

Abstract
From epidemiological studies it is known that diabetes patients display increased risk of developing dementia. Moreover, cognitive impairment and Alzheimer’s disease (AD) are also accompanied by impaired glucose homeostasis and insulin signalling. Although there is plenty of evidence for a connection between insulin-resistant diabetes and AD, definitive linking mechanisms remain elusive. Cerebrovascular complications of diabetes, alterations in glucose homeostasis and insulin signalling, as well as recurrent hypoglycaemia are the factors that most likely affect brain function and structure. While difficult to study in patients, the mechanisms by which diabetes leads to brain dysfunction have been investigated in experimental models that display phenotypes of the disease. The present article reviews the impact of diabetes and AD on brain structure and function, and discusses recent findings from translational studies in animal models that link insulin resistance to metabolic alterations that underlie brain dysfunction. Such modifications of brain metabolism are likely to occur at early stages of neurodegeneration and impact regional neurochemical profiles and constitute non-invasive biomarkers detectable by magnetic resonance spectroscopy (MRS).
Keywords: Neurodegeneration, Diabetes, Alzheimer’s disease, Insulin, Metabolism
Diabetes mellitus can be defined as a chronic metabolic disorder characterized by hyperglycaemia, resulting from inappropriate insulin secretion and/or action [1]. The vast majority of the diabetes cases are included in two main categories, classified according to the underlying cause, which are type 1 diabetes (T1D) or insulin-dependent diabetes, generally caused by an autoimmune reaction to antigens of pancreatic β-cells, leading to impaired insulin production, and type 2 diabetes (T2D) or insulin-resistant diabetes, characterised by inefficiency of insulin action. While T1D is mainly observed in children and adolescents, T2D is more common among adults, accounting for more than 90% of the diabetes cases worldwide. The prevalence of diabetes and impaired glucose tolerance achieved now epidemic proportions, respectively at 6.9% and 8.3% of the world population, and predicted to raise to 8.0% and 10.1% in 2035 [2]. Moreover, diabetes is nowadays a leading cause of death accounting for 8.4% of global mortality in people aged between 20 and 79 years [2]. The main contributor for the high prevalence of diabetes is the rise of obesity, related to the combination of ample food availability and a sedentary lifestyle, contrasting to less abundant food supplies and higher physical activity observed until a century ago.
Diabetes is associated with the occurrence of well described microvascular complications that affect different organs, leading most commonly to retinopathy, nephropathy and peripheral neuropathy, which development is dependent on the duration of the disease and glycaemia control [3]. When the control of the disorder allowed patients to live longer and without these traditional complications, diabetes-associated brain dysfunction was observable. The concept that diabetes affects the central nervous system (CNS) was recognised in 1922, when evidence appeared for diabetes-induced cognitive dysfunction [4], and the term “diabetic encephalopathy” was introduced in 1950 to describe the complication of diabetes resulting on brain dysfunction and leading to cognitive impairment [5].
Several clinical studies reported lower performance on several cognitive domains in T1D patients, when compared to the general healthy population, including notably learning and memory impairment [6-10]. The magnitude of these cognitive deficits is often mild but severe cases can occur [11, 12]. Along with these deficits on brain function, diabetes was also reported to induce structural alterations, neuronal loss, demyelination and gliosis [13]. Accordingly, magnetic resonance imaging (MRI) and computed tomography showed general brain atrophy and increased occurrence of white mater hyper-intensities that are thought to result from small infarcts [14-16]. In T2D patients, impaired cognitive function was observed in particular when solving complex cognitive tasks [17-22]. Cognitive dysfunction is further accentuated in elderly T2D patients with reduced diabetes control [23, 24]. In summary, diabetes causes cognitive dysfunction that is moderated at younger age but can be accentuated in elderly diabetic patients and by poor control over blood glucose levels, and hampers daily functioning, thus reducing the quality of life. The neurochemical mechanisms underlying the development of diabetes-associated cognitive decline are poorly understood. Animal experimentation has begun aiming to understand the mechanistic processes leading to diabetic encephalopathy, but greater efforts must still be taken to achieve this goal.
Cognitive impairment in diabetes and AD
The prevalence of neurodegenerative disorders leading to dementia is raising worldwide due to aging of the population (increased lifespan), a situation that represents an enormous social burden [25]. While Alzheimer’s disease (AD) is the most common type of dementia, accounting for 60-80% of cases [26], about 8% of all cases of dementia can be attributable to diabetes mellitus [27] and this tends to increase with the growing prevalence of diabetes. The high incidence of Alzheimer’s disease in T2D is linked to insulin resistance, hyperinsulinemia and hyperglycemia, as well as commonly accompanying hypercholesterolemia, hypertension and obesity [28, 29]. Cognitive dysfunction in patients with diabetes particularly impairs the ability to control glycaemia and thus is also associated with enhanced risk of severe hypoglycaemia episodes and cardiovascular diseases, relative to patients without cognitive abnormalities [30, 31]. Thus, preventing these events and their associated morbidity is of clinical importance. For that, the mechanisms of diabetic encephalopathy must be understood, and the discovery of reliable disease biomarkers and design of preventive strategies are necessary.
Diabetic encephalopathies in T1D and T2D differ in the underlying mechanisms and the nature of resulting cognitive deficits. Encephalopathy in T1D occurs in younger patients and involves impaired learning abilities, intelligence development and memory retrieval. Although uncontrolled sustained hyperglycaemia leads to brain alterations and functional deterioration due to degeneration of the microvasculature, another major underlying component leading to encephalopathy in T1D patients appears to be insulin deficiency with downstream effects on the expression of neurotrophic factors, on the efficiency of neurotransmission, and on oxidative and apoptotic stressors, resulting in loss of neuronal integrity and connectivity defects in the still developing brain [29]. Furthermore, T1D patients are at risk for developing hypoglycaemia unawareness and associated complications (see section “Hypoglycaemia and hypoglycaemia unawareness”).
In contrast to T1D, clinical research over the two last decades has established a striking link between encephalopathy in T2D and AD. T2D is associated with an increased risk of cognitive dysfunction and dementia, affecting the brain by mechanisms that include: (1) metabolic and vascular risk factors within the metabolic syndrome such as dyslipidaemia, hypercholestrolemia and hypertension, which were found to be associated with cerebrovascular disease, accelerated cognitive decline and dementia; (2) hyperglycaemia that has adverse effects on the brain and its vasculature; (3) insulin that can directly modulate energy metabolism, synaptic plasticity and learning and memory, as well as regulate the metabolism of the β-amyloid peptide (Aβ) and Tau protein, the building blocks of amyloid plaques and neurofibrillary tangles [28, 32, 33].
Brain metabolism and cognitive performance
Glucose catabolism is the major pathway of energy production in the mature brain, thus constituting the bioenergetic basis for neurotransmission. Therefore, increases in glucose metabolism during brain activation have been used for functional mapping by 2-deoxy-glucose autoradiography and positron-emission tomography (PET), and indirectly influence hemodynamic changes observed with functional MRI. Moreover, pathways of brain glucose oxidation have been suggested to be linked to the glutamine-glutamate/GABA cycling between astrocytes and neurons, i.e. the rates of glutamatergic and GABAergic neurotransmission [34]. For the sake of example, this was demonstrated by measuring a reduction of extracellular glucose in the hippocampus during a hippocampal-dependent spatial memory test [35, 36]. Moreover, blockade of brain glucose transport or astrocytic glucose metabolism inhibits memory consolidation (see [37] and references therein). In line with this, somatosensory stimulation increases the rate of glycogenolysis [38, 39] and thus reduces brain glycogen levels [40, 41], the glucose storage in the brain. Thus, impaired brain glucose homeostasis may compromise learning processes and memory consolidation, and this could constitute a link between diabetes and brain alterations leading to encephalopathy.
Brain metabolism in aging
Mitochondrial production of reactive oxygen species (ROS) is the main drive for accumulated cellular stress that primarily leads to aging-associated impairments of mitochondrial functions such as regulation of Ca2+ homeostasis and ATP synthesis, that is energy production (reviewed in [42]). In turn, defects in energy metabolism at the level of the tricarboxylic acid (TCA) cycle, electron transport chain and oxidative phosphorylation further enhance the ROS production and affects amino acid homeostasis (see [43] and references therein). Moreover, faulty mitochondrial ATP synthesis triggers higher demand from glycolysis. A 13C MRS study in the human brain demonstrated that the neuronal TCA cycle is indeed reduced in elderly subjects, resulting in deficient glutamate-glutamine cycling between neurons and astrocytes [44]. In contrast, glial oxidative metabolism was increased probably due to astrogliosis [44]. A large longitudinal MRS study in C57Bl/6 mice over 2 years further confirmed the age-induced impairment in amino acid homeostasis, with substantial reduction in the levels of neuro-active amino acids including the major excitatory and inhibitory neurotransmitters, respectively glutamate and GABA [43]. These metabolic modifications that occur in the brain due to cellular damage accumulated during lifespan are crucial for functional deterioration in aged subjects [45] and are further accentuated in neurodegenerative disorders [42].
Brain metabolism in AD
Impaired cerebral energy metabolism is known to occur in AD patients. PET with [18F]fluorodeoxyglucose in AD patients revealed progressive reduction in glucose metabolism, namely in parietal and temporal lobes [46]. This method has thus been employed to identify mild cognitive impairment and early stages of AD in living patients. Accordingly, after glucose administration, glucose was observed in the brain of AD patients at higher levels than healthy subjects [47], likely reflecting reduced glucose metabolism. Indeed, recent studies suggested that hypometabolism of glucose in the brain of AD patients is closely linked to cognitive decline [48] and can be indicative of larger subsequent cerebral atrophy [49]. Although cerebral atrophy is not generally related to the regional binding of the Pittsburgh compound B (measuring levels of Aβ aggregation), a relationship between Aβ deposition and atrophy was observed in early AD [49, 50]. Interestingly, it was also recently suggested that brain areas of high resting glucose metabolism (reflecting high neuronal activity) in healthy elderly individuals generally correspond to those where the level of regional Pittsburgh compound B uptake is greater in AD patients [51]. This suggests that Aβ deposition may be dependent on neuronal activity, probably because neuronal activity-associated high mitochondrial metabolism is prone to oxidative stress.
It is known that Aβ neurotoxicity involves induction of oxidative stress with concomitant damage to biomolecules including membrane lipids, proteins and nucleic acids, which promotes synaptic and mitochondrial dysfunction and eventually leads to apoptosis. These events are also promoted by intracellular neurofibrillary tangles that are formed by hyperphosphorylation of the microtubule-associated protein Tau. In brief, mitochondrial dysfunction involves electron transport chain dysfunction, opening of the mitochondrial permeability transition pore that disrupts oxidative phosphorylation, impaired mitochondrial dynamics that regulates axonal transport, and reduction of mitochondrial creatine kinase activity that decreases the energetic buffering effect of phosphocreatine [42, 52]. This then leads to synaptic dysfunction mainly due to insufficient energy supply for neurotransmitter recycling and for maintenance of cellular electrochemical gradients (Ca2+ and Na+ homeostasis). However, little is known about AD-induced specific alterations of neuronal and astrocytic metabolism involved in neurotransmission. Studies in vivo in AD patients [53] and in rodent models of the disease [54, 55] using 13C MRS pointed towards a reduction of glucose oxidative metabolism, namely in neurons with concomitant increased lactate production, and a reduction in the rate of glial pyruvate carboxylation that is necessary for de novo synthesis of amino acids, namely glutamine, glutamate, GABA and aspartate. Decreased glutamate levels were recently reported in the brain of AD patients [56]. In addition, Penner et al. found that glutamate concentration was increased in the right hippocampus of AD patients after four months of galantamine treatment relative to pre-treatment levels, and those variations in glutamate levels were related to variations in N-acetylaspartate (NAA) content and cognitive performance [57]. Interestingly, it has been recurrently demonstrated that the regional levels of NAA and glutamate in the brain are positively associated since both are mainly located within neuronal cells (see [43] and references there in). NAA is believed to be produced in neuronal mitochondria and then used for myelin synthesis in olygodendrocytes [58]. Thus it is plausible that dysfunctional mitochondria are unable to produce sufficient NAA for adequate maintenance of myelin sheaths around long axonal fibres.
The majority of the clinical studies using 1H MRS consistently revealed that the brain of AD patients contains less NAA and more myo-inositol, compared to cognitively healthy elderly subjects, and that the concentration of these two neurochemicals is correlated to the decline in cognitive performance [42]. In general these modifications of NAA and myo-inositol are interpreted as loss of neuronal integrity and gliosis, respectively [58]. Consistent with neurodegeneration, hippocampal NAA levels were found to be positively associated to hippocampal volume across subjects presenting mild to moderate severity AD and age-matched healthy controls [59], i.e. loss of NAA correlates with atrophy of the cerebral tissue. Furthermore, the modifications of NAA and myo-inositol in the posterior cingulate of AD patients were associated to reduction of diffusion parameters measured by diffusion tensor imaging, namely mean diffusivity and fractional anisotropy [60], consistent with astrogliosis and/or axonal degeneration.
Insulin signalling defects in aging and AD
Insulin receptors are ubiquitously expressed in the brain, especially in the hippocampus, cortex, hypothalamus, olfactory bulb and pituitary, with particular high density in neurons. Insulin in the brain can reach local levels one order of magnitude greater than in plasma (due to local production) and exert its actions not only through insulin receptors but also via insulin-like growth factor 1 (IGF-1) receptors that are as well widely distributed throughout the brain [61, 62]. The insulin/IGF-1-mediated activation of complex signalling cascades serves the regulation a plethora of cellular processes including growth, differentiation and metabolism [63]. In the CNS, insulin exerts further control over neurotransmission, synaptic plasticity and cognitive processes, cellular proliferation and antiapoptotic mechanisms, and antioxidant defence [64, 65]. Therefore, defects in brain insulin signalling pathways result in altered brain function.
Cellular challenges during normal aging lead to many homeostatic alterations and ultimately result in neurodegeneration, impaired neuronal function and reduced cognitive performance [45]. Epigenetic factors modulated by aging and a sedentary lifestyle have been described to induce a metabolic shift by attenuating mitochondrial metabolism and increasing reliance on glycolysis [66]. This alteration starts a damaging cycle involving oxidized membrane receptors, signalling molecules, transcription factors, and epigenetic transcriptional regulators [62, 66]. Accordingly, decrease in insulin receptor levels and impaired insulin signalling was observed in the hippocampus, cortex and choroid plexus [62], and insulin resistance was reported to be associated with cognitive decline in non-demented aging [67].
Impaired insulin signalling is believed to be associated with extracellular Aβ deposition and with hyperphosphorylation of the microtubule-associated protein Tau and formation of neurofibrillary tangles, which are hallmarks of AD. Through the phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) pathway, insulin stimulates amyloid precursor protein metabolism, promotes Aβ release from neurons preventing its intracellular accumulation, and increases insulin-degrading enzyme (IDE) protein levels [68]. Thus, insulin resistance may contribute to increase intracellular Aβ and its extracellular accumulation. Since IDE degrades insulin and Aβ, as well as other peptides, altered insulin levels can also interfere with the clearance of Aβ [69]. On the other hand, insulin binding and receptor autophosphorylation are inhibited by Aβ [70]. Tau phosphorylation is inhibited by insulin, and impaired insulin signalling in a neuron-specific insulin receptor knockout mouse resulted in reduced phosphorylation of Akt and GSK3β, leading to increased Tau phosphorylation [71].
AD is in fact associated with impaired expression of insulin receptors [33] and altered insulin and IGF-1 levels [32, 72], which affects the cAMP response element-binding protein (CREB) pathway that controls memory formation, and the PI3K/Akt pathway signalling that regulates glucose homeostasis, thus increasing glucose levels and potentiating the formation of advanced glycation end-products [61, 73]. In line with this, intranasal insulin administration has been effective in enhancing cognitive performance in healthy humans and some patients with AD [74, 75] and in improving memory formation in rodents [76], which may include regulation of glucose uptake and/or metabolism [77]. In contrast, decreases in insulin or IGF-1 signalling in rodent models of AD triggered reductions in Aβ deposition and in cognitive performance [78-81]. In summary, insulin signalling appears involved in the pathophysiology of AD, but the link between T2D, AD and cognitive dysfunction remains unclear.
Neurodegeneration in animal models of diabetes
Elderly-associated mild memory and cognitive impairment and AD are accompanied by atrophy of hippocampal formation [82-85]. When compared to healthy subjects, individuals with T2D display deficits in hippocampal-based memory performance with preservation of other cognitive domains, and show reduction of hippocampal volume but not other brain areas [86]. Cognitive dysfunction during diabetes is therefore particularly associated with significant changes in the integrity of the hippocampus, a brain region considered to mediate memory formation in animals, and electrophysiological analyses indicate that diabetes induces defects on long-term potentiation (LTP) in hippocampal slices, a form of synaptic plasticity considered to reflect learning and memory processes in mammals [87, 88]. Based on this, the vast majority of translational studies in animal models of diabetes were dedicated to the study of hippocampal structure and function. Impaired hippocampal-dependent spatial learning and memory have been demonstrated in different animal models of diabetes using the Morris water maze [89-93] and the Y-maze [94, 95].
Insulin-sensitive diabetes
Diabetic encephalopathy can derive from cellular damage caused by both glucose neurotoxicity upon hyperglycaemia and defective insulin signalling by either insulin deficiency or receptor desensitization [96]. Most translational/pre-clinical studies aiming to identify mechanisms of diabetic encephalopathy have investigated animal models characterised by insulin deficiency (being sensitive to insulin administration) and chronic hyperglycaemia. Such experimental diabetic conditions cause deficits in spatial learning and synaptic plasticity [90], synaptic degeneration [97-99], increased astrocyte reactivity and proliferation [98, 100, 101], oxidative stress [102] and altered metabolism [98, 103]. Furthermore, chronic hyperglycaemia and hypoinsulinemia in an animal model of T1D (streptozotocin-treated rats) lead to impairment of multiple neuromodulation systems in the hippocampus that control metabolism and synaptic activity [97, 98, 104, 105]. This may contribute to altered synaptic plasticity and memory performance, and constitute a potential target for neuroprotection. Given the close link between brain function and energy metabolism [34], metabolic alterations have been described in the brain of experimental models of T1D. These could involve both substrate delivery through the blood-brain-barrier (BBB) and intermediary metabolism in neurons and astrocytes.
Structural alterations were reported in the BBB of diabetic animals [106], consistent with increased BBB permeability observed in individuals with diabetes [107], suggesting that loss of BBB integrity may be involved in CNS dysfunction in diabetes. However, permeability of the BBB to glucose does not seem to be altered in the hippocampus and cortex of streptozotocin-induced T1D rats [98, 103]. Therefore, hyperglycaemia in uncontrolled diabetes results in proportionally increased glucose concentration in the brain. How the higher levels of glucose in diabetes are handled by neurons and astrocytes has not been clearly demonstrated.
Reduced hippocampal neurogenesis was reported to occur in two models of T1D, the non-obese diabetic (NOD) mice [108] and the streptozotocin-induced diabetic rats [109, 110]. Compared to non-diabetic controls, neuronal loss was observed in streptozotocin-induced diabetic rats [111] and BB/Wor diabetic rats [112]. Before the loss of neurons, synaptic deterioration was observed in the brain of diabetic Chinese hamster [113] and streptozotocin-induced diabetic rats [93, 97, 98, 114, 115]. In line with this, the efficiency of neurotransmission was found altered in diabetic brains. In particular, glutamatergic neurotransmission is strongly impaired in diabetes [88] and is probably associated with a reduction in energy metabolism supporting the glutamate-glutamine cycle [34]. Cortical slices but not synaptosomes prepared from the brain of streptozotocin-induced diabetic rats displayed reduced uptake of glutamate [116], suggesting impaired glutamate clearance by astrocytes that can result in higher levels of glutamate in the synaptic cleft and induce excitotoxycity. Also the content of dopamine, norepinephrine and serotonin was reported to be altered in certain brain regions in alloxan-induced [117] and streptozotocin-induced [114, 115] diabetic rats, which may be prevented by insulin treatment [115].
These structural and neurochemical alterations result in functional anomalies. The latencies of auditory and visual potentials were found to be prolonged in streptozotocin-induced diabetic rats [118, 119] and type 1 BB/Wor diabetic rats [120, 121] in relation to controls. Likewise, in hippocampal slices from streptozotocin-induced diabetic rats, LTP is impaired, whereas long-term depression (LTD) is enhanced when compared with control rats [90, 91, 122, 123]. These altered hippocampal synaptic transmission and plasticity were paralleled by reduced spatial learning and memory [91] that was prevented by glycaemia control with insulin treatment [122].
In insulin-deficient mice, intranasal insulin treatment was shown to ameliorate synaptic degeneration and deficits in learning and memory, without preventing hyperglycaemia [124]. This suggests that impairment of central insulin signalling is an important factor for diabetes-induced brain injury. However, in contrast to hypoinsulinemia and associated hyperglycaemia, the mechanisms of impaired insulin sensitivity leading to altered brain homeostasis, synaptic activity and memory performance are much less understood.
Insulin-resistant diabetes
Animal models of insulin resistance displayed lower insulin levels in the brain. Namely, obese (fa/fa) and lean heterozygous (Fa/fa) Zucker rats had lower immunoreactive insulin in the olfactory bulb, hypothalamus, hippocampus, cerebral cortex, amygdala, midbrain and hindbrain, in comparison to lean homozygous (Fa/Fa) Zucker rats [125]. Reduction in insulin levels was also found in the cerebrospinal fluid of high-fat fed, insulin-resistant dogs and were suggested to result mainly from impaired transport from plasma across the BBB [126]. However, insulin has been proposed to be produced in the brain [62] and could possibly counterbalance an eventual reduction in the uptake from blood. Nevertheless, aberrant insulin regulation in the brain will certainly have consequences for brain function. In pre-diabetes or T2D, insulin resistance is directly associated with reduced cortical glucose consumption and cognitive impairment [127]. Furthermore, like in AD, deficient insulin-PI3K/Akt signalling in animal models of diabetes may further induce hyperphosphorylation of the Tau protein and favour Aβ deposition [61] that leads to formation of neurofibrillary tangles and amyloid plaques.
In a mouse model of obesity-associated T2D, NONcNZO10/LtJ mice under an 11% fat diet, we recently reported impaired hippocampal-dependent spatial memory as indicated by decreased spontaneous alternation in the Y-maze, which was accompanied by decreased density of nerve terminal proteins (synaptophysin, SNAP25), glutamatergic markers (vesicular glutamate transporters), increased astrogliosis (immunoreactivity to glial fibrillary acidic protein, GFAP) and altered adenosinergic modulation system in the hippocampus compared to controls (NON/LtJ mice) kept under the same diet [95]. Interestingly, we failed to identify diabetes-induced neuronal loss in brain sections stained either cresyl violet or FluoroJade-C, which was corroborated by the lack of altered microtubule-associated protein 2 (MAP2) immunoreactivity, a marker of neuronal density [95]. This indicates that T2D primarily causes synaptic deterioration without neuronal death, which resembles the events occurring in aging [128] and models mimicking early AD [129, 130], thus corroborating the association of insulin resistance and T2D with increased incidence of AD. Moreover, compared to non-diabetic Wistar rats, also non-obese insulin-resistant Goto-Kakizyki (GK) rats displayed reduced spatial memory, which was accompanied by reduced pre-synaptic markers and increased glial specific proteins in the hippocampus [131]. Altered expression of genes involved in neurotransmission (among others) was found in the cortex of GK rats [132], indicating that diabetes-induced synaptic alterations are not region specific. A recent publication also reported that, relative to controls, GK rats display learning and memory deficits in a Morris water maze that are proportional to the grade of insulin resistance, reduction in dendritic spine density and neuronal viability in CA1 of the hippocampus, and impaired insulin-stimulated Akt/CREB phathway [133], consistent with AD-like pathology. Furthermore, synaptosomal preparations from the brain of GK rats show impaired mitochondrial function and increased vulnerability to oxidative stress and Aβ toxicity [134, 135], suggesting impaired intermediary metabolism at the level of the synapse. Although the brain of diabetic GK rats is more susceptible to Aβ toxicity and oxidative stress than control Wistar rats, it is not known if Aβ accumulation, Tau hyperphosphorylation and formation of neurofibrillary tangles occur in the brain of this model for non-obese insulin-resistant diabetes. In obese BBZDR/Wor rats that develop insulin resistance with hyperinsulinemia and hyperglycaemia, neuronal degeneration, reduced presynaptic densities and gliosis was reported in the frontal cortex, as well as down regulation of IGF-1 receptors and increased APP, β-secretase, Aβ and phosphorylated Tau [29, 136]. Insulin receptor substrate 2 knockout mice, a model of insulin-resistant diabetes, also displayed hyperphosphorylated Tau in the hippocampus [137].
In addition, glutamatergic and GABAergic neurons may adapt differently to diabetic encephalopathy, since there was a reduction in vesicular glutamate transporters (VGLUT1/2) in the hippocampus of NONcNZO10/LtJ mice compared to non-diabetic NON/LtJ mice, and a lack of diabetes-induced modification of vesicular GABA transporters [95]. The preservation of GABAergic nerve terminals in diabetes is also corroborated by Galanopoulus et al. who found reduced glutamate decarboxylase activity in cortical slices but not synaptosomes prepared from brains of streptozotocin-induced diabetic rats [116], which suggests that diabetes may impact the metabolism of GABA in glial cells but not in inhibitory nerve terminals. Interestingly, glutamatergic nerve terminals are also more susceptible to Aβ-induced toxicity than GABAergic terminals, namely in the hippocampus [130]. Indeed, it is generally accepted that mainly glutamatergic and cholinergic pathways are disrupted in AD [138], and there is a growing body of evidence that GABAergic function is preserved in early phases of AD but becomes impaired when the disease is well established [139, 140].
In summary, although the mechanistic link from impaired oxidative glucose metabolism caused by insulin resistance to altered neurotransmission and impaired brain function remains to be fully elucidated, it may involve increased metabolic rates in glial cells due to astrogliosis and impaired neuronal metabolism, associated to synaptodegeneration. Probably, these metabolic alterations are caused by mitochondrial dysfunction that causes oxidative stress and affects neurotransmission efficiency at glutamatergic (excitatory) rather than GABAergic (inhibitory) nerve terminals, and which may occur much before appearance of neurofibrillary tangles/amyloid plaques. Insulin resistance-induced alterations of neuronal and glial energy metabolism remain to be investigated. Moreover, efforts should be devoted to the study of experimental models displaying insulin-resistance without the confounder of severe hyperglycaemia that causes microvasculature deterioration.
Neurochemical profiling in models of diabetes by MRS
Proton MRS became a tool of choice to investigate metabolic alterations induced by neurological disorders in a non-invasive manner [58] and it has also been employed to study brain metabolism in experimental models of diabetes. Diabetic rats under chronic hyperglycemia, induced by streptozotocin administration, display a plethora of metabolic alterations in the hippocampus and cortex, most of which are normalized upon acute restoration of euglycaemia [98, 103]. Some of the metabolites more affected by hyperglycemia were myo-inositol, taurine and creatine, which are considered major organic osmolytes regulating brain osmotic adaptation. Similar results were obtained in Goto-Kakizaki rats, an experimental model of insulin resistance and T2D [131]. High concentration of myo-inositol was also reported in the hippocampus of Zucker diabetic fatty rats compared to controls [141] and in the brain of diabetes patients [142, 143]. Thus, the study of the neurochemical profile supports the hypothesis that diabetes-induced hippocampal dysfunction involves deregulation of the osmotic balance. In contrast to AD, NAA levels do not appear altered in the diabetic brain, suggesting preservation of neuronal integrity, despite synaptic degeneration.
Similar to what is observed in AD patients, double transgenic mice expressing human mutant APP and human mutated PS1 (APP/PS1) display decreased glutamate and NAA concentrations in the hippocampus, that were observed at the time where hippocampal volume was already reduced but amyloid plaques were not yet present [144-146]. When amyloid deposits became detectable, these mice also presented increased brain myo-inositol levels, when compared to wild type mice [147], which were accompanied by astrogliosis [148]. Increased myo-inositol and glutamine and decreased NAA and glutamate were reported to occur in the cortex of these mice and, once amyloid plaques are observed, NAA levels were inversely associated to the area of cortex occupied by plaques [144].
Dedeoglu et al. but not Marjanska et al. reproduced these findings in mice possessing only the mutated APP gene. Both authors reported, however, higher cerebral taurine to creatine ratio in the brain of APP transgenic mice compared to wild type mice [145, 149]. These results were confirmed by MRS of brain extracts, in which glutathione levels were additionally found to be lower than in controls [149], consistent with redox deregulation [52].
Hypoglycaemia and hypoglycaemia unawareness
Postprandial hyperglycaemia periods are often prevented by insulin administration, principally in cases of T1D. However, episodes of severe hypoglycaemia are a major complication of this glycaemia control. Diminished brain function during acute hypoglycaemia leads to potential physical danger and, moreover, recurrent hypoglycaemia imposes long-lasting damaging effects on brain function [150, 151]. Cognitive performance is altered during experimentally induced hypoglycaemia [152-158], and some studies in patients with T1D suggested an association between recurrent hypoglycaemia and cognitive impairment [11, 16, 159-162].
During severe hypoglycaemia, the absence of sufficient glucose supply to the brain leads to neuroglycopenia that results in energy deprivation and thus other substrates are mobilised for ATP synthesis. Hypoglycaemia causes a reduction in brain glycogen and lactate levels and then depletes metabolic intermediates, namely glucose-6-phosphate, pyruvate, citrate, 2-oxoglutarate and malate [163], and leads to progressive reduction of brain glutamate, glutamine, GABA and alanine with concomitant increase in aspartate that can efficiently act on NMDA receptors [164-168]. The transition to encephalographic silence in hypoglycaemia (i.e. cessation of neural activity) is accompanied by a rapid decline in levels of phosphocreatine, nucleoside triphosphate, and an increase in inorganic orthophosphate, as measured in vivo by 31P MRS [166]. In this isoelectric state, most glycolytic and citric acid cycle intermediates are very low and glucose-6-phosphate and pyruvate are virtually absent [163]. Only when glutamate and glutamine become substantially exhausted, aspartate becomes an important energy source [168]. Glucose administration to recover from hypoglycaemia allows for metabolic recovery and nearly restores amino acids to pre-hypoglycemia levels [166, 168].
The occurrence of recurrent hypoglycaemia episodes impacts the body’s defence mechanisms against hypoglycaemia episodes. Many diabetes patients display a progressive decay in the counter-regulatory response over time, resulting in reduction of hypoglycaemia awareness [169, 170], where the glycaemia threshold for counter-regulation symptoms and cognitive dysfunction is shifted to lower plasma glucose levels and may be detected only upon severe neuroglycopenia [171]. Together, the hypoglycaemia-induced decrease in the sympathoadrenal response to blood glucose variations and the hypoglycaemia unawareness are termed hypoglycaemia-associated autonomic failure (HAAF). HAAF in diabetes patients leads to an increased risk for the occurrence of severe hypoglycaemia episodes.
Beyond the precise mechanisms that sense blood glucose and link the sensory machinery to the autonomic and endocrine responses which alert the patient to the low blood glucose levels and ultimately restore glucose homeostasis, very little is known about the mechanisms responsible for hypoglycemia unawareness [172]. It has been proposed that adaptation to recurrent hypoglycaemia involves increased fuel availability to the brain from the blood stream, namely glucose [173-175] and acetate [176, 177]. In line with this, from studies in both humans and rodents, it has been proposed that recurrent hypoglycaemia leads to enhanced expression and density of glucose carrier proteins [178-180]. In addition to increased substrate uptake from the blood, hypoglycaemia unawareness could be linked to the so-called glycogen “supercompensation”, in which the brain adapts to hypoglycaemia by increasing glucose storage in the form of extra glycogen content [181, 182].
In neurons, the machinery for glycogen synthesis is believed to be physiologically inactive and glycogen phosphorylase nearly absent, rendering astrocytes the primary glycogen storage [183]. However, a recent study reported that cultured neurons have measurable glycogen levels and that its metabolism is important upon hypoxia [184]. Brain glycogen concentration lies between 3 and 7 μmol/g and has a turnover time of 5-10 hours [34]. For these concentration and turnover, a 10% reduction of glucose supply and a resting glucose consumption of 0.4-0.5 μmol/g/min would lead to depletion of glial glycogen in a couple of hours [185], emphasizing its role in buffering substrate delivery to the brain. Brain glycogen content is reduced upon insulin-induced hypoglycaemia due to increased glycogenolysis and, in contrast, it increases with circulating insulin and with brain glucose concentration [34]. Brain glycogen may also have a role in buffering fuel demands upon increased neuronal activity: it accumulates under deep anaesthesia but somatosensory stimulation upregulates glycogenolysis leading to a decrease in brain glycogen levels [40, 41]. Although suggesting a direct role of glycogen in brain function, this was not observed under visual stimulation in either rodents [41] or humans [186].
Using 13C MRS to measure enrichment from 13C-labelled glucose, Choi and co-authors submitted rats to hypoglycaemia and found that the rat brain displayed a 153% increase in glycogen compared to baseline after recovering for 7 hours under hyperglycaemia [181]. In mice, 6 hours after recovery at euglycaemia from insulin-induced acute hypoglycaemia caused a 25% increase in whole brain glycogen content [182]. However, 27 hours after the hypoglycaemia period, brain glycogen levels had return to baseline levels. The authors observed similar results when animals were submitted to daily recurrent hypoglycaemia insults for 9 days. In line with the effect of systemic hypoglycaemia, neuroglycopenia induced by repetitive intracere-broventricular injection of 2-deoxy-D-glucose for 3 days induced increases in brain glycogen content in the hypothalamus and cortex by 69% and 153%, respectively, in comparison to saline-injected rats [187]. Such glycogen modifications were associated to 5′-adenosine monophosphate-activated protein kinase (AMPK), a glucose sensor that is suppressed by high glucose [188] and activated by low glucose concentrations [189]. In contrast, Herzog et al. failed to detect glycogen supercompensation after acute or recurrent hypoglycaemia in the rat cerebellum, cortex and hypothalamus [190]. In line with this, after recovery from chronic and sustained hypoglycaemia bellow 3 mM for 2 weeks, brain glycogen levels and turnover were not different from control rats [191]. A recent study on patients with T1D did not support the glycogen supercompensation hypothesis [192]. Interestingly, brain glycogen supercompensation was also observed in rats after depletion upon exhaustive exercise [193]. In this study, brain glycogen levels were decreased with exhaustive exercise and were supercompensated at 6 hours after exercise in several brain areas, and it was sustained until 24 hours after exercise in both cortex and hippocampus (long-lasting supercompensation).
Increased glial glycogen stores can eventually sustain neuronal activity for a longer period [194] during a subsequent hypoglycaemia episode and, therefore, contribute to the shift in the glycaemia threshold at which counter-regulation initiates. However, it remains to be addressed whether excessive content of glycogen can effectively buffer brain glucose supply to glucose-sensing neurons for longer periods.
All together, these data suggest that glycogen content and metabolism may have an important role in hypoglycaemia unawareness, even though very little is known on brain glycogen metabolism in diabetes conditions. Animal models of diabetes characterised by insulin deficiency and chronic hyperglycaemia show unaltered glucose transport at the blood-brain-barrier [98], thus leading to high brain glucose concentration at hyperglycaemia. In chronic, sustained hyperglycaemia, the rodent brain does not display evident alteration of brain glycogen levels [195-198]. On the other hand, it is not clear whether insulin regulates whole brain glycogen deposition.
Whether hypoglycaemia-induced brain glycogen supercompensation occurs in diabetes conditions remains to be clearly demonstrated. Recently, a study on a small sample of patients with insulin-dependent diabetes and hypoglycaemia unawareness suggested that brain glycogen concentration is even slightly lower than in healthy subjects (although not significantly different) [192]. However this was never studied in well controlled experimental models of diabetes, and the effects of severity and duration of the hypoglycaemia insult, as well as the conditions of recovery from hypoglycaemia, on the degree of glycogen supercompensation remain to be ascertained. Moreover, a possible involvement of glycogen metabolism in impaired insulin signalling in T2D was never investigated.
In summary, in addition to diabetes itself, the occurrence of recurrent hypoglycaemia episodes due to treatment are also responsible for CNS injury. While some studies reported that cognitive decline in diabetic patients treated with insulin can be largely attributed to recurrent episodes of hypoglycaemia rather than to hyperglycaemia, others do not support this notion [199, 200]. Because, T1D patients without hypoglycaemia episodes also display a decline in cognitive function [201, 202], insulin signalling defects and glucose neurotoxicity remain the most important contributors for the development of dementia.
Conclusion
Uncontrolled hyperglycaemia in diabetes mellitus leads to well described microvascular complications that affect many organs. While the development and severity of these complications is mostly dependent on the degree of glycaemia control and duration of the disease, treatment with insulin frequently results in hypoglycaemia episodes. Both hyper- and hypoglycaemia affect brain function and may lead to cognitive dysfunction and dementia, but impaired insulin signalling appears as a very important factor driving neurodegeneration. Experimental diabetic conditions cause deficits in spatial learning and synaptic plasticity, synaptic degeneration and increased astrocyte reactivity and proliferation particularly in the hippocampus and cortical areas involved in learning and memory processing (Figure 1). These structural and functional modifications are likely to involve alterations of cerebral metabolism and, because they occur in both insulin deficiency and insulin resistance, the lack of adequate cerebral insulin signalling seems to be crucial for the development of diabetic encephalopathy and AD, with which diabetes shares cellular features.
Figure 1.
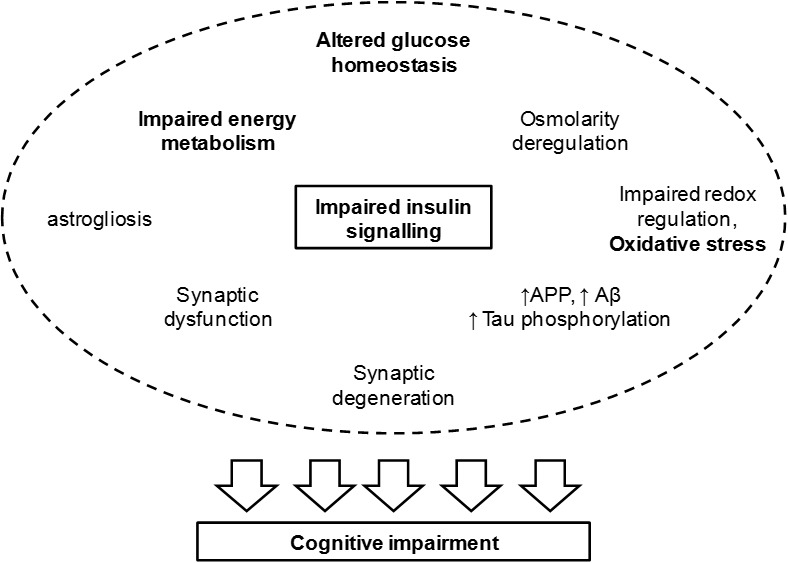
Events associated to impaired insulin signalling and leading to cognitive deterioration. Insulin regulates synaptic activity and glucose metabolism. Thus impaired insulin signalling leads to synaptic dysfunction and altered glucose homeostasis that impacts energy metabolism, osmolarity and redox balance. Furthermore, Aβ clearance and tau phosphorylation are under control of insulin/IGF-1 receptors. Hence, T2D leads to increased amount of amyloid precursor protein (APP), Aβ accumulation and tau hyperphosphorylation, leading to the formation of neurofibrillary tangles. Increased oxidative stress upon redox imbalance further affects mitochondrial metabolism and favours protein aggregation. In fact, advanced glycation end-products (AGE) in oxidative stress lead to a number of protein modifications that have functional consequences on metabolic pathways for signaling and energy production. While impaired energy metabolism may directly impact synaptic efficiency due to impaired membrane repolarisation and neurotransmitter synthesis/recycling, neurofibrillary tangles resulting from protein aggregation lead to degeneration of nerve terminals. This leads to cognitive impairment and is accompanied by astrogliosis and possibly by neuroinflamation.
Acknowledgments
This work was supported by the Swiss National Science Foundation (grant 148250) and by the Centre d’Imagerie BioMédicale (CIBM) of the UNIL, UNIGE, HUG, CHUV, EPFL, the Leenaards and Jeantet Foundations.
References
- [1].World Health Organization (1999). Definition, diagnosis and classification of diabetes mellitus and its complications: report of a WHO consultation. Geneva: World Health Organization. [Google Scholar]
- [2].International Diabetes Federation (2013). Diabetes Atlas, 6th ed. Brussels, Belgium: International Diabetes Federation. [Google Scholar]
- [3].Malecki MT (2004). Type 2 Diabetes mellitus and its complications: from the molecular biology to the clinical practice. Rev Diabet Stud, 1: 5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Miles WR,Root HF (1922). Psychologic tests applied to diabetic patients. Arch Int Med, 30: 767-777. [Google Scholar]
- [5].DeJong RN (1950). The nervous system complications in diabetes mellitus with special reference to cerebrovascular changes. J Nerv Ment Dis, 111: 181-206. [Google Scholar]
- [6].Brands AMA,Biessels GJ,de Haan EHF,Kappelle LJ,Kessels RPC (2005). The effects of type 1 diabetes on cognitive performance. Diabetes Care, 28: 726-735. [DOI] [PubMed] [Google Scholar]
- [7].Holmes CS,Richman LC (1985). Cognitive profiles of children with insulin-dependent diabetes. J Dev Behav Pediatr, 6: 323-326. [PubMed] [Google Scholar]
- [8].Ryan CM,Williams TM (1993). Effects of insulin-dependent diabetes on learning and memory efficiency in adults. J Clin Exp Neuropsychol, 15: 685-700. [DOI] [PubMed] [Google Scholar]
- [9].Ryan CM,Williams TM,Finegold DN,Orchard TJ (1993). Cognitive dysfunction in adults with type 1 insulin-dependent diabetes mellitus of long duration: effects of recurrent hypoglycaemia and other chronic complications. Diabetologia, 36: 329-334. [DOI] [PubMed] [Google Scholar]
- [10].Ryan CM (1988). Neurobehavioral complications of type I diabetes. Examination of possible risk factors. Diabetes Care, 11: 86-93. [DOI] [PubMed] [Google Scholar]
- [11].Deary IJ,Crawford JR,Hepburn DA,Langan SJ,Blackmore LM,Frier BM (1993). Severe hypoglycemia and intelligence in adult patients with insulin-treated diabetes. Diabetes, 42: 341-344. [DOI] [PubMed] [Google Scholar]
- [12].Gold AE,Deary IJ,Jones RW,O’Hare JP,Reckless JP,Frier BM (1994). Severe deterioration in cognitive function and personality in five patients with long-standing diabetes: a complication of diabetes or a consequence of treatment? Diabet Med, 11: 499-505. [DOI] [PubMed] [Google Scholar]
- [13].DeJong RN (1977). CNS manifestations of diabetes mellitus. Postgrad Med, 61, 101-107. [DOI] [PubMed] [Google Scholar]
- [14].Araki Y,Nomura M,Tanaka H,Yamamoto H,Yamamoto T,Tsukaguchi I,Nakamura H (1994). MRI of the brain in diabetes mellitus. Neuroradiology, 36: 101-103. [DOI] [PubMed] [Google Scholar]
- [15].Lunetta M,Damanti AR,Fabbri G,Lombardo M,Di Mauro M,Mughini L (1994). Evidence by magnetic resonance imaging of cerebral alterations of atrophy type in young insulin-dependent diabetic patients. J Endocrinol Invest, 17: 241-245. [DOI] [PubMed] [Google Scholar]
- [16].Perros P,Deary IJ,Sellar RJ,Best JJ,Frier BM (1997). Brain abnormalities demonstrated by magnetic resonance imaging in adult IDDM patients with and without a history of recurrent severe hypoglycemia. Diabetes Care, 20: 1013-1018. [DOI] [PubMed] [Google Scholar]
- [17].Gradman TJ,Laws A,Thompson LW,Reaven GM (1993). Verbal learning and/or memory improves with glycemic control in older subjects with non-insulin-dependent diabetes mellitus. J Am Geriatr Soc, 41: 1305-1312. [DOI] [PubMed] [Google Scholar]
- [18].Perlmuter LC,Hakami MK,Hodgson-Harrington C,Ginsberg J,Katz J,Singer DE,Nathan DM (1984). Decreased cognitive function in aging non-insulin-dependent diabetic patients. Am J Med, 77: 1043-1048. [DOI] [PubMed] [Google Scholar]
- [19].Reaven GM,Thomson LW,Nahum P,Haskins E (1990). Relationship between hyperglycemia and cognitive junction in older NIDDM patients. Diabetes Care, 13: 16-21. [DOI] [PubMed] [Google Scholar]
- [20].Ryan CM,Geckle M (2000). Why is learning and memory dysfunction in Type 2 diabetes limited to older adults?. Diabetes Metab Res Rev, 16: 308-315. [DOI] [PubMed] [Google Scholar]
- [21].Strachan MW,Deary IJ,Ewing FM,Frier BM (1997). Is type II diabetes associated with an increased risk of cognitive dysfunction? A critical review of published studies. Diabetes Care, 20, 438-445. [DOI] [PubMed] [Google Scholar]
- [22].Worrall G,Moulton N,Briffett E (1993). Effect of type II diabetes mellitus on cognitive function. J Fam Pract, 36: 639-643. [PubMed] [Google Scholar]
- [23].Sinclair AJ,Girling AJ,Bayer AJ (2000). Cognitive dysfunction in older subjects with diabetes mellitus: impact on diabetes self-management and use of care services. All Wales Research into Elderly AWARE Study. Diabetes Res Clin Pract, 50: 203-212. [DOI] [PubMed] [Google Scholar]
- [24].Tun PA,Nathan DM,Perlmuter LC (1990). Cognitive and affective disorders in elderly diabetics. Clin Geriatr Med, 6: 731-746. [PubMed] [Google Scholar]
- [25].Cummings JL (2004). Alzheimer’s disease. N Engl J Med, 351: 56-67. [DOI] [PubMed] [Google Scholar]
- [26].Alzheimer’s Association (2012). 2012 Alzheimer’s disease facts and figures. Alzheimers Dement, 8: 131-168. [DOI] [PubMed] [Google Scholar]
- [27].Kloppenborg RP,van den Berg E,Kappelle LJ,Biessels GJ (2008). Diabetes and other vascular risk factors for dementia: which factor matters most? A systematic review. Eur J Pharmacol, 585: 97-108. [DOI] [PubMed] [Google Scholar]
- [28].Biessels GJ,Kappelle LJ; Utrecht Diabetic Encephalopathy Study Group (2005). Increased risk of Alzheimer’s disease in Type II diabetes: insulin resistance of the brain or insulin-induced amyloid pathology? Biochem Soc Trans, 33(Pt 5): 1041-1044. [DOI] [PubMed] [Google Scholar]
- [29].Sima AA (2010) Encephalopathies: the emerging diabetic complications. Acta Diabetol, 47: 279-293. [DOI] [PubMed] [Google Scholar]
- [30].de Galan BE,Zoungas S,Chalmers J,Anderson C,Dufouil C,Pillai A,Cooper M,Grobbee DE,Hackett M,Hamet P,Heller SR, et al. (2009). Cognitive function and risks of cardiovascular disease and hypoglycaemia in patients with type 2 diabetes: the Action in Diabetes and Vascular Disease: Preterax and Diamicron Modified Release Controlled Evaluation (ADVANCE) trial. Diabetologia, 52: 2328-2336. [DOI] [PubMed] [Google Scholar]
- [31].Punthakee Z,Miller ME,Launer LJ,Williamson JD,Lazar RM,Cukierman-Yaffee T,Seaquist ER,Ismail-Beigi F,Sullivan MD,Lovato LC, et al. (2012). Poor cognitive function and risk of severe hypoglycemia in type 2 diabetes: post hoc epidemiologic analysis of the ACCORD trial. Diabetes Care, 35: 787-793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Craft S,Peskind E,Schwartz MW,Schellenberg GD,Raskind M,Porte D Jr (1998). Cerebrospinal fluid and plasma insulin levels in Alzheimer’s disease: relationship to severity of dementia and apolipoprotein E genotype. Neurology, 50(1): 164-8. [DOI] [PubMed] [Google Scholar]
- [33].Steen E,Terry BM,Rivera EJ,Cannon JL,Neely TR,Tavares R,Xu XJ,Wands JR,de la Monte SM (2005). Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease--is this type 3 diabetes? J Alzheimers Dis, 7(1): 63-80. [DOI] [PubMed] [Google Scholar]
- [34].Lanz B,Gruetter R,Duarte JMN (2013). Metabolic flux and compartmentation analysis in the brain in vivo. Front Endocrinol, 4: 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].McNay EC,Fries TM,Gold PE (2000). Decreases in rat extracellular hippocampal glucose concentration associated with cognitive demand during a spatial task. Proc Natl Acad Sci USA, 97: 2881-2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].McNay EC,McCarty RC,Gold PE (2001). Fluctuations in brain glucose concentration during behavioral testing: dissociations between brain areas and between brain and blood. Neurobiol Learn Mem, 75(3): 325-337. [DOI] [PubMed] [Google Scholar]
- [37].Gibbs ME,Hutchinson D,Hertz L (2008). Astrocytic involvement in learning and memory consolidation. Neurosci Biobehav Rev, 32: 927-944. [DOI] [PubMed] [Google Scholar]
- [38].Swanson RA,Morton MM,Sagar SM,Sharp FR (1992). Sensory stimulation induces local cerebral glycogenolysis: demonstration by autoradiography. Neuroscience, 51: 451-461. [DOI] [PubMed] [Google Scholar]
- [39].Harley CW,Milway JS,Fara-On M (1995). Medial forebrain bundle stimulation in rats activates glycogen phosphorylase in layers 4, 5b and 6 of ipsilateral granular neocortex. Brain Res, 685: 217-223. [DOI] [PubMed] [Google Scholar]
- [40].Cruz NF,Dienel GA (2002). High glycogen levels in brains of rats with minimal environmental stimuli: implications for metabolic contributions of working astrocytes. J Cereb Blood Flow Metab, 22: 1476-1489. [DOI] [PubMed] [Google Scholar]
- [41].Dienel GA,Ball KK,Cruz NF (2007). A glycogen phosphorylase inhibitor selectively enhances local rates of glucose utilization in brain during sensory stimulation of conscious rats: implications for glycogen turnover. J Neurochem, 102: 466-478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Duarte JMN,Schuck PF,Wenk GL,Ferreira GC (2014). Metabolic disturbances in diseases with neurological involvement. Aging and Disease, 5(4): 238-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Duarte JMN,Do KQ,Gruetter R (2014). Longitudinal neurochemical modifications in the aging mouse brain measured in vivo by 1H MRS. Neurobiol Aging, 35(7): 1660-1668. [DOI] [PubMed] [Google Scholar]
- [44].Boumezbeur F,Mason GF,de Graaf RA,Behar KL,Cline GW,Shulman GI,Rothman DL,Petersen KF (2010). Altered brain mitochondrial metabolism in healthy aging as assessed by in vivo magnetic resonance spectroscopy. J Cereb Blood Flow Metab 30(1): 211-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Bizon JL,Foster TC,Alexander GE andGlisky EL (2012). Characterizing Cognitive Aging of Working Memory and Executive Function in Animal Models. Front Ag Neurosci, 4: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Friedland RP,Budinger TF,Ganz E,Yano Y,Mathis CA,Koss B,Ober BA,Huesman RH,Derenzo SE (1983). Regional cerebral metabolic alterations in dementia of the Alzheimer type: positron emission tomography with [18F]fluorodeoxyglucose. J Comput Assist Tomogr, 7(4): 590-598. [DOI] [PubMed] [Google Scholar]
- [47].Haley AP,Knight-Scott J,Simnad VI,Manning CA (2006). Increased glucose concentration in the hippocampus in early Alzheimer’s disease following oral glucose ingestion. Magn Reson Imaging, 24: 715-720. [DOI] [PubMed] [Google Scholar]
- [48].Landau SM,Mintun MA,Joshi AD,Koeppe RA,Petersen RC,Aisen PS,Weiner MW,Jagust WJ; Alzheimer’s Disease Neuroimaging Initiative (2012). Amyloid deposition, hypometabolism, and longitudinal cognitive decline. Ann Neurol, 72(4): 578-586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Adriaanse SM,van Dijk KR,Ossenkoppele R,Reuter M,Tolboom N,Zwan MD,Yaqub M,Boellaard R,Windhorst AD,van der Flier WM,Scheltens P,Lammertsma AA,Barkhof F,van Berckel BN (2014). The effect of amyloid pathology and glucose metabolism on cortical volume loss over time in Alzheimer’s disease. Eur J Nucl Med Mol Imaging, In press [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Chételat G,Villemagne VL,Bourgeat P,Pike KE,Jones G,Ames D,Ellis KA,Szoeke C,Martins RN,O’Keefe GJ,Salvado O,Masters CL,Rowe CC; Australian Imaging Biomarkers and Lifestyle Research Group (2010). Relationship between atrophy and beta-amyloid deposition in Alzheimer disease. Ann Neurol, 67(3): 317-324. [DOI] [PubMed] [Google Scholar]
- [51].Shin J,Tsui W,Li Y,Lee SY,Kim SJ,Cho SJ,Kim YB,de Leon MJ (2011). Resting-state glucose metabolism level is associated with the regional pattern of amyloid pathology in Alzheimer’s disease. Int J Alzheimers Dis, 2011: 759780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Luque-Contreras D,Carvajal K,Toral-Rios D,Franco-Bocanegra D,Campos-Peña V (2014). Oxidative Stress and Metabolic Syndrome: Cause or Consequence of Alzheimer’s Disease? Oxid Med Cell Longev. 2014: 497802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lin AP,Shic F,Enriquez C,Ross BD (2003). Reduced glutamate neurotransmission in patients with Alzheimer’s disease - an in vivo 13C magnetic resonance spectroscopy study. MAGMA. 16(1): 29-42. [DOI] [PubMed] [Google Scholar]
- [54].Nilsen LH,Witter MP,Sonnewald U (2014). Neuronal and astrocytic metabolism in a transgenic rat model of Alzheimer’s disease. J Cereb Blood Flow Metab, 34(5): 906-914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Tiwari V,Patel AB (2014). Pyruvate Carboxylase and Pentose Phosphate Fluxes are Reduced in AβPP-PS1 Mouse Model of Alzheimer’s Disease: A 13C NMR Study. J Alzheimers Dis, 41(2): 387-399. [DOI] [PubMed] [Google Scholar]
- [56].Fayed N,Modrego PJ,Rojas-Salinas G,Aguilar K (2011). Brain glutamate levels are decreased in Alzheimer’s disease: a magnetic resonance spectroscopy study. Am J Alzheimers Dis Other Demen, 26(6): 450-456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Penner J,Rupsingh R,Smith M,Wells JL,Borrie MJ,Bartha R (2010). Increased glutamate in the hippocampus after galantamine treatment for Alzheimer disease. Prog Neuropsychopharmacol Biol Psychiatry 34(1): 104-10. [DOI] [PubMed] [Google Scholar]
- [58].Duarte JMN,Lei H,Mlynárik V,Gruetter R (2012). The neurochemical profile quantified by in vivo 1H NMR spectroscopy. NeuroImage, 61(2): 342-362 [DOI] [PubMed] [Google Scholar]
- [59].Schuff N,Amend D,Ezekiel F,Steinman SK,Tanabe J,Norman D,Jagust W,Kramer JH,Mastrianni JA,Fein G,Weiner MW (1997). Changes of hippocampal N-acetyl aspartate and volume in Alzheimer’s disease. A proton MR spectroscopic imaging and MRI study. Neurology, 49(6): 1513-1521. [DOI] [PubMed] [Google Scholar]
- [60].Ding B,Chen KM,Ling HW,Zhang H,Chai WM,Li X,Wang T (2008). Diffusion tensor imaging correlates with proton magnetic resonance spectroscopy in posterior cingulate region of patients with Alzheimer’s disease. Dement Geriatr Cogn Disord, 25(3): 218-225. [DOI] [PubMed] [Google Scholar]
- [61].Candeias E,Duarte AI,Carvalho C,Correia SC,Cardoso S,Santos RX,Plácido AI,Perry G,Moreira PI (2012). The impairment of insulin signaling in Alzheimer’s disease. IUBMB Life, 64(12): 951-957. [DOI] [PubMed] [Google Scholar]
- [62].Duarte AI,Moreira PI,Oliveira CR (2012). Insulin in central nervous system: more than just a peripheral hormone. J Aging Res, 2012: 1-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Musi N,Goodyear LJ (2006). Insulin resistance and improvements in signal transduction. Endocrine, 29: 73-80. [DOI] [PubMed] [Google Scholar]
- [64].Wada A,Yokoo H,Yanagita T,Kobayashi H (2005). New twist on neuronal insulin receptor signaling in health, disease, and therapeutics. J Pharmacol Sci, 99: 128-143. [DOI] [PubMed] [Google Scholar]
- [65].Mielke JG,Wang YT (2011). Insulin, synaptic function, and opportunities for neuroprotection. Prog Mol Biol Transl Sci, 98: 133-186. [DOI] [PubMed] [Google Scholar]
- [66].Brewer GJ (2010). Epigenetic oxidative redox shift (EORS) theory of aging unifies the free radical and insulin signaling theories. Exp Gerontol, 45: 173-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Burns JM,Honea RA,Vidoni ED,Hutfles LJ,Brooks WM,Swerdlow RH (2012). Insulin is differentially related to cognitive decline and atrophy in Alzheimer’s disease and aging. Biochim Biophys Acta, 1822(3): 333-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Zhao L,Teter B,Morihara T,Lim GP,Ambegaokar SS,Ubeda OJ,Frautschy SA,Cole GM (2004). Insulin-degrading enzyme as a downstream target of insulin receptor signaling cascade: implications for Alzheimer’s disease intervention. J Neurosci, 24(49): 11120-11126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Farris W,Mansourian S,Chang Y,Lindsley L,Eckman EA,Frosch MP,Eckman CB,Tanzi RE,Selkoe DJ,Guenette S (2003). Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci U S A, 100: 4162-4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Xie L,Helmerhorst E,Taddei K,Plewright B,Van Bronswijk W,Martins R (2002). Alzheimer’s beta-amyloid peptides compete for insulin binding to the insulin receptor. J Neurosci, 22(10): RC221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Schubert M,Gautam D,Surjo D,Ueki K,Baudler S,Schubert D,Kondo T,Alber J,Galldiks N,Küstermann E,Arndt S,Jacobs AH,Krone W,Kahn CR,Brüning JC (2004). Role for neuronal insulin resistance in neurodegenerative diseases. Proc Natl Acad Sci U S A, 101(9): 3100-3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Trueba-Sáiz A,Cavada C,Fernandez AM,Leon T,González DA,Fortea Ormaechea J,Lleó A,Del Ser T,Nuñez A,Torres-Aleman I (2013). Loss of serum IGF-I input to the brain as an early biomarker of disease onset in Alzheimer mice. Transl Psychiatry, 3:e330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Talbot K,Wang HY,Kazi H,Han LY,Bakshi KP,Stucky A,Fuino RL,Kawaguchi KR,Samoyedny AJ,Wilson RS,Arvanitakis Z,Schneider JA,Wolf BA,Bennett DA,Trojanowski JQ,Arnold SE (2012). Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest, 122(4): 1316-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Benedict C,Frey WH 2nd,Schiöth HB,Schultes B,Born J,Hallschmid M (2011). Intranasal insulin as a therapeutic option in the treatment of cognitive impairments. Exp Gerontol, 46: 112-115. [DOI] [PubMed] [Google Scholar]
- [75].Reger MA,Watson GS,Green PS,Wilkinson CW,Baker LD,Cholerton B,Fishel MA,Plymate SR,Breitner JC,DeGroodt W,Mehta P,Craft S (2008). Intranasal insulin improves cognition and modulates beta-amyloid in early AD. Neurology, 70(6): 440-448 [DOI] [PubMed] [Google Scholar]
- [76].Park CR,Seeley RJ,Craft S,Woods SC (2000). Intracerebroventricular insulin enhances memory in a passive-avoidance task. Physiol Behav, 68: 509-514. [DOI] [PubMed] [Google Scholar]
- [77].Bingham EM,Hopkins D,Smith D,Pernet A,Hallett W,Reed L,Marsden PK,Amiel SA (2002). The role of insulin in human brain glucose metabolism: an 18fluoro-deoxyglucose positron emission tomography study. Diabetes, 51: 3384-3390. [DOI] [PubMed] [Google Scholar]
- [78].Freude S,Hettich MM,Schumann C,Stöhr O,Koch L,Köhler C,Udelhoven M,Leeser U,Müller M,Kubota N,Kadowaki T,Krone W,Schröder H,Brüning JC,Schubert M (2009). Neuronal IGF-1 resistance reduces Abeta accumulation and protects against premature death in a model of Alzheimer’s disease. FASEB J, 23(10): 3315-3324. [DOI] [PubMed] [Google Scholar]
- [79].Killick R,Scales G,Leroy K,Causevic M,Hooper C,Irvine EE,Choudhury AI,Drinkwater L,Kerr F,Al-Qassab H,Stephenson J,Yilmaz Z,Giese KP,Brion JP,Withers DJ,Lovestone S (2009). Deletion of Irs2 reduces amyloid deposition and rescues behavioural deficits in APP transgenic mice. Biochem Biophys Res Commun, 386(1): 257-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Cohen E1,Paulsson JF,Blinder P,Burstyn-Cohen T,Du D,Estepa G,Adame A,Pham HM,Holzenberger M,Kelly JW,Masliah E,Dillin A (2009). Reduced IGF-1 signaling delays age-associated proteotoxicity in mice. Cell, 139(6): 1157-1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Stöhr O,Schilbach K,Moll L,Hettich MM,Freude S,Wunderlich FT,Ernst M,Zemva J,Brüning JC,Krone W,Udelhoven M,Schubert M (2013). Insulin receptor signaling mediates APP processing and β-amyloid accumulation without altering survival in a transgenic mouse model of Alzheimer’s disease. Age (Dordr), 35(1): 83-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].de la Monte SM (1989). Quantitation of cerebral atrophy in preclinical and end-stage Alzheimer’s disease. Ann Neurol, 25: 450-459. [DOI] [PubMed] [Google Scholar]
- [83].Convit A,De Leon MJ,Tarshish C,De Santi S,Tsui W,Rusinek H,George A (1997). Specific hippocampal volume reductions in individuals at risk for Alzheimer’s disease. Neurobiol Aging, 18: 131-138. [DOI] [PubMed] [Google Scholar]
- [84].De Leon MJ,George AE,Golomb J,Tarshish C,Convit A,Kluger A,De Santi S,McRae T,Ferris SH,Reisberg B,Ince C,Rusinek H,Bobinski M,Quinn B,Miller DC,Wisniewski HM (1997). Frequency of hippocampal formation atrophy in normal aging and Alzheimer’s disease. Neurobiol Aging, 18: 1-11. [DOI] [PubMed] [Google Scholar]
- [85].Pennanen C,Kivipelto M,Tuomainen S,Hartikainen P,Hänninen T,Laakso MP,Hallikainen M,Vanhanen M,Nissinen A,Helkala EL,Vainio P,Vanninen R,Partanen K,Soininen H (2004). Hippocampus and entorhinal cortex in mild cognitive impairment and early AD. Neurobiol Aging, 25: 303-310. [DOI] [PubMed] [Google Scholar]
- [86].Gold SM,Dziobek I,Sweat V,Tirsi A,Rogers K,Bruehl H,Tsui W,Richardson S,Javier E,Convit A (2007). Hippocampal damage and memory impairments as possible early brain complications of type 2 diabetes. Diabetologia, 50: 711-719. [DOI] [PubMed] [Google Scholar]
- [87].Biessels GJ,van der Heide LP,Kamal A,Bleys RL,Gispen WH (2002). Ageing and diabetes: implications for brain function. Eur J Pharmacol, 441: 1-14. [DOI] [PubMed] [Google Scholar]
- [88].Trudeau F,Gagnon S,Massicotte G (2004). Hippocampal synaptic plasticity and glutamate receptor regulation: influences of diabetes mellitus. Eur J Pharmacol, 490: 177-186. [DOI] [PubMed] [Google Scholar]
- [89].Flood JF,Mooradian AD,Morley JE (1990). Characteristics of learning and memory in streptozocin-induced diabetic mice. Diabetes, 39: 1391-1398. [DOI] [PubMed] [Google Scholar]
- [90].Biessels GJ,Kamal A,Ramakers GM,Urban IJ,Spruijt BM,Erkelens DW,Gispen WH (1996). Place learning and hippocampal synaptic plasticity in streptozotocin-induced diabetic rats. Diabetes, 45: 1259-1266. [DOI] [PubMed] [Google Scholar]
- [91].Kamal A,Biessels GJ,Duis SE,Gispen WH (2000). Learning and hippocampal synaptic plasticity in streptozotocin-diabetic rats: interaction of diabetes and ageing. Diabetologia, 43: 500-506. [DOI] [PubMed] [Google Scholar]
- [92].Luesse HG,Schiefer J,Spruenken A,Puls C,Block F,Kosinski CM (2001). Evaluation of R6/2 HD transgenic mice for therapeutic studies in Huntington’s disease: behavioral testing and impact of diabetes mellitus. Behav Brain Res, 126: 185-195. [DOI] [PubMed] [Google Scholar]
- [93].Li X,Aou S,Hori T,Oomura Y (2002). Spatial memory deficit and emotional abnormality in OLETF rats. Physiol Behav, 75: 15-23. [DOI] [PubMed] [Google Scholar]
- [94].Nitta A,Murai R,Suzuki N,Ito H,Nomoto H,Katoh G,Furukawa Y,Furukawa S (2002). Diabetic neuropathies in brain are induced by deficiency of BDNF. Neurotoxicol Teratol, 24: 695-701. [DOI] [PubMed] [Google Scholar]
- [95].Duarte JMN,Agostinho PM,Carvalho RA,Cunha RA (2012). Caffeine consumption prevents diabetes-induced memory impairment and synaptotoxicity in the hippocampus of NONcNZO10/LtJ mice. PLoS ONE, 7: e21899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Rönnemaa E,Zethelius B,Sundelöf J,Sundström J,Degerman-Gunnarsson M,Berne C,Lannfelt L,Kilander L (2008). Impaired insulin secretion increases the risk of Alzheimer disease. Neurology, 71: 1065-1071. [DOI] [PubMed] [Google Scholar]
- [97].Duarte JMN,Oliveira CR,Ambrosio AF &Cunha RA (2006). Modification of adenosine A1 and A2A receptor density in the hippocampus of streptozotocin-induced diabetic rats. Neurochem Int, 48: 144-150. [DOI] [PubMed] [Google Scholar]
- [98].Duarte JMN,Carvalho RA,Cunha RA,Gruetter R (2009). Caffeine consumption attenuates neurochemical modifications in the hippocampus of streptozotocin-induced diabetic rats. J Neurochem, 111: 368-379. [DOI] [PubMed] [Google Scholar]
- [99].Grillo CA,Piroli GG,Wood GE,Reznikov LR,McEwen BS,Reagan LP (2005). Immunocytochemical analysis of synaptic proteins provides new insights into diabetes-mediated plasticity in the rat hippocampus. Neuroscience, 136: 477-486. [DOI] [PubMed] [Google Scholar]
- [100].Baydas G,Nedzvetskii VS,Tuzcu M,Yasar A,Kirichenko SV (2003). Increase of glial fibrillary acidic protein and S-100B in hippocampus and cortex of diabetic rats: effects of vitamin E. Eur J Pharmacol, 462: 67-71. [DOI] [PubMed] [Google Scholar]
- [101].Saravia FE,Revsin Y,Gonzalez Deniselle MC,Gonzalez SL,Roig P,Lima A,Homo-Delarche F,De Nicola AF (2002). Increased astrocyte reactivity in the hippocampus of murine models of type 1 diabetes: the nonobese diabetic (NOD) and streptozotocin-treated mice. Brain Res, 957: 345-353. [DOI] [PubMed] [Google Scholar]
- [102].Ates O,Cayli SR,Yucel N,Altinoz E,Kocak A,Durak MA,Turkoz Y,Yologlu S (2007). Central nervous system protection by resveratrol in streptozotocin-induced diabetic rats. J Clin Neurosci, 14: 256-260. [DOI] [PubMed] [Google Scholar]
- [103].Wang WT,Lee P,Yeh HW,Smirnova IV,Choi IY (2012). Effects of acute and chronic hyperglycemia on the neurochemical profiles in the rat brain with streptozotocin-induced diabetes detected using in vivo ¹H MR spectroscopy at 9.4 T. J Neurochem, 121(3): 407-417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Duarte JMN,Oses JP,Rodrigues RJ,Cunha RA (2007). Modification of purinergic signalling in the hippocampus of streptozotocin-induced diabetic rats. Neuroscience, 149: 382-391 [DOI] [PubMed] [Google Scholar]
- [105].Duarte JMN,Nogueira C,Mackie K,Oliveira CR,Cunha RA,Köfalvi A (2007). Increase of cannabinoid CB1 receptor density in the hippocampus of streptozotocin-induced diabetic rats. Experimental Neurology, 204: 479-484 [DOI] [PubMed] [Google Scholar]
- [106].Mooradian AD (1997). Central nervous system complications of diabetes mellitus - a perspective from the blood-brain barrier. Brain Res Brain Res Rev, 23: 210-218. [DOI] [PubMed] [Google Scholar]
- [107].Starr JM,Wardlaw J,Ferguson K,MacLullich A,Deary IJ,Marshall I (2003). Increased blood-brain barrier permeability in type II diabetes demonstrated by gadolinium magnetic resonance imaging. J Neurol Neurosurg Psychiatry, 74: 70-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Beauquis J,Saravia F,Coulaud J,Roig P,Dardenne M,Homo-Delarche F,De Nicola A (2008). Prominently decreased hippocampal neurogenesis in a spontaneous model of type 1 diabetes, the nonobese diabetic mouse. Exp Neurol, 210: 359-367. [DOI] [PubMed] [Google Scholar]
- [109].Beauquis J,Roig P,Homo-Delarche F,De Nicola A,Saravia F (2006). Reduced hippocampal neurogenesis and number of hilar neurones in streptozotocin-induced diabetic mice: reversion by antidepressant treatment. Eur J Neurosci, 23: 1539-1546. [DOI] [PubMed] [Google Scholar]
- [110].Stranahan AM,Arumugam TV,Cutler RG,Lee K,Egan JM,Mattson MP (2008). Diabetes impairs hippocampal function through glucocorticoid-mediated effects on new and mature neurons. Nat Neurosci, 11: 309-317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Jakobsen J,Sidenius P,Gundersen HJ,Østerby R (1987). Quantitative changes of cerebral neocortical structure in insulin-treated long-term streptozocin-induced diabetes in rats. Diabetes, 36: 597-601. [DOI] [PubMed] [Google Scholar]
- [112].Li ZG,Zhang W,Grunberger G,Sima AAF (2002). Hippocampal neuronal apoptosis in type 1 diabetes. Brain Res, 946: 221-231. [DOI] [PubMed] [Google Scholar]
- [113].Luse SA (1970). The ultrastructure of the brain in the diabetic Chinese hamster with special reference to synaptic abnormalities. Electroencephalogr Clin Neurophysiol, 29: 410. [PubMed] [Google Scholar]
- [114].Trulson ME,Jacoby JH,MacKenzie RG (1986). Streptozotocin-induced diabetes reduces brain serotonin synthesis in rats. J Neurochem, 46: 1068-1072. [DOI] [PubMed] [Google Scholar]
- [115].Barber M,Kasturi BS,Austin ME,Patel KP,MohanKumar SM,MohanKumar PS (2003). Diabetes-induced neuroendocrine changes in rats: role of brain monoamines, insulin and leptin. Brain Res, 964: 128-135. [DOI] [PubMed] [Google Scholar]
- [116].Galanopoulos E,Lellos V,Papadakis M,Philippidis H,Palaiologos G (1998). Effects of fasting and diabetes on some enzymes and transport of glutamate in cortex slices or synaptosomes from rat brain. Neurochem Res, 13(3): 243-248. [DOI] [PubMed] [Google Scholar]
- [117].Kulikov AV,Arkhipova LV,Tretyak TM,Bragin AG (1986). Serotonin and norepinephrine content in brain structures of rats with experimental and transplantation-compensated diabetes. J Hirnforsch, 27: 495-499. [PubMed] [Google Scholar]
- [118].Biessels GJ,Cristino NA,Rutten GJ,Hamers FP,Erkelens DW,Gispen WH (1999). Neurophysiological changes in the central and peripheral nervous system of streptozotocin-diabetic rats. Course of development and effects of insulin treatment. Brain, 122 Pt 4: 757-768. [DOI] [PubMed] [Google Scholar]
- [119].Rubini R,Biasiolo F,Fogarolo F,Magnavita V,Martini A,Fiori MG (1992). Brainstem auditory evoked potentials in rats with streptozotocin-induced diabetes. Diabetes Res Clin Pract, 16: 19-25. [DOI] [PubMed] [Google Scholar]
- [120].Chakrabarti S,Zhang WX,Sima AAF (1991). Optic neuropathy in the diabetic BB-rat. Adv Exp Med Biol, 291: 257-264. [DOI] [PubMed] [Google Scholar]
- [121].Sima AAF,Zhang WX,Cherian PV,Chakrabarti S (1992). Impaired visual evoked potential and primary axonopathy of the optic nerve in the diabetic BB/W-rat. Diabetologia, 35: 602-607. [DOI] [PubMed] [Google Scholar]
- [122].Biessels GJ,Kamal A,Urban IJ,Spruijt BM,Erkelens DW,Gispen WH (1998). Water maze learning and hippocampal synaptic plasticity in streptozotocin-diabetic rats: effects of insulin treatment. Brain Res, 800: 125-135. [DOI] [PubMed] [Google Scholar]
- [123].Kamal A,Biessels GJ,Urban IJ,Gispen WH (1999). Hippocampal synaptic plasticity in streptozotocin-diabetic rats: impairment of long-term potentiation and facilitation of long-term depression. Neuroscience, 90: 737-745. [DOI] [PubMed] [Google Scholar]
- [124].Francis GJ,Martinez JA,Liu WQ,Xu K,Ayer A,Fine J,Tuor UI,Glazner G,Hanson LR,Frey WH 2nd,Toth C (2008). Intranasal insulin prevents cognitive decline, cerebral atrophy and white matter changes in murine type I diabetic encephalopathy. Brain, 131: 3311-3334. [DOI] [PubMed] [Google Scholar]
- [125].Baskin DG,Stein LJ,Ikeda H,Woods SC,Figlewicz DP,Porte D Jr,Greenwood MR,Dorsa DM (1985). Genetically obese Zucker rats have abnormally low brain insulin content. Life Sci, 36(7): 627-633. [DOI] [PubMed] [Google Scholar]
- [126].Kaiyala KJ,Prigeon RL,Kahn SE,Woods SC,Schwartz MW (2000). Obesity induced by a high-fat diet is associated with reduced brain insulin transport in dogs. Diabetes, 49(9): 1525-1533. [DOI] [PubMed] [Google Scholar]
- [127].Baker LD,Cross D,Minoshima S,Belongia D,Watson GS,Craft S (2011). Insulin resistance is associates with Alzheimer-like reductions in regional cerebral glucose metabolism for cognitively impaired normal adults with pre-diabetes or early type 2 diabetes. Arch Neurol, 68: 51-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Canas PM,Duarte JMN,Rodrigues RJ,Köfalvi A,Cunha RA (2009). Modification upon aging of the density of presynaptic modulation systems in the hippocampus. Neurobiology of Aging, 30: 1877-1884 [DOI] [PubMed] [Google Scholar]
- [129].Canas PM,Porciúncula LO,Cunha GM,Silva CG,Machado NJ,Oliveira JM,Oliveira CR,Cunha RA (2009). Adenosine A2A receptor blockade prevents synaptotoxicity and memory dysfunction caused by beta-amyloid peptides via p38 mitogen-activated protein kinase pathway. J Neurosci, 29: 14741-14751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Canas PM,Simões AP,Rodrigues RJ,Cunha RA (2014). Predominant loss of glutamatergic terminal markers in a β-amyloid peptide model of Alzheimer’s disease. Neuropharmacology, 76 Pt A: 51-56. [DOI] [PubMed] [Google Scholar]
- [131].Duarte JMN,Carvalho RA,Cunha RA,Gruetter R (2009). Effect of long-term caffeine consumption on glucose transport and osmolarity alterations in the hippocampus of STZ-induced and Goto-Kakizaki diabetic rats: in vivo 1H MRS study at 9.4 T. Proc Intl Soc Mag Reson Med, 17: 1084. [Google Scholar]
- [132].Abdul-Rahman O,Sasvari-Szekely M,Ver A,Rosta K,Szasz BK,Kereszturi E,Keszler G (2012). Altered gene expression profiles in the hippocampus and prefrontal cortex of type 2 diabetic rats. BMC Genomics, 13: 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Li XH,Xin X,Wang Y,Wu JZ,Jin ZD,Ma LN,Nie CJ,Xiao X,Hu Y,Jin MW (2013). Pentamethylquercetin Protects Against Diabetes-Related Cognitive Deficits in Diabetic Goto-Kakizaki Rats. J Alzheimers Dis, 34(3): 755-767. [DOI] [PubMed] [Google Scholar]
- [134].Pereira C,Moreira P,Seiça R,Santos MS,Oliveira CR (2000). Susceptibility to beta-amyloid-induced toxicity is decreased in goto-kakizaki diabetic rats: involvement of oxidative stress. Exp Neurol, 161: 383-391. [DOI] [PubMed] [Google Scholar]
- [135].Moreira PI,Santos MS,Moreno AM,Seiça R,Oliveira CR (2003). Increased vulnerability of brain mitochondria in diabetic (Goto-Kakizaki) rats with aging and amyloid-beta exposure. Diabetes, 52: 1449-1456. [DOI] [PubMed] [Google Scholar]
- [136].Li ZG,Zhang W,Sima AA (2007). Alzheimer-like changes in rat models of spontaneous diabetes. Diabetes, 56(7): 1817-1824. [DOI] [PubMed] [Google Scholar]
- [137].Schubert M,Brazil DP,Burks DJ,Kushner JA,Ye J,Flint CL,Farhang-Fallah J,Dikkes P,Warot XM,Rio C,Corfas G,White MF (2003). Insulin receptor substrate-2 deficiency impairs brain growth and promotes tau phosphorylation. J Neurosci, 23(18): 7084-7092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Selkoe DJ (2002). Alzheimer’s disease is a synaptic failure. Science, 298(5594): 789-791. [DOI] [PubMed] [Google Scholar]
- [139].Krantic S,Isorce N,Mechawar N,Davoli MA,Vignault E,Albuquerque M,Chabot JG,Moyse E,Chauvin JP,Aubert I,McLaurin J,Quirion R (2012). Hippocampal GABAergic neurons are susceptible to amyloid-β toxicity in vitro and are decreased in number in the Alzheimer’s disease TgCRND8 mouse model. J Alzheimers Dis, 29(2): 293-308. [DOI] [PubMed] [Google Scholar]
- [140].Limon A,Reyes-Ruiz JM,Miledi R (2012). Loss of functional GABAA receptors in the Alzheimer diseased brain. Proc Natl Acad Sci U S A, 109(25): 10071-10076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].van Der Graaf M,Janssen SW,van Asten JJ,Hermus AR,Sweep CG,Pikkemaat JA,Martens GJ,Heerschap A (2004). Metabolic profile of the hippocampus of Zucker Diabetic Fatty rats assessed by in vivo 1H magnetic resonance spectroscopy. NMR Biomed, 17: 405-410. [DOI] [PubMed] [Google Scholar]
- [142].Geissler A,Frund R,Scholmerich J,Feuerbach S,Zietz B (2003). Alterations of cerebral metabolism in patients with diabetes mellitus studied by proton magnetic resonance spectroscopy. Exp Clin Endocrinol Diabetes, 111: 421-427. [DOI] [PubMed] [Google Scholar]
- [143].Kreis R,Ross BD (1992). Cerebral metabolic disturbances in patients with subacute and chronic diabetes mellitus: detection with proton MR spectroscopy. Radiology, 184: 123-130. [DOI] [PubMed] [Google Scholar]
- [144].Choi JK,Jenkins BG,Carreras I,Kaymakcalan S,Cormier K,Kowall NW,Dedeoglu A (2010). Anti-inflammatory treatment in AD mice protects against neuronal pathology. Exp Neurol, 223: 377-384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Marjanska M,Curran GL,Wengenack TM,Henry PG,Bliss RL,Poduslo JF,Jack CR Jr,Ugurbil K,Garwood M (2005). Monitoring disease progression in transgenic mouse models of Alzheimer’s disease with proton magnetic resonance spectroscopy. Proc Natl Acad Sci U S A, 102: 11906-11910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Oberg J,Spenger C,Wang FH,Andersson A,Westman E,Skoglund P,Sunnemark D,Norinder U,Klason T,Wahlund LO,Lindberg M (2008). Age related changes in brain metabolites observed by 1H MRS in APP/PS1 mice. Neurobiol Aging, 29:1423-1433. [DOI] [PubMed] [Google Scholar]
- [147].Jack CR Jr,Marjanska M,Wengenack TM,Reyes DA,Curran GL,Lin J,Preboske GM,Poduslo JF,Garwood M (2007). Magnetic resonance imaging of Alzheimer’s pathology in the brains of living transgenic mice: a new tool in Alzheimer’s disease research. Neuroscientist, 13: 38-48. [DOI] [PubMed] [Google Scholar]
- [148].Chen SQ,Wang PJ,Ten GJ,Zhan W,Li MH,Zang FC (2009). Role of myo-inositol by magnetic resonance spectroscopy in early diagnosis of Alzheimer’s disease in APP/PS1 transgenic mice. Dement Geriatr Cogn Disord, 28: 558-566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Dedeoglu A,Choi JK,Cormier K,Kowall NW,Jenkins BG (2004). Magnetic resonance spectroscopic analysis of Alzheimer’s disease mouse brain that express mutant human APP shows altered neurochemical profile. Brain Res, 1012: 60-65. [DOI] [PubMed] [Google Scholar]
- [150].Ferguson SC,Blane A,Wardlaw J,Frier BM,Perros P,McCrimmon RJ,Deary IJ (2005). Influence of an early-onset age of type 1 diabetes on cerebral structure and cognitive function. Diabetes Care, 28: 1431-1437. [DOI] [PubMed] [Google Scholar]
- [151].Warren RE,Frier BM (2005) Hypoglycemia and cognitive function. Diabetes Obes Metab 7: 493-503. [DOI] [PubMed] [Google Scholar]
- [152].Cox DJ,Gonder-Frederick LA,Schroeder DB,Cryer PE,Clarke WL (1993). Disruptive effects of acute hypoglycemia on speed of cognitive and motor performance. Diabetes Care, 16: 1391-1393. [DOI] [PubMed] [Google Scholar]
- [153].Deary IJ,Sommerfield AJ,McAulay V,Frier BM (2003). Moderate hypoglycaemia obliterates working memory in humans with and without insulin treated diabetes. J Neurol Neurosurg Psychiatry, 74: 278-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Draelos MT,Jacobson AM,Weinger K,Widom B,Ryan CM,Finkelstein DM,Simonson DC (1995). Cognitive function in patients with insulin-dependent diabetes mellitus during hyperglycemia and hypoglycaemia. Am J Med, 98: 135-144. [DOI] [PubMed] [Google Scholar]
- [155].Gold AE,Deary IJ,MacLeod KM,Thomson KJ,Frier BM (1995). Cognitive function during insulin-induced hypoglycemia in humans: short-term cerebral adaptation does not occur. Psychopharmacology (Berl), 119: 325-333. [DOI] [PubMed] [Google Scholar]
- [156].Maran A,Lomas J,Macdonald IA,Amiel SA (1995). Lack of preservation of higher brain function during hypoglycaemia in patients with intensively-treated IDDM. Diabetologia, 38: 1412-1418. [DOI] [PubMed] [Google Scholar]
- [157].Oberg J,Spenger C,Wang FH,Andersson A,Westman E,Skoglund P,Sunnemark D,Norinder U,Klason T,Wahlund LO,Lindberg M (2008). Age related changes in brain metabolites observed by 1H MRS in APP/PS1 mice. Neurobiol Aging, 29: 1423-1433. [DOI] [PubMed] [Google Scholar]
- [158].McAulay V,Deary IJ,Ferguson SC,Frier BM (2001). Acute hypoglycemia in humans causes attentional dysfunction while nonverbal intelligence is preserved. Diabetes Care, 24: 1745-1750. [DOI] [PubMed] [Google Scholar]
- [159].Sommerfield AJ,Deary IJ,McAulay V,Frier BM (2003). Short-term, delayed, and working memory are impaired during hypoglycemia in individuals with type 1 diabetes. Diabetes Care, 26: 390-396. [DOI] [PubMed] [Google Scholar]
- [160].Fanelli CG,Paramore DS,Hershey T,Terkamp C,Ovalle F,Craft S,Cryer PE (1998). Impact of nocturnal hypoglycemia on hypoglycemic cognitive dysfunction in type 1 diabetes. Diabetes, 47: 1920-1927. [DOI] [PubMed] [Google Scholar]
- [161].Langan SJ,Deary IJ,Hepburn DA,Frier BM (1991). Cumulative cognitive impairment following recurrent severe hypoglycaemia in adult patients with insulin-treated diabetes mellitus. Diabetologia, 34: 337-344. [DOI] [PubMed] [Google Scholar]
- [162].Lincoln NB,Faleiro RM,Kelly C,Kirk BA,Jeffcoate WJ (1996). Effect of long-term glycemic control on cognitive function. Diabetes Care, 19: 656-658. [DOI] [PubMed] [Google Scholar]
- [163].Wredling R,Levander S,Adamson U,Lins PE (1990). Permanent neuropsychological impairment after recurrent episodes of severe hypoglycaemia in man. Diabetologia, 33: 152-157. [DOI] [PubMed] [Google Scholar]
- [164].Lewis LD,Ljunggren B,Norberg K,Siesjö BK (1974). Changes in carbohydrate substrates, amino acids and ammonia in the brain during insulin-induced hypoglycemia. J Neurochem, 23: 659-671. [DOI] [PubMed] [Google Scholar]
- [165].Engelsen B,Fonnum F (1993). Effects of hypoglycemia on the transmitter pool and the metabolic pool of glutamate in rat brain. Neurosci Lett, 42: 317-322. [DOI] [PubMed] [Google Scholar]
- [166].Dawson RM (1950). Studies on the glutamine and glutamic acid content of the rat brain during insulin hypoglycaemia. Biochem J, 47: 386-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Behar KL,den Hollander JA,Petroff OA,Hetherington HP,Prichard JW,Shulman RG (1985). Effect of hypoglycemic encephalopathy upon amino acids, high-energy phosphates, and pHi in the rat brain in vivo: detection by sequential 1H and 31P NMR spectroscopy. J Neurochem, 44: 1045-1055. [DOI] [PubMed] [Google Scholar]
- [168].Davis JM,Himwich WA,Pederson VC (1970). Hypoglycemia and developmental changes in free amino acids of rat brain. J Appl Physiol, 29: 219-222. [DOI] [PubMed] [Google Scholar]
- [169].Rao R,Ennis K,Long JD,Ugurbil K,Gruetter R,Tkac I (2010). Neurochemical changes in the developing rat hippocampus during prolonged hypoglycemia. J Neurochem, 114: 728-738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Dagogo-Jack SE,Craft S,Cryer PE (1993). Hypoglycemia-associated autonomic failure in insulin-dependent diabetes mellitus. Recent antecedent hypoglycemia reduces autonomic responses to, symptoms of, and defense against subsequent hypoglycemia. J Clin Invest, 91, 819-828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Cryer PE (2004). Diverse causes of hypoglycemia-associated autonomic failure in diabetes. N Engl J Med, 350: 2272-2279. [DOI] [PubMed] [Google Scholar]
- [172].Fanelli CG,Pampanelli S,Porcellati F,Bolli GB (1998). Shift of glycaemic thresholds for cognitive function in hypoglycemia unawareness in humans. Diabetologia, 41: 720-723. [DOI] [PubMed] [Google Scholar]
- [173].Marty N,Dallaporta M,Thorens B (2007). Brain glucose sensing, counterregulation, and energy homeostasis. Physiology (Bethesda), 22: 241-251 [DOI] [PubMed] [Google Scholar]
- [174].Boyle PJ,Nagy RJ,O’Connor AM,Kempers SF,Yeo RA,Qualls C (1994). Adaptation in brain glucose uptake following recurrent hypoglycemia. Proc Natl Acad Sci USA, 91: 9352-9356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Criego AB,Tkáč I,Kumar A,Thomas W,Gruetter R,Seaquist ER (2005). Brain glucose concentrations in patients with type 1 diabetes and hypoglycemia unawareness. J Neurosci Res, 79: 42-47. [DOI] [PubMed] [Google Scholar]
- [176].McCall AL,Fixman LB,Fleming N,Tornheim K,Chick W,Ruderman NB (1986). Chronic hypoglycemia increases brain glucose transport. Am J Physiol Endocrinol Metab, 251: E442-E447. [DOI] [PubMed] [Google Scholar]
- [177].Gulanski BI,De Feyter HM,Page KA,de Aguiar RB,Mason GF,Rothman DL,Sherwin RS (2013). Increased Brain Transport and Metabolism of Acetate in Hypoglycemia Unawareness. J Clin Endocrinol Metab, 98(9): 3811-3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Mason GF,Petersen KF,Lebon V,Rothman DL,Shulman GI (2006). Increased brain monocarboxylic acid transport and utilization in type 1 diabetes. Diabetes, 55: 929-934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Kumagai AK,Kang YS,Boado RJ,Pardridge WM (1995). Upregulation of blood-brain barrier GLUT1 glucose transporter protein and mRNA in experimental chronic hypoglycemia. Diabetes, 44: 1399-1404. [DOI] [PubMed] [Google Scholar]
- [180].Simpson IA,Appel NM,Hokari M,Oki J,Holman GD,Maher F,Koehler-Stec EM,Vannucci SJ,Smith QR (1999). Blood-brain barrier glucose transporter: effects of hypo- and hyperglycemia revisited. J Neurochem, 72: 238-247. [DOI] [PubMed] [Google Scholar]
- [181].Choi IY,Seaquist ER,Gruetter R (2003). Effect of hypoglycemia on brain glycogen metabolism in vivo. J Neurosci Res, 72: 25-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Canada SE,Weaver SA,Sharpe SN,Pederson BA (2011). Brain glycogen supercompensation in the mouse after recovery from insulin-induced hypoglycemia. J Neurosci Res, 89: 585-591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Brown AM,Ransom BR (2007). Astrocyte glycogen and brain energy metabolism. Glia, 55: 1263-1271. [DOI] [PubMed] [Google Scholar]
- [184].Saez I,Duran J,Sinadinos C,Beltran A,Yanes O,Tevy MF,Martínez-Pons C,Milán M,Guinovart JJ (2014). Neurons have an active glycogen metabolism that contributes to tolerance to hypoxia. J Cereb Blood Flow Metab, 34: 945-955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Gruetter R (2003). Glycogen: the forgotten cerebral energy store. J Neurosci Res, 74(2): 179-183. [DOI] [PubMed] [Google Scholar]
- [186].Oz G,Seaquist ER,Kumar A,Criego AB,Benedict LE,Rao JP,Henry PG,Van De Moortele PF,Gruetter R (2007). Human brain glycogen content and metabolism: implications on its role in brain energy metabolism. Am J Physiol Endocrinol Metab, 292(3): E946-951. [DOI] [PubMed] [Google Scholar]
- [187].Alquier T,Kawashima J,Tsuji Y,Kahn BB (2007). Role of hypothalamic adenosine 5′-monophosphate-activated protein kinase in the impaired counterregulatory response induced by repetitive neuroglucopenia. Endocrinology, 148: 1367-1375 [DOI] [PubMed] [Google Scholar]
- [188].Minokoshi Y,Alquier T,Furukawa N,Kim YB,Lee A,Xue B,Mu J,Foufelle F,Ferre P,Birnbaum MJ,Stuck BJ,Kahn BB (2004). AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature, 428: 569-574 [DOI] [PubMed] [Google Scholar]
- [189].Han SM,Namkoong C,Jang PG,Park IS,Hong SW,Katakami H,Chun S,Kim SW,Park JY,Lee KU,Kim MS (2005). Hypothalamic AMP-activated protein kinase mediates counter-regulatory responses to hypoglycemia in rats. Diabetologia, 48: 2170-2178 [DOI] [PubMed] [Google Scholar]
- [190].Herzog RI,Chan O,Yu S,Dziura J,McNay EC,Sherwin RS (2008). Effect of acute and recurrent hypoglycemia on changes in brain glycogen concentration. Endocrinology, 149: 1499-1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Lei H,Gruetter R (2006). Effect of chronic hypoglycemia on glucose concentration and glycogen content in rat brain: A localized 13C NMR study. J Neurochem, 99: 260-268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Oz G,Tesfaye N,Kumar A,Deelchand DK,Eberly LE,Seaquist ER (2012). Brain glycogen content and metabolism in subjects with type 1 diabetes and hypoglycemia unawareness. J Cereb Blood Flow Metab, 32(2): 256-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Matsui T,Ishikawa T,Ito H,Okamoto M,Inoue K,Lee MC,Fujikawa T,Ichitani Y,Kawanaka K,Soya H (2012). Brain glycogen supercompensation following exhaustive exercise. J Physiol, 590(Pt 3): 607-616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Suh SW,Bergher JP,Anderson CM,Treadway JL,Fosgerau K,Swanson RA (2007). Astrocyte glycogen sustains neuronal activity during hypoglycemia: studies with the glycogen phosphorylase inhibitor CP-316,819 ([R-R*,S*]-5-chloro-N-[2-hydroxy-3-(methoxymethylamino)-3-oxo-1-(phenylmethyl)propyl]-1H-indole-2-carboxamide). J Pharmacol Exp Ther, 321: 45-50 [DOI] [PubMed] [Google Scholar]
- [195].Nelson SR,Schulz DW,Passonneau JV,Lowry OH (1968). Control of glycogen levels in brain. J Neurochem, 15: 1271-1279. [DOI] [PubMed] [Google Scholar]
- [196].Wagner SR 4th,Lanier WL (1994). Metabolism of glucose, glycogen, and high-energy phosphates during complete cerebral ischemia. A comparison of normoglycemic, chronically hyperglycemic diabetic, and acutely hyperglycemic nondiabetic rats. Anesthesiology, 81: 1516-1526. [DOI] [PubMed] [Google Scholar]
- [197].Sánchez-Chávez G,Hernández-Berrones J,Luna-Ulloa LB,Coffe V,Salceda R (2008). Effect of diabetes on glycogen metabolism in rat retina. Neurochem Res, 33: 1301-1308. [DOI] [PubMed] [Google Scholar]
- [198].Lanier WL,Hofer RE,Gallagher WJ (1996). Metabolism of glucose, glycogen, and high-energy phosphates during transient forebrain ischemia in diabetic rats: effect of insulin treatment. Anesthesiology, 84: 917-925. [DOI] [PubMed] [Google Scholar]
- [199].Deary IJ,Frier BM (1996). Severe hypoglycemia and cognitive impairment in diabetes: link not proven. Br Med J, 313: 767-780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Austin EJ,Deary IJ (1999). Effects of repeated hypoglycaemia on cognitive function. A psychometrically validated reanalysis of the Diabetes Control and Complications Trial data. Diabetes Care, 22: 1273-1277. [DOI] [PubMed] [Google Scholar]
- [201].Kramer L,Fasching P,Madl C,Schneider B,Damjancic P,Waldhäusl W,Irsigler K,Grimm G (1998). Previous episodes of hypoglycemic coma are not associated with permanent cognitive brain dysfunction in IDDM patients on intensive insulin treatment. Diabetes, 47: 1909-1914. [DOI] [PubMed] [Google Scholar]
- [202].Schoenle EJ,Schoenle D,Molinari L,Largo RH (2002). Impaired intellectual development in children with Type I diabetes: association with HbA1c, age at diagnosis and sex. Diabetologia, 45: 108-114. [DOI] [PubMed] [Google Scholar]