

Abstract
Alzheimer’s disease (AD) is a progressive neurodegenerative disease characterized in the brain by the formation of amyloid-beta (Aβ)-containing plaques and neurofibrillary tangles containing the microtubule-associated protein tau. Neuroinflammation is another feature of AD and astrocytes are receiving increasing attention as key contributors. Although some progress has been made, the molecular mechanisms underlying the pathophysiology of AD remain unclear. Interestingly, some of the main proteins involved in AD, including amyloid precursor protein (APP) and tau, have recently been shown to be SUMOylated. The post-translational modification by SUMO (small ubiquitin-like modifier) has been shown to regulate APP and tau and may modulate other proteins implicated in AD. Here we present an overview of recent studies suggesting that protein SUMOylation might be involved in the underlying pathogenic mechanisms of AD and discuss how this could be exploited for therapeutic intervention.
Keywords: Alzheimer’s disease, neuroinflammation, post-translational protein modification, SUMOylation, SUMO
Alzheimer’s disease
Alzheimer’s disease (AD) is recognised as the most common cause of chronic dementia among the aging population. It was estimated that 35.6 million people lived with dementia worldwide in 2010, with numbers expected to almost double every 20 years, to 65.7 million in 2030 and 115.4 million in 2050 [1]. Increased incidence of AD occurs with increased life expectancy, and subsequently incurs an escalating economic and social burden. No effective treatment for AD exists; therefore, the understanding of the molecular mechanisms of cellular damage involved is a critically important area of research to find ways of tackling this highly debilitating condition.
The onset of AD is characterised by a progressive decline in cognitive function whereby mild impairments in memory are surpassed by increasingly significant higher cognitive deficits in language, recognition and skilled movements [2]. Pathological studies have indicated that certain areas of the brain are predisposed to exhibiting senile plaques and neurofibrillary tangles, which, combined with the severe cognitive impairment, are hallmarks of AD [3]. The senile plaques are primarily comprised of aggregated amyloid beta (Aβ) peptide fragments and the neurofibrillary tangles are aggregates of a hyperphosphorylated form of the microtubule-associated protein tau [4]. The combination of these features leads to severe synaptic dysfunction.
The exact molecular mechanisms involved in AD are still unknown. The synaptic loss and death of specific neuronal populations in AD are consequence of a cascade of multiple deleterious molecular and cellular events rather than a single pathogenic factor. Several lines of evidence support the hypothesis that accumulation of Aβ in the brain is the primary factor driving AD, whereas the formation of neurofibrillary tangles is proposed to result from an imbalance between Aβ production and clearance [5]. In addition to these classical neuropathological features, activation of glial cells has been documented in the AD brain, suggesting a contribution of neuroinflammation to the pathogenesis of AD [6, 7].
Aβ generation
After synthesis in the endoplasmic reticulum, amyloid precursor protein (APP) is glycosylated, tyrosyl sulphated and sialyted in the Golgi complex [8]. The full-length protein has a relatively short half-life: both during and after its trafficking through the secretary pathway, it undergoes proteolytic cleavage to release its products into the vesicle lumen and extracellular space. It is not known when APP cleavage occurs, but evidence suggests that it is located near the cell surface [9].
Three protagonist protein complexes have been identified as cleaving APP and its products: α-, β- and γ-secretases. Enzymatic cleavage of APP by α-secretase occurs at the lysine 16-leucine 17 bond of APP, precluding the formation of Aβ. The resulting large amino-terminal fragment of APP is secreted from cultured cells and found in human cerebral spinal fluid, suggesting a constitutive presence. β-secretase activity combines the activities of two complimentary aspartyl proteases, β-site APP-cleaving enzyme (BACE)-1 and -2. β-secretase cleaves APP 16 residues n-terminal to the α-secretase cleavage site resulting in a smaller ectodomain and leaving a 99-residue stump in the membrane. The C-terminal membrane fragments after α- or β-secretase cleavage can be further processed by a third enzyme, γ-secretase. This is thought to be a tetramer complex located in the membrane, one component of which is the presinillin (PS)-1 protein, characteristic of the London Familial mutation. γ-secretase cuts in the middle of the transmembrane domain of APP [10].
Following α-secretase cleavage, γ-secretase activity results in secretion of a 3 kDa fragment (p3), it has been observed in cerebral spinal fluid and plasma, suggesting that these cleavage pathways are happening constitutively and are not aberrant processes. Sequential cleavage of APP by β- and γ-secretases results in the production of the 39-42 amino acid protein Aβ (amyloidogenic proteolytic pathwhay). The proportion of the 1-42 peptide appears to be increased in transgenic AD mice possessing a familial mutation which causes increased β-secretase cleavage, suggesting an imbalance of fragments [11].
Neurofibrillary Tangles and Tau
Neurofibrillary tangles result from the intracellular accumulation of cytoskeletal elements and consist mainly of paired helical filaments, which are aggregates of the hyperphosphorylated form of the microtubule-associated protein tau [9]. The tangles disrupt the transport of cellular components, such as nerve growth factor, contributing to neurodegeneration. It is significant that tau accumulation has been related to cognitive impairment [12], suggesting that the disease process is sequential involving first the build up of toxic Aβ followed by that of tau, which leads to neurodegeneration.
Plaques and tangles can occur independently of each other. For example, in Lewy body disease plaques of Aβ are seen with virtual absence of tau, and paired helical fragments of tau can be seen in less common neurodegenerative diseases such as Kuf’s disease and progressive supranuclear palsy [9]. This suggests that the mechanisms are functionally distinct, but are related in AD.
Neuroinflammation in AD
Neuroinflammation is commonly observed in various CNS conditions including neurodegenerative disorders such as AD [13]. The Aβ peptide triggers microglial and astrocytic gliosis, which plays several roles in the neurodegenerative process [14, 15]. Some of these roles include phagocytizing cell debris after synaptic rewiring and memory formation and the release of pro-inflammatory chemokines and cytokines. These pro-inflammatory cytokines can cause a vicious cycle of inflammation that has been characterized in a number of neurodegenerative diseases including AD [7, 16].
Astrocytes are the most numerous cell-type in the human brain and are indispensable for maintaining a healthy CNS. They are receiving increasing attention as key contributors to neurodegenerative disease, including AD [17]. Loss of function in astrocytes, associated with astrocyte reactivity, is considered to have an important role in neurodegeneration [18]. In human patients, and mouse models of AD, there is increased astrocyte reactivity, assessed by up-regulation of the intermediate filament protein, glial fibrillary acidic protein (GFAP), and morphological changes in astrocytes [19, 20]. AD-related Aβ peptides cause astrocyte reactivity and the expression of GFAP is strongly associated with AD disease severity and cognitive decline [21, 22]. Specifically, Aβ-treated reactive astrocytes have been shown to be involved in inflammatory responses and to exacerbate neurotoxicity [23, 24, 25]. Astrocytes activated by Aβ produce chemokines and cytokines that may result in neuronal damage [26]. Receptor binding of chemokines and cytokines stimulates a variety of intracellular signaling pathways that have been implicated in AD, including the activation of protein kinase C (PKC), c-Jun N-terminal kinase (JNK), p38 mitogen-activated protein kinase (p38/MAPK), PI3 kinase (PI3-K), extracellular signaling-related kinase (ERK), as well as activation of caspase-1 and caspase-3 [27, 28].
Protein SUMOylation
Protein SUMOylation is the covalent attachment of small ubiquitin-like modifier (SUMO) proteins to specific lysine residues in target proteins. This post-translational modification regulates many aspects of normal protein function, including interactions, subcellular localization, activity, stability and partnering, and it has been shown to modulate an increasing number of cellular pathways [29, 30]. Three major SUMO isoforms, SUMO-1, SUMO-2 and SUMO-3, are expressed in cells, with SUMO-2 and SUMO-3 being very similar to each other (96% identical in amino acid sequence), but different from SUMO-1. SUMO-4 has been described, but it is not yet clear whether it is endogenously expressed in cells. A gene for SUMO-4 has been described although the expression of SUMO-4 protein in cells is still controversial [31].
Like ubiquitin, SUMO isoforms are expressed as precursor proteins and need to be matured before they enter a multi-step enzymatic pathway that is comparable to but distinct from ubiquitination. First, the SUMO protein is cleaved by one of the specific SUMO proteases called SENPs to expose the C-terminal diglycine motif required for conjugation to targets. In an ATP-dependent reaction, the mature SUMO is covalently attached to a cysteine in the SUMO E1 activating enzyme. The SUMO is then transferred from the E1 to the SUMO E2 conjugating enzyme, Ubc9, which attaches the SUMO to a lysine in the target protein that is typically, but not always found within the consensus sequence ψKX(E/D) (ψ represents a hydrophobic residue, K the lysine for SUMO conjugation, X any amino acid and E/D an acidic glutamate or aspartate residue). SUMO E3 ligating enzymes stimulate protein SUMOylation by associating with Ubc9 and substrates to increase the efficiency of the reaction (Figure 1) [31].
Figure 1.
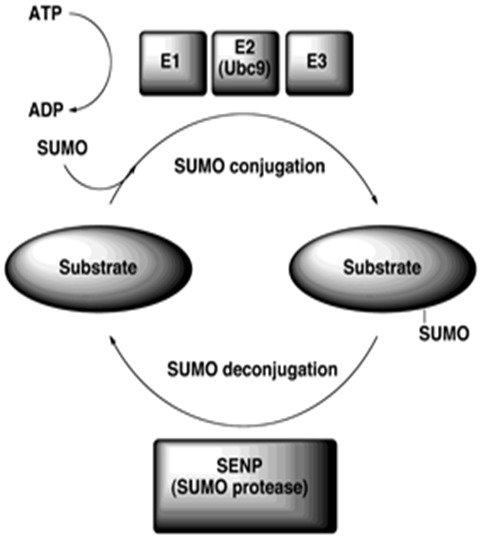
The SUMO conjugation pathway. SUMO is expressed as a precursor protein and processed by a SUMO protease (SENP). Mature SUMO is activated in an ATP-dependent manner by the SUMO activating enzyme (E1) and is transferred through a transesterification process to the unique SUMO conjugating enzyme (E2) Ubc9. SUMO is next conjugated to the target lysine of a substrate (consensus sequence ΨKXE/D). Conjugation, in some instances, is facilitated by a SUMO E3 ligase. The sumoylation pathway is reversible as specific proteases can remove and recycle SUMO from modified substrates.
SUMOylation is a highly dynamic and reversible process as the same proteases involved in the maturation of the precursor proteins are also responsible for the removal and recycling of SUMOs from their substrates. In contrast to ubiquitin, which mainly tags proteins for degradation, SUMOylation is important for the normal functions of proteins in the cell. However, over the past few years, several studies have indicated that SUMOylation also has a role in aging and neurodegenerative diseases, including AD (Table 1) [32, 33].
Table 1.
The involvement of SUMOylation in Alzheimer’s disease, aging and neuroinflammation.
| Study (author and year) | SUMO effect |
|---|---|
| Li et al., 2003 [41] | SUMO-3 overexpression reduces Aβ production via regulation of APP processing in HEK293 cells. Monosumoylation has a positive effect on Aβ production as opposed to polysumoylation, which negatively regulates Aβ production. |
| Dorval and Fraser, 2006 [34] | Inhibition of phosphatases by okadaic acid increased SUMO1-tau levels in HEK293 cells. |
| Dorval et al., 2007 [42] | SUMO-3 overexpression increases Aβ40/Aβ42 secretion and up-regulates the expression of BACE. Suppression of SUMO-1 or SUMO-2 does not affect APP levels or Aβ production in HEK293 cells. |
| Takahashi et al., 2008 [44] | SUMO-1 and phospho-tau colocalization is observed in degenerating neurites around neuritic amyloid plaques in the cortex of aged mice. |
| Zhang and Sarge, 2008 [43] | Sumoylation of lysines 587 and 595 of APP is associated with decreased levels of Aβ aggregates in HeLa cells. |
| Ahn et al., 2009 [55] | Genetic variations of Ubc9 gene might be associated with a risk of AD in the Korean population. |
| Akar and Feinstein, 2009 [47] | LPS decreases mRNA levels of SUMO-1, Ubc9 and SENP1 in primary astrocytes and a similar decrease occurred during normal aging in brain. |
| Lee et al., 2009 [49] | SUMOylation is required for the suppression of STAT1-dependent inflammatory responses by LXRs in IFN-γ-stimulated brain astrocytes. |
| McMillan et al., 2011 [36] | Changes in individual bands of SUMO-1 or SUMO-2/3 conjugation are apparent in the hippocampus, cortex and cerebellum, although global levels were unaltered in Tg2576 mice (9 months). |
| Fang et al., 2011 [56] | BACE gene promoter activity is inhibited by the overexpression of SUMO proteins. |
| Yang et al., 2012 [39] | Free unconjugated SUMO-3 increases in the hippocampus may be correlated with spatial learning ability in old C57BL/6 mice. |
| Yun et al., 2013 [38] | SUMO-1 free protein is elevated compared to wild-type mice in APP/PS1 double transgenic mice. SUMO-1 modulates Aβ generation via BACE1 accumulation. |
| Hoppe et al., 2013 [48] | SUMO-1 overexpression abrogates Aβ-induced increase in GFAP in primary astrocytes. |
| Nisticò et al., 2013 [40] | SUMO-1 conjugation is increased in Tg2576 mice in cortex and hippocampus (3 and 6 months) and this is paralleled by increased expression levels of Ubc9 and SENP1 in both brain regions. SUMO-2-ylation is decreased in old transgenic mice (17 months), concomitant with an elevated amyloid deposition, which occurs at this stage in AD mice model. |
Abbreviations: APP (Amyloid Precursor Protein), PS1 (Presenilin-1), HEK (Human Embryonic Kidney), STAT1 (Signal Transducers and Activators of Transcription-1), LXR (Liver X receptor), IFN-γ (Interferon-γ), Ubc9 (SUMO-conjugating enzyme), SENP1 (SUMO-specific protease 1), BACE (β-secretase 1).
Neurodegenerative diseases are characterized by an area-specific extensive neuronal loss: a recurring feature of these diseases appears to be aggregation and accumulation of misfolded proteins. A role of protein SUMOylation has been investigated in a number of neurodegenerative diseases, including brain ischemia and AD [33, 34], with variable effects on protagonist proteins involved in disease pathogenesis. Important proteins in the pathogenesis of AD, such as APP and tau are reported SUMO substrates [35].
SUMOylation in AD
Models to investigate the potential role of protein SUMOylation in AD include in vitro systems, such as primary neuronal and organotypic hippocampal slice cultures treated with Aβ and in vivo AD transgenic mouse models. While the first enable researchers to manipulate the SUMOylation pathway, by overexpression or knockdown of relevant proteins, the second provide a more physiological model that allows the observation of global levels of SUMOylation in different brain regions.
Global SUMOylation in AD has been explored in different brain regions of an APP transgenic mouse model of AD. In this model, global levels of SUMO-1 or SUMO-2/3 were not significantly altered between 9-month-old Tg2576 and wild-type mice. However, on examination of individual bands within these global SUMO blots, several SUMO-2/3 bands were considerably decreased in the Tg2576 mice compared to controls. This suggested that, at least at this disease stage, subtle changes in SUMOylation by SUMO-2/3 may occur [36]. In fitting with a role of SUMO-2/3 in Aβ-mediated cellular stress, an unbiased proteomic study of artificially generated amyloidogenic proteins pulled down SUMO-2 as an interacting protein amyloidogenic surface. Thus, SUMO-2 may be down-regulated through its binding to amyloid proteins, such as Aβ and thus functionally impaired [37].
Recently, a study has been shown that over-expression of the three SUMO isorforms (SUMO-1 to -3), in a neuroglioma cell line, up-regulated β-secretase (BACE-1) levels, and consequently resulted in altered APP processing and Aβ generation, independent of covalent SUMO conjugation. Moreover, SUMO-1 depletion decreased BACE-1 and Aβ generation [38]. In the same study, an increase in free SUMO-1 levels, but not in SUMO-2/3 protein, was observed in 18-month-old APP double transgenic mouse compared to wild-type animals. SUMO-1 immunoreactivity increased in the brains of transgenic mice and was co-localized with amyloid plaques. In another study a significant increase in free unconjugated SUMO-2/3 levels was observed in the hippocampus of 25-month-old C57BL/6 mice compared to 7-month-old mice [39].
More recently, another study performed an age-related analysis on the expression levels of global protein SUMOylation and SUMO-related enzymes in the Tg2576 mouse model of AD. Significant differences in the SUMOylation/de-SUMOylation balance were observed at an early stage of the pathology. At 3 months, SUMO-1 conjugation, but not SUMO-2/3 conjugation, was augmented both in cortical and hippocampal tissue of Tg2576 mice compared to wild-type mice. They also demonstrated that the increase of SUMOylation in Tg2576 brain corresponds to a high expression level of Ubc9 and SENP-1 at the same time point [40].
Due to the conflicting results from these studies, it remains unclear and controversial how SUMOylation might be affected with aging and AD. Further studies are necessary to elucidate the contradictory and unresolved issues, including by which mechanisms SUMOylation regulates Aβ processing in neuronal cell types and in in vivo AD models.
APP and SUMOylation
Published interest in APP SUMOylation stems from an unbiased screen of a mammalian fetal cDNA library to explore potential modulators of APP metabolism. This identified full length SUMO-2, initially noted as SUMO-3 and later corrected, as causing altered APP processing patterns. However, subsequent work produced inconclusive results: SUMO-2 increased both amyloidogenic and non-amyloidogenic processing of APP, but was not co-immunoprecipitated with APP, suggesting SUMO-2 did not bind APP directly. Effects were validated in human embryonic kidney 293 (HEK293) cells and a human neuroblastoma cell line. In vivo work found subcellular restriction of SUMO-2 to the neuronal soma. Overall, no convincing role for SUMO-2 was established, though findings were suggestive of a potential role in APP processing which merited further work [41].
Another study investigated overexpression and RNAi knockdown of SUMO-1, -2 and -3. Co-transfection of HEK293 cells with HA-tagged SUMO-1, 2 and 3 plus APP695 followed by examination of extracellular Aβ levels showed no SUMO-1 or -2 effects. However, overexpression of SUMO-3 increased Aβ40 and Aβ42 levels, as well as the levels of APP. The observed APP degradation at a slower rate and the increase in BACE led to the idea of a SUMO-3-specific effect on BACE and APP. Using SUMO-3 conjugation and polymerization-deficient mutants resulted in no differences in APP cleavage from the active, polymerizing form, suggesting that the effect is due to SUMO-3 monomers and is a non-covalent effect or that some compensation is occurring. A decrease in SUMOylated substrates was observed after SUMO knockdown by RNAi, but no parallel changes in the amyloidogenic processing of APP were observed suggesting endogenous SUMO was not an essential regulatory factor and played an indirect role in APP processing and production of Aβ [42].
In contrast with these observations, another study identified a ψKx(E/D) SUMOylation motif around lysines 587 and 595 of APP. It is important to note that these lysines are adjacent to the β-secretase cleavage site of APP and lysine 595 is mutated in the APP found in the Swedish early-onset familial form of the disease. Using a His-tag pull-down of APP found it to be conjugated to SUMO-1 and -2, supporting the view that APP is SUMOylated. In addition, transfecting non-SUMOylatable mutants at these lysines resulted in increased levels of Aβ. Mouse brain APP immunoprecipitation also indicated SUMO-1 and SUMO-2 conjugation. By up-regulating Ubc9, increased APP SUMOylation and decreased Aβ aggregates were observed. These data point towards the idea of a direct SUMOylation of APP to down-regulate the amyloidogenic pathway in AD [43].
These mixed results from different groups may be explained by different experimental approaches. Factors to consider are both covalent and non-covalent interactions of SUMO, compensatory factors of different SUMO proteins due to overexpression or knockdown of other SUMO proteins. In addition, the role of the proteasomal degradation pathway and interplay with ubiquitination may also have a compensatory role. None of the above studies look at direct effects in neuronal cells and are restricted mainly to heterologous expression systems. However, they imply a role of SUMOylation in APP processing by secretases, which merits further investigation.
Tau and SUMOylation
Another obvious target for SUMOylation studies in AD is tau. Co-transfection of HEK 293 cells with His-tagged SUMO-1, -2 and -3 with tau showed conjugation of SUMO-1 to tau [34]. Potential SUMOylation sites were investigated and lysine 340 was one of the major acceptor sites identified. The correlation between phosphorylation states of tau was also investigated and SUMO was found to bind to dephosphorylated tau much more significantly than to phosphorylated tau, implicating a potential function of SUMO in binding to microtubule-bound tau (non-phosphorylated) to prevent its proteasomal degradation. Pharmacological decreases in phosphorylation or increases in microtubule stabilization up-regulated tau SUMOylation, suggesting preffered binding of SUMO-1 to soluble (mislocalised) tau. Pharmacological proteasomal inhibition significantly decreased tau SUMOylation and increased ubiquitination. Proteasomal inhibition is a common feature in aging and other neurodegenerative disorders and may implicate a mechanism of disruption of the SUMO pathway in tau tangle formation.
SUMOylation was later investigated in in vivo models of AD. Immunohistochemical staining of APP, Aβ, phospho-tau and ubiquitin in cortical slices from Tg2576 mice revealed ubiquitin-APP co-localization occurred in neurites. However, few phospho-tau granules in the neuritic plaques were ubiquitin positive, whereas all phospho-tau granules and punctuate deposits were SUMO-1 positive. No SUMO-1 positive deposits were observed in non-transgenic mice [44]. Therefore, the results of this study correlate with Dorval and Fraser’s (2006) findings of proteasome failure and interplay of SUMO-1 and ubiquitin conjugation to tau in AD pathogenesis [34].
The above studies utilize either observational in vivo or in vitro cell line models for investigation of protein interactions in AD. Although they provide preliminary evidence for a role of SUMO proteins in AD pathogenesis involving APP and tau, validation in physiological systems such as neuronal cultures or organotypic slice cultures and further in vivo studies would increase the relevance of these results. In addition, although tau and APP are the most highly associated proteins with AD, and thus are obvious targets for investigation of SUMOylation, a plethora of proteins are implicated in AD and a more global approach could merit further investigation.
SUMOylation and neuroinflammation
Although several roles for SUMOylation in the brain have been reported, the involvement of SUMOylation in neuroinflammation is a relatively new topic and there are limited studies concerning glial SUMOylation. Growing evidence suggests that glial SUMOylation can impact cellular processes that are relevant under pathological conditions. SUMOylation can have pro- or anti-inflammatory actions, depending on the specific SUMO isoform that is conjugated, the target protein and cell-type [45, 46].
There is little data on SUMOylation in astrocytes. The first report confirmed the existence and functionality of this important signaling pathway in primary astrocytes showing that the mRNA levels for SUMO-1, the conjugating Ubc9 and the de-conjugating SENP-1 are decreased following treatment of astrocytes with lipopolysaccharide (LPS). This study suggested that loss of the SUMO pathway in astrocytes under inflammatory conditions is associated with pathophysiological processes [47]. In astrocytes, nitric oxide synthase 2 (NOS2/iNOS) is induced by inflammatory stimuli and the nitric oxide (NO) generated by iNOS contributes to disease progression in a variety of neurological diseases, including AD. Akar and Feinstein (2009) demonstrated that SUMOylation regulates iNOS expression in astrocytes. Overexpression of the SUMO components was able to reduce NOS2 promoter activation, possibly involving SUMOylation of the C/EBPβ transcription factor [47].
A more recent study implicated SUMO in the inflammatory signaling mediated by Aβ in primary astrocytes [48]. This study found that concomitant with astrogliosis, Aβ exposure down-regulates SUMO-1 conjugated proteins, as well as the conjugating enzyme, Ubc9, in astrocytes. The JNK inhibition, by the natural compound curcumin or a JNK inhibitor, prevented Aβ-mediated reduction of SUMO-1 conjugation. Furthermore, overexpression of constitutively active SUMO-1, but not its inactive mutant, abrogated Aβ-induced GFAP up-regulation and morphological reactivity in astrocytes, suggesting an anti-inflammatory role for SUMO-1 in the brain.
Although not much is known about the roles of SUMOylation in astrocytes, under pro-inflammatory conditions induced by LPS or Aβ, these studies reinforce the role of the SUMO pathway in regulating astrocyte function and indicate that glial SUMOylation may have anti-inflammatory therapeutic benefits. Besides, the potential roles of SUMOylation in glia-mediated processes in the brain emerge as a new aspect of interest in neurodegenerative diseases, such as AD.
It is therapeutically useful to regulate inflammatory processes underlying neurodegenerative diseases, and another study has shown that SUMOylation is required for suppression of STAT1-dependent inflammatory responses by liver X receptors (LXRs) in IFN-γ-stimulated brain astrocytes [49]. While not yet directly shown, it is probable that such SUMO regulation of inflammatory signaling also occurs in the microglia. In cultured macrophages, peroxisome proliferator-activated receptor-gamma (PPAR-γ) and LXRs have been found to be SUMOylated, which regulates the transrepression of inflammatory response genes [50].
SUMOylation is usually associated with a cytoprotective response to cellular stressors. However, we note that SUMOylation of certain proteins in astrocytes, namely the C-terminal proteolytic fragment of the glutamate transporter (EAAT2) has been associated with gliotoxicity [51]. EAAT2 is a predominantly astroglial glutamate transporter responsible for the majority of synaptic glutamate clearance in the mammalian CNS and its dysfunction has been linked with many neurological disorders. SUMOylation of this astroglial EAAT2 fragment governs its intracellular compartmentalization and accumulation in astrocytes where it triggers astrocyte-mediated neurotoxic effects, whereas non-fragmented EAAT2 resides on the plasma membrane with increase in EAAT2-mediated glutamate uptake [52]. The same study has also shown evidence that the entire EAAT2 protein can be a SUMO target, however, the consequences and fate of the SUMOylated transporter remain to be elucidated.
α-Synuclein (α-syn) is the major constituent of Lewy bodies and glial cytoplasmic inclusions, which are pathological hallmarks of neurodegenerative disorders like Parkinson’s disease or multiple system atrophy (MSA), respectively. In oligodendroglial cells, SUMO-1 has been implicated in protein aggregation and identified as a constituent in inclusion bodies in MSA [53, 54].
Although still a relatively underexplored topic, the involvement of SUMOylation in glial cells warrant additional study and could become a potential therapeutic target in brain diseases, including AD as well as other neurological diseases.
Conclusions
Since its discovery, SUMOylation is emerging as an important post-translational modification for the regulation of nuclear cytosolic and membrane proteins in the cell. An emerging trend is that this modification does not act alone but interplays actively with other modifications, such as phosphorylation, acetylation, and ubiquitination, to form integrated modification programs for coordinated regulation of various proteins in response to diverse signaling cues. Overall, the studies strongly suggest that there is potentially dysregulation of SUMOylation under AD pathology and that such dysregulation could involve changes in the SUMO enzymes and conjugation profile. There is currently an increasing interest on and discovery of neuronal and glial SUMO substrates to understand its role in the progression and pathology of AD. In addition, components of the SUMOylation pathway should now be more deeply investigated as not only potential neurodegenerative diseases risk factors but also therapeutic targets for drug discovery.
Acknowledgements
Juliana Bender Hoppe was a recipient of CNPq postdoctoral fellowship. We thank Dr Ulrich Mayer for his assistance with the manuscript.
References
- [1].Prince M,Bryce R,Albanese E,Wimo A,Ribeiro W,Ferri CP (2013). The global prevalence of dementia: a systematic review and metaanalysis. Alzheimers Dement, 9:63-75. [DOI] [PubMed] [Google Scholar]
- [2].Blennow K,De Leon MJ,Zetterberg H (2006). Alzheimer’s disease. Lancet, 368: 387-403. [DOI] [PubMed] [Google Scholar]
- [3].Tiraboschi P,Hansen LA,Thal LJ,Corey-Bloom J (2004). The importance of neuritic plaques and tangles to the development and evolution of AD. Neurology, 62: 1984-9. [DOI] [PubMed] [Google Scholar]
- [4].LaFerla FG,Green KN,Oddo S (2007). Intracellular amyloid-beta in Alzheimer’s disease. Nat Rev Neurosci, 8: 499-509. [DOI] [PubMed] [Google Scholar]
- [5].Hardy J,Selkoe D (2002). The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science, 297: 353-356. [DOI] [PubMed] [Google Scholar]
- [6].McGeer EG,McGeer PL (2010). Neuroinflammation in Alzheimer’s disease and mild cognitive impairment: a field in its infancy. J Alzheimers Dis, 19: 355-61. [DOI] [PubMed] [Google Scholar]
- [7].Garwood CJ,Pooler AM,Atherton J,Hanger DP,Noble W (2011). Astrocytes are important mediators of Aβ-induced neurotoxicity and tau phosphorylation in primary culture. Cell Death, 2:1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Dawbarn D,Allen SJ (1995). Neurobiology of Alzheimer’s Disease. Bios Scientific. [Google Scholar]
- [9].Selkoe D (2001). Alzheimer’s Disease: Genes, Proteins, and Therapy. Physiol Rev, 81: 741-766. [DOI] [PubMed] [Google Scholar]
- [10].Allen S (2007). Alzheimer’s disease: a hundred years of investigation. Neurobiology of Alzheimer’s disease. Oxford, Oxford University Press: 1-36. [Google Scholar]
- [11].Haass C,Schlossmacher MG,Hung AY,Vigo-Pelfrey C,Mellon A,Ostaszewski BL,Lieberburg I,Koo EH,Schenk D,Teplow DB,Selkoe DJ (1992). Amyloid beta-peptide is produced by cultured cells during normal metabolism. Nature, 359: 322-325. [DOI] [PubMed] [Google Scholar]
- [12].Robertson E (2007). Reducing Endogenous Tau Ameliorates Amyloid b-Induced Deficits in an Alzheimer’s Disease Mouse Model. Science, 316: 750-754. [DOI] [PubMed] [Google Scholar]
- [13].Amor S,Puentes F,Baker F,van der Valk P (2010). Inflammation in neurodegenerative diseases. Immunology, 129:154-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lue LF,Kuo YM,Beach T,Walker DG (2010). Microglia activation and anti-inflammatory regulation in Alzheimer’s disease. Mol Neurobiology, 41:115-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ralay Ranaivo H,Craft JM,Hu W,Guo L,Wing LK,Van Eldik LJ (2006). Glia as a therapeutic target: selective suppression of human amyloid-beta-induced upregulation of brain proinflammatory cytokine production attenuates neurodegeneration. J Neurosci, 26:662-670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Drew P,Xu J,Storer P,Chavis J,Racke M (2006). Peroxisome proliferator-activated receptor agonist regulation of glial activation: Relevance to CNS inflammatory disorders. Neurochem Int, 49:183-189. [DOI] [PubMed] [Google Scholar]
- [17].Parpura P,Heneka MT,Montana V,Oliet SH,Schousboe A,Haydon PG,Stout RF,Spray DC Jr.,Reichenbach A,Pannicke T,Pekny M,Pekna M,Zorec R,Verkhratsky A (2012). Glial cells in (patho) physiology. J Neurochem, 121:4-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Verkhratsky A,Sofroniew MV,Messing A,deLanerolle NC,Rempe D,Rodriguez JJ,Nedergaard M (2012). Neurological diseases as primary gliopathies: a reassessment of neurocentrism. ASN Neuro, 4: e00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Olabarria M,Noristani HN,Verkhratsky A,Rodriguez JJ (2010). Concomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer’s disease. Glia, 58:831-838. [DOI] [PubMed] [Google Scholar]
- [20].Simpson JE,Ince PG,Lace G,Forster G,Shaw PJ,Matthews F,Savva G,Brayne C,Wharton SB (2010). M.R.C. Cognitive Function Ageing Neuropathology Study Group, Astrocyte phenotype in relation to Alzheimer-type pathology in the ageing brain. Neurobiol Aging, 31:578-590. [DOI] [PubMed] [Google Scholar]
- [21].Da Rocha-Souto B,Scotton TC,Coma M,Serrano-Pozo A,Hashimoto T,Sereno L,Rodriguez M,Sanchez B,Hyman BT,Gomez-Isla T (2011). Brain oligomeric beta-amyloid but not total amyloid plaque burden correlates with neuronal loss and astrocyte inflammatory response in amyloid precursor protein/tau transgenic mice. J Neuropathol Exp Neurol, 70:360-376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Itagaki S,McGeer PL,Akiyama H,Zhu S,Selkoe D (1989). Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J Neuroimmunol, 24:173-182. [DOI] [PubMed] [Google Scholar]
- [23].Abeti R,Abramov AY,Duchen MR (2011). Beta-amyloid activates PARP causing astrocytic metabolic failure and neuronal death. Brain, 134:1658-1672. [DOI] [PubMed] [Google Scholar]
- [24].Abramov AY,Canevari L,Duchen MR (2004). Beta-amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J Neurosci, 24:565-575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Allaman I,Gavillet M,Belanger M,Laroche T,Viertl D,Lashuel HA,Magistretti PJ (2010). Amyloid-beta aggregates cause alterations of astrocytic metabolic phenotype: impact on neuronal viability. J Neurosci, 30:3326-3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Johnstone M,Gearing AJ,Miller KM (1999). A central role for astrocytes in the inflammatory response to beta-amyloid: chemokines, cytokines and reactive oxygen species are produced. J Neuroimmunol, 93:182-193. [DOI] [PubMed] [Google Scholar]
- [27].Van Eldik LJ,Thompson WL,Ralay Ranaivo H,Behanna HA,Martin Watterson D (2007). Glia proinflammatory cytokine upregulation as a therapeutic target for neurodegenerative diseases: function-based and target-based discovery approaches. Int Rev Neurobiol, 82:277-96. [DOI] [PubMed] [Google Scholar]
- [28].Anisman H (2009). Cascading effects of stressors and inflammatory immune system activation: implications for major depressive disorder. J Psychiatry Neurosci, 34:4-20. [PMC free article] [PubMed] [Google Scholar]
- [29].Gareau JR,Lima CD (2010). The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nat Rev Mol Cell Biol, 11: 861-871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Martin S,Wilkinson KA,Nishimune A,Henley JM (2007). Emerging extranuclear roles of protein SUMOylation in neuronal function and dysfunction. Nat Rev Neurosci 8: 948-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bohren KM,Gabbay KH,Owerbach D (2007). Affinity chromatography of native SUMO proteins using His-tagged recombinant UBC9 bound to Co2+-charged talon resin. Protein Expr Purif. 54: 289-94. [DOI] [PubMed] [Google Scholar]
- [32].Dorval V,Fraser PE (2007). SUMO on the road to neurodegeneration. Biochim Biophys Acta, 1773: 694-706. [DOI] [PubMed] [Google Scholar]
- [33].Lee L,Sakurai M,Matsuzaki S,Arancio O,Fraser P (2013). SUMO and Alzheimer’s disease. Neuromolecular Med, 15: 720-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Silveirinha V,Stephens GJ,Cimarosti H (2013). Molecular targets underlying SUMO-mediated neuroprotection in brain ischemia. J Neurochem. 127: 580-91. [DOI] [PubMed] [Google Scholar]
- [35].Dorval V,Fraser PE (2006). Small ubiquitin-like modifier (SUMO) modification of natively unfolded proteins tau and alpha-synuclein. J Biol Chem, 281: 9919-24. [DOI] [PubMed] [Google Scholar]
- [36].McMillan LE,Brown JT,Henley JM,Cimarosti H (2011). Profiles of SUMO and ubiquitin conjugation in an Alzheimer’s disease model. Neurosci Lett, 502: 201-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Olzscha H,Schermann SM,Woerner AC,Pinkert S,Hecht MH,Tartaglia GG,Vendruscolo M,Hayer-Hartl M,Hartl FU,Vabulas RM (2011). Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell, 144: 67-78. [DOI] [PubMed] [Google Scholar]
- [38].Yun SM,Cho SJ,Song JC,Song SY,Jo SA,Jo C,Yoon K,Tanzi RE,Choi EJ,Koh YH (2013). SUMO1 modulates Aβ generation via BACE1 accumulation. Neurobiol Aging, 34:650-62. [DOI] [PubMed] [Google Scholar]
- [39].Yang QG,Wang F,Zhang Q,Xu WR,Chen YP,Chen GH (2012). Correlation of increased hippocampal Sumo3 with spatial learning ability in old C57BL/6 mice. Neurosci Lett, 518:75-9. [DOI] [PubMed] [Google Scholar]
- [40].Nisticò R,Ferraina C,Marconi V,Blandini F,Negri L,Egebjerg J,Feligioni M (2014). Age-related changes of protein SUMOylation balance in the AβPP Tg2576 mouse model of Alzheimer’s disease. Frontiers in Pharmacology, 5: 1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Li Y,Wang H,Wang S,Quon D,Liu YW,Cordell B (2003). Correction to ‘Positive and negative regulation of APP amyloidogenesis by sumoylation’. Proc Natl Acad Sci USA 100:259-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Dorval VMJ,Mazzella MJ,Mathews PM,Hay RT,Fraser PE (2007). Modulation of Abeta generation by small ubiquitin-like modifiers does not require conjugation to target proteins. Biochem J, 404: 309-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zhang YQ,Sarge KD (2008). Sumoylation of amyloid precursor protein negatively regulates Abeta aggregate levels. Biochem Biophys Res Commun, 374: 673-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Takahashi KM,Ishida M,Komano H,Takahashi H (2008). SUMO-1 immunoreactivity co-localizes with phospho-Tau in APP transgenic mice but not in mutant Tau transgenic mice. Neurosci Lett, 441: 90-3. [DOI] [PubMed] [Google Scholar]
- [45].Jennewein C1,Kuhn AM,Schmidt MV,Meilladec-Jullig V,von Knethen A,Gonzalez FJ,Brüne B (2008). Sumoylation of peroxisome proliferator-activated receptor gamma by apoptotic cells prevents lipopolysaccharide-induced NCoR removal from kappaB binding sites mediating transrepression of proinflammatory cytokines. J Immunol, 181:5646-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Schneider Aguirre R,Karpen SJ (2013). Inflammatory mediators increase SUMOylation of retinoid X receptor α in a c-Jun N-terminal kinase-dependent manner in human hepatocellular carcinoma cells. Mol Pharmacol, 84:218-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Akar CA,Feinstein DL. Modulation of inducible nitric oxide synthase expression by sumoylation (2009). J Neuroinflammation, 6:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Hoppe JB,Rattray M,Tu H,Salbego CG,Cimarosti H (2013). SUMO-1 conjugation blocks beta-amyloid-induced astrocyte reactivity. Neurosci Lett, 546:51-56. [DOI] [PubMed] [Google Scholar]
- [49].Lee JH,Park SM,Kim OS,Lee CS,Woo JH,Park SJ,Joe E,Jou I (2009). Differential SUMOylation of LXRα and LXRβ mediates transrepression of STAT1 inflammatory signaling in IFN-γ-stimulated brain astrocytes. Mol Cell, 35:806-817. [DOI] [PubMed] [Google Scholar]
- [50].Pascual-García M,Rué L,León T,Julve J,Carbó JM,Matalonga J,Auer H,Celada A,Escolà-Gil JC,Steffensen KR,Pérez-Navarro E,Valledor AF (2013). Reciprocal negative cross-talk between liver X receptors (LXRs) and STAT1: effects on IFN-γ-induced inflammatory responses and LXR-dependent gene expression. J Immunol, 190:6520-32. [DOI] [PubMed] [Google Scholar]
- [51].Foran E,Bogush A,Goffredo M,Roncaglia P,Gustincich S,Pasinelli P,Trotti D (2011). Motor neuron impairment mediated by a sumoylated fragment of the glial glutamate transporter EAAT2. Glia, 59:1719-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Foran E,Rosenblum L,Bogush A,Pasinelli P,Trotti D (2014). Sumoylation of the astroglial glutamate transporter EAAT2 governs its intracellular compartmentalization. Glia, Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Riedel M,Goldbaum O,Wille M,Richter-Landsberg C (2011). Membrane lipid modification by docosahexaenoic acid (DHA) promotes the formation of α-synuclein inclusion bodies immunopositive for SUMO-1 in oligodendroglial cells after oxidative stress. J Mol Neurosci, 43:290-302. [DOI] [PubMed] [Google Scholar]
- [54].Wong MB,Goodwin J,Norazit A,Meedeniya AC,Richter-Landsberg C,Gai WP,Pountney DL (2013). SUMO-1 is associated with a subset of lysosomes in glial protein aggregate diseases. Neurotox Res, 23:1-21. [DOI] [PubMed] [Google Scholar]
- [55].Ahn K,Song JH,Kim DK,Park MH,Jo SA,Koh YH (2009). Ubc9 gene polymorphisms and late-onset Alzheimer’s disease in the Korean population: a genetic association study. Neurosci Lett, 465: 272-5. [DOI] [PubMed] [Google Scholar]
- [56].Fang H,Du X,Meng FT,Zhou JN (2011). SUMO negatively regulates BACE expression. Neuro Endocrinol Lett, 32: 313-6. [PubMed] [Google Scholar]