

Abstract
We investigated the role of periostin, an extracellular matrix protein, in the pathophysiology of osteoarthritis (OA). In OA, dysregulated gene expression and phenotypic changes in articular chondrocytes culminate in progressive loss of cartilage from the joint surface. The molecular mechanisms underlying this process are poorly understood. We examined periostin expression by immunohistochemical analysis of lesional and nonlesional cartilage from human and rodent OA knee cartilage. In addition, we used small interfering (si)RNA and adenovirus transduction of chondrocytes to knock down and up-regulate periostin levels, respectively, and analyzed its effect on matrix metalloproteinase (MMP)-13, a disintegrin and MMP with thrombospondin motifs (ADAMTS)-4, and type II collagen expression. We found high periostin levels in human and rodent OA cartilage. Periostin increased MMP-13 expression dose [1–10 µg/ml (EC50 0.5–1 μg/ml)] and time (24–72 h) dependently, significantly enhanced expression of ADAMTS4 mRNA, and promoted cartilage degeneration through collagen and proteoglycan degradation. Periostin induction of MMP-13 expression was inhibited by CCT031374 hydrobromide, an inhibitor of the canonical Wnt/β-catenin signaling pathway. In addition, siRNA-mediated knockdown of endogenous periostin blocked constitutive MMP-13 expression. These findings implicate periostin as a catabolic protein that promotes cartilage degeneration in OA by up-regulating MMP-13 through canonical Wnt signaling.—Attur, M., Yang, Q., Shimada, K., Tachida, Y., Nagase, H., Mignatti, P., Statman, L., Palmer, G., Kirsch, T., Beier, F., Abramson, A. B. Elevated expression of periostin in human osteoarthritic cartilage and its potential role in matrix degradation via matrix metalloproteinase-13.
Keywords: posttraumatic osteoarthritis, chondrocytes, Wnt signaling, ADAMTS
Osteoarthritis (OA) is characterized by progressive loss of cartilage from the articulating surfaces of diarthrodial joints. Although the etiology of the disease is unknown, it is widely accepted that these degenerative changes arise from an imbalance of synthetic and degradative pathways that control cartilage extracellular matrix (ECM) metabolism (1–4). Progress in the development of disease-modifying treatments for OA depends on increased understanding of the molecular changes that lead to progressive disease within the joint tissues, including articular chondrocytes, bone, and synovium (5). Recent investigations have identified matrix proteins, proteinases, chemokines, mitochondrial proteins, and other molecules that are differentially expressed in diseased OA cartilage, as compared with non-OA controls (6, 7).
Chondrocyte-mediated production, assembly, and turnover of the ECM are essential in maintaining cartilage homeostasis. However, during the progression of OA, the cartilage ECM is actively remodeled by chondrocytes in response to abnormal mechanical force and inflammatory conditions. Recent studies have shown the emerging role of the matricellular and ECM proteins, including tenascin, F-spondin (Spon1), thrombospondin, osteopontin, and perlecan, as well as collagen fragments, which regulate prominent chondrocyte functions, such as apoptosis and activation of latent TGF-β, pro-MMP, and a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS) (8–10). To identify the genes involved in OA pathogenesis, we performed Affymetrix (Santa Clara, CA, USA) gene expression analyses of nondiseased and OA cartilage, to characterize functional molecules that are up-regulated in OA (11–13). In the current study, human OA cartilage expressed high levels of the ECM protein, periostin [osteoblast-specific factor 2 or fasciclin (FAS)-1-like protein], which promotes degradation of proteoglycans and collagen in cartilage via induction of ECM-degrading enzymes, suggesting a previously unrecognized role in the pathogenesis of osteoarthritis. Periostin expression is induced by TGF-β and signals through integrin αvβ3 in osteoblast and fibroblast cells (14, 15).
We therefore examined the effect of periostin on activation of signaling pathways in chondrocytes. Furthermore, the integrin and matrix protein interactions are shown to activate MAPKs, leading to increased NF-κB signaling. Gene expression studies in surgically induced rodent models of OA have shown that these degradative pathways are linked to dysregulation of Wnt, hedgehog, and TGF-β signaling, thought to play an important role in OA progression. In addition, integrins can activate Wnt signaling via crosstalk with other surface receptors (16, 17). In our current study, periostin-induced MMP-13 expression was inhibited by CCT031374 hydrobromide, an inhibitor of the canonical Wnt/β-catenin signaling pathway. Thus, periostin warrants further investigation as a valuable therapeutic target for treatment of OA.
MATERIALS AND METHODS
Reagents
All culture media and fetal bovine serum (FBS) were purchased from Life Technologies (Gaithersburg, MD, USA); recombinant IL-1β was from PeproTech (Rocky Hill, NJ, USA); the human periostin ELISA kit was from Adipogen International, Inc. (San Diego, CA, USA); carrier-free recombinant periostin expressed in NSO mouse myeloma cells (cat. no. 3548-F2), MMP-13 ELISA kits, the Wnt inhibitor CCT031374 hydrobromide (cat. no. 4675/10), and the NF-кB MG pathway inhibitor 132 (cat. no. 1748) were from R&D Systems (Minneapolis, MN, USA); the MEK1/2 [ERK1/2-PD98059 (cat. no. P215)] and p38 SB202190 (cat. no. S7067) inhibitors, rabbit anti-periostin antibody (SAB4300696 for Western blot analysis), and HPA012306 for immunohistochemistry were from Sigma-Aldrich (St. Louis, MO, USA); anti-β-catenin antibodies (cat. no. 9562), phospho-MAPK family antibodies [pERK1/2, p38, and pJNK (cat. no. 9910S)], pAKT (cat. no. 9271), and pan-AKT (cat. no. C67E7) were from Cell Signaling Technology, Inc. (Danvers, MA, USA); and the C1,C2 collagen fragment ELISA kit was from IBEX (Montréal, QC, Canada).
Human knee cartilage procurement
Normal human knee cartilage (subjects aged 50–88 yr, 50% female) samples were procured from the National Disease Research Interchange (NDRI, Philadelphia, PA, USA). Human OA cartilage was obtained under the guidelines of the Institutional Review Board of New York University (NYU) School of Medicine for use of surgically discarded human tissues, from the knees of patients with a diagnosis of advanced OA (patients aged 50–85 yr, 75% female) who underwent knee replacement surgery. OA knee cartilage samples were obtained from the operating room within 2–4 h of surgery and normal cartilage within 2–16 h of death. Non-OA cartilage is defined as cartilage with no histologic evidence of degeneration, procured from donors with no history of joint diseases. All normal cartilage samples were obtained from the knee, and no tissue processing was performed on the samples before their use in the study. OA specimens obtained at the time of surgery were examined by the authors and confirmed to have gross evidence of OA (thinning or localized loss of cartilage and focal eburnation) and the histologic diagnosis of OA or normal cartilage was confirmed by pathologists at NYU Hospital for Joint Diseases. For RNA expression studies, the specimens were immediately snap-frozen at −140°C.
Isolation and culture of primary human and bovine chondrocytes
Chondrocytes were isolated from human OA knee cartilage and from bovine cartilage, obtained from young calves’ hooves, by digestion with collagenase P (0.1%) for 12–16 h in Ham’s F-12 medium. The cells were cultured as monolayers in chondrocyte growth medium (Cell Application, Inc., San Diego, CA, USA) for 3 d before initiation of the experiments. To avoid complications associated with chondrocyte dedifferentiation, all experiments were performed on nonpassaged, primary cells within 5 d of cell harvest. For gene and protein expression studies, we performed 8–72 h stimulation studies. For matrix (collagen and aggrecan) degradation we collected samples at 3–7 d after stimulation.
Explant organ culture of cartilage and analysis of mediators
Femoral heads were isolated from 6-wk-old C57BL/6 mice, and human knee articular cartilage was obtained as surgical discards from patients undergoing knee replacement surgery. Organ culture was performed as described (18, 19). In brief, cartilage was cut into 3 mm discs, and 4–6 discs (∼100 mg) were placed in 2 ml Ham’s F-12 medium supplemented with 0.1% human albumin, with or without modulators for 24–72 h. The conditioned medium was collected at regular intervals for measurement of markers associated with cartilage degradation. ELISA kits were used to measure secreted pro-MMP-13 and periostin. ProMMP-13 and periostin concentrations were determined with the respective standard curves, according to the manufacturer’s instructions. In addition, the supernatants were analyzed for levels of C1,2C proteolytic fragments of collagen and sulfated glycosaminoglycans (sGAGs).
Chondrocyte activation and gene expression
Freshly isolated human chondrocytes were seeded into 6-well plates (5 × 105 cells/well) in chondrocyte medium and, after 48 h, were incubated in serum-free Ham’s F-12 medium containing 0.2% LPS-free human albumin for additional 24 h. The cells were treated with recombinant periostin (1–10 µg/ml) or IL-1β (10 ng/ml) in fresh serum-free medium for 4–24 h. The supernatants were collected and stored for measurement of MMP-13 and periostin. The cells were lysed in Trizol, and total RNA was isolated with RNeasy (Qiagen, Valencia, CA, USA), according to the manufacturer’s instructions.
Cartilage protein extraction and Western blot analysis
Cartilage samples were ground into a fine powder with Freezer Mill (Thomas Scientific, Swedesboro, NJ, USA), and proteins were extracted with 10 mM Tris buffer containing 0.2 M lithium bromide, 1% deoxycholate, 0.2% Triton X-100, 1 mM EDTA, and protease inhibitor cocktail (Calbiochem, San Diego, CA, USA) for 4 h at 4°C. The extracts were centrifuged briefly to generate clear supernatants and dialyzed against PBS for 24 h to remove the salts. Proteins were extracted from monolayer chondrocytes by using the M-PER reagent (Pierce, Rockford, IL, USA). All protein concentrations were measured with the bicinchoninic acid (BCA) reagent (Pierce). For electrophoresis, 100 µg of cartilage extract protein or 20–100 µg of cell extract protein was electrophoresed in an SDS 10% polyacrylamide gel. The proteins were transferred onto nitrocellulose membranes, and the blots were probed with the appropriate antibodies. The antigen–antibody complexes were visualized with an ECL detection kit (Thermo Fisher Scientific, Rockford, IL, USA).
Determination of sGAG release
The amount of sGAG released into the culture medium from human OA cartilage or mouse femoral head explants was measured by the 1,9-dimethylmethylene blue (DMB) detection method (20). Absolute amounts were determined by interpolation to a standard curve of chondroitin sulfate and the results expressed as micrograms of sGAG per gram of cartilage.
Human cartilage RNA labeling and hybridization of microarrays
Five micrograms of total RNA was used for double-stranded (ds) cDNA synthesis by the Gibco BRL SuperScript Choice System (Life Technologies). Purified ds cDNA was used for synthesis of biotin-labeled cRNA with the ENZO BioArray High-yield RNA transcript labeling kit (Affymetrix). The cRNA was purified with the RNeasy kit (Qiagen), fragmented at 95°C for 35 min for target preparation, and hybridized against the human U133A array according to the manufacturer’s instructions, and the expression levels were normalized (9).
Differential gene expression analysis in rat anterior cruciate ligament and medial meniscectomy OA models
All experimental procedures were performed in accordance with the in-house guidelines of the Institutional Animal Care and Use Committee (IACUC) of Daiichi Sankyo Co., Ltd.. Surgical procedures were performed on male 8-wk-old Sprague-Dawley rats under general anesthesia (21–24). For the medial meniscectomy (MM) model, the medial collateral ligament was transected, and the medial meniscus was removed completely. For the anterior cruciate ligament (ACL) model, only the ACL was transected. The sham operation was performed by using the same approach, but without ligament transection or meniscectomy. On d 3 and wk 2, 4, 6, 8, and 10, cartilage was harvested from the surgically altered knees on the medial side of the tibial plateau (MTP) and femoral condyle of 5 rats, immersed immediately in Trizol reagent (Life Technologies) at 4°C, and stored at −80°C until RNA extraction.
Total RNA from rat cartilage samples was extracted as recommended by the manufacturer and further purified with the RNeasy MinElute Cleanup Kit (Qiagen). Five rats were used for each experimental model (sham surgery, ACL, and MM). We prepared cDNA from each animal independently, and performed GeneChip analysis with the pooled cDNAs from the same experimental model. Each data point represents one time point from one experimental model. Total RNA quality was verified with a Bioanalyzer 2100 (Agilent, Santa Clara, CA, USA). Two rounds of amplification and labeling were performed and 10 μg labeled and fragmented cRNAs were hybridized onto a GeneChip Rat Genome 230 2.0 Array (Affymetrix). Posthybridization staining and washing were conducted, and arrays were scanned with a GeneChip Scanner 3000 (Affymetrix). Probe set signals were calculated by using the Affymetrix MAS5 algorithm from files scaled to a target signal value of 100.
Expression of periostin, MMP-13, and GAPDH in medial and lateral cartilage of normal (no surgery; n = 4), sham surgery (n = 5), and MM (n = 4) rats was assessed by real-time quantitative PCR (RT-qPCR), performed with predesigned TaqMan primers from Life Technologies-Applied Biosystems (Foster City, CA, USA).
Differential gene expression analysis in an ACL transection/partial medial meniscectomy OA model
An additional OA model was performed in the laboratory of F.B. at the University of Western Ontario (London, ON, Canada), in accordance with protocols approved by the Animal Care and Use Committee at the University of Western Ontario. OA was surgically induced in the right knees of 300–325 g Sprague-Dawley rats by performing ACL transection combined with partial medial meniscectomy (ACLT/PMX), followed by forced mobilization, according to the methods described by Appleton et al. (25). Articular cartilage samples were harvested from surgically altered and sham surgery control joints and processed for RNA extraction and microarray analysis (26).
Periostin and MMP-13 immunostaining of OA cartilage in a PMX mouse model
Periostin and MMP-13 localization in murine OA cartilage was determined after surgical induction of OA in 12-wk-old male (C57BL/6) mice by medial collateral ligament transection and PMX (27). In brief, the animals were placed under general anesthesia, the medial collateral ligament was transected, and the medial meniscus was partially removed, aided by a surgical microscope. A sham operation was performed on the contralateral knee joint, taking the same approach but with no ligament transection or meniscectomy. The animals were then allowed unrestricted activity and food and water ad libitum. At 8 wk after surgery, the mice were euthanized, and the knee joints were harvested and fixed in 4% paraformaldehyde, decalcified in 0.2 mM EDTA, and embedded in paraffin. Sections (4 μm thick) perpendicular to the cartilage surface were incubated with sheep testicular hyaluronidase (2 mg/ml; Sigma-Aldrich) in PBS (pH 7.5), for 30 min at 37°C and immunostained with the Histostain SP Kit (Zymed Laboratories, Inc., San Francisco, CA, USA) as recommended by the manufacturer. Control sections were incubated with nonimmune rabbit serum.
Preparation of adenoviral vectors and transduction of primary chondrocytes
Full-length human periostin cDNA was PCR amplified and cloned into the pAdCMV-V5-DEST shuttle vector (Life Technologies). The adenovirus was propagated in human embryonic kidney (HEK)293 cells grown in DMEM supplemented with 10% FBS and antibiotics. The empty vector was used to generate a control virus. Two days after infection, the HEK293 cells were harvested for cell lysate preparation. The cells were frozen and thawed 3 times and centrifuged at 15,000 g for 10 min. The crude viral lysate containing the recombinant adenovirus was filtered through a 0.2 μm pore size filter and stored at −70°C. With control β-galactosidase adenovirus, we observed a 15–20% transduction efficiency in OA chondrocyte cultures. Before infection, primary chondrocytes were grown in monolayer cultures for 48 h, washed twice with serum-free DMEM, and transduced with the adenovirus encoding periostin (Ad-periostin) or the control adenovirus devoid of periostin cDNA (Ad-mock) for 4 h at 37°C, followed by washing twice with serum-free DMEM. The cells were grown in serum-free medium for an additional 24–48 h, after which the culture supernatant was collected, and the cells were lysed in Trizol for RNA expression analysis.
Periostin knockdown
In primary chondrocyte cultures, prevalidated periostin small interfering (si)RNAs (cat no. 4392420; Life Technologies) and control (scrambled) siRNAs (cat no. 4390843) were delivered into primary OA chondrocytes with TransIT-siQUEST Transfection Reagent, according to the manufacturer’s instructions (Mirus Bio, LLC, Madison, WI, USA). The cells were transfected with 2.5 nM siRNA oligos for 12 h in serum-free medium and grown in fresh serum-free medium for an additional 48–72 h. The culture supernatants were collected, and the cells were lysed in TRI reagent for RNA isolation to confirm periostin knockdown. ELISA of the supernatants was used to determine MMP-13 and periostin levels.
MMP-13 assay
After viral infection, culture supernatants were incubated with assay buffer [50 mM TrisCl, 10 mM CaCl2, 150 mM NaCl, and 0.05% Brij-35 (pH 7.5)] supplemented with 1 mM p-aminophenylmercuric acetate (APMA) for 2 h at 37°C. The samples (20 µl) were then incubated with the MMP-13-specific fluorogenic substrate MOCAc-Pro-Cha-Gly-Nva-His-Ala-Dap(Dnp)-NH2 [(7-methoxycoumarin-4-yl)acetyl-l-prolyl-l-cyclohexylalanyl-glycyl-l-norvalyl-l-histidinyl-l-alanyl-Nβ-(2,4-initrophenyl)-l-2,3-diaminopropionic amide] (cat. no. SMO-3682-PI; Peptides International, Louisville, KY, USA) at 37°C for 1 h, and fluorescence was measured with a Synergy HT (Biotek, Winooski, VT, USA) microplate reader at 340 nm (excitation) and 405 nm (emission).
Gene expression analysis by RT-qPCR
RNA was extracted from monolayer cultures of chondrocytes with TRI reagent (Life Technologies) and further purified with a micro RNeasy column (Qiagen) according to the manufacturer’s instructions. One microgram of total RNA was primed with oligo (dT)-18 primers, and cDNA was synthesized according to the manufacturer’s instructions (Clontech, Mountain View, CA, USA). Predesigned TaqMan primers for RT-qPCR were purchased from Life Technologies-Applied Biosystems. RT-qPCR reactions were run on an ABI Prism 7300 sequence detection system (Life Technologies-Applied Biosystems). Relative gene expression levels were calculated using the 2−ΔΔCt method (28). Target mRNA levels were normalized to GAPDH mRNA.
T-cell factor reporter plasmid assay
Human C28/I2 chondrocyte cells were transiently transfected with the TOPflash luciferase reporter plasmid (EMD Millipore, Billerica, MA, USA), which contains T-cell factor (TCF) response elements, and a secreted form of human placental alkaline phosphatase (pSEAP) control vector for transfection efficiency. Transfected cells were incubated with periostin (5 µg/ml) for 24 h, and the relative luciferase activity of cell lysates was measured with a luciferase assay kit (Promega, Madison, WI, USA) on a Tristar LB 941 microplate luminometer (Berthold Technologies, Oak Ridge, TN, USA). pSEAP activity in the supernatants was measured with the TROPIX chemiluminescence CSPD assay kit (Life Technologies-Applied Biosystems).
Cell metabolic activity assay
Primary OA chondrocytes were grown in 24-well tissue culture plates for 18–24 h and treated with a Wnt inhibitor (CCT031374 hydrobromide) or vehicle control for an additional 24 h. The cells were washed once with PBS and incubated in DMEM containing 0.1 μg/ml of 1 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Sigma-Aldrich) for 1 h, after which the medium was removed, and 100 μl DMSO (Sigma-Aldrich) was added to each well. After thorough mixing on an orbital shaker for 1 min, absorbance was measured at 540 nm with a SpectraMax Plus microplate reader (Molecular Devices, Sunnyvale, CA, USA). Triplicate samples were assayed.
Statistical analysis
Each experiment was performed with 3–5 independent chondrocyte preparations from 4–5 individual cartilage samples, and each experimental condition was analyzed in duplicate or triplicate samples. Data are expressed as means ± sd. All statistical analyses were performed with GraphPad Prism, ver. 4.0. Nonparametric 2-tailed Student’s t test for pairwise comparisons between groups, 1-way ANOVA with post-Bonferroni multiple comparison test, and postlinear trend analysis were used as appropriate, with significance set at P < 0.05.
RESULTS
Expression of periostin in human OA cartilage
We investigated the relative expression levels of periostin mRNA in normal and OA cartilage by gene expression profiling. Comparison of RNA from pooled samples of OA and normal cartilage using U95Av2 and U133A Affymetrix microarray revealed significant (∼2- to 4-fold) up-regulation of periostin in diseased tissue (P < 0.0043; Fig. 1A). We further validated the elevated expression of periostin mRNA by RT-qPCR analysis of 19 individual OA cartilage samples vs. 13 non-OA controls (P = 0.0003; Fig. 1B). Periostin mRNA was up-regulated more than 21-fold (mean) in OA cartilage relative to non-OA cartilage samples (Fig. 1B). A 4-fold up-regulation of periostin protein levels was also observed in OA cartilage by ELISA (P = 0.04; Fig. 1C). Western blotting analysis of the same protein samples with periostin monoclonal antibody showed a band with an apparent Mr of 90,000, consistent with the observed size of periostin. In nondiseased cartilage (N), periostin was absent in 3 of 4 samples; conversely, in OA cartilage, periostin was abundant in 5 of 6 samples (Fig. 1D).
Figure 1.
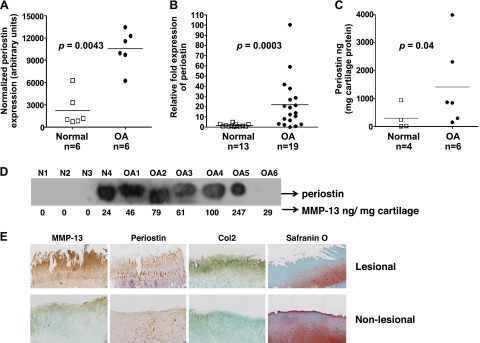
Elevated expression of periostin in OA cartilage. A) Total RNA was isolated from 6 pools of normal and OA cartilage. For each pool, 5–10 individual cartilage samples (1 g each) were pooled. RNA was hybridized against human U133A arrays. After normalization, periostin expression was expressed in arbitrary units. B) RT-qPCR analysis of periostin mRNA in 13 normal and 19 OA cartilage specimens from human knee joints. ELISA (C) and Western blot analysis (D) of periostin levels in protein extracts from 6 OA and 4 non-OA cartilage samples. ELISA periostin values were normalized to total cartilage protein. Horizontal lines: mean values. E) Immunohistochemical analysis of lesional and nonlesional OA knee cartilage identified cell-associated periostin in lesional areas. OA knee cartilage from a total knee replacement was sectioned and stained for periostin with Vectastain reagents (Vector Laboratories, Burlingame, CA, USA). A, B) Mann-Whitney test. C) Nonparametric 2-tailed Student’s t test.
Further evidence of periostin up-regulation in human OA was provided by immunohistochemical staining of OA cartilage sections with anti-periostin antibody. Pericellular and cell-associated staining of periostin was observed in the superficial, middle, and deep zones of lesional OA cartilage, whereas only superficial zone staining was observed in the nonlesional areas of the same specimen (Fig. 1E).
Periostin gene expression is increased in rat articular cartilage after surgical induction of OA
We performed gene profiling studies to assess differential mRNA expression in established rodent models of OA. OA was induced by either ACLT or MM, and medial and lateral articular cartilages were harvested and analyzed for global gene expression using Affymetrix RAE230_2.0 GeneChips. Relative to the nonsurgical sham controls, periostin mRNA levels increased in both medial (Fig. 2A) and lateral (Fig. 2B) articular cartilage in both surgical models. In both cases, elevation of periostin mRNA levels appeared to reflect disease severity.
Figure 2.

Periostin is up-regulated in rat knee cartilage after induction of OA. OA was induced by MM and ACL surgery. At the indicated times, periostin expression in the articular cartilage of surgical knees and sham surgery controls was determined by microarray analysis of samples from both OA models. Higher levels of periostin mRNA were detected in both the medial (A) and lateral (B) articular cartilage of surgical animals than in those of sham surgery controls in both surgical models.
In the MM model, in which joints rapidly develop OA within 4 wk, periostin expression reached its peak level 1 wk after surgery, faster than in the ACL model, which requires up to 12 wk for cartilage degeneration (21, 22). We also confirmed elevated expression of periostin and MMP-13 in the MM model by RT-qPCR (Fig. 3). The results showed that both periostin and MMP-13 increased significantly with OA progression during the early period after surgery (1–2 wk). Furthermore, the relative expression levels of MMP-13 and periostin were higher in the medial cartilage (Fig. 3A, B) compared with the lateral (Fig. 3C, D) cartilage. In this MM model, periostin expression correlated significantly with MMP-13 expression (r = 0.71; P = 0.0005).
Figure 3.

RT-qPCR validation of MMP-13 (A, C) and periostin (B, D) expression in medial (A, B) and lateral (C, D) cartilage of rats in the normal (n = 4), sham (n = 5), and MM (n = 4) groups. The graphs show MMP-13/GAPDH and periostin/GAPDH ratios. *P < 0.05; **P < 0.01 by the nonparametric Mann-Whitney U test for pair-wise comparisons between the sham and MM groups.
In the slower ACL model, peak levels of periostin were observed at 3–6 wk, suggesting a correlation between periostin level and disease progression. Periostin up-regulation was accompanied by increased expression of established hypertrophic chondrocyte markers, including MMP-13, alkaline phosphatase, Col10A1, and Spon1 (data not shown).
We also confirmed elevated expression of periostin in a third surgical OA model in rats, ACLT/PMX, whereby OA is induced by a combination of ACL transection, partial meniscectomy, and forced mobilization (25, 26). Figure 4 shows a 3- to 4-fold increase of periostin expression in the surgically altered (ipsilateral) joints relative to that in nonsurgical (contralateral) and sham control joints 4 wk after surgery. Thus, altogether, these data show that periostin expression is elevated in both human OA cartilage and in rodent surgical models of OA.
Figure 4.

In a third surgical model (ACLT/PMX), OA was induced in Sprague-Dawley rats by a combination of ACLT, PMX, and forced mobilization. The knees of sham surgery animals and the nonsurgical/contralateral knees of the ACLT/PMX rats served as controls. Rats underwent forced mobilization (3 times, 30 min/wk) on a rotator (25). Periostin expression in the articular cartilage of ipsilateral (surgical), contralateral, and sham surgery controls was determined by Affymetrix microarray analysis 4 wk after surgery. Horizontal lines: mean values. Mann-Whitney U test.
Periostin and MMP-13 protein expression is elevated in mice after surgical induction of OA
Periostin expression was also examined in a murine model of PMX (27). Eight weeks after PMX surgery, the MTP and femoral condyles of the knee joints showed a major loss of articular cartilage, consistent with a previous report (27) and reduced safranin O staining in the remaining cartilage relative to the sham surgery control cartilage. Increased MMP-13 and periostin immunostaining was evident in the articular cartilage of the MTP and femoral condyles 8 wk after surgery compared with that in the controls (Fig. 5). Periostin staining was localized mostly around the superficial zone of articular cartilage, whereas MMP-13 staining extended from the superficial to the middle zones. In contrast, the articular cartilage of sham surgery knee joints showed no detectable periostin or MMP-13 immunostaining. Because periostin staining was prevalent in lesional OA cartilage and partially overlapped with immunostaining of MMP-13, we next investigated whether periostin elicits catabolic effects in OA chondrocytes.
Figure 5.
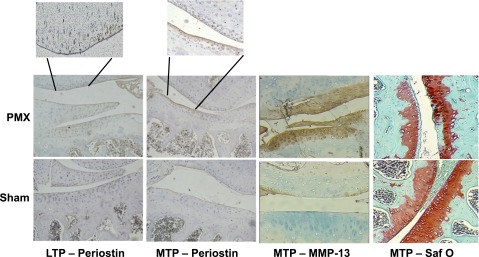
PMX OA model in 12-wk-old C57BL/6 male mice. The contralateral knee joints of these mice underwent control sham surgery. Red-brown indicates positive staining for periostin. Intense periostin immunostaining was evident in the articular cartilage of both the medial (MTP) and lateral tibial plateau (LTP) and in the femoral condyles 8 wk after surgery. Intense MMP-13 staining was also evident in the LTP. Conversely, the articular cartilage of sham surgery knee joints showed no MMP-13 or periostin immunostaining. The rightmost panels show cartilage degeneration in surgical joints at 8 wk after surgery, as evidenced by safranin O staining.
Periostin promotes catabolic effects in human cartilage explant cultures
Periostin aggravates atherosclerotic valve degeneration by inducing angiogenesis and MMP expression, particularly MMP-13 (29). Both processes have been implicated in the degeneration of OA cartilage. To evaluate the effect of periostin on cartilage, we cultured human OA cartilage explants with recombinant periostin and measured collagen degradation and sGAG release in culture supernatants. For this purpose, we used periostin concentrations (1–5 µg/ml) within the range of those used in published in vitro studies (0.25–5 μg/ml) (15, 29–33).
Collagen degradation, measured by ELISA of the proteolytic fragment C1,2C in culture supernatants, was increased approximately 2-fold after periostin treatment for 72 h (Fig. 6A; P < 0.05, 1 μg/ml periostin vs. control; P < 0.01, 5 μg/ml periostin vs. control). Likewise, periostin treatment led to a significant (∼20–30%) dose-dependent increase in sGAG release compared with that in the untreated controls (Fig. 6B; P < 0.05, 1 μg/ml periostin vs. control; P < 0.01, 5 μg/ml periostin vs. control).
Figure 6.

Periostin augments collagen and proteoglycan degradation in OA cartilage explant cultures. Human OA cartilage explants from 4 independent patients were incubated with recombinant periostin (1 and 5 µg/ml) in duplicate wells of 24-well plates in serum-free medium. Medium was changed after 48 h and analyzed 72 h after addition of periostin. A) Type II collagen degradation (n = 4) assayed using the C1,2C-based ELISA. B) sGAG release (n = 4) measured by the DMB assay. IL-1 was used as a positive control for induction of matrix degradation. By post-ANOVA linear trend test between control and periostin treatment, the correlation was R2 = 0.195; P = 0.0024 (C1,2C), and R2 = 0.173; P = 0.0009 (sGAG), respectively. *P < 0.05; **P < 0.01.
Periostin (1–10 µg/ml) was as potent an inducer of catabolic activity as 1 ng/ml IL-1β, which was used as a positive control. Thus, these findings indicate that periostin promotes collagen and proteoglycan degradation in OA cartilage.
Periostin induces MMP-13 expression and proteoglycan loss in mouse femoral head explant cultures
To investigate whether periostin elicits degradative effects on intact, nondiseased joint cartilage in situ, we used an explant model of femoral head cartilage from 6-wk-old C57BL/6 mice. Femoral head explants were incubated with periostin (1 μg/ml), and the conditioned medium was analyzed for MMP-13 levels after 72 h by immunoblotting and for sGAG release after 144 h, as evidence of cartilage loss. Periostin increased MMP-13 secretion and sGAG release >25-fold (Fig. 7; P = 0.0001).
Figure 7.

Periostin induces MMP-13 and matrix degradation in mouse femoral head explant culture. Femoral heads were incubated in 0.1 ml of serum-free DMEM in 96-well plates (2 heads/well), with or without 1 µg/ml of recombinant periostin. Four cultures were assayed for (A) MMP-13 expression 3 d after addition of periostin and (B) sGAG release into the medium 7 d after addition. Nonparametric 2-tailed Student's t test.
Exogenous periostin and adenovirus-mediated overexpression of periostin induce expression of ECM-degrading enzymes
Because periostin increases collagen degradation in OA cartilage, we examined its effect on the production of catabolic/proinflammatory mediators. In human OA cartilage explant culture, addition of periostin to the culture medium (1, 5, 10 µg/ml) increased pro-MMP-13 secretion 2- to 8-fold (Fig. 8A) in a dose-dependent manner. We next analyzed periostin-induced MMP-13 expression in human OA chondrocytes and found that periostin (1 and 10 µg/ml) also increased pro-MMP-13 levels in culture supernatants in a time- and dose-dependent manner (Fig. 8B, C).
Figure 8.

Periostin augments MMP-13 secretion in OA cartilage explants and chondrocyte cultures. A) OA MMP-13 concentration in the culture supernatant of 3 independent cartilage explants incubated with periostin (1–10 µg/ml) for 24 h. By post-ANOVA linear trend test, the correlation was R2 = 0.476; P = 0.0003. B, C) ELISA of MMP-13 levels in 3 independent primary OA chondrocyte cultures in duplicate wells incubated with each concentration of periostin (1–10 μg/ml) in serum-free medium for 8 h (B) or 24 h (C). By post-ANOVA linear trend test between control and periostin treatment, the correlation was R2 = 0.476; P = 0.0003 (8 h) and R2 = 0.394; P = 0.0003 (24 h). **P < 0.01.
We then investigated whether adenovirally expressed endogenous periostin cDNA induces a similar effect. For this purpose, we transduced normal bovine and OA human chondrocytes with Ad-periostin or, as a control, Ad-mock, and measured periostin and MMP-13 in the conditioned medium. In Ad-periostin-transduced OA chondrocytes, periostin expression was 1.5- to 5-fold higher than in Ad-mock-transduced controls (Fig. 9A). Elevated periostin expression was paralleled by increased mRNA and protein levels of MMP-13 (2- to 3-fold) (Fig. 9A, B), as well as ADAMTS-4 (4-fold) and ADAMTS-5 (1.5-fold), whereas COL2A1 levels decreased (Fig. 9B). Similar results were obtained with Ad-periostin transduction of nondiseased bovine chondrocytes (Fig. 9C, D). A fluorescent peptide-based MMP-13 activity assay was used to determine the effect of adenovirus-mediated periostin expression on MMP-13 levels in bovine chondrocytes. Total MMP-13 activity was >10-fold higher in Ad-periostin- than in control Ad-mock-transduced cells (Fig. 9D). Therefore, these data showed that exogenous periostin or adenovirus-mediated periostin expression up-regulate MMP-13 and ADAMTS in both OA and normal chondrocytes.
Figure 9.

Periostin induces MMP-13 and ADAMTS-4 and decreases type II collagen expression in human OA and bovine chondrocytes. Three independent human OA (A, B) and bovine (C, D) chondrocytes were transduced in triplicate with Ad-mock or Ad-periostin for 4 h, and incubated in serum-free medium for 5 d. MMP-13, ADAMTS4, ADAMTS5, and Col 2 were also measured by RT-qPCR. Relative gene expression levels were calculated by the 2−ΔΔCt method (28). E) siRNA-mediated downregulation of endogenous periostin reduces constitutive MMP-13 expression in OA chondrocytes. MMP-13 and periostin levels in OA chondrocyte culture supernatants after transfection of periostin siRNA or control siRNA. Data are means ± sd of duplicate samples from 2 independent cultures.
Knockdown of endogenous periostin attenuates constitutive MMP-13 expression
Because periostin overexpression augmented MMP-13 in OA chondrocytes (Fig. 9A, B), we examined whether siRNA-mediated silencing of endogenous periostin blocks constitutive MMP-13 expression. Transfection of human OA chondrocytes with siRNA to periostin decreased levels of both endogenous periostin and MMP-13 by 50% (Fig. 9E), showing that endogenous periostin regulates MMP-13 expression. Therefore, elevated expression of periostin accelerates cartilage loss and OA progression.
Induction of MMP-13 by recombinant periostin in chondrocytes is dose dependent and sustained up to 72 h
To understand whether periostin’s effect on MMP-13 expression is transient or sustained, we treated human OA chondrocytes with different concentrations (1–10 μg/ml) of recombinant periostin and analyzed MMP-13 levels (24–72 h) in the culture supernatant. Periostin induced MMP-13 expression in OA chondrocytes in a dose- and time-dependent manner (Fig. 10).
Figure 10.

Dose- and time-dependent induction of MMP-13 expression by periostin in OA chondrocyte cultures. MMP-13 concentration in the culture supernatant of 3 independent OA chondrocyte cultures incubated with periostin (1–10 µg/ml) for 24–72 h. Data are means ± sd. *P < 0.05, by 2-tailed Student’s t test, treated sample vs. corresponding control.
We used normal human and mouse chondrocytes to determine the EC50 of periostin induction of MMP-13. We performed experiments with normal human chondrocytes and found that MMP-13 expression and activity increased with higher periostin concentration (Table 1). These findings indicate sustained catabolic effects of periostin on chondrocytes in vitro. Periostin increased MMP-13 activity in normal human chondrocytes significantly at 500 and 1000 ng/ml, but not at 10 or 100 ng/ml. These results indicate that 500–1000 ng/ml periostin administered as a recombinant protein is sufficient to induce MMP-13 in normal chondrocytes.
TABLE 1.
Effects of periostin on MMP-13 expression and activity in human chondrocytes
| Treatment group | pro-MMP-13 ELISA (pg/ml) |
MMP-13 activity (FU) |
|||
|---|---|---|---|---|---|
| Mean | ±sd | Mean | ±sd | P | |
| Control | 17.05 | 0.68 | 650.13 | 64.81 | N/A |
| Periostin (ng/ml) | |||||
| 10 | 17.53 | 0.06 | 555.77 | 266.52 | 0.606 |
| 100 | 18.69 | 0.37 | 681.42 | 293.12 | 0.872 |
| 500 | 20.99 | 4.50 | 1011.70 | 52.979 | 0.002* |
| 1000 | 23.81 | 3.04 | 1088.30 | 150.84 | 0.024* |
| 5000 | 28.98 | 0.57 | 1594.40 | 200.62 | 0.009* |
Normal human chondrocytes were isolated from knee cartilage. The chondrocytes (2 × 105 cells/well) were grown in 24-well plates and treated with different concentrations of recombinant periostin (10–5000 ng/ml) in 0.5 ml serum-free medium. The culture supernatants were collected and activated with 1 μM APMA, and total MMP-13 activity was measured in triplicate (11). The MMP-13 level in the supernatant was determined by ELISA. Data are expressed as fluorescence units (FU), from individual experiments (performed in triplicate) as well as means ± SD of each set of 3 experiments. *P < 0.05, by 2-tailed Student’s t test; treated vs. unstimulated cultures (controls).
Periostin induces MMP13 expression via Wnt signaling
Dysregulated Wnt signaling pathways are known to play an important role in OA development and progression (34). Periostin is involved in the activation of Wnt-β-catenin signaling in osteoblasts in response to parathyroid hormone (15). Therefore, we investigated whether periostin similarly activates canonical Wnt signaling in human OA chondrocytes. Western blot analysis of β-catenin showed that treatment of human OA chondrocytes with periostin (1 and 5 µg/ml) stabilized total β-catenin levels relative to those in untreated controls in all 3 OA chondrocyte populations (Fig. 11A). However, periostin did not induce phosphorylation of ERK1/2, p38, or JNK under the same culture conditions in contrast to IL-1β (10 ng/ml), used as a positive control, which induces all 3 kinases.
Figure 11.

Periostin activates β-catenin signaling in human chondrocytes. A) Western blot analysis of β-catenin in human articular chondrocytes treated with periostin (1–5 µg/ml) for the indicated times. Tubulin is shown as a loading control. IL-1, a known activator of the MAPK and Wnt signaling pathways, was used as a positive control. Representative blots of 3 independent chondrocyte cultures are shown. Age, sex, and basal periostin levels of the 3 OA chondrocyte cultures are indicated above the blots. B) Human OA chondrocytes from 3 independent patients pretreated with inhibitors of either MEK1/2 (ERK1/2-PD98059), p38 (SB202190), or NF-кB (MG-132) (10 µM) or control vehicle (DMSO) for 2 h were incubated with recombinant periostin (5 µg/ml) in serum free-medium for an additional 24 h, and the supernatant was analyzed for MMP-13 by ELISA. For control vs. periostin-treated culture supernatants, P < 0.01 by 1-way ANOVA with post-Bonferroni multiple comparison test. C) TOPflash activity in vehicle- and periostin-treated human C28/I2 chondrocytes. Cells were transfected with TOPflash luciferase reporter and cotransfected with pSEAP vector as a control for transfection efficiency. Means ± sd of 3 independent experiments are shown. Nonparametric Student's 2-tailed t test. D) Human OA chondrocytes (n = 3) pretreated with the Wnt inhibitor CCT031374 hydrobromide (10–50 µM) or control vehicle for 2 h were incubated with recombinant periostin (5 µg/ml) in serum free-medium for an additional 24 h. The supernatant was analyzed for MMP-13, and the cells were used for total RNA isolation. Data are means ± sd from 3 independent chondrocyte cultures. P = 0.025, control vs. periostin-treated; P = 0.042, periostin-treated with Wnt inhibitor vs. without Wnt inhibitor. Nonparametric Student's 2-tailed t test. E) Human OA chondrocytes from 3 patients were pretreated with the Wnt inhibitor CCT031374 (50 µM) or control vehicle (DMSO) for 2 h. Recombinant periostin (5 µg/ml) was added in serum free-medium for 24 h. Total RNA was isolated and analyzed for MMP-13, ADAMTS4, ADAMTS5, and Col2A1 expression by RT-qPCR. *P < 0.05.
We performed additional experiments with chondrocyte cultures to determine the effects of MEK1/2 (ERK1/2-PD98059), p38 (SB202190), and NF-кB (MG-132) inhibitors on periostin’s (5 µg/ml) induction of MMP-13. As expected, periostin significantly up-regulated MMP-13 expression, and none of the inhibitors significantly reduced the effect (Fig. 11B). However, as expected, all 3 inhibitors significantly inhibited IL-1- induced MMP-13 expression (data not shown). This finding is consistent with immunoblotting results, which showed that periostin did not activate MAPKs.
These data indicated that periostin selectively activates Wnt signaling in human OA chondrocytes. To further investigate the role of periostin in activation of Wnt signaling, we used the TOPflash reporter assay (EMD Millipore). Addition of recombinant periostin (5 µg/ml) to the medium of chondrocyte cultures strongly induced TCF-mediated luciferase activity (Fig. 11C). Further, periostin-induced expression of MMP-13 in human OA chondrocytes was significantly reduced by the Wnt pathway inhibitor CCT031374 in a dose-dependent manner (35) (Fig. 11D). To ensure that this effect was not caused by inhibitor-induced cell toxicity, chondrocyte viability and proliferation were tested over a range of inhibitor concentrations by using the MTT assay, and no toxic effects were observed (data not shown). Therefore, these findings show that periostin induces MMP-13 expression in human chondrocytes via activation of canonical Wnt signaling.
Finally, we characterized the effect of the Wnt inhibitor CCT031374 on periostin-induced target gene expression. CCT031374 significantly blocked periostin induction of MMP-13 and ADAMTS4 and partly reversed periostin-mediated inhibition of Col2 expression, but had no effect on ADAMTS5 expression (Fig. 11E).
DISCUSSION
ECM degradation, a central process in OA pathogenesis, leads to loss of joint functionality. Abundant evidence indicates that OA chondrocytes undergo phenotypic alterations and express genes that are not expressed in normal cartilage. We have shown that the latent TGF-β1-activating ECM protein, Spon1, is highly expressed in both human OA cartilage and a rodent surgical model of OA and induces MMP-13 expression in cultured human chondrocytes (9). Age-related changes also occur in chondrocytes and cartilage ECM, thereby contributing to the development of OA (9, 36–39). It is notable that altered TGF-β signaling or TGF-β target genes expressed in development or in adult mice have also been shown to augment progression of experimental OA in rodents (40–42). In this article we show that a downstream target of TGF-β, periostin, has catabolic activity in chondrocytes and that its overexpression in cartilage and subchondral bone in different rodent OA models indicates a unique treatment approach for OA.
Periostin is a homodimeric protein that belongs to the FAS domain superfamily of molecules. It is synthesized by smooth muscle cells, fibroblasts, and osteoblasts, as well as in periosteum and periodontal ligaments. It interacts with other ECM proteins such as fibronectin, collagen, and heparin (43). A mesenchymal matricellular protein, periostin is TGFβ-inducible and vitamin K dependently γ-carboxylated and serves as both an adhesion molecule and a tumor suppressor. Vitamin K deficiency is associated with incident knee OA (44), and dietary vitamin K inadequacy/deficiency induced by chronic anticoagulant (warfarin) therapy has been linked to age-associated conditions including OA (45). We found that TGFβ-1 induces periostin expression in a time-dependent manner, whereas IL-1β inhibits periostin expression (data not shown).
In the present work we report that periostin is overexpressed in lesions of both human OA and multiple surgical models of rodent OA, where it can act in situ to induce MMP-13 and ADAMTS4 expression. These results are consistent with those of Karlsson et al. (46), who detected periostin among multiple differentially expressed genes in cartilage by microarray analysis. Our ex vivo (cartilage explant) and in vitro (chondrocyte culture) studies showed that effective recombinant periostin concentrations required to induce significant catabolic activities range from 0.5 to 5 µg/ml. These concentrations are in the range of concentrations used in published studies (15, 29–33). Lourido et al. (33) have shown recently that, in addition to cartilage, OA synovial fluid contains elevated levels of periostin relative to non-OA controls. Therefore, it is conceivable that, during OA progression, local periostin concentrations are sufficiently high to stimulate MMP-13 expression.
Analysis of periostin expression levels by qPCR and immunoblotting analysis showed periostin up-regulation in OA cartilage from individual patients but with considerable variation, which may reflect disease heterogeneity. For these studies, whole tissue was harvested because the amount of cartilage available within the diseased knee joint was often limiting. It is thus possible that regional differences in OA progression incorporate multiple disease states with local variations in periostin expression.
The reported catabolic activity of periostin in diseases such as polycystic kidney disease and cardiac valve complex degeneration is also consistent with its role as a catabolic mediator in OA (47). Recently, periostin has been shown to play an essential role in the progression of cardiac valve complex degeneration by inducing angiogenesis and expression of MMP-2 and -13 (29). In bone, periostin has been implicated in osteoblast recruitment, attachment, and spreading, and periostin deficiency has been shown to alter bone properties and local modeling and remodeling processes in response to fractures (48). Periostin is also significantly elevated in cartilage from mice with surgically induced OA (49). In addition, 2 groups have independently shown increased expression of periostin in subchondral bone affected by OA (50, 51). In an elegantly performed mouse destabilized medial meniscus (DMM) OA model, Loeser et al. (52) used both histologic and gene expression analyses to show a phasic process of OA development, with an early phase (2 and 4 wk after surgery) characterized by expression of genes active in ECM remodeling, including periostin, chemokines (CCL21, CXCR7) and Wnt pathway regulators. The expression of periostin in a posttraumatic (DMM) model of OA has recently been validated by Gardiner et al. (53). Furthermore, progressive increases in subchondral bone area and thickness were observed from the early phase onward in this model. These findings suggest a plausible role of elevated periostin expression as a promoter of OA progression through increased remodeling processes in cartilage and subchondral bone, although confirmation must be sought via a mouse model of OA in Postn-deficient mice.
The MAPK, PI3K, and Wnt signaling pathways are identified as dysregulated in OA chondrocytes and cartilage through global expression profiling. Many recent studies have shown crosstalk among TGF-β, bone morphogenetic proteins, hypoxia-inducible factors, Wnt/β-catenin, NF-κB, MAPK, and Indian hedgehog pathways in chondrocytes (54). Wnt/β-catenin signaling plays a critical role in the development and function of articular chondrocytes, and recent studies have also implied the involvement of dysregulated or activated Wnt pathway regulators in the etiology and progression of OA (34, 55). Moreover, inhibitors of Wnt signaling attenuate or inhibit IL-1β induced cartilage degeneration (56). Therefore, we examined the effect of periostin on activation of these signaling pathways. We found that periostin activates canonical Wnt/β-catenin signaling but does not induce ERK1/2, p38, NF-кB, or JNK activation in OA chondrocytes, indicating a selective effect on signaling pathway activation. Altogether, our findings provide evidence that periostin contributes to cartilage degeneration through induction of MMP-13 expression via Wnt signaling. MMP-13 has been shown to be critical for OA progression, and pharmacologic inhibition of MMP-13 slows articular cartilage degeneration in a murine model of surgically induced knee OA (57, 58). In a preclinical in vivo murine model of human melanoma, periostin promotes angiogenesis by interacting with integrins αvβ3 and αvβ5 via its FAS1-2 domain (59). In addition, it induces MMP-2 expression via the αvβ3 integrin/ERK pathway in human periodontal ligament cells (60). Wnt/MAPK/PI3K are all activated downstream of integrins (certainly in osteoblasts); therefore (although not demonstrated in this study), integrin interactions are likely essential in the transmission of periostin effects in OA chondrocytes. Further investigation of the role of integrin αvβ3 in periostin-mediated catabolic effects in chondrocytes is warranted.
In conclusion, the current study shows that periostin is a catabolic factor that promotes collagen and proteoglycan degradation through activation of the canonical Wnt pathway and downstream induction of MMP-13 and ADAMTS4 expression. In addition, periostin, previously reported as an osteoblast-specific factor, is up-regulated in both human and rodent OA. These observations point to periostin as a novel therapeutic target to prevent OA progression.
Acknowledgments
This work was supported in part by U.S. National Institutes of Health (NIH) National Institute of Arthritis and Musculoskeletal and Skin Diseases Grant R01-AR054817 (to S.B.A.) and U.S. NIH National Cancer Institute Grant R01-CA136715 (to P.M.); and by a research grant from Daiichi Sankyo Co., Ltd. (Tokyo, Japan). K.S., Y.T., and H.N. are employees of Daiichi Sankyo Co., Ltd. The authors declare no conflicts of interests.
Glossary
- ACL
anterior cruciate ligament
- ACLT/PMX
anterior cruciate ligament transection/partial medial meniscectomy
- ADAMTS
a disintegrin and metalloproteinase with thrombospondin motifs; Ad-mock, adenovirus devoid of periostin cDNA; Ad-periostin, adenovirus encoding periostin
- AKT
protein kinase B
- APMA
p-aminophenylmercuric acetate
- DMB
1,9-dimethylmethylene blue
- DMM
destabilized medial meniscus
- ds cDNA
double-stranded cDNA
- ECM
extracellular matrix
- FAS
fasciclin
- HEK
human embryonic kidney
- LTP
lateral tibial plateau
- MM
medial meniscectomy
- MMP
matrix metalloproteinase
- MTP
medial tibial plateau
- OA
osteoarthritis
- PMX
partial medial meniscectomy
- pSEAP
secreted form of human placental alkaline phosphatase
- RT-qPCR
real-time quantitative PCR
- sGAG
sulfated glycosaminoglycan
- siRNA
small interfering RNA
- Spon1
F-spondin
- TCF
T-cell factor
REFERENCES
- 1.Loeser R. F. (2006) Molecular mechanisms of cartilage destruction: mechanics, inflammatory mediators, and aging collide. Arthritis Rheum. 54, 1357–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abramson S. B., Attur M., Yazici Y. (2006) Prospects for disease modification in osteoarthritis. Nat. Clin. Pract. Rheumatol. 2, 304–312 [DOI] [PubMed] [Google Scholar]
- 3.Goldring M. B., Goldring S. R. (2007) Osteoarthritis. J. Cell. Physiol. 213, 626–634 [DOI] [PubMed] [Google Scholar]
- 4.Attur M., Krasnokutsky-Samuels S., Samuels J., Abramson S. B. (2013) Prognostic biomarkers in osteoarthritis. Curr. Opin. Rheumatol. 25, 136–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Houard X., Goldring M. B., Berenbaum F. (2013) Homeostatic mechanisms in articular cartilage and role of inflammation in osteoarthritis. Curr. Rheumatol. Rep. 15, 375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sandell L. J. (2007) Modern molecular analysis of a traditional disease: progression in osteoarthritis. Arthritis Rheum. 56, 2474–2477 [DOI] [PubMed] [Google Scholar]
- 7.Lotz M. (2012) Osteoarthritis year 2011 in review: biology. Osteoarthritis Cartilage 20, 192–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Attur M. G., Dave M. N., Stuchin S., Kowalski A. J., Steiner G., Abramson S. B., Denhardt D. T., Amin A. R. (2001) Osteopontin: an intrinsic inhibitor of inflammation in cartilage. Arthritis Rheum. 44, 578–584 [DOI] [PubMed] [Google Scholar]
- 9.Attur M. G., Palmer G. D., Al-Mussawir H. E., Dave M., Teixeira C. C., Rifkin D. B., Appleton C. T., Beier F., Abramson S. B. (2009) F-spondin, a neuroregulatory protein, is up-regulated in osteoarthritis and regulates cartilage metabolism via TGF-beta activation. FASEB J. 23, 79–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maldonado M., Nam J. (2013) The role of changes in extracellular matrix of cartilage in the presence of inflammation on the pathology of osteoarthritis. Biomed. Res. Int. 2013, 284873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Amin A. R., Di Cesare P. E., Vyas P., Attur M., Tzeng E., Billiar T. R., Stuchin S. A., Abramson S. B. (1995) The expression and regulation of nitric oxide synthase in human osteoarthritis-affected chondrocytes: evidence for up-regulated neuronal nitric oxide synthase. J. Exp. Med. 182, 2097–2102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amin A. R., Attur M., Patel R. N., Thakker G. D., Marshall P. J., Rediske J., Stuchin S. A., Patel I. R., Abramson S. B. (1997) Superinduction of cyclooxygenase-2 activity in human osteoarthritis-affected cartilage. Influence of nitric oxide. J. Clin. Invest. 99, 1231–1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Attur M. G., Dave M. N., Tsunoyama K., Akamatsu M., Kobori M., Miki J., Abramson S. B., Katoh M., Amin A. R. (2002) “A system biology” approach to bioinformatics and functional genomics in complex human diseases: arthritis. Curr. Issues Mol. Biol. 4, 129–146 [PubMed] [Google Scholar]
- 14.Wen W., Chau E., Jackson-Boeters L., Elliott C., Daley T. D., Hamilton D. W. (2010) TGF-ß1 and FAK regulate periostin expression in PDL fibroblasts. J. Dent. Res. 89, 1439–1443 [DOI] [PubMed] [Google Scholar]
- 15.Bonnet N., Conway S. J., Ferrari S. L. (2012) Regulation of beta catenin signaling and parathyroid hormone anabolic effects in bone by the matricellular protein periostin. Proc. Natl. Acad. Sci. USA 109, 15048–15053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loeser R. F. (2014) Integrins and chondrocyte-matrix interactions in articular cartilage. Matrix Biol. 39, 11–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marie P. J., Haÿ E., Saidak Z. (2014) Integrin and cadherin signaling in bone: role and potential therapeutic targets. Trends Endocrinol. Metab. 25, 567–575 [DOI] [PubMed] [Google Scholar]
- 18.Attur M., Millman J. S., Dave M. N., Al-Mussawir H. E., Patel J., Palmer G., Abramson S. B. (2011) Glatiramer acetate (GA), the immunomodulatory drug, inhibits inflammatory mediators and collagen degradation in osteoarthritis (OA) cartilage. Osteoarthritis Cartilage 19, 1158–1164 [DOI] [PubMed] [Google Scholar]
- 19.Minashima T., Zhang Y., Lee Y., Kirsch T. (2014) Lithium protects against cartilage degradation in osteoarthritis. Arthritis Rheumatol. 66, 1228–1236 [DOI] [PubMed] [Google Scholar]
- 20.Handley C. J., Buttle D. J. (1995) Assay of proteoglycan degradation. Methods Enzymol. 248, 47–58 [DOI] [PubMed] [Google Scholar]
- 21.Bendele A. M. (2001) Animal models of osteoarthritis. J. Musculoskelet. Neuronal Interact. 1, 363–376 [PubMed] [Google Scholar]
- 22.Bove S. E., Laemont K. D., Brooker R. M., Osborn M. N., Sanchez B. M., Guzman R. E., Hook K. E., Juneau P. L., Connor J. R., Kilgore K. S. (2006) Surgically induced osteoarthritis in the rat results in the development of both osteoarthritis-like joint pain and secondary hyperalgesia. Osteoarthritis Cartilage 14, 1041–1048 [DOI] [PubMed] [Google Scholar]
- 23.Kamekura S., Kawasaki Y., Hoshi K., Shimoaka T., Chikuda H., Maruyama Z., Komori T., Sato S., Takeda S., Karsenty G., Nakamura K., Chung U. I., Kawaguchi H. (2006) Contribution of runt-related transcription factor 2 to the pathogenesis of osteoarthritis in mice after induction of knee joint instability. Arthritis Rheum. 54, 2462–2470 [DOI] [PubMed] [Google Scholar]
- 24.Nagase H., Nagasawa Y., Tachida Y., Sakakibara S., Okutsu J., Suematsu N., Arita S., Shimada K. (2013) Deiodinase 2 upregulation demonstrated in osteoarthritis patients cartilage causes cartilage destruction in tissue-specific transgenic rats. Osteoarthritis Cartilage 21, 514–523 [DOI] [PubMed] [Google Scholar]
- 25.Appleton C. T., McErlain D. D., Pitelka V., Schwartz N., Bernier S. M., Henry J. L., Holdsworth D. W., Beier F. (2007) Forced mobilization accelerates pathogenesis: characterization of a preclinical surgical model of osteoarthritis. Arthritis Res. Ther. 9, R13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Appleton C. T., Pitelka V., Henry J., Beier F. (2007) Global analyses of gene expression in early experimental osteoarthritis. Arthritis Rheum. 56, 1854–1868 [DOI] [PubMed] [Google Scholar]
- 27.Campbell K. A., Minashima T., Zhang Y., Hadley S., Lee Y. J., Giovinazzo J., Quirno M., Kirsch T. (2013) Annexin A6 interacts with p65 and stimulates NF-κB activity and catabolic events in articular chondrocytes. Arthritis Rheum. 65, 3120–3129 [DOI] [PubMed] [Google Scholar]
- 28.Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 29.Hakuno D., Kimura N., Yoshioka M., Mukai M., Kimura T., Okada Y., Yozu R., Shukunami C., Hiraki Y., Kudo A., Ogawa S., Fukuda K. (2010) Periostin advances atherosclerotic and rheumatic cardiac valve degeneration by inducing angiogenesis and MMP production in humans and rodents. J. Clin. Invest. 120, 2292–2306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang L., Serada S., Fujimoto M., Terao M., Kotobuki Y., Kitaba S., Matsui S., Kudo A., Naka T., Murota H., Katayama I. (2012) Periostin facilitates skin sclerosis via PI3K/Akt dependent mechanism in a mouse model of scleroderma. PLoS ONE 7, e41994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shih C. H., Lacagnina M., Leuer-Bisciotti K., Pröschel C. (2014) Astroglial-derived periostin promotes axonal regeneration after spinal cord injury. J. Neurosci. 34, 2438–2443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kikuchi Y., Kunita A., Iwata C., Komura D., Nishiyama T., Shimazu K., Takeshita K., Shibahara J., Kii I., Morishita Y., Yashiro M., Hirakawa K., Miyazono K., Kudo A., Fukayama M., Kashima T. G. (2014) The niche component periostin is produced by cancer-associated fibroblasts, supporting growth of gastric cancer through ERK activation. Am. J. Pathol. 184, 859–870 [DOI] [PubMed] [Google Scholar]
- 33.Lourido L., Calamia V., Mateos J., Fernández-Puente P., Fernández-Tajes J., Blanco F. J., Ruiz-Romero C. (2014) Quantitative proteomic profiling of human articular cartilage degradation in osteoarthritis. J. Proteome Res. 13, 6096–6106 [DOI] [PubMed] [Google Scholar]
- 34.Olex A. L., Turkett W. H., Fetrow J. S., Loeser R. F. (2014) Integration of gene expression data with network-based analysis to identify signaling and metabolic pathways regulated during the development of osteoarthritis. Gene 542, 38–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ewan K., Pajak B., Stubbs M., Todd H., Barbeau O., Quevedo C., Botfield H., Young R., Ruddle R., Samuel L., Battersby A., Raynaud F., Allen N., Wilson S., Latinkic B., Workman P., McDonald E., Blagg J., Aherne W., Dale T. (2010) A useful approach to identify novel small-molecule inhibitors of Wnt-dependent transcription. Cancer Res. 70, 5963–5973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Attur M., Ben-Artzi A., Yang Q., Al-Mussawir H. E., Worman H. J., Palmer G., Abramson S. B. (2012) Perturbation of nuclear lamin A causes cell death in chondrocytes. Arthritis Rheum. 64, 1940–1949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lotz M., Loeser R. F. (2012) Effects of aging on articular cartilage homeostasis. Bone 51, 241–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Loeser R. F. (2013) Aging processes and the development of osteoarthritis. Curr. Opin. Rheumatol. 25, 108–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li Y., Wei X., Zhou J., Wei L. (2013) The age-related changes in cartilage and osteoarthritis. Biomed. Res. Int. 2013, 916530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van der Kraan P. M., Blaney Davidson E. N., van den Berg W. B. (2010) A role for age-related changes in TGFbeta signaling in aberrant chondrocyte differentiation and osteoarthritis. Arthritis Res. Ther. 12, 201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shen J., Li J., Wang B., Jin H., Wang M., Zhang Y., Yang Y., Im H. J., O’Keefe R., Chen D. (2013) Deletion of the transforming growth factor β receptor type II gene in articular chondrocytes leads to a progressive osteoarthritis-like phenotype in mice. Arthritis Rheum. 65, 3107–3119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhen G., Wen C., Jia X., Li Y., Crane J. L., Mears S. C., Askin F. B., Frassica F. J., Chang W., Yao J., Carrino J. A., Cosgarea A., Artemov D., Chen Q., Zhao Z., Zhou X., Riley L., Sponseller P., Wan M., Lu W. W., Cao X. (2013) Inhibition of TGF-β signaling in mesenchymal stem cells of subchondral bone attenuates osteoarthritis. Nat. Med. 19, 704–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Merle B., Garnero P. (2012) The multiple facets of periostin in bone metabolism. Osteoporos. Int. 23, 1199–1212 [DOI] [PubMed] [Google Scholar]
- 44.Misra D., Booth S. L., Tolstykh I., Felson D. T., Nevitt M. C., Lewis C. E., Torner J., Neogi T. (2013) Vitamin K deficiency is associated with incident knee osteoarthritis. Am. J. Med. 126, 243–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McCann J. C., Ames B. N. (2009) Vitamin K, an example of triage theory: is micronutrient inadequacy linked to diseases of aging? Am. J. Clin. Nutr. 90, 889–907 [DOI] [PubMed] [Google Scholar]
- 46.Karlsson C., Dehne T., Lindahl A., Brittberg M., Pruss A., Sittinger M., Ringe J. (2010) Genome-wide expression profiling reveals new candidate genes associated with osteoarthritis. Osteoarthritis Cartilage 18, 581–592 [DOI] [PubMed] [Google Scholar]
- 47.Conway S. J., Izuhara K., Kudo Y., Litvin J., Markwald R., Ouyang G., Arron J. R., Holweg C. T., Kudo A. (2014) The role of periostin in tissue remodeling across health and disease. Cell. Mol. Life Sci. 71, 1279–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bonnet N., Gineyts E., Ammann P., Conway S. J., Garnero P., Ferrari S. (2013) Periostin deficiency increases bone damage and impairs injury response to fatigue loading in adult mice. PLoS ONE 8, e78347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Loeser R. F., Olex A. L., McNulty M. A., Carlson C. S., Callahan M. F., Ferguson C. M., Chou J., Leng X., Fetrow J. S. (2012) Microarray analysis reveals age-related differences in gene expression during the development of osteoarthritis in mice. Arthritis Rheum. 64, 705–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang R., Fang H., Chen Y., Shen J., Lu H., Zeng C., Ren J., Zeng H., Li Z., Chen S., Cai D., Zhao Q. (2012) Gene expression analyses of subchondral bone in early experimental osteoarthritis by microarray. PLoS ONE 7, e32356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chou C. H., Lee C. H., Lu L. S., Song I. W., Chuang H. P., Kuo S. Y., Wu J. Y., Chen Y. T., Kraus V. B., Wu C. C., Lee M. T. (2013) Direct assessment of articular cartilage and underlying subchondral bone reveals a progressive gene expression change in human osteoarthritic knees. Osteoarthritis Cartilage 21, 450–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Loeser R. F., Olex A. L., McNulty M. A., Carlson C. S., Callahan M., Ferguson C., Fetrow J. S. (2013) Disease progression and phasic changes in gene expression in a mouse model of osteoarthritis. PLoS ONE 8, e54633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gardiner M. D., Vincent T. L., Driscoll C., Burleigh A., Bou-Gharios G., Saklatvala J., Nagase H., Chanalaris A. (2015) Transcriptional analysis of micro-dissected articular cartilage in post-traumatic murine osteoarthritis. Osteoarthritis Cartilage 23, 616–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mariani E., Pulsatelli L., Facchini A. (2014) Signaling pathways in cartilage repair. Int. J. Mol. Sci. 15, 8667–8698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Blom A. B., van Lent P. L., van der Kraan P. M., van den Berg W. B. (2010) To seek shelter from the WNT in osteoarthritis? WNT-signaling as a target for osteoarthritis therapy. Curr. Drug Targets 11, 620–629 [DOI] [PubMed] [Google Scholar]
- 56.Yuasa T., Otani T., Koike T., Iwamoto M., Enomoto-Iwamoto M. (2008) Wnt/beta-catenin signaling stimulates matrix catabolic genes and activity in articular chondrocytes: its possible role in joint degeneration. Lab. Invest. 88, 264–274 [DOI] [PubMed] [Google Scholar]
- 57.Fosang A. J., Beier F. (2011) Emerging frontiers in cartilage and chondrocyte biology. Best Pract. Res. Clin. Rheumatol. 25, 751–766 [DOI] [PubMed] [Google Scholar]
- 58.Wang M., Sampson E. R., Jin H., Li J., Ke Q. H., Im H. J., Chen D. (2013) MMP13 is a critical target gene during the progression of osteoarthritis. Arthritis Res. Ther. 15, R5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Orecchia P., Conte R., Balza E., Castellani P., Borsi L., Zardi L., Mingari M. C., Carnemolla B. (2011) Identification of a novel cell binding site of periostin involved in tumour growth. Eur. J. Cancer 47, 2221–2229 [DOI] [PubMed] [Google Scholar]
- 60.Watanabe T., Yasue A., Fujihara S., Tanaka E. (2012) Periostin regulates MMP-2 expression via the αvβ3 integrin/ERK pathway in human periodontal ligament cells. Arch. Oral Biol. 57, 52–59 [DOI] [PubMed] [Google Scholar]