largest human miRNA cluster, also known as the C14MC (Chr 14 mega cluster) that contains 56 miRNAs. This cluster was of particular interest, because many of its miRNAs have been reported to be glucose sensitive and are expressed in pancreatic islets (28, 29). Furthermore, a Genome Wide Association Study has found that the 14q32 miRNA cluster is associated with altered susceptibility to T1D (30). Specifically, the most strongly associated SNP rs941576, located within this region (GRCh37.p5: 101306045), has been found to be involved with a paternally inherited risk of T1D (30).
To evaluate the significance of the observation that 32 of 56 miRNAs in the cluster are predicted totarget at least 1 of the 3 autoantigens, we reasoned that there are 1881 miRNA precursors across the human genome (as reported by miRBase). In addition to the 14q32 cluster, the only other cluster of size >32 is located on chromosome 19 and contains 49 miRNA precursors. The observation that 32 of 419 miRNAs, predicted to target at least 1 of the 3 autoantigens, reside in the 14q32 has a 1-sided P < 2e-09, by χ2 test. Furthermore, a more cautious simulation that treats as a positive event any random assignment of the miRNAs to clusters, in which 32 or more miRNAs of 419 fall in any large cluster (chromosome 14 or 19), yielded an empirical P < 8e-08, by simulation with 1 billion replicates. We therefore decided to focus on this miRNA cluster.
Analysis (Fig. 1A; Supplemental Table 1) revealed that 12 miRNAs from this cluster are predicted to target PTPRN2 (Fig. 1A, green ovals); 6 to target GAD2 (pink ovals), 2 to target PTPRN (blue ovals), 7 to target both PTPRN and PTPRN2 (half blue/half green oval), 3 to target both PTPRN2 and GAD2 (half green/half pink ovals), 1 to target both PTPRN and GAD2 (half blue/half pink oval), and 1 to target all 3 autoantigens (green/pink/ blue oval). We then prioritized these miRNAs based on their target conservation between human and mouse (Supplemental Table 1), autoantigenic target prediction rate, and possible relevance to T1D or autoimmunity in general. To confirm their interaction with Ptprn, Ptprn2, and Gad2, we focused on 12 of these miRNAs for further analyses.
Figure 1.
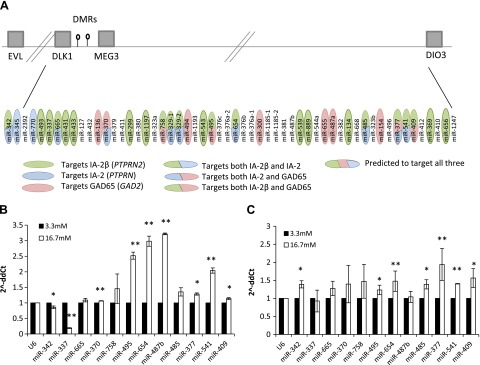
A) The 14q32 miRNA cluster. Gray boxes represent some of the genes on the cluster, lollipops represent the 2 DMRs (IG-DMR and MEG3-DMR). Distances and locations are not drawn to scale. Ovals represent miRNAs predicted to target PTPRN2 (green), PTPRN (blue), GAD2 (pink), or both PTPRN2 and PTPRN (half green/blue ovals), PTPRN and GAD2 (half blue/pink ovals), PTPRN2 and GAD2 (half green/pink ovals), or all 3 (green/blue/pink oval) based on TargetScan and miRWalk target prediction algorithms. B) miRNA expression after glucose stimulation in mouse islets. qReal-time PCR analysis of the 12 miRNAs in the 14q32 cluster (predicted to target Ptprn2, Ptprn, and Gad2) after glucose stimulation for 72 h (16.7 mM; white) compared with exposure to 3.3 mM glucose for 72 h (black) in freshly isolated mouse islets. *P < 0.04, **P < 0.005. C) miRNA expression after glucose stimulation in the MIN6 cell line. qReal-time PCR analysis of the same 12 miRNAs after glucose stimulation for 72 h (16.7 mM glucose, white vs. 3.3 mM, black) in the mouse insulinoma cell line MIN6. *P < 0.04, **P < 0.01. Fold change is presented as 2−ΔΔCt, relative to the small nuclear RNA gene U6. Vertical lines represent sd. Experiments were repeated at least 3 times.
Using qReal-time PCR we analyzed the expression levels of the 12 miRNAs in mouse islets (Fig. 1B) and in the MIN6 cell line (Fig. 1C). Upon stimulation of mouse islets with high glucose, mir-370, -495, -654, -487b, -377, -541, and -409 showed significant overexpression, whereas miR-342 and -337 showed a significant reduction in their expression (Fig. 1B). Some of these changes were also observed, although to a lesser extent, in MIN6 cells (Fig. 1C), with the exception of miR-342, which showed an increase in MIN6 cells. This discrepancy in expression levels could result from a lower sensitivity to glucose in the MIN6, which is a transformed cell line, as compared to freshly isolated mouse islets, which include a heterogeneous cell population. In addition, all 12 miRNAs gave detectable PCR products at a relatively low cycle number (17–26 cycles) in mouse islets and MIN6 cells. These results show that the 12 miRNAs from the 14q32 cluster, predicted to target the major T1D autoantigens, are highly expressed in islets and that at least 10 of the 12 miRNAs are sensitive to glucose levels in mouse islets and the MIN6 cell line.
To determine whether miRNAs play a role in autoantigen regulation, we transiently transfected MIN6 cells with different siRNAs designed to target mouse Dicer1 (NM_148948), which mediates the final process of miRNA maturation (31). We chose to focus on the short-term effect of transfection to avoid the involvement of downstream and possible feedback loop pathways on gene expression. At 48 h after transfection with Dicer1 siRNA, Dicer1 levels were found to be reduced by ∼40% and a significant reduction also was observed in the mRNA level of all 3 autoantigens, as compared to scrambled and mock transfection controls (Fig. 2A). These results show that miRNAs play a role in the regulation of the 3 T1D autoantigens, either directly or indirectly.
Figure 2.
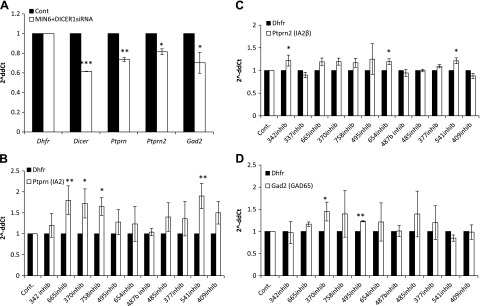
Endogenous target expression after transfection with Dicer siRNA and with the different miRNA inhibitors in MIN6 cells. qReal-time PCR analysis of expression. A) Transient transfection with siRNA for Dicer1, relative to transfection with a negative scrambled sequence and mock transfection. *P < 0.05, **P < 0.002, P < 1.3e-05. B) Ptprn expression levels after transfection with miRNA inhibitors for 72 h. *P < 0.03, **P < 0.002. C) Ptprn2 expression levels after transfection with miRNA inhibitors for 72 h. *P < 0.04. D) Gad2 expression levels after transfection with miRNA inhibitors for 72 h. *P < 0.04, **P < 0.002. Fold change is presented as 2−ΔΔCt relative to a transfection control samples and normalized to Dhfr or β-actin. Vertical lines represent sd. All experiments were repeated at least 3 times.
Ptprn, Ptprn2, and Gad2 levels are increased by miRNAs inhibitors in MIN6 cells
To assess whether the 12 miRNAs affect the expression of their predicted target genes (i.e., the genes coding the 3 major autoantigens IA-2, IA-2β, and GAD65) we used specific miRNA inhibitors for the respective miRNAs. After transfection of MIN6 cells with the different miRNA inhibitors, we analyzed the endogenous target gene expression. Ptprn levels showed an overall increase in expression with significant increases after transfection with miR-665, -370, -758, or -541 inhibitors (Fig. 2B). Ptprn2 showed a similarly significant increase in its expression after transfection with the miR-342, -654 or -541 inhibitors (Fig. 2C). Finally, Gad2 showed a significant increase in expression after transfection with miR-370 or -495 inhibitors (Fig. 2D). As a control, miR-487b, which was predicted not to target any of the 3 autoantigens, was included in the 12 miRNAs used for the inhibitor screen, and no change in the expression of Ptprn, Ptprn2, or Gad2 was observed after transfection with the miR-487b inhibitor.
Ptprn, Ptprn2, and Gad2 levels are decreased by miRNAs mimics in MIN6 cells
Based on the initial miRNA inhibitor screen, we focused on 5 of the miRNAs that showed a significant effect on at least 1 of their target genes. As seen in Fig. 3A, overexpression of miR-665 with its mimic led to a significant reduction in the endogenous level of Ptprn. Overexpression of miR-342 with its mimic resulted in a significant decrease in the expression of both Ptprn (Fig. 3A) and Ptprn2 (Fig. 3B). Likewise, overexpression of the miR-370 mimic led to a significant reduction in the expression of Gad2 (Fig. 3C). Because both miR-342 and -665 mimics led to a significant reduction in Ptprn and are also predicted to target Ptprn2, we asked whether transfection with both of these mimics would result in an even greater reduction in the levels of their targets. Indeed, the combination of miR-342 and -665 mimics led to an ∼70% reduction in Ptprn and ∼55% reduction in Ptprn2 levels, whereas no significant change was observed with Gad2, which was not a predicted target (Fig. 3D). The change in Ptprn and Ptprn2 showed that the combination of mimics was more effective than either mimic alone.
Figure 3.
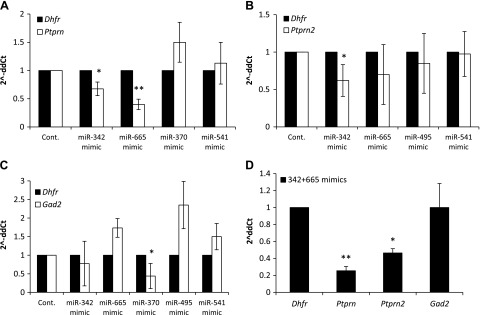
Endogenous target levels after overexpressing different miRNAs in MIN6 cells by using mimic transfections. A) Ptprn expression levels 72 h after transfection with miRNA mimics. *P < 0.05, **P < 0.01. B) Ptprn2 expression levels 72 h after transfection with miRNA mimics. *P < 0.04. C) Gad2 expression levels 72 h after transfection with miRNA mimics. *P < 0.05. D) mRNA expression level of all 3 autoantigens 72 h after transfection with a combination of miR-342 and -665 mimics. *P < 0.004, **P < 0.002. Analyses were performed by qReal-time PCR. Fold change is presented as 2−ΔΔCt relative to a transfection control samples and normalized to Dhfr or β-actin. Vertical lines represent sd. All experiments were repeated at least 3 times.
Mir-342 affects IA-2β through direct interaction with its mRNA in RNA-induced silencing complexes
To analyze in greater depth the interaction between a miRNA and its target gene, miR-342 was chosen for further study. MIN6 cells were transfected with the miR-342 mimic, and protein was extracted. Equal amounts of protein extracts were used for Western blot analysis with IA-2β antibody or α-tubulin (Fig. 4A, left) Our results (for all blots), quantified by Image J (Fig. 4A, right) showed an ∼50% reduction in IA-2β antigen levels after miR-342 overexpression.
Figure 4.

A) Effect of miR-342 mimic on IA-2β. A) IA-2β protein level 72 h after transfection with the miR-342 mimic in MIN6 cells. A representative Western blot of the endogenous IA-2β and α-tubulin, with protein extracts from MIN6 cells 72 h after transfection with a control sequence or miR-342 mimic (left). Average of adjusted density values after Image J quantification on 3 different blots normalized to α-tubulin (right). Experiments were repeated 3 times, and similar results were obtained. Error bar, sd. *P < 0.015. B) RIP PCR analysis, with anti-Ago2 antibody or anti-IgG (background control) used to assess the changes in enrichment of miRNA:mRNA in silencing complexes after miR-342 over expression for 48–72 h in MIN6 cells with Ptprn-, Ptprn-, and Gad2-specific primers, relative to the input fraction (10% of material before immunoprecipitation). Enrichment was quantified with qReal-time PCR. Fold change is presented as bound fraction over input fraction, normalized to Gapdh or β-actin. Error bars, sd. Experiments were repeated at least 3 times.
To confirm that there is a direct interaction between miR-342 and its targets, we used the RIP assay (32, 33), with an antibody that recognizes Ago2, a core component of the RNA-induced silencing complex (RISC) (33, 34), followed by reverse transcription and qReal-time PCR with primers specific for Ptprn2, Ptprn, and Gad2, to determine their levels of enrichment within the RISCs before and after overexpression of miR-342 in MIN6 cells. Our results showed that Ptprn, Ptprn2, and Gad2 (Fig. 4B) were present in the Ago2-IP (bound) fraction in MIN6 cells, as compared to their background levels in the anti-IgG-IP (bound) fraction, and that overexpression of miR-342 in these cells led to a significant enrichment of Ptprn mRNA and a modest but significant enrichment of Ptprn2 mRNA within the RISCs. As expected, since Gad2 is not a predicted target of miR-342, overexpression of miR-342 did not lead to an increase in the enrichment of Gad2 mRNA in the RISCs (Fig. 4B). These findings confirm that the mRNAs encoding IA-2 and IA-2β are direct targets of miR-342 and provide a further proof that IA-2, IA-2β, and GAD65 autoantigens are regulated by miRNAs.
DISCUSSION
In this study, we searched for miRNAs that are predicted to target the mRNAs of 1 or more of the major human T1D autoantigens (i.e., IA-2, IA-2β, and GAD65) and found that 32 such miRNAs are located in the same miRNA cluster (Fig. 1A). Many of these targets also are conserved between human and mouse (Supplemental Table 1). Although our search for clustered miRNA organization yielded several smaller clusters with miRNAs predicted to target these autoantigens, we chose to focus on the largest one, 14q32 (also known as C14MC), for several reasons. First, it had, by far, the largest number of miRNAs predicted to target the T1D autoantigens, many of its miRNAs were known to be expressed in pancreatic islets, and some of them were also known to be glucose-sensitive (29). Second, this cluster has been found to be associated with a paternally inherited risk for T1D (30) and several of its members have been found to be differentially expressed in patients with T1D (35, 36). Third, several of the 14q32 miRNAs target important genes involved in pancreatic development and regeneration (35, 37, 38) and are altered in immune responses and also in some immune-related diseases and disease models (39–41).
We then confirmed that the expression level of 12 of these miRNAs is altered in response to glucose stimulation in both freshly isolated mouse islets and in MIN6 cells (Fig. 1B, C) and also confirmed the effect of some of these miRNAs on the endogenous levels of their predicted autoantigenic targets by transfection with miRNA inhibitors and mimics (Figs. 2, 3). Our experiments further showed a bidirectional effect of 3 of these miRNAs on the mRNA of their targets: miR-665 on Ptprn, miR-342 on Ptprn2, and miR-370 on Gad2. Furthermore, transfection with a combination of miR-342 and -665 mimics showed a more robust effect on the mRNA of both Ptprn and Ptprn2. At the protein level, transfection with miR-342 mimic led to an ∼50% reduction in IA-2β protein and to a lesser extent after miR-665 and -541 mimics (data not shown). We were not able to detect significant changes in protein levels after transfection with the various miRNA inhibitors, suggesting that their effect may be too mild to detect in Western blot analysis. In fact, many miRNAs act more as rheostats than as on–off switches, and their downstream effects often are determined by the tissue context and the sum of effects on their specific set of target genes expressed in that tissue. However, a more robust effect of miRNAs is observed in certain situations, such as cancer, in which specific miRNAs may play a major role in the pathologic processes, and, depending on the type of tumor and set of target genes expressed, some miRNAs could have a dual life/death role as tumor suppressors or oncogenes (42). Nevertheless, our Dicer knockdown (Fig. 2A) and RIP (Fig. 4B) analyses showed that the mRNA levels of all 3 T1D autoantigens—Ptprn, Ptprn2 and Gad2—are regulated by miRNAs, both directly and indirectly.
Of the 12 miRNAs that we focused on, in the 14q32 cluster, inhibitors and mimics confirmed that 5 of them target Ptprn, 3 target Ptprn2, 2 target Gad2, and many more of them are predicted to target at least 1 autoantigen (Fig. 5). Thus, the 14q32 cluster represents a major network of miRNAs that target 3 of the major auotantigens in T1D. Because IA-2, IA-2β, and GAD65 all increase in response to glucose (11–13) and because most of the miRNAs in this study also showed an increase in response to glucose, we postulate that in normal conditions, these miRNAs play a role in keeping the level of the major T1D autoantigens from going out of balance as a result of alterations in glucose levels. In addition to glucose, a previous report notably showed that exposure of rat pancreatic islets to a combination of proinflammatory cytokines (IL-1β, IFN-γ, and TNF-α) leads to changes in the expression of several miRNAs, including 4 from the 14q32 cluster: miR-541, -337, -370, and -127 (43).
Figure 5.
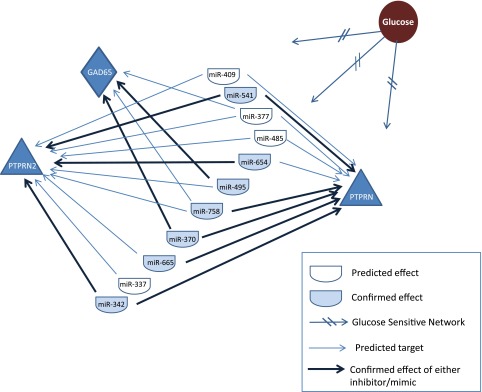
A glucose-sensitive miRNA network. Glucose (brown circle) affects the expression levels of (at least) 11 different miRNAs from the 14q32 cluster (half ovals) that in turn target the genes Ptprn, Ptprn2, and Gad2, which encode, respectively, 3 of the major T1D autoantigens: IA-2, IA-2β, and GAD65. Arrows: miRNA targets. Some of these miRNAs target more than 1 gene (2 or 3 arrows originating from the same miRNA). Dark arrows: confirmed targets.
Although the 32 miRNAs in the 14q32 cluster are only a fraction of the total miRNAs in the genome that are predicted to target the major T1D autoantigens, they reside within 1 cluster and are affected by similar stimuli, which increases their potential impact as a regulatory unit and their likelihood of being involved in complex phenotypes such as T1D or related autoimmune diseases, as compared to individual miRNAs scattered throughout the genome.
Of particular interest, the14q32 miRNA cluster is imprinted on the paternal chromosome and is expressed exclusively from the maternal chromosome with the exception of miR-342 which is embedded in the EVL gene. Experiments to evaluate the imprinting status of miR-342 under normal conditions have been inconclusive (44). Genomic imprinting is an epigenetic phenomenon in which a subset of mammalian autosomal genes are expressed in a parent-of-origin–specific manner. Most imprinted genes are clustered within large chromosomal domains. The imprinting of the 14q32 cluster is controlled, at least in part, by a germline-derived primary intergenic differentially methylated region (IG-DMR) and a postfertilization-derived secondary differentially methylated region [maternally expressed 3 (MEG3)-DMR], both are located between the paternally expressed delta like 1 homolog (DLK1) gene and the MEG3 gene (Fig. 1A) (44, 45). In addition, imprinting defects for various imprinted regions have been described in transient neonatal diabetes (46) and in several syndromes characterized by obesity or other diabetic features, such as Prader-Willi syndrome (47). Thus, inappropriate miRNA responses, possibly caused by aberrant methylation or imprinting defects affecting the 14q32 miRNA cluster, have the potential to provoke out-of-balance responses to glucose or other stimuli that could lead to aberrant autoantigen expression. miR-342 has been shown to be altered in regulatory T cells from patients with T1D (36), and 3 genetic variations in a putative promoter of miR-541 have been identified in patients with T1D by direct sequencing (35), suggesting a clinical link between miRNAs within the 14q32 cluster and T1D.
Because it is well known that many patients with T1D are at greater risk of development of other autoimmune diseases, we hypothesized that different miRNAs from the same14q32 cluster would target other disease-related autoantigens. Using the TargetScan and miRWalk algorithms, we found multiple miRNAs in the 14q32 cluster that are predicted to target transglutaminase 2 (TGM2), thyroid peroxidase (TPO), thyroglobulin (TG), and 21-hydroxylase (CYP21A2), which are the major autoantigens in celiac disease, autoimmune thyroid disease, and Addison’s disease, respectively (Table 1). Further investigation of this region could shed light on the mechanisms leading to the development of several autoimmune diseases in some patients with T1D, whereas these diseases do not develop in other patients with T1D. The fact that so many miRNAs in the 14q32 cluster are predicted to target autoantigens in T1D and related autoimmune diseases suggests that this network regulates autoimmune processes and that T1D and its related autoimmune diseases may share a common epigenetic mechanism.
TABLE 1.
miRNAs in the 14q32 cluster predicted to target autoantigens in T1D-related autoimmune diseases
| Disease | Autoantigen/associated auto-immune disease | 14q32 miRNAs predicted to target the autoantigen | 14q32 miRNA predicted to target IA-2, IA-2β, and GAD65 |
|---|---|---|---|
| Celiac disease | TGM2 | 342, 345, 770, 665, 431, 432, 370, 379, 299, 1197, 654, 1185-1, 1185-2, 539, 134, 485, 323b, 377, 541 | 342, 345, 770, 665, 431, 370, 299, 1197, 654, 539, 134, 485, 377, 541 |
| Autoimmune thyroid disease | TPO | 127, 370, 543, 544a | 370, 543 |
| TG | 493 | 493 | |
| Idiopathic Addison disease | CYP21A2 | 342, 127, 380, 494, 654, 539, 487a, 485, 377, 541, 412 | 342, 494, 654, 539, 487a, 485, 377, 541 |
Acknowledgments
The authors thank Dr. John Tsang for advice and meaningful discussions. This work was supported by the Intramural Research Program of the National Institute of Dental and Craniofacial Research, and by the Intramural Research Program of the National Library of Medicine, U.S. National Institutes of Health (Bethesda, MD, USA). L.A. and A.L.N. conceived of the project, designed and performed experiments, and wrote the manuscript. H.X., G.T., and A.B. performed experiments, participated in the discussions and reviewed the manuscript. A.A.S. performed the simulation, advised on other aspects of the bioinformatics and reviewed the manuscript. The authors declare no conflicts of interest.
Glossary
- Ago2
argonaute 2
- C14MC
chromosome 14 mega cluster
- CYP21A2
21-hydroxylase
- DCV
dense core vesicles
- Dicer1
ribonuclease type III gene
- GAD65
glutamic acid decarboxylase
- IA
islet antigen
- IG-DMR
intergenic differentially methylated region
- MEG3-DMR
maternally expressed gene 3 differentially methylated region
- MIN6
mouse insulinoma (cell line)
- miRNA
microRNA
- PTPRN
protein tyrosine phosphatase receptor, type N
- qReal-time PCR
quantitative real-time PCR
- RIP
RNA immunoprecipitation
- RISC
RNA-induced silencing complex
- siRNA
short interfering RNA
- T1D
type 1 diabetes
- TG
thyroglobulin
- TGM
transglutaminase
- TPO
thyroid peroxidase
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Pihoker C., Gilliam L. K., Hampe C. S., Lernmark A. (2005) Autoantibodies in diabetes. Diabetes 54(Suppl 2), S52–S61 [DOI] [PubMed] [Google Scholar]
- 2.Cai T., Hirai H., Zhang G., Zhang M., Takahashi N., Kasai H., Satin L. S., Leapman R. D., Notkins A. L. (2011) Deletion of Ia-2 and/or Ia-2β in mice decreases insulin secretion by reducing the number of dense core vesicles. Diabetologia 54, 2347–2357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harashima S. I., Harashima C., Nishimura T., Hu Y., Notkins A. L. (2007) Overexpression of the autoantigen IA-2 puts beta cells into a pre-apoptotic state: autoantigen-induced, but non-autoimmune-mediated, tissue destruction. Clin. Exp. Immunol. 150, 49–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caromile L. A., Oganesian A., Coats S. A., Seifert R. A., Bowen-Pope D. F. (2010) The neurosecretory vesicle protein phogrin functions as a phosphatidylinositol phosphatase to regulate insulin secretion. J. Biol. Chem. 285, 10487–10496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kubosaki A., Nakamura S., Notkins A. L. (2005) Dense core vesicle proteins IA-2 and IA-2beta: metabolic alterations in double knockout mice. Diabetes 54(Suppl 2), S46–S51 [DOI] [PubMed] [Google Scholar]
- 6.Saeki K., Zhu M., Kubosaki A., Xie J., Lan M. S., Notkins A. L. (2002) Targeted disruption of the protein tyrosine phosphatase-like molecule IA-2 results in alterations in glucose tolerance tests and insulin secretion. Diabetes 51, 1842–1850 [DOI] [PubMed] [Google Scholar]
- 7.Wang C., Mao R., Van de Casteele M., Pipeleers D., Ling Z. (2007) Glucagon-like peptide-1 stimulates GABA formation by pancreatic beta-cells at the level of glutamate decarboxylase. Am. J. Physiol. Endocrinol. Metab. 292, E1201–E1206 [DOI] [PubMed] [Google Scholar]
- 8.Sorenson R. L., Garry D. G., Brelje T. C. (1991) Structural and functional considerations of GABA in islets of Langerhans: beta-cells and nerves. Diabetes 40, 1365–1374 [DOI] [PubMed] [Google Scholar]
- 9.Arvan P., Pietropaolo M., Ostrov D., Rhodes C. J. (2012) Islet autoantigens: structure, function, localization, and regulation. Cold Spring Harb. Perspect. Med. 2, a007658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tian J., Dang H., Chen Z., Guan A., Jin Y., Atkinson M. A., Kaufman D. L. (2013) γ-Aminobutyric acid regulates both the survival and replication of human β-cells. Diabetes 62, 3760–3765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Björk E., Kämpe O., Karlsson F. A., Pipeleers D. G., Andersson A., Hellerström C., Eizirik D. L. (1992) Glucose regulation of the autoantigen GAD65 in human pancreatic islets. J. Clin. Endocrinol. Metab. 75, 1574–1576 [DOI] [PubMed] [Google Scholar]
- 12.Hagopian W. A., Karlsen A. E., Petersen J. S., Teague J., Gervassi A., Jiang J., Fujimoto W., Lernmark A. (1993) Regulation of glutamic acid decarboxylase diabetes autoantigen expression in highly purified isolated islets from Macaca nemestrina. Endocrinology 132, 2674–2681 [DOI] [PubMed] [Google Scholar]
- 13.Mandemakers W., Abuhatzira L., Xu H., Caromile L. A., Hébert S. S., Snellinx A., Morais V. A., Matta S., Cai T., Notkins A. L., De Strooper B. (2013) Co-regulation of intragenic microRNA miR-153 and its host gene Ia-2 β: identification of miR-153 target genes with functions related to IA-2β in pancreas and brain. Diabetologia 56, 1547–1556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Planas R., Carrillo J., Sanchez A., de Villa M. C., Nuñez F., Verdaguer J., James R. F., Pujol-Borrell R., Vives-Pi M. (2010) Gene expression profiles for the human pancreas and purified islets in type 1 diabetes: new findings at clinical onset and in long-standing diabetes. Clin. Exp. Immunol. 159, 23–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bartel D. P. (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–297 [DOI] [PubMed] [Google Scholar]
- 16.Guay C., Regazzi R. (2013) Circulating microRNAs as novel biomarkers for diabetes mellitus. Nat. Rev. Endocrinol. 9, 513–521 [DOI] [PubMed] [Google Scholar]
- 17.Melkman-Zehavi T., Oren R., Kredo-Russo S., Shapira T., Mandelbaum A. D., Rivkin N., Nir T., Lennox K. A., Behlke M. A., Dor Y., Hornstein E. (2011) miRNAs control insulin content in pancreatic β-cells via downregulation of transcriptional repressors. EMBO J. 30, 835–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu Y., You W., Wang H., Li Y., Qiao N., Shi Y., Zhang C., Bleich D., Han X. (2013) MicroRNA-24/MODY gene regulatory pathway mediates pancreatic β-cell dysfunction. Diabetes 62, 3194–3206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ceribelli A., Satoh M., Chan E. K. (2012) MicroRNAs and autoimmunity. Curr. Opin. Immunol. 24, 686–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iborra M., Bernuzzi F., Invernizzi P., Danese S. (2012) MicroRNAs in autoimmunity and inflammatory bowel disease: crucial regulators in immune response. Autoimmun. Rev. 11, 305–314 [DOI] [PubMed] [Google Scholar]
- 21.Kapsogeorgou E. K., Gourzi V. C., Manoussakis M. N., Moutsopoulos H. M., Tzioufas A. G. (2011) Cellular microRNAs (miRNAs) and Sjögren’s syndrome: candidate regulators of autoimmune response and autoantigen expression. J. Autoimmun. 37, 129–135 [DOI] [PubMed] [Google Scholar]
- 22.Pauley K. M., Cha S., Chan E. K. (2009) MicroRNA in autoimmunity and autoimmune diseases. J. Autoimmun. 32, 189–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Altuvia Y., Landgraf P., Lithwick G., Elefant N., Pfeffer S., Aravin A., Brownstein M. J., Tuschl T., Margalit H. (2005) Clustering and conservation patterns of human microRNAs. Nucleic Acids Res. 33, 2697–2706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baskerville S., Bartel D. P. (2005) Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. RNA 11, 241–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grün D., Wang Y. L., Langenberger D., Gunsalus K. C., Rajewsky N. (2005) microRNA target predictions across seven Drosophila species and comparison to mammalian targets. PLOS Comput. Biol. 1, e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ravier M. A., Rutter G. A. (2010) Isolation and culture of mouse pancreatic islets for ex vivo imaging studies with trappable or recombinant fluorescent probes. Methods Mol. Biol. 633, 171–184 [DOI] [PubMed] [Google Scholar]
- 27.Lakey J. R., Young A. T., Pardue D., Calvin S., Albertson T. E., Jacobson L., Cavanagh T. J. (2001) Nonviral transfection of intact pancreatic islets. Cell Transplant. 10, 697–708 [PubMed] [Google Scholar]
- 28.Hennessy E., Clynes M., Jeppesen P. B., O’Driscoll L. (2010) Identification of microRNAs with a role in glucose stimulated insulin secretion by expression profiling of MIN6 cells. Biochem. Biophys. Res. Commun. 396, 457–462 [DOI] [PubMed] [Google Scholar]
- 29.Tang X., Muniappan L., Tang G., Ozcan S. (2009) Identification of glucose-regulated miRNAs from pancreatic beta cells reveals a role for miR-30d in insulin transcription. RNA 15, 287–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wallace C., Smyth D. J., Maisuria-Armer M., Walker N. M., Todd J. A., Clayton D. G. (2010) The imprinted DLK1-MEG3 gene region on chromosome 14q32.2 alters susceptibility to type 1 diabetes. Nat. Genet. 42, 68–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hammond S. M. (2005) Dicing and slicing: the core machinery of the RNA interference pathway. FEBS Lett. 579, 5822–5829 [DOI] [PubMed] [Google Scholar]
- 32.Keene J. D., Komisarow J. M., Friedersdorf M. B. (2006) RIP-ChIP: the isolation and identification of mRNAs, microRNAs and protein components of ribonucleoprotein complexes from cell extracts. Nat. Protoc. 1, 302–307 [DOI] [PubMed] [Google Scholar]
- 33.Tan L. P., Seinen E., Duns G., de Jong D., Sibon O. C., Poppema S., Kroesen B. J., Kok K., van den Berg A. (2009) A high throughput experimental approach to identify miRNA targets in human cells. Nucleic Acids Res. 37, e137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang W. X., Wilfred B. R., Hu Y., Stromberg A. J., Nelson P. T. (2010) Anti-argonaute RIP-ChIP shows that miRNA transfections alter global patterns of mRNA recruitment to microribonucleoprotein complexes. RNA 16, 394–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Han B., Shi X., Peng Q., Gao W. (2012) Study on genetic bariance of miR-541 in type 1 diabetes. ISRN Endocrinol. 2012, 630861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hezova R., Slaby O., Faltejskova P., Mikulkova Z., Buresova I., Raja K. R., Hodek J., Ovesna J., Michalek J. (2010) microRNA-342, microRNA-191 and microRNA-510 are differentially expressed in T regulatory cells of type 1 diabetic patients. Cell. Immunol. 260, 70–74 [DOI] [PubMed] [Google Scholar]
- 37.Joglekar M. V., Parekh V. S., Hardikar A. A. (2007) New pancreas from old: microregulators of pancreas regeneration. Trends Endocrinol. Metab. 18, 393–400 [DOI] [PubMed] [Google Scholar]
- 38.Simion A., Laudadio I., Prévot P. P., Raynaud P., Lemaigre F. P., Jacquemin P. (2010) MiR-495 and miR-218 regulate the expression of the Onecut transcription factors HNF-6 and OC-2. Biochem. Biophys. Res. Commun. 391, 293–298 [DOI] [PubMed] [Google Scholar]
- 39.Dai Y., Huang Y. S., Tang M., Lv T. Y., Hu C. X., Tan Y. H., Xu Z. M., Yin Y. B. (2007) Microarray analysis of microRNA expression in peripheral blood cells of systemic lupus erythematosus patients. Lupus 16, 939–946 [DOI] [PubMed] [Google Scholar]
- 40.Fernandez-Valverde S. L., Taft R. J., Mattick J. S. (2011) MicroRNAs in β-cell biology, insulin resistance, diabetes and its complications. Diabetes 60, 1825–1831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang L., Qin Y., Tong L., Wu S., Wang Q., Jiao Q., Guo Z., Lin L., Wang R., Zhao W., Zhong Z. (2012) MiR-342-5p suppresses coxsackievirus B3 biosynthesis by targeting the 2C-coding region. Antiviral Res. 93, 270–279 [DOI] [PubMed] [Google Scholar]
- 42.Liu H., Kohane I. S. (2009) Tissue and process specific microRNA-mRNA co-expression in mammalian development and malignancy. PLoS One 4, e5436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bravo-Egana V., Rosero S., Klein D., Jiang Z., Vargas N., Tsinoremas N., Doni M., Podetta M., Ricordi C., Molano R. D., Pileggi A., Pastori R. L. (2012) Inflammation-mediated regulation of microRNA expression in transplanted pancreatic islets. J. Transplant. 2012, 723614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seitz H., Royo H., Bortolin M. L., Lin S. P., Ferguson-Smith A. C., Cavaillé J. (2004) A large imprinted microRNA gene cluster at the mouse Dlk1-Gtl2 domain. Genome Res. 14, 1741–1748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kagami M., O’Sullivan M. J., Green A. J., Watabe Y., Arisaka O., Masawa N., Matsuoka K., Fukami M., Matsubara K., Kato F., Ferguson-Smith A. C., Ogata T. (2010) The IG-DMR and the MEG3-DMR at human chromosome 14q32.2: hierarchical interaction and distinct functional properties as imprinting control centers. PLoS Genet. 6, e1000992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Temple I. K., Shield J. P. (2010) 6q24 transient neonatal diabetes. Rev. Endocr. Metab. Disord. 11, 199–204 [DOI] [PubMed] [Google Scholar]
- 47.Shemer R., Hershko A. Y., Perk J., Mostoslavsky R., Tsuberi B., Cedar H., Buiting K., Razin A. (2000) The imprinting box of the Prader-Willi/Angelman syndrome domain. Nat. Genet. 26, 440–443 [DOI] [PubMed] [Google Scholar]