

Abstract
Prolonged immobilization (IM) causes skeletal muscle atrophy characterized by mitochondrial deterioration and proteolysis. Muscle remobilization (RM) increases reactive oxygen species generation, proinflammatory cytokine expression, and oxidative stress, preventing muscle from quick recovery. Thus, we hypothesized that overexpression of peroxisome proliferator-activated receptor γ coactivator 1-α (PGC-1α) via in vivo transfection would promote mitochondrial biogenesis and antioxidant defense, thus ameliorating the aforementioned deteriorations in a mouse model with 14-d IM followed by 5-d RM. PGC-1α transfection in tibialis anterior muscle resulted in a 7.2- and 4-fold increase in PGC-1α content in cytosol and nucleus, respectively. Mitochondrial biogenic (cytochrome c, mitochondrial transcription factor A), morphologic (mitochondrial density, mDNA/nDNA ratio), and functional (cytochrome c oxidase activity, ATP synthesis rate) markers, as well as fiber cross-sectional area, significantly increased in IM-RM muscle by PGC-1α overexpression. These effects were accompanied by an 18% decrease in H2O2, 30% decrease in nuclear factor-κB-DNA binding, and 25% reduction of IL-1β and-6 production in IM-RM muscle. There was a 34% increase in superoxide dismutase-2 activity, along with a 3.5-fold increase in NAD-dependent deacetylase sirtuin-3 expression caused by enhanced PGC-1α-estrogen-related receptor α binding. Our findings highlighted the importance of PGC-1α in protecting muscle from metabolic and redox disturbances caused by IM.—Kang, C., Goodman, C. A., Horberger, T. A., Ji, L. L. PGC-1α overexpression by in vivo transfection attenuates mitochondrial deterioration of skeletal muscle caused by immobilization.
Keywords: atrophy, inflammation, oxidative stress
Loss of skeletal muscle mass occurs because of denervation, bed rest, microgravity, and aging, which leads to a predictable decline of mobility and quality of life (1, 2). Numerous studies suggest that muscle immobilization (IM) enhances proteolysis, oxidative stress, inflammation, and metabolic disturbance (3–5). Two parallel events occur during muscle IM: protein synthesis is decreased because of the diminished stimulation from the IGF-AKT-mTOR axis, whereas protein degradation is enhanced caused by activation of the ubiquitin-proteolytic pathway controlled by the redox-sensitive NF-κB, and the lysosomal-autophagy controlled mainly by the forkhead box protein O (FOXO) pathway (4). Two end point enzymes responsible for the activation of protein degradation are the muscle-specific E3 ubiquitin ligases, namely muscle atrophy F-box (also known as Atrogin-1) and muscle ring finger-1 (6, 7). Atrogin-1 and muscle ring finger-1 control the ubiquitination and degradation of both regulatory (e.g., calcineurin and MyoD) and structural (e.g., myosin and troponin I) proteins (6, 8–11). Interestingly, remobilization (RM) of a muscle previously immobilized for an extended period of time does not always reverse the aforementioned pathogenesis quickly. Although some investigators showed mitochondrial morphologic and functional improvements and reduced atrogen-1 and muscle ring finger-1 activation after IM muscle was reloaded (12), others found clear signs of oxidative stress following muscle RM, such as increased reactive oxygen species (ROS) generation, lipid peroxidation, and disturbance of glutathione status, resulting in reduced muscle function and early fatigue (13–15). Furthermore, the IM-RM-activated NF-κB pathway stimulates the expression of proinflammatory cytokines such as TNF-α, IL-1β, and IL-6 that escalate inflammation and further oxidative stress (16).
The peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) is a key transcriptional coactivator that regulates mitochondrial biogenesis, metabolic function, fiber type composition, and antioxidant gene expression (17, 18). Furthermore, PGC-1α has demonstrated anti-inflammatory effects in skeletal muscle. For example, PGC-1α knockout mice showed higher mRNA levels of TNF-α and IL-6, as well as higher serum IL-6 level than wild-type (19, 20). Conversely, life-long PGC-1α overexpression has been shown to suppress proinflammatory cytokine expression in old mice (21). We recently demonstrated that after 14 d of IM and 5 d of RM, mouse tibialas anterior (TA) muscle significantly decreased PGC-1α protein content, accompanied by decreased nuclear respiratory factor (Nrf)1 and 2, mitochondrial transcription factor A (Tfam), inner membrane protein marker cytochrome c (cyt c), mitochondrial to nuclear DNA ratio (mDNA:nDNA), and ATP generation rate (16). Although a cause-effect relationship cannot be established, the down-regulation of PGC-1α appears to be a critical cellular mechanism leading to the deterioration of muscle phenotypic changes following IM-RM. Thus, preserving and increasing PGC-1α level and signaling seem to be a plausible strategy to ameliorate muscle integrity and function when it is subjected to IM-RM.
In the current study, we tested the hypothesis that PGC-1α overexpression via in vivo transfection would up-regulate mitochondrial protein content and gene expression of antioxidant enzymes and reduce inflammatory responses to IM-RM. Our data clearly demonstrated the critical role of PGC-1α in the defined experimental paradigm.
MATERIALS AND METHODS
Animals and experimental design
Female FVB/N mice (age 8–10 wk; body weight, 21–23 g) were housed individually in the animal facilities at the University of Wisconsin-Madison and University of Minnesota Twin Cities in temperature-controlled rooms (22°C), on a reverse 12-h light/dark cycle. All animals were fed a chow diet and tap water ad libitum. The animal use protocol was approved by the University of Wisconsin-Madison and University of Minnesota Twin Cities Research Animal Resource Center. After a 1 wk acclimation, mice were randomly assigned to 6 groups: 7-d immobilization (IM-7), 14 d IM (IM-14), 19 days IM (IM-19), IM-14 followed by 5 d remobilization (RM) (IM-14/RM-5), IM-14 followed by 10-d RM (IM-14/RM-10), and sham controls (Con). For the PGC-1α in vivo transfection study, the mice were randomly divided into 4 groups: control (Con), subject to green fluorescent protein (GFP) transfection (Con-GFP); Con, subject to PGC-1α in vivo transfection (Con-PGC-1α); 5 days remobilization after 2 wk immobilization (IM-RM), with GFP transfection (IM-RM-GFP); or IM-RM and PGC-1α in vivo transfection group (IM-RM-PGC-1α).
Mouse hindlimb IM
Details of mouse hindlimb IM were described previously (16). Briefly, mice were anesthetized with 100 mg/kg ketamine plus 10 mg/kg xylazine via intraperitoneal injection. IM and IM-RM mice in this study were immobilized wherein one of the hindlimbs was randomly selected to be fixed in knee extension and ankle plantarflexion. This was accomplished by cutting a 1.5 ml microtube longitudinally and placing the limb between 2 halves. The 2 pipette tip halves were secured around the limb via a medical adhesive bandage. The animals were free to move, eat, and drink ad libitum. The IM procedure prevented movement of the immobilized leg alone.
PGC-1α in vivo transfection
Flag or GFP-tagged PGC-1α was obtained from Addgene (kindly provided by Dr. Bruce M. Spiegelman). Briefly, wild-type PGC-1α inserts (NM_008904) were cloned into the pcDNA3.1 myc-His A or pEGFP-C1 vector. For the electroporation protocol, mice were anesthetized with 100 mg/kg ketamine plus 10 mg/kg xylazine, and a small incision was made through the skin covering the TA muscle. A 27-gauge needle was used to inject plasmid DNA solution (2.5 μg/μl GFP, 2.7 μg/μl Flag-PGC-1α, or 2.5 μg/μl GFP-PGC-1α) into the proximal (6 μl) and distal (6 μl) ends of the muscle belly. Following the injections, electric pulses were applied through 2 stainless steel pin electrodes (1 cm gap; Harvard Apparatus, Holliston, MA, USA) laid on top of the proximal and distal myotendinous junctions. Eight 20 ms square-wave electric pulses at a frequency of 1 Hz were delivered with an ECM 830 electroporation unit (BTX-Harvard Apparatus) at a field strength of 180 V/cm. Following the electroporation procedure, the incision was closed with Vetbond surgical glue (3M, St. Paul, MN, USA).
Western blot analysis
After the mouse was killed with injection of sodium pentobartital (120 mg/kg body weight), a portion of the TA muscle was immediately frozen in liquid nitrogen, and frozen muscles were homogenized with a tissue grinder (Tekmar, Mason, OH, USA) for 15 s in ice-cold buffer. The NE-PER kit (Thermo Scientific, Waltham, MA, USA) was used according to manufacturer’s description to separate nuclear and cytosolic extracts in the presence of protease inhibitor cocktails (Roche, Basel, Switzerland) during nuclear and cytoplasmic extraction. Proteins were separated by SDS-PAGE, transferred to a PVDF membrane (Millipore, Billerica, MA, USA), and blocked with 5% of either bovine serum albumin or skim milk prior to primary antibody incubation. A list of antibodies used in Western blotting is provided in Table 1. Following secondary antibody incubation, the membranes were developed using peroxide and ECL Plus Western Blotting Detection Reagents (GE Healthcare, Little Chalfont, Buckinghamshire, United Kingdom) to expose Hyperfilm ECL (GE Healthcare) and analyzed using ImageJ software (version 1.42q; National Institutes of Health, Bethesda, MD, USA).
TABLE 1.
List of primary antibodies
| Antibody name | Provider |
|---|---|
| PGC-1α | Calbiochem (ST1202) |
| Atrogin-1 | ECM Bioscience (AP2041)a |
| MuRF-1 | ECM Bioscience (MP3401)a |
| Nrf1 | Santa Cruz (sc-23624) |
| Nrf2 | Santa Cruz (sc-13032) |
| Cyt c | Abcam (ab53056) |
| COX IV | Abcam (ab16056) |
| Tfam | Abcam (ab131607) |
| Flag M2 | Sigma-Aldrich (F-1804) |
| p-IKK | Cell Signaling (#2078) |
| p-IkBα | Cell Signaling (#9246) |
| IkBα | Cell Signaling (#9242) |
| NFκB p65 | Cell Signaling (#3034) |
| SIRT3 | Santa Cruz (sc-99143) |
| SOD2 | Santa Cruz (sc-30080) |
| Acetyl-lysine | Cell Signaling (#9441) |
| Catalase | Abcam (as16731) |
| GPx1 | Abcam (ab22604) |
| 4-HNE | Abcam (ab46545) |
| α-tubulin | Abcam (ab18251) |
| FoxO3 | Abcam (ab12162) |
| p-FoxO3 | Santa Cruz (sc-101681) |
| VDAC1/porin | Abcam (ab15895) |
| Histone H2B | Abcam (ab1790) |
ECM Bioscience, Versailles, KY, USA.
Muscle fiber cross-sectional area and electron microscopy
Samples of ∼2 mm3 size were placed in a fixative solution of 3% paraformaldehyde, 1.5% glutaraldehyde, and 2.5% sucrose in 0.1 M sodium cacodylate buffer with 5 mM calcium chloride and 5 mM magnesium chloride (pH 7.4) for 8 h to overnight at 4°C, rinsed in buffer, then placed in 1% osmium tetroxide and 0.1 M sodium cacodylate buffer for ≥4 h. Specimens were rinsed in ultrapure water, dehydrated in an ethanol series, and embedded in Embed 812 resin (Electron Microscopy Sciences, Hatfield, PA, USA). Thick sections 1–2 µm thick were cut with a Leica Ultracut UCT microtome using a diamond blade, collected on glass slides, stained with 1% tolouidine blue, and examined with a Nikon Ti-S microscope system for measuring muscle cross-sectional area (CSA). Ultrathin sections of 80–100 nm thickness were cut and poststained with 3% uranyl acetate followed by triple-lead stain. Sections were examined with an Phillips CM 12 transmission electron microscope (FEI, Hillsboro, OR, USA) operating at 60 kV. Images were recorded with a Maxim DL digital capture system (Diffraction Ltd., Ottawa, ON, Canada).
Immunoprecipitation
Proteins from mouse tissues were extracted in lysis buffer (50 mM Tris-Cl, pH 7.5, 150 mM NaCl, 10% glycerol, 2 mM MgCl2, 1 mM DTT, and 1% NP40) supplemented with a complete protease inhibitor cocktail (Roche). Protein extracts were subjected to centrifugation at 8000 g for 10 min. Protein lysates were precleared with protein A beads for 30 min before immunoprecipitation with specified antibodies for 4 h. After the incubation, the beads were pelleted by centrifugation at 8000 g for 30 s and washed with ice-cold buffer 3 times. After washes, the pellets were eluted to 2× sample buffer, vortexed, and boiled at 100°C for 5 min. Thereafter, the mixtures were centrifuged at 8000 g for 30 s, and supernatant was loaded onto an acrylamide gel.
Chromatin immunoprecipitation assays
Chromatin immunoprecipitation (ChIP) assay was performed according to the manufacturer’s protocols (Millipore). Immunoprecipitation was carried out with anti-estrogen-related receptor α (ERRα; Abcam, Cambridge, United Kingdom) or M2 anti-FLAG antibody or normal mouse IgG (Sigma-Aldrich, St. Louis, MO, USA), followed by incubation with protein G Sepharose. After ChIP, DNA was purified using PCR purification kit (Qiagen, Venlo, The Netherlands). Quantitative real-time PCR analysis was performed with input (2% of total immunoprecipitation) and immunoprecipitated DNA. The primers were designed to amplify the region of −491 to −319 bp of the mouse SIRT3 promoter containing an ERRα binding site: 5′-AGCGTCCCACTAGCCTCACGGGTTG-3′ (forward) and 5′-GAGGACCCAAGTCTGCAGGCTTGAG-3′ (reverse). The quantitative RT-PCR thermal cycling conditions were 50°C for 2 min, 95°C for 10 min, 40 cycles of 95°C for 15 s, and 60°C for 1 min. Tenfold serial dilutions of standard were included in each experiment to generate the standard curve.
RNA extraction and real-time quantitative PCR
Total RNA was extracted according to the manufacturer's instructions (RNeasy Mini Kit; Qiagen). The RNA quantification and purity were estimated by spectrophotometer absorption readings at 260 and 280 nm, and cDNA was synthesized by SuperScript II Reverse Transcriptase kit (Invitrogen, Carlsbad, CA, USA). Ppargc1a, Esrra, Nrf1, Creb1, Cycs, Cox4i1, Tfam, Tnf, Il1a, Il1b, Il6, Rela, Chuk, and Ccl2 levels were quantified using SYBR Green analysis with a StepOnePlus real time PCR system (Applied Biosystems, Foster City, CA, USA). Forward and reverse primers for the aforementioned genes are provided in Table 2, and the relative mRNA levels were calculated by cycle threshold values, which were normalized to the internal control glyceraldehyde 3-phosphate dehydrogenase mRNA.
TABLE 2.
Primer sequences for real-time PCR
| Target | Primer sequence | |
| Forward | Reverse | |
| Ppargc1a (NM_008904) | 5′-TGTAGCGACCAATCGGAAAT-3′ | 5′-TGAGGACCGCTAGCAAGTTT-3′ |
| Esrra (NM_007953) | 5′-GGAGTACGTCCTGCTGAAAGCT-3′ | 5′-CACAGCCTCAGCATCTTCAATG-3′ |
| Nrf1 (NM_010938) | 5′-CTTACAAGGTGGGGGACAGA-3′ | 5′-CCGATATCCTGGTGGTCACT-3′ |
| Creb1 (NM_133828) | 5′-AGAAGCAGCACGGAAGAGAG-3′ | 5′-TTTCAAGCACTGCCACTCTG-3′ |
| Cycs (NM_007808) | 5′-GGACGTCTGTCTTCGAGTCC-3′ | 5′-AACAGACCGTGGAGATTTGG-3′ |
| Cox4i1 (NM_009941) | 5′-GTTGGCTACCAGGGCACTTA-3′ | 5′-CACATCAGGCAAGGGGTAGT-3′ |
| Tfam (NM_009360) | 5′-TGGAAAGCATACAAAGAAGCTG-3′ | 5′-GACAGATTTTTCCAAGCCTCA-3′ |
| Tnf (NM_013693) | 5′-CCACCACGCTCTTCTGTCTAC-3′ | 5′-AGGGTCTGGGCCATAGAACT-3′ |
| Il1a (NM_010554) | 5′-CCAGAAGAAAATGAGGTCGG-3′ | 5′-AGCGCTCAAGGAGAAGACC-3′ |
| Il1b (NM_008361) | 5′-AAGAGCTTCAGGCAGGCAGTATCA-3′ | 5′-TGCAGCTGTCTAGGAACGTCA-3′ |
| Il6 (NM_031168) | 5′-ACCAGAGGAAATTTTCAATAGGC-3′ | 5′-TGATGCACTTGCAGAAAACA-3′ |
| Rela (NM_009045) | 5′-TAGGTCCTTTTGCGCTTCTC-3′ | 5′-GCTCCTGTTCGAGTCTCCAT-3′ |
| Chuk (NM_007700) | 5′-TTTCGGGAACGTCAGTCTGT-3′ | 5′-GCACCATCGCTCTCTGTTTT-3′ |
| Ccl2 (NM_011333) | 5′-ATTGGGATCATCTTGCTGGT-3′ | 5′-CCTGCTGTTCACAGTTGCC-3′ |
| Gapdh (NM_008084) | 5′-CGTCCCGTAGACAAAATGGT-3′ | 5′-TTGATGGCAACAATCTCCAC-3′ |
Mitochondrial isolation
After weighing out muscle tissue, 5 ml ice-cold isolation medium for skeletal muscle (IMSM) 1 (0.25 M sucrose, 1 mM EDTA, 5 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 0.2% bovine serum albumin, 13 U/10 ml collagenase) was added and minced using scissors making the pieces as small as possible. IMSM 1 was then added up to a 1:10 ratio, and the mixture was kept on ice for 30 min. Using a Potter-Elvehjem homogenizer, the mixture was further homogenized. The homogenate was filtered through 2 layers of sterile gauze. The homogenate was spun at 4°C for 10 min at 700 g. The supernatant was saved on ice, and the pellet was resuspended in a 1:10 ratio of IMSM 1 (added slowly to allow further manual homogenization) and was spun again under the same conditions (4°C for 10 min at 700 g). The supernatant was combined with the previous supernatant, and the pellet was discarded. The combined supernatants were spun at 4°C for 10 min at 12,000 g. The supernatant was poured off and discarded. Fat on the centrifuge tube was wiped off with a sterile cotton-tipped applicator, and the pellet was resuspended using a smooth-headed Potter-Elvehjem pestle (Sigma-Aldrich) in 15 ml (added slowly to allow resuspension) IMSM 2 (0.25 M sucrose and 1 mM EGTA). The mixture was again spun under the same conditions (4°C for 10 min at 12,000 g), and the supernatant was discarded. The supernatant was poured off and discarded. Fat on the centrifuge tube was wiped off with a sterile cotton-tipped applicator, 200 μl IM2 was added, and the mitochondria were carefully resuspended using a smooth-headed Potter-Elvehjem pestle. The mitochondria were transferred to a beaker and kept on ice until use.
Quantitative analysis of mitochondrial DNA
Mitochondria were lysed in the presence of 0.5% SDS and 0.2 mg/ml proteinase K in 10 mM Tris-HCl, 0.15 M NaCl, and 0.005 M EDTA. Mitochondrial DNA (mtDNA) was then purified by a DNA purification kit (Qiagen). Total DNA was isolated using standard protocols. To quantify the amount of mtDNA present per nuclear genome, we used the following primers: mtDNA (NADH dehydrogenase 1; ND1) forward primer, CCTATCACCCTTGCCATCAT; mtDNA reverse primer, GAGGCTGTTGCTTGTGTGAC; nuclear DNA (Pecame gene on chromosome 6) forward primer, ATGGAAAGCCTGCCATCATG; nuclear DNA reverse primer, TCCTTGTTGTTCAGCATCAC. Quantification of relative copy number differences was carried out using analysis of the difference in threshold amplification between mtDNA and nuclear DNA (ΔΔCt method). The quantitative RT-PCR thermal cycling conditions were 50°C for 2 min, 95°C for 10 min, 40 cycles of 95°C for 15 s, and 60°C for 1 min.
Mitochondrial ATP production rate
After the mitochondria were isolated from TA muscle, the pellet was resuspended in buffer (180 mM sucrose, 35 mM KH2PO4, 10 mg acetate, 5 mM EDTA) and kept on ice. The reaction mixture included a luciferin-luciferase ATP monitoring reagent (Enliten ATP Assay System; Promega, Madison, WI, USA), 35 μg ADP, and substrates; 1 mM pyruvate and 1 mM malate were added to the reaction mixture as substrate for oxidation phosphorylation. When ATP was the limiting component in the luciferase reaction, the intensity of the emitted light was proportional to ATP concentration. Measurement of the light intensity using a luminometer permitted direct quantitation of ATP. ATP production reactions were monitored at 25°C for 20–25 min with a luminometer.
Mitochondrial ROS production
Mitochondrial ROS production was determined using Amplex Red (Invitrogen). The Amplex Red reagent, in combination with horseradish peroxidase, has been used to detect H2O2 released from biologic samples, and it is used to measure H2O2 as an indicator of superoxide production. SOD was added at 40 U/ml to convert all superoxide into H2O2. The Amplex Red reagent reacted with H2O2 in a 1:1 stoichiometry to produce the red fluorescent oxidation product, resorufin. Resorufin formation at an excitation wavelength of 551 nm and an emission wavelength of 581 nm was determined using a fluorescence spectrophotometer (model F-2000; Hitachi, Tokyo, Japan).
Proinflammatory cytokine and 8-isoprostane measurements
TNF-α, IL-6 (BD Bioscience, San Jose, CA, USA), and IL-1β (Abcam) were measured in TA muscle homogenate (Tris-HCl buffer, pH 7.4) using an ELISA assay kit. 8-Isoprostane was measured in TA muscle homogenate using a chemiluminescence competitive ELISA assay kit following manufacturer’s instructions (Cayman Chemical, Ann Arbor, MI, USA).
NF-κB p65 DNA binding assay
NF-κB p65 transcription factor assay kit (Thermo Scientific) was used to detect active forms of NF-κB p65. The consensus sequence for NF-κB binding (5′-ACAGTTGAGGGGACTTTCCCAGGC-3′) was mixed with nuclear extract, and the active transcription factor bound to the consensus sequence was incubated with p65 primary antibody and then with a secondary horseradish peroxidase-conjugated antibody. The resulting signal was detected using a luminometer (Bio-Tek, Winooski, VT, USA).
Reporter gene assay
C2C12 myoblasts or TA muscles were cotransfected with either a firefly luciferase expression vector (pGL3 basic; Promega) driven by the cyt c promoter sequence (a kind donation from Dr. Rudolf J. Wiesner) or a 3× (NF-κB)tk-Luc, a firefly luciferase reporter construct containing 3 repeated NF-κB-responsive elements and a Renilla luciferase expression vector (kindly provided by Dr Huifang Cheng). After C2C12 myoblast or TA muscles were collected, firefly luciferase activities were determined using the Dual-Luciferase reporter assay kit (Promega) with a Synergy H1 luminometer (Bio-Tek) as described in the manufacturer's instructions. To control transfection efficiency, firefly activity was normalized to Renilla activity, and each experiment was prepared in triplicate.
Measurement of reduced and oxidized glutathione levels, GPx, glutathione reductase, and catalase activities
A glutathione assay kit (Cayman Chemical) was used to measure the reduced glutathione (GSH) and oxidized glutathione (GSSG) levels in muscle. The reaction between GSH and 5,5′-dithio-bis-2-nitrobenzoic acid results a colored product (5-thio-2-nitrobenzoic acid). The absorbance of 5-thio-2-nitrobenzoic acid was measured at the wavelength of 405 nm with a BioTek synergy H1 reader. Glutathione peroxidase, glutathione reductase, and catalase enzyme activities were measured using Cayman assay kit at a wavelength of 340 or 540 nm.
Immunohistochemical analysis
Muscles were excised and fixed in 4% PFA for 4 h, 15% sucrose for 4 h, and 30% sucrose overnight before frozen sections were prepared. The samples were submerged in optimal cutting temperature compound (Tissue-Tek; Sakura, Torrance, CA, USA) and frozen in liquid nitrogen-chilled isopentane. Cross sections (10 μm in thickness) from the midbelly of the muscle were obtained with a cryostat and fixed in 20°C acetone for 10 min. Under gentle rocking, the rehydrated sections were incubated in PBS for 15 min followed by a 20-min incubation in solution B (PBS containing 0.5% bovine serum albumin and 0.5% Triton X-100). Sections were then incubated with the anti-laminin 2α primary antibody (#ab11575; Abcam) dissolved in solution B for 1 h at room temperature. Sections were washed with PBS and then incubated with the Alexa Fluor 594-conjugated secondary antibody dissolved in solution B for 1 h at room temperature. Finally, the sections were washed with PBS and mounted with Shandon-Mount mounting medium (Thermo Scientific) and a coverslip. Transfected fibers (GFP or GFP-tagged PGC-1α) and laminin were identified in dual-fluorescent images and captured with a Nikon Eclipse Ti-S fluorescent microscope and analyzed using NIS-Elements 4.11 (Nikon). The staining intensity or CSA of 40–90 randomly selected transfected and nontransfected fibers per sample was measured by tracing the periphery of individual fibers.
Enzyme activity
Skeletal muscles were homogenized with a tissue grinder (Tekmar, Vernon, BC Canada) with a 1:10 (w:v) dilution in ice-cold lysis buffer containing 50 mM Tris (pH 7.4 at 4°C), 150 mM NaCl, 1% Triton X-100, 1% IGEPAL (NP-40), 1 mM PMSF, and protease inhibitor cocktail (Roche). Afterward, the homogenate was incubated on ice for 10 min and centrifuged at 1000 g for 5 min at 4°C, and then the supernatant was separated into aliquots and stored at −80°C. SOD2 activity was measured by monitoring the auto-oxidation of epinephrine to adrenochrome at 320 nm to measure activity following KCN addition.
Citrate synthase (CS) activity was measured in whole tissue lysates or mitochondria as recommended by manufacture’s protocol (Sigma-Aldrich), and cytochrome c oxidase (COX) activity was measured in mitochondria using Sigma assay kit, which used an optimized colorimetric assay based on observation of the decrease in absorbance of ferrocytochrome c measured at 550 nm, which is caused by its oxidation to ferricytochrome c by COX.
Determination of xanthine oxidase and nicotinamide adenine dinucleotide phosphate oxidase activity
Xanthine oxidase activity
Xanthine oxidase (XO) activity was measured in muscle homogenate using a fluorometric method according to the manufacturer’s protocols (Amplex Red Xanthine/Xanthine Oxidase Assay Kit; Invitrogen). The reactions were started by adding 50 μL of the Amplex Red reagent/horseradish peroxidase or xanthine working solution to the samples and controls. The mixture was incubated for 30 min at 37°C, and the fluorescence at an excitation wavelength of 560 nm and an emission wavelength of 590 nm was measured. For each point, background fluorescence or absorbance was corrected by subtracting the values derived from the noxanthine oxidase control.
Nicotinamide adenine dinucleotide phosphate oxidase activity
The oxidation of 2',7'-dichlorfluorescein (DCFH) at 37°C was monitored in duplicate at 488 nm excitation and 525 nm emission via fluorescence spectrophotometer (model F-2000: Hitachi). Buffers were used to measure muscle homogenate ROS generation: 0.1 mM FeCl3, 1.7 mM ADP, 0.1 mM NADPH, and 5 nM DCF-DA (Sigma-Aldrich) in 0.1 M PBS, pH 7.4, to target nicotinamide adenine dinucleotide phosphate oxidase (NOX). Following the addition of homogenate and 5 nM DCFH-diacetate, the cuvettes were inverted 5 times and placed in a 37°C water bath for 15 min to allow the DCFH-diacetate to enter the cells. Next, each cuvette was maintained at 37°C and read every 4 min for a total of 40 min.
Cell culture and transient transfection
C2C12 myoblasts were maintained in DMEM in the presence of 20% fetal bovine serum and 1% penicillin-streptomycin. Transient transfection was performed at a confluence of ∼75% using Lipofectamine 2000 (Life Technologies, Grand Island, NY, USA) with plasmid DNA. Lysates from transfected cells were prepared 36–48 h after transfection and assayed for immunoblot assay or luciferase activity. All transfection experiments were performed in triplicate.
Mitochondria superoxide production in single muscle fiber
Single muscle fibers were enzymatically isolated from the TA muscle. Excised TA muscles were incubated in dissociation medium containing 0.4% (w/v) type I collagenase (Sigma-Aldrich), DMEM (Life Technologies), 50 IU penicillin, 50 μg/ml streptomycin, and 10% fetal bovine serum (Life Technologies) for 1.5–2 h at 37°C. Fibers were plated in 2 ml Dulbecco’s PBS containing MitoSox red for 10 min at 37°C in a tissue culture incubator. Fluorescence images were captured using Nikon Eclipse Ti-S fluorescent microscope and analyzed using NIS-Elements 4.11 (Nikon).
Statistical analysis
Experimental data were expressed as means ± se, and group comparisons were made by Student’s t test (quantitative RT-PCR time course) or 2-way ANOVA (PGC-1α transfecton study). The Tukey-Kramer method was used as a post hoc test when ANOVA reached significance (P < 0.05).
RESULTS
IM-RM suppressed gene expressions of mitochondrial biogenesis and induced inflammatory cytokines in mouse muscle
To examine how IM and RM affect mitochondrial biogenesis and proinflammatory pathways, we first screened the changes of mRNA levels in response to 7, 14, and 19 d of IM (IM-7, IM-14, IM-19), and 5 and 10 d of RM following 14 d of IM (RM-5, RM-10). mRNA analysis of mitochondria biogenesis genes revealed that expressions of PGC-1α (Ppargc1a), ERRα (Esrra), Nrf1, Creb1, cyt c (Cycs), COX IV (Cox4i1), and Tfam were gradually diminished as the duration of muscle IM increased to 19 d (Fig. 1A). Noticeably, the mRNA levels at RM-5 were still significantly lower than control and recovered only after RM-10, indicating 5 d of muscle reloading was not sufficient to restore mitochondrial biogenesis. It is interesting to note that all inflammatory markers, including TNF-α (Tnf), IL-1α (Il1a), IL-1β (Il1b), IL-6 (Il6), p65 (Rela), IKKα (Chuk), and monocyte chemoattractant protein-1 (Ccl2) were significantly elevated after IM-14, whereas peak levels for TNF-α, IL-1α, IL-1β, IKKα, and monocyte chemoattractant protein-1 were reached at RM-5 (Fig. 1B), confirming our early reports (16). Except for IL-1α and p65, the expressions of most inflammatory markers declined at RM-10. Taken together, the early stage of recovery from muscle IM triggered dramatic inflammatory responses, as well as a suppression of mitochondria biogenesis machinery.
Figure 1.

Early stage RM following IM elicits attenuated mitochondria biogenesis and increased inflammatory factors in mouse TA muscle. To access mRNA expressions of (A) mitochondria biogenesis and (B) inflammatory factors, quantitative RT-PCR was performed on RNA extracted from TA of sham control (Con), 7-d immobilization (IM-7), 14-d IM (IM-14), 19-d IM (IM-19), IM-14 followed by 5-d RM (RM-5), and IM-14 followed by 10-d RM (RM-10). One of the hindlimbs was randomly selected to be fixed in knee extension and ankle plantarflexion. Values are the means ± sem (n = 7). *P < 0.05 vs. Con; **P < 0.01 vs. Con; Student's t test.
PGC-1α overexpression protected muscle mitochondria from deterioration during IM-RM
In vivo transfection of PGC-1α in the TA muscle was used to assess whether PGC-1α overexpression could ameliorate muscle response to IM-RM (5-d RM following 14-d IM). First, we confirmed that TA muscles injected with a plamid DNA encoding flag-tagged PGC-1α expressed a significantly higher level of PGC-1α protein compared with the control TA muscle and the myocardium (Supplemental Fig. S1). PGC-1α protein was significantly lowered by IM-RM in TA muscle treated with GFP (P < 0.05; Fig. 2A), consistent with the reduced mRNA level (Fig. 1A). PGC-1α transfection markedly increased PGC-1α protein levels in both Con and IM-RM groups by >7.2-fold compared with GFP groups (P < 0.01; Fig. 2B). The increased muscle PGC-1α expression was paralleled by an ∼4-fold increase in nuclear PGC-1α expression (P < 0.01; Fig. 2C).
Figure 2.

Effect of PGC-1α overexpression on major components of the PGC-1α signaling pathway. GFP or PGC-1α was overexpressed with in vivo DNA electroporation in the TA muscle after 14 d of IM followed by 5 days of RM (IM-RM). Same procedures of DNA injection were also applied to control (Con). A) Western blot analysis for protein contents of PGC-1α in whole lysates (B) and nuclear extracts (C) (n = 7). D) Representative Western blot images and quantification of Nrf1 (E) and Nrf2 (F) protein contents from nuclear extracts. (n = 7). G) Coimmunoprecipitation of PGC-1α with Nrf1 in the IM-RM group. TA muscles were transfected with flag-tagged PGC-1α and subjected to immunoprecipitation with anti-flag, anti-Nrf1, or control IgG followed by immunoblotting with antibodies as indicated. H) Luciferase activity driven by the PGC-1α and cytochrome c promoter assayed in C2C12 (n = 4) and mouse TA muscle (n = 7) with IM-RM. Firefly and Renilla luciferase activities were measured by the dual-luciferase reporter assay. Values are the means ± sem. *P < 0.05 vs. Con; **P < 0.01 vs. Con; ++P < 0.05 PGC-1α vs. GFP; 2-way ANOVA with Tukey-Kramer’s post hoc test.
Nrf1 content was decreased by 60% in IM-RM vs. Con (P < 0.01), but was not affected by PGC-1α overexpression (Fig. 2D, E). Nrf2 level was not affected by either IM-RM or PGC-1α (Fig. 2F). However, co-immunoprecipitation assay showed that PGC-1α overexpression increases PGC-1α/Nrf1 interaction in TA muscle with IM-RM (Fig. 2G). In addition, the luciferase reporter gene assay with cotransfections of PGC-1α and cyt c promoter revealed an increase in cyt c promoter activity in both C2C12 myoblast (P < 0.01) and TA muscle with IM-RM (P < 0.05; Fig. 2H).
To further assess the effect of PGC-1α overexpression on nuclear encoded mitochondrial proteins, cyt c, COX IV, and Tfam protein contents were quantified in both cytoplasm and mitochondria (Fig. 3A). Cytoplasmic cyt c content was decreased by 70% (P < 0.01) in IM-RM (Fig. 3B), whereas mitochondria cyt c level was not affected (Fig. 3C). PGC-1α transfection increased cytoplasmic and mitochondria cyt c content by 2.7∼5-fold (P < 0.01) in both Con and IM-RM groups. IM-RM group displayed a 70% lower cytoplasmic COX IV levels (P < 0.01), whereas PGC-1-overexpressing mice exhibited a 3.2-fold (P < 0.01) increase, thus completely recovering COX IV to Con levels (Fig. 3D). In contrast, mitochondrial COX IV content was not affected by either IM-RM or PGC-1α transfection (Fig. 3E). Cytoplasmic and mitochondrial Tfam contents were decreased by 50% (P < 0.01) and 21% (P < 0.05), respectively, in IM-RM (Fig. 3F, G). With PGC-1α overexpression, there was an ∼2.9-fold increase in cytoplasmic Tfam in both the Con and IM-RM groups (P < 0.01) and an ∼70% increase in mitochondrial Tfam in both Con and IM-RM groups (P < 0.01). It is important to note that in all the abovementioned protein markers, except mitochondrial COX IV, PGC-1α transfected IM-RM muscle showed higher levels than those in Con–GFP muscle, indicating a complete reversal of declined protein profiles.
Figure 3.

Effect of PGC-1α overexpression on nuclear encoded mitochondria proteins in mouse TA muscle. Conditions of IM-RM and Con are described in Fig. 2. A) Representative Western blot images of cyt c, COX IV, Tfam in cytoplasmic, and mitochondria fractions. The quantifications of cyt c (B: cytoplasm; C: mitochondria), COX IV (D: cytoplasm; E: mitochondria), and Tfam (F: cytoplasm; G: mitochondria). Values are the means ± sem (n = 7). *P < 0.05 vs. Con; **P < 0.01 vs. Con; +P < 0.05 PGC-1α vs. GFP; ++P < 0.05 PGC-1α vs. GFP; 2-way ANOVA with Tukey-Kramer’s post hoc test.
PGC-1α overexpression improved mitochondrial function in IM-RM muscle
We next evaluated whether muscle mitochondrial density was affected by IM-RM and ameliorated by PGC-1α overexpression. A quantitative real-time PCR assay was performed to measure mtDNA level using nuclear DNA as a reference. Although the mtDNA/nDNA ratio was decreased by 72% (P < 0.01) with IM-RM, a 2-fold increase was seen with PGC-1α overexpression (P < 0.05) in IM-RM (Fig. 4A). To gain some insight into mitochondrial morphology, transmission electron microscopy was used to quantitate mitochondrial density in TA muscle (Fig. 4B). Although the IM-RM group showed ∼50% lower mitochondrial density than Con (P < 0.05) with GFP treatment, a 25% (P < 0.05) increase in mitochondrial density was observed in IM-RM vs. Con with PGC-1α transfection (Fig. 4C).
Figure 4.
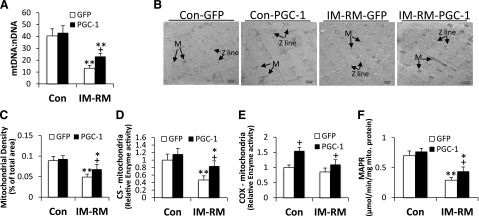
PGC-1α overexpression reversed mitochondrial dysfunction caused by IM-RM. Conditions of IM-RM and Con are described in Fig. 2. A) Quantifications of mtDNA by analyzing of the difference in threshold amplification between mtDNA and nuclear DNA using quantitative RT-PCR (ΔΔCt method; n = 7). B) Representative transmission electronic microscopy images of mitochondria in TA muscle. M, mitochondria; scale bars, 1 µm. C) Mitochondrial density analysis. (n = 5). D) Mitochondrial CS activity (n = 7). E) Mitochondrial cytochrome c oxidase (COX) activity (n = 7). F) Mitochondrial ATP production rate (MAPR) (n = 10). Values are the means ± sem. *P < 0.05 vs. Con; **P < 0.01 vs. Con; +P < 0.05 PGC-1α vs. GFP; 2-way ANOVA with Tukey-Kramer’s post hoc test.
To examine whether PGC-1α in vivo transfection could increase mitochondria functional capacity, CS and COX activity, as well as MAPR, were measured. IM-RM resulted in a 53% decrease in mitochondria CS activity (P < 0.01) vs. Con; with PGC-1α transfection, CS activity was increased by 44% (P < 0.05) in IM-RM muscle (Fig. 4D), whereas there was no effect by PGC-1α in tissue homogenates (Supplemental Fig. S2). In addition, PGC-1α overexpression increased mitochondrial COX activity by 55% in Con and ∼25% in IM-RM (P < 0.05; Fig. 4E). As a direct index of mitochondrial function, MAPR showed an ∼58% decrease in IM-RM vs. Con (P < 0.01). PGC-1α did not increase MAPR in Con but increased MAPR by 52% (P < 0.05) in the IM-RM group (Fig. 4F).
PGC-1α overexpression mitigated IM-RM-induced oxidative stress in mitochondria
We previously showed that mouse TA muscle subjected to an IM-RM regimen identical to that of the present study demonstrated clear signs of oxidative stress (16). Here, we found that IM-RM increased mitochondrial H2O2 generation by ∼80% (P < 0.01), whereas this level was 18% lower (P < 0.05) with PGC-1α transfection (Fig. 5A). To identify other potential sources of ROS generation, we measured cytosolic XO and NOX activity in various groups of mice. XO activity was not different between groups, whereas there was a 2.3-fold increase (P < 0.01) in NOX activity in IM-RM vs. Con (Supplemental Fig. S3). However, neither XO nor NOX activity was affected by PGC-1α overexpression. Levels of superoxide were determined using MitoSox within mitochondria of single muscle fibers (Fig. 5B). There was a 70% decrease in mitochondria superoxide level with PGC-1α overexpression compared with GFP control in the IM-RM group (P < 0.01; Fig. 5C).
Figure 5.

Mouse skeletal muscle overexpressing PGC-1α is protected from mitochondria oxidative stress and damage. Conditions of IM-RM and Con are described in Fig. 2. A) ROS production by isolated mitochondria from TA muscle (n = 10). B) Images of single muscle fibers loaded with the mitochondrial ROS indicator, MitoSOX Red, isolated from TA muscles transfected by either GFP (i, ii, iii) or GFP-PGC-1α (iv, v, vi) in the IM-RM group. C) Relative MitoSox fluorescence intensity in IM-RM (n = 6). D) Cytoplasmic and (E) mitochondrial 8-isoprostanes (n = 7). F, G) Levels of cytoplasmic and mitochondrial 4-hydroxynonenal protein adducts (n = 7). Values are the means ± sem. *P < 0.05 vs. Con; **P < 0.01 vs. Con; +P < 0.05 PGC-1α vs. GFP; ++P < 0.05 PGC-1α vs. GFP; 2-way ANOVA with Tukey-Kramer’s post hoc test.
As a muscle oxidative damage marker, 8-isoprostane content was elevated 3.9-and 3.5-fold (P < 0.01) in cytosol and mitochondria, respectively, in IM-RM vs. Con (Fig. 5D, E). PGC-1α treatment did not affect cytosolic 8-isoprostane level but reduced mitochondria 8-isoprostane content by 30% (P < 0.05) in IM-RM muscle. There was a 1.5-and 3.3-fold increase in cytoplasmic and mitochondrial 4-HNE modified proteins, respectively, in IM-RM vs. Con (P < 0.01; Fig. 5F, G). PGC-1α overexpression resulted in a 40% (P < 0.01) decrease in IM-RM-induced 4-HNE modified proteins mitochondria.
PGC-1α overexpression attenuated inflammatory responses induced by IM-RM
To assess the effect of PGC-1α overexpression on muscle inflammatory status, major elements of the NF-κB pathway were determined by Western blot (Fig. 6A). Phosphorylation of IKK was increased by 50% (P < 0.05; Fig. 6B), whereas phosphorylated (p)-IκB:IκB ratio was increased by 8.8-fold (P < 0.01) with IM-RM (Fig. 6C). There was a 2.7- to 3.7-fold increase in cytoplasmic p65 (P < 0.05) and nuclear p65 (P < 0.01), respectively, in IM-RM vs. Con (Fig. 6D, E). Furthermore, there was a 3.7-fold increase in NF-κB-dependent promoter activity (P < 0.01; Fig. 6F) and a 5.4-fold increase in p65 NF-κB-DNA binding activity with IM-RM (P < 0.01; Fig. 6G). PGC-1α treatment had no effect on the increased protein levels of NF-κB pathway; however, NF-κB-dependent promoter activity and NF-κB-DNA binding were reduced by 54% (P < 0.01) and 30% (P < 0.05), respectively, in the PGC-1α overexpression vs. Con groups.
Figure 6.

PGC-1α overexpression decreased NF-κB activity and proinflammatory cytokines in IM-RM muscle. Conditions of IM-RM and Con are described in Fig. 2. Western blot analysis (A) was performed to access the effect of PGC-1α on p-IKK (B), p-IκB:IκB ratio (C), cytosolic p65 (D), and nuclear p65 (E). F) NF-κB-dependent transcriptional activity measured by NF-κB-dependent promoter luciferase reporter gene assay. G) NF-κB p65 DNA binding activity. Proinflammatory cytokine TNF-α (H), IL-6 (I), and IL-1β (J) levels were determined by ELISA assay. Values are the means ± sem (n = 7). *P < 0.05 vs. Con; **P < 0.01 vs. Con; +P < 0.05 PGC-1α vs. GFP; ++P < 0.05 PGC-1α vs. GFP; 2-way ANOVA with Tukey-Kramer’s post hoc test.
IM-RM increased IL-6, IL-1β, and TNF-α levels by 3.7- (P < 0.01), 2.5- (P < 0.05), and 2.5-fold (P < 0.01), respectively, compared with Con (Fig. 6H–J). PGC-1α transfection decreased IL-6 and IL-1β by 25% and 26% (P < 0.05), respectively, but had no effect on TNF-α in IM-RM. None of the inflammatory cytokines was affected by PGC-1α overexpression in Con mice.
Effects of PGC-1α transfection on TA muscle weight, fiber size, and proteolytic pathway
Body weight of mice was not affected by IM-RM regimen or in vivo DNA transfection procedures. TA to body weight ratio was decreased by ∼10.4% (P < 0.05) with IM-RM in both GFP and PGC-1α transfection groups (Supplemental Table S1). In separate groups of animals not subjected to in vivo transfection procedures, we found no significant difference in the magnitude of TA/body weight ratio loss after 14 and 19 d of IM (Supplemental Table S2).
The CSAs of TA muscle fibers were found to decrease by 40% in IM-RM vs. Con (P < 0.01). However, when TA muscle fiber size of PGC-1α-transfected and GFP-transfected mice were compared, there was no significant difference (Fig. 7A, B).
Figure 7.

Effects of PGC-1α overexpression on muscle morphology and protein metabolism. Conditions of IM-RM and Con are described in Fig. 2. A) Representative microscopy images of TA muscle sections. Scale bars represent 100 µm. B) Quantitation of TA muscle cross-sectional area for each group (n = 7). C) Immunostaining for laminin (red) of cross section of TA muscle electroporated with GFP or GFP-tagged PGC-1α plasmid. D) Average cross-sectional area and (E) distribution of cross-sectional area of transfected fibers from GFP or GFP-PGC-1α transfected muscels. F) Representative protein expressions of p-FoxO3a and FoxO3a, and ratio of p-FoxO3a to FoxO3a (n = 7). Activity of the ubiquitin-proteasome pathway was assessed by atrogin-1 (G) and MuRF1 (H) mRNA levels (n = 7). Values are the means ± sem. *P < 0.05 vs. Con; **P < 0.01 vs. Con; +P < 0.05 PGC-1α vs. GFP; 2-way ANOVA with Tukey-Kramer’s post hoc test.
Because the in vivo PGC-1α transfection procedure derives ∼60% transfection efficiency and thus the transfected TA muscle still contained ∼40% nontransfected fibers, we further investigated the CSA of TA muscle fibers only in GFP or GFP-tagged PGC-1α-transferred fibers, using an immunohistochemical analysis method. The average CSA of GFP transfected fibers in TA was decreased by 47% in IM-RM vs. Con (P < 0.01). There was no difference in CSA between GFP- and GFP-PGC-1α-transferred fibers in Con, whereas in the IM-RM groups, CSA of GFP-PGC-1α-transferred fibers showed a 30% increase compared with that of GFP-transferred fibers (P < 0.05; Fig. 7C, D). The CSA histograms shows the IM-RM related differences between the GFP- and GFP-PGC-1α-transferred fibers in their fiber size distribution. The fibers with large CSAs tend to decrease in the IM-RM group compared with Con, whereas GFP-PGC-1α in IM-RM group shows a rightward shift to bigger fibers across the entire range of fiber size compared with GFP-transferred fibers (Fig. 7E).
To gain some insight into the mechanism that might influence muscle fiber size, we examined the extent of Foxo3 phosphorylation, which could have an impact on autophagic and proteasomal degradation pathways. The ratio of phosphorylated to nonphosphorylated Foxo3a content was decreased by IM-RM (P < 0.01), whereas PGC-1α transfection significantly reversed the decline of the ratio (P < 0.01; Fig. 7F).
We next evaluated the impact of IM-RM and PGC-1α transfection on the gene expression of 2 muscle-specific E3 ubiquitin ligases, as markers of protein degradation potential. Atrogin-1 and muscle ring finger-1 mRNA levels were significantly increased by 4- (P < 0.01) and 2.5-fold (P < 0.05), respectively, in IM-RM vs. Con (Fig. 7G, H). PGC-1α overexpression decreased Atrogin-1 and muscle ring finger-1 mRNA levels by 46 and 36%, respectively, in the IM-RM groups (P < 0.05) but not in Con.
PGC-1α transfection increased SOD2 activity caused by SIRT3-mediated deacetylation
The effect of PGC-1α transfection on muscle antioxidant defense was examined. As shown in Table 3, SOD2 and catalase activities were not different between IM-RM and Con in GFP animals, but increased by PGC-1α transfection by 25% (P < 0.05) and ∼2.1-fold (P < 0.05), respectively, in both Con and IM-RM groups. Glutathione peroxidase activity decreased 55% (P < 0.01) with IM-RM, but was increased by 27% (P < 0.05) in Con and 51% (P < 0.01) with PGC-1α transfection. Glutathione reductase activity was decreased by 25% (P < 0.05) with IM-RM, but was unaffected with PGC-1α treatment. Despite the changes in enzyme activity, protein contents of SOD2, catalase, and glutathione peroxidase were not affected with IM-RM or PGC-1α (Supplemental Fig. S4).
TABLE 3.
Effect of PGC-1α overexpression on antioxidant enzyme activities in mouse TA muscle
| Con | IM-RM | |||
| Enzyme | GFP | PGC-1 | GFP | PGC-1 |
| SOD2 | 22.1 ± 4.8 | 28.1 ± 4.7a | 19.7 ± 4.4a | 26.3 ± 4.7 |
| Catalase | 17.9 ± 3.8 | 37.6 ± 6.1a | 23.3 ± 3.9 | 44.8 ± 6.3a |
| GPx | 132.1 ± 17.2 | 167.8 ± 20.3a | 59.5 ± 8.6b | 121.5 ± 9.8c |
| GR | 42.5 ± 5.1 | 47.6 ± 6.4 | 31.8 ± 5.5d | 34.9 ± 5.9d |
| GSH | 8.5 ± 0.7 | 8.7 ± 0.6 | 6.9 ± 0.7d | 7.4 ± 0.8d |
| GSSG | 0.12 ± 0.01 | 0.14 ± 0.03 | 0.42 ± 0.03b | 0.31 ± 0.02b,c |
| GSSG/GSH | 0.014 | 0.016 | 0.061b | 0.042a,b |
Enzyme activities of SOD 2 (units/min/mg protein), catalase (mmol/min/mg protein), glutathione peroxidase (GPx) (nmol/min/mg protein), and glutathione reductase (GR) (nmol/min/mg protein). Abundance of oxidized (GSSG) (µmol/g protein) and reduced (GSH) glutathione (µmol/g protein), and GSSG/GSH ratio. Values are the means ± sem. IM-RM, 5 d of RM following 14 d of IM.
P < 0.05 PGC-1α vs. GFP;
P < 0.01 vs. Con;
P < 0.05 PGC-1α vs. GFP;
P < 0.05 vs. Con; 2-way ANOVA with Tukey’s post hoc test.
The GSH content in TA muscle showed an ∼18% decrease in IM-RM (P < 0.05), whereas GSSG level was increased by 3.5-fold (P < 0.01), resulting in a 5-fold increase in GSSG/GSH ratio (P < 0.01; Table 3). These data indicated a severe oxidative stress caused by IM-RM. PGC-1α overexpression decreased GSSG by 26% (P < 0.01) and GSSG/GSH ratio by 33% (P < 0.05) in the IM-RM but not in the GFP groups.
Because SIRT3 is an important deacetylase that could exert extensive effects on enzymes at the posttranslational level, we evaluated the SIRT3 protein content with Western blot. SIRT3 content was not affected by IM-RM; however, PGC-1α overexpression resulted in a 3.5- and 2.9-fold increase in SIRT3 content in Con and IM-RM (P < 0.01), respectively (Fig. 8A, B). Previously, PGC-1α has been shown to induce ERRα-mediated transcription (22). Thus, we further performed a ChIP assay to elucidate whether PGC-1α enhances the ERRα interaction to the SIRT3 promoter. As shown in Fig. 8C, PGC-1α mediated a significant increase in ERRα-SIRT3 promoter interaction by 5.2-fold in Con (P < 0.05) and 4.9-fold in IM-RM (P < 0.05). Furthermore, interaction of flag-tagged PGC-1α protein with the SIRT3 promoter was enhanced by 59% in IM-RM vs. Con (P < 0.05).
Figure 8.

PGC-1α-induced SIRT3 activates SOD2 by deacetylation. Conditions of IM-RM and Con are described in Fig. 2. A) Representative protein expression in isolated mitochondria from TA muscle. B) Quantification of SIRT3 protein normalized to mitochondria loading control VDAC1/porin (n = 7). C) ChIP analysis of TA muscles transfected with GFP or flag-tagged PGC-1α. DNA immunoprecipitated by antibodies to anti-ERRα, anti-flag, or IgG (control) was amplified by real-time PCR. D) Reciprocal coimmunoprecipitation assays to detect interaction between SIRT3 and SOD2. E) Endogenous SOD2 immunopurified with anti-SOD2 antibody followed by Western blotting with acetyl-lysine antibody (n = 7). Values are the means ± sem. *P < 0.05 vs. Con; +P < 0.05 PGC-1α vs. GFP; ++P < 0.05 PGC-1α vs. GFP; 2-way ANOVA with Tukey-Kramer’s post hoc test.
To determine whether PGC-1α-induced SIRT3 overexpression increases SOD2 activity caused by deacetylation, SIRT3 and SOD2 binding activity was determined. As shown in Fig. 8D, PGC-1α enhanced SIRT3-SOD2 binding in both Con and IM-RM. To assess acetylation levels of SOD2, endogenous proteins were immunoprecipitated with anti-SOD2 antibody and analyzed by Western blot analysis using acetyl-lysine antibody. Basal acetylation level was increased by 57% in IM-RM, and PGC-1α decreased SOD2 acetylation by 30% in IM-RM (P < 0.05; Fig. 8E). Thus, it appears that the increased SOD2 activity without change in enzyme protein level may be explained by decreased acetylation.
DISCUSSION
Muscle disuse atrophy is characterized by decreased muscle fiber CSA, reduced muscle strength, increased fatigability, and insulin resistance (5, 13, 23, 24). Mitochondrial dysfunction plays a critical role in causing these phenotypical changes because of increased oxidative stress, increased inflammatory response, and activation of biodegrative pathways (25). Seeking biologic mechanisms and interventions focusing on improving mitochondrial function can be an effective approach to ameliorate IM-induced disruption of muscle morphologic and functional integrity. In the current study, we focused on the early stage of RM after an extended period of IM because of our previous findings that most mitochondrial genes were still suppressed to below resting levels; there was an apparent increase in the gene expression of inflammatory cytokines; and NF-κB, the most important pathway promoting oxidative stress and inflammation, was activated (16). Here we show that mitochondria malfunction during the early stage of muscle recovery from IM and increased inflammation and oxdative damage after RM was reversed by PGC-1α overexpression, whereas mitochondrial biogenesis and function, as well as antioxidant defense, were enhanced.
PGC-1α overexpression ameliorated mitochondrial dysfunction caused by IM-RM
PGC-1α is known to be a major player that integrates multiple cell signaling pathways in determination of mitochondrial protein content and oxidative capacity (26, 27), antioxidant gene expression (18), and exercise-induced adaptations (22). In the current study, PGC-1α content was lowered by half after 14 and 19 d of IM, which is consistent with previous reports that decreased PGC-1α expression accompanies denervation and muscular inactivity (2, 28). Under several different experimental conditions, the decrease in PGC-1α expression took place at the early stage of intervention preceding protein degradation and weight loss. For example, a large fall of PGC-1α mRNA was observed just 1 d after nerve section in rat gastrocnemius muscle (29). Recently, Liu et al. reported that reloading rat soleus muscle unloaded for 3 wk resulted a partial reversal of fiber CSA and mitochondrial deterioration, as well as prevention of unloading-induced muscle ring finger-1 and Atrogin-1, and these findings were accompanied by an elevation of PGC-1α and NRF-1 (12). Because of the differences in species (rat vs. mouse), unloading model (hindlimb suspension vs. banding), and muscle phenotype (soleus vs. TA), the results of that study cannot be directly compared with our data. However, both studies raised the possibility that PGC-1α signaling plays a key role in maintaining mitochondria content and integrity and can even impact on preservation of muscle protein content and fiber size during IM and RM. Thus, in the current study, we first hypothesize that elevation of PGC-1α levels via the in vivo transfection procedure might protect mitochondrial dysfunction caused by immobilization.
Our data clearly indicated that mitochondrial content and function were improved with PGC-1α overexpression in the IM-RM muscle. Tricarboxylic acid cycle enzyme marker CS and electron transport chain marker COX activities in mitochondria were significantly increased, followed by enhanced MAPR after IM-RM. Furthermore, PGC-1α overexpression increased mitochondrial volume density and the mDNA/nDNA ratio, possibly because of the PGC-1α-induced increase in Tfam expression.
PGC-1α overexpression reduced inflammatory responses caused by IM-RM
Inflammation constitutes an important mechanism in the pathogenesis of muscle disuse atrophy, during which the production of several proinflammatory cytokines are elevated. TNF-α, the most potent activator of the NF-κB pathway, is well documented to increase its expression in association with muscle waste, cancer, and aging (30–32). IM per se appeared to activate NF-κB and proinflammatory cytokine expression to some extent (Fig. 1B), but significant increases of TNF-α, IL-1, IL-6, and and monocyte chemoattractant protein did not take place until TA muscle was remobilized. The patterns of mRNA expression were consistent with our previous findings that protein contents of proinflammatory cytokines were elevated only after the banding of mouse hindlimb was removed (16). In the present study, muscle TNF-α, IL-6, and IL-1β levels were dramatically increased in IM-RM mice. In addition, p-IKK and p-IκB:IκB ratio were elevated, along with increased cytoplasmic and nuclear p65 contents, confirming the critical role of NF-κB in IM-RM-induced muscle inflammation (33, 34).
Previous research has revealed that PGC-1α has anti-inflammatory effects. PGC-1α knockout mice had a higher basal mRNA expression of inflammatory markers such as TNF-α and IL-6 and a higher serum IL-6 level (19, 20), whereas PGC-1α overexpressing mice showed a lower expression rate of these inflammatory cytokines in skeletal muscle compared with their wild-type counterparts (21). However, it is unclear whether the attenuation of inflammation in these transgenic studies was caused by systemic effects or higher PGC-1α level in the muscle. In the current study, increased PGC-1α gene expression by in vivo muscle transfection did not affect IM-RM-inflicted IKK activation, IκB phosphorylation, or p65 increase, but NF-κB-DNA binding was significantly reduced and so were protein contents of 2 proinflammatory cytokines, IL-1β and IL-6. The mechanism for the anti-inflammatory effect of PGC-1α is still unclear. However, a previous study indicated that p65 could directly repress PGC-1α activity in cardiac cells through physical interaction, because the LXXLL domains of PGC-1α are involved in its association with p65 (35). Thus, reduced nuclear p65-DNA binding caused by PGC-1α overexpression found in our study could underlie a major mechanism for the anti-inflammatory role of PGC-1α in remobilized muscle because of its inhibition on NF-κB activation. However, direct evidence for protein-protein interaction between PGC-1α and p65 has yet to be provided in studies.
PGC-1α overexpression attenuated oxidative stress and up-regulated antioxidant defense
Until the early 1990s, it was widely believed that ROS production was limited to contracting skeletal muscle, whereas oxidative injury does not occur in inactive muscles. However, many studies thereafter have shown that ROS generation and oxidative stress are a major etiologic mechanism for disuse atrophy of locomotive muscles (14), as well as the unloaded diaphragm during prolonged mechanical ventilation (36). In the present study, we demonstrated that mitochondria are the main source of H2O2 production in IM-RM TA muscle, whereas NADPH oxidase activation also played a major role. We further showed that oxidative stress in IM-RM muscle was reduced because of PGC-1α overexpression, evidenced by decreased mitochondrial H2O2 production, lipid peroxidation level (marked by 8-isoprostane and 4-HNE), and glutathione oxidation. Our data suggest that reduced oxidative damage seen in the endurance training studies may be conferred via elevated intracellular PGC-1α level (37, 38). When PGC-1α level was suppressed or silenced in muscle, training failed to derive expected adaptations seen under physiologic conditions (39, 40).
One of the important reasons for PGC-1α overexpression to mitigate ROS and oxidative stress could be related to increased antioxidant defense in TA muscle caused by in vivo transfection. PGC-1α has been identified as a crucial nuclear cofactor in the gene expression of several antioxidant enzymes such as SOD2, GPx, catalase, and uncoupling protein-2 (18, 41). Decreased expression of antioxidant genes in muscle-specific PGC-1α knockout mice is likely a major contributing factor in the observed increase in proinflammatory cytokine expression (20). Several studies have demonstrated decreased expression of SOD1, SOD2, and/or GPx1 mRNA, as well as SOD2 protein content in the skeletal muscle of PGC-1α-null mice (20, 40), whereas PGC-1α overexpression was shown to enhance SOD2 gene expression in aged muscle (21). In the present study, PGC-1α overexpression did not affect SOD2, GPx1, or catalase protein content in IM-RM muscle; however, PGC-1α overexpression resulted in an increase in SOD2, GPx, and catalase enzyme activities. The up-regulation of these enzymes could directly influence the mitochondrial and cytosolic ROS levels and thus provide an explanation for decreased NF-κB activation and inflammation as discussed above. It could also reduce intracellular oxidative stress including less disturbance of redox status, which could influence a wide range of cellular events in the pathogenesis of muscle atrophy.
Up-regulation of SIRT3 by PGC-1α deacetylated and activated SOD2
We further investigated the mechanism by which PGC-1α overexpression enhanced SOD2 activity in the IM-RM muscle. A recent study indicated that SOD2 is acetylated at Lys 68, which decreases SOD2 catalytic activity, whereas mitochondrial deacetylase SIRT3 binds to, deacetylates, and activates SOD2 (42). SIRT3-deficient animals exhibit striking mitochondrial protein hyperacetylation, suggesting that SIRT3 is a major mitochondrial deacetylase (43). PGC-1α activates mouse SIRT3 promoter mediated by an ERR binding element mapped to the promoter region (44) Inhibition of ERRα has been shown to attenuate PGC-1α-mediated SIRT3 and its target gene expression and total cellular respiration (44). Remarkably, basal ATP production in SIRT3-null mice was found to reduce >50% (45). The exact molecular mechanism by which SIRT3 affects oxidative metabolism in skeletal muscle is still elusive. Previous studies have shown that SIRT3 can serve as a broad-ranging mitochondrial deacetylase targeting many different enzymes (43). Our data demonstrated that SIRT3 protein level was increased by PGC-1α transfection in both control and IM-RM muscles. We further showed that PGC-1α-ERRα binding on the SIRT3 promoter was enhanced (Fig. 8C). As a result, the acetylation level of SOD2 enzyme molecules was decreased, which explains the enhanced SOD2 enzyme activity in PGC-1α-overexpressing animals. As a deacetylase present mainly in mitochondria (with a possible small fraction in the nucleus), SIRT3 has other potential effects on a broad range of metabolic enzymes (such as acetyl-CoA synthetase), uncoupling proteins, and on PGC-1α. Indeed, deacetylation is one of the major mechanisms by which PGC-1α enhances its co-activating capacity. Thus, SIRT3 and PGC-1α constitute 2 of the most effective regulators in the cell to control metabolic and antioxidant gene expression that could impact on the recovery of immobilized muscle from prolonged disturbance and stress.
Fourteen days of IM-RM significantly decreased TA muscle weight proportional to body weight. The loss of muscle mass was thought to be mainly a result of decreased protein content as shown by the present study and previous investigators (16, 28). Although both decreased protein synthesis and increased protein degradation take place in muscle disuse atrophy, the latter plays a predominate role mainly because of the activation of 2 muscle-specific E3 lygases, Atrogin-1 and muscle ring finger-1 (6, 7). Both enzymes are highly overexpressed in the animal model of acute muscle atrophy and control the ubiquitination and degradation of both regulatory (e.g., calcineurin and MyoD) and structural (e.g., myosin and troponin I) proteins (3, 46). Our data showing a 2.5- to 4-fold increase in muscle ring finger-1 and Atrogen-1 mRNA levels were consistent with our previous report using a similar IM-RM mouse model (16). The present study clearly indicates that a higher PGC-1α level suppresses the expression of these 2 enzymes by 40–50% and facilitates augment of CSA during muscle recovery (Fig. 7). These effects were probably caused by decreased NF-κB signaling and oxidative stress, primary activators of the ubiquitin-proteolysis system.
In conclusion, our data suggest that a futile cycle may exist among ROS generation, NF-κB activation, and inflammation that results in sustained muscle proteolysis and mitochondrial deterioration during the early stage of IM-RM muscle in the current experimental model. The futile cycle may have prevented the muscle from quick recovery from IM (≥5 d). PGC-1α plays an important role in counteracting oxidative stress and ameliorating oxidative-antioxidant balance (Fig. 9). Thus, PGC-1α overexpression may have a significant therapeutic potential in counteracting deleterious consequence of muscle immobilization and facilitating its recovery.
Figure 9.

Schematic overview of the effects of PGC-1α on muscle IM-RM. IM-RM elicits increased mitochondrial ROS generation that activates NF-κB pathway, a major pathway to promote inflammatory cytokine production. PGC-1α enhances mitochondrial biogenesis and function by up-regulating nuclear encoded mitochondrial proteins and SIRT3, a positive regulator of antioxidant enzymes especially SOD2 with deacetylation. Ub, protein ubiquitination; Ac, acetylation.
Acknowledgments
This project was supported in part by grants from the Wisconsin Alumni Research Foundation and the University of Minnesota Office of Vice-President for Research (to L.L.J.), and a U.S. National Institutes of Health, National Institute of Arthritis and Musculoskeletal and Skin Diseases Grant AR057347 (to T.A.H.). The authors declare no conflicts of interest.
Glossary
- ChIP
chromatin immunoprecipitation
- COX
cytochrome c oxidase
- CREB
cyclic AMP response element binding protein
- CS
citrate synthase
- CSA
cross-sectional area
- cyt c
cytochrome c
- DCFH
2',7'-dichlorfluorescein
- ERRα
estrogen-related receptor α
- FOXO
forkhead box protein O
- GFP
green fluorescent protein
- GSH
glutathione
- GSSG
oxidized glutathione
- IM
immobilization
- IMSM
isolation medium for skeletal muscle
- MAPR
mitochondria ATP production rate
- mtDNA
mitochondrial DNA
- NOX
nicotinamide adenine dinucleotide phosphate oxidase
- NRF-1
nuclear respiratory factor-1
- p-IκB
phosphorylated IκB
- PGC-1α
peroxisome proliferator-activated receptor γ coactivator 1-α
- RM
remobilization
- ROS
reactive oxygen species
- SIRT3
sirtuin-3
- SOD
superoxide dismutase
- TA
tibialis anterior
- Tfam
mitochondrial transcription factor A
- XO
xanthine oxidase
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
References
- 1.Thomason D. B., Booth F. W. (1990) Atrophy of the soleus muscle by hindlimb unweighting. J. Appl. Physiol. 68, 1–12 [DOI] [PubMed] [Google Scholar]
- 2.Timmons J. A., Norrbom J., Schéele C., Thonberg H., Wahlestedt C., Tesch P. (2006) Expression profiling following local muscle inactivity in humans provides new perspective on diabetes-related genes. Genomics 87, 165–172 [DOI] [PubMed] [Google Scholar]
- 3.Krawiec B. J., Frost R. A., Vary T. C., Jefferson L. S., Lang C. H. (2005) Hindlimb casting decreases muscle mass in part by proteasome-dependent proteolysis but independent of protein synthesis. Am. J. Physiol. Endocrinol. Metab. 289, E969–E980 [DOI] [PubMed] [Google Scholar]
- 4.Jackman R. W., Kandarian S. C. (2004) The molecular basis of skeletal muscle atrophy. Am. J. Physiol. Cell Physiol. 287, C834–C843 [DOI] [PubMed] [Google Scholar]
- 5.Kandarian S. C., Jackman R. W. (2006) Intracellular signaling during skeletal muscle atrophy. Muscle Nerve 33, 155–165 [DOI] [PubMed] [Google Scholar]
- 6.Bodine S. C., Latres E., Baumhueter S., Lai V. K., Nunez L., Clarke B. A., Poueymirou W. T., Panaro F. J., Na E., Dharmarajan K., Pan Z. Q., Valenzuela D. M., DeChiara T. M., Stitt T. N., Yancopoulos G. D., Glass D. J. (2001) Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 294, 1704–1708 [DOI] [PubMed] [Google Scholar]
- 7.Gomes M. D., Lecker S. H., Jagoe R. T., Navon A., Goldberg A. L. (2001) Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc. Natl. Acad. Sci. USA 98, 14440–14445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li H. H., Kedar V., Zhang C., McDonough H., Arya R., Wang D. Z., Patterson C. (2004) Atrogin-1/muscle atrophy F-box inhibits calcineurin-dependent cardiac hypertrophy by participating in an SCF ubiquitin ligase complex. J. Clin. Invest. 114, 1058–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tintignac L. A., Lagirand J., Batonnet S., Sirri V., Leibovitch M. P., Leibovitch S. A. (2005) Degradation of MyoD mediated by the SCF (MAFbx) ubiquitin ligase. J. Biol. Chem. 280, 2847–2856 [DOI] [PubMed] [Google Scholar]
- 10.Kedar V., McDonough H., Arya R., Li H. H., Rockman H. A., Patterson C. (2004) Muscle-specific RING finger 1 is a bona fide ubiquitin ligase that degrades cardiac troponin I. Proc. Natl. Acad. Sci. USA 101, 18135–18140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clarke B. A., Drujan D., Willis M. S., Murphy L. O., Corpina R. A., Burova E., Rakhilin S. V., Stitt T. N., Patterson C., Latres E., Glass D. J. (2007) The E3 Ligase MuRF1 degrades myosin heavy chain protein in dexamethasone-treated skeletal muscle. Cell Metab. 6, 376–385 [DOI] [PubMed] [Google Scholar]
- 12.Liu J., Peng Y., Feng Z., Shi W., Qu L., Li Y., Liu J., Long J. (2014) Reloading functionally ameliorates disuse-induced muscle atrophy by reversing mitochondrial dysfunction, and similar benefits are gained by administering a combination of mitochondrial nutrients. Free Radic. Biol. Med. 69, 116–128 [DOI] [PubMed] [Google Scholar]
- 13.Caron A. Z., Drouin G., Desrosiers J., Trensz F., Grenier G. (2009) A novel hindlimb immobilization procedure for studying skeletal muscle atrophy and recovery in mouse. J. Appl. Physiol. 106, 2049–2059 [DOI] [PubMed] [Google Scholar]
- 14.Kondo H., Nakagaki I., Sasaki S., Hori S., Itokawa Y. (1993) Mechanism of oxidative stress in skeletal muscle atrophied by immobilization. Am. J. Physiol. 265, E839–E844 [DOI] [PubMed] [Google Scholar]
- 15.Powers S. K., Kavazis A. N., McClung J. M. (2007) Oxidative stress and disuse muscle atrophy. J. Appl. Physiol. 102, 2389–2397 [DOI] [PubMed] [Google Scholar]
- 16.Kang C., Ji L. L. (2013) Muscle immobilization and remobilization downregulates PGC-1α signaling and the mitochondrial biogenesis pathway. J. Appl. Physiol. 115, 1618–1625 [DOI] [PubMed] [Google Scholar]
- 17.Scarpulla R. C. (2008) Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev. 88, 611–638 [DOI] [PubMed] [Google Scholar]
- 18.St-Pierre J., Drori S., Uldry M., Silvaggi J. M., Rhee J., Jäger S., Handschin C., Zheng K., Lin J., Yang W., Simon D. K., Bachoo R., Spiegelman B. M. (2006) Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 127, 397–408 [DOI] [PubMed] [Google Scholar]
- 19.Handschin C., Chin S., Li P., Liu F., Maratos-Flier E., Lebrasseur N. K., Yan Z., Spiegelman B. M. (2007) Skeletal muscle fiber-type switching, exercise intolerance, and myopathy in PGC-1α muscle-specific knock-out animals. J. Biol. Chem. 282, 30014–30021 [DOI] [PubMed] [Google Scholar]
- 20.Handschin C., Choi C. S., Chin S., Kim S., Kawamori D., Kurpad A. J., Neubauer N., Hu J., Mootha V. K., Kim Y. B., Kulkarni R. N., Shulman G. I., Spiegelman B. M. (2007) Abnormal glucose homeostasis in skeletal muscle-specific PGC-1α knockout mice reveals skeletal muscle-pancreatic β cell crosstalk. J. Clin. Invest. 117, 3463–3474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wenz T., Rossi S. G., Rotundo R. L., Spiegelman B. M., Moraes C. T. (2009) Increased muscle PGC-1α expression protects from sarcopenia and metabolic disease during aging. Proc. Natl. Acad. Sci. USA 106, 20405–20410 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22.Schreiber S. N., Emter R., Hock M. B., Knutti D., Cardenas J., Podvinec M., Oakeley E. J., Kralli A. (2004) The estrogen-related receptor (ERR) functions in PPAR coactivator 1 (PGC-1)-induced mitochondrial biogenesis. Proc. Natl. Acad. Sci. USA 101, 6472–6479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.LeBlanc, A. D., Schneider, V. S., Evans, H. J., Pientok, C., Rowe, R., and Spector, E. (1992) Regional changes in muscle mass following 17 weeks of bed rest. J. Appl. Physiol. (1985) 73, 2172–2178 [DOI] [PubMed]
- 24.Stuart C. A., Shangraw R. E., Prince M. J., Peters E. J., Wolfe R. R. (1988) Bed-rest-induced insulin resistance occurs primarily in muscle. Metabolism 37, 802–806 [DOI] [PubMed] [Google Scholar]
- 25.Powers S. K., Wiggs M. P., Duarte J. A., Zergeroglu A. M., Demirel H. A. (2012) Mitochondrial signaling contributes to disuse muscle atrophy. Am. J. Physiol. Endocrinol. Metab. 303, E31–E39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.St-Pierre J., Lin J., Krauss S., Tarr P. T., Yang R., Newgard C. B., Spiegelman B. M. (2003) Bioenergetic analysis of peroxisome proliferator-activated receptor γ coactivators 1α and 1β (PGC-1α and PGC-1β) in muscle cells. J. Biol. Chem. 278, 26597–26603 [DOI] [PubMed] [Google Scholar]
- 27.Wu Z., Puigserver P., Andersson U., Zhang C., Adelmant G., Mootha V., Troy A., Cinti S., Lowell B., Scarpulla R. C., Spiegelman B. M. (1999) Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98, 115–124 [DOI] [PubMed] [Google Scholar]
- 28.Sandri M., Lin J., Handschin C., Yang W., Arany Z. P., Lecker S. H., Goldberg A. L., Spiegelman B. M. (2006) PGC-1α protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc. Natl. Acad. Sci. USA 103, 16260–16265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sacheck J. M., Hyatt J. P., Raffaello A., Jagoe R. T., Roy R. R., Edgerton V. R., Lecker S. H., Goldberg A. L. (2007) Rapid disuse and denervation atrophy involve transcriptional changes similar to those of muscle wasting during systemic diseases. FASEB J. 21, 140–155 [DOI] [PubMed] [Google Scholar]
- 30.Guttridge D. C., Mayo M. W., Madrid L. V., Wang C. Y., Baldwin A. S. Jr (2000) NF-kappaB-induced loss of MyoD messenger RNA: possible role in muscle decay and cachexia. Science 289, 2363–2366 [DOI] [PubMed] [Google Scholar]
- 31.Li Y. P., Reid M. B. (2000) NF-kappaB mediates the protein loss induced by TNF-alpha in differentiated skeletal muscle myotubes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 279, R1165–R1170 [DOI] [PubMed] [Google Scholar]
- 32.Tisdale M. J. (1997) Biology of cachexia. J. Natl. Cancer Inst. 89, 1763–1773 [DOI] [PubMed] [Google Scholar]
- 33.Sandri M. (2008) Signaling in muscle atrophy and hypertrophy. Physiology (Bethesda) 23, 160–170 [DOI] [PubMed] [Google Scholar]
- 34.Li H., Malhotra S., Kumar A. (2008) Nuclear factor-kappa B signaling in skeletal muscle atrophy. J. Mol. Med. 86, 1113–1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Álvarez-Guardia D., Palomer X., Coll T., Davidson M. M., Chan T. O., Feldman A. M., Laguna J. C., Vázquez-Carrera M. (2010) The p65 subunit of NF-kappaB binds to PGC-1α, linking inflammation and metabolic disturbances in cardiac cells. Cardiovasc. Res. 87, 449–458 [DOI] [PubMed] [Google Scholar]
- 36.Falk D. J., Deruisseau K. C., Van Gammeren D. L., Deering M. A., Kavazis A. N., Powers S. K. (2006) Mechanical ventilation promotes redox status alterations in the diaphragm. J. Appl. Physiol. 101, 1017–1024 [DOI] [PubMed] [Google Scholar]
- 37.Baar K., Wende A. R., Jones T. E., Marison M., Nolte L. A., Chen M., Kelly D. P., Holloszy J. O. (2002) Adaptations of skeletal muscle to exercise: rapid increase in the transcriptional coactivator PGC-1. FASEB J. 16, 1879–1886 [DOI] [PubMed] [Google Scholar]
- 38.Irrcher I., Adhihetty P. J., Sheehan T., Joseph A. M., Hood D. A. (2003) PPARgamma coactivator-1α expression during thyroid hormone-and contractile activity-induced mitochondrial adaptations. Am. J. Physiol. Cell Physiol. 284, C1669–C1677 [DOI] [PubMed] [Google Scholar]
- 39.Leick L., Hellsten Y., Fentz J., Lyngby S. S., Wojtaszewski J. F., Hidalgo J., Pilegaard H. (2009) PGC-1alpha mediates exercise-induced skeletal muscle VEGF expression in mice. Am. J. Physiol. Endocrinol. Metab. 297, E92–E103 [DOI] [PubMed] [Google Scholar]
- 40.Leick L., Lyngby S. S., Wojtaszewski J. F., Pilegaard H. (2010) PGC-1α is required for training-induced prevention of age-associated decline in mitochondrial enzymes in mouse skeletal muscle. Exp. Gerontol. 45, 336–342 [DOI] [PubMed] [Google Scholar]
- 41.Valle I., Álvarez-Barrientos A., Arza E., Lamas S., Monsalve M. (2005) PGC-1α regulates the mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovasc. Res. 66, 562–573 [DOI] [PubMed] [Google Scholar]
- 42.Chen Y., Zhang J., Lin Y., Lei Q., Guan K. L., Zhao S., Xiong Y. (2011) Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep. 12, 534–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lombard D. B., Alt F. W., Cheng H. L., Bunkenborg J., Streeper R. S., Mostoslavsky R., Kim J., Yancopoulos G., Valenzuela D., Murphy A., Yang Y., Chen Y., Hirschey M. D., Bronson R. T., Haigis M., Guarente L. P., Farese R. V. Jr., Weissman S., Verdin E., Schwer B. (2007) Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell. Biol. 27, 8807–8814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kong X., Wang R., Xue Y., Liu X., Zhang H., Chen Y., Fang F., Chang Y. (2010) Sirtuin 3, a new target of PGC-1α, plays an important role in the suppression of ROS and mitochondrial biogenesis. PLoS ONE 5, e11707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ahn B. H., Kim H. S., Song S., Lee I. H., Liu J., Vassilopoulos A., Deng C. X., Finkel T. (2008) A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. USA 105, 14447–14452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stevenson E. J., Giresi P. G., Koncarevic A., Kandarian S. C. (2003) Global analysis of gene expression patterns during disuse atrophy in rat skeletal muscle. J. Physiol. 551, 33–48 [DOI] [PMC free article] [PubMed] [Google Scholar]