

Abstract
The central nervous system (CNS) is protected by a complex blood-brain barrier system; however, a broad diversity of virus, bacteria, fungi, and protozoa can gain access and cause illness. As pathogens replicate, they release molecules that can be recognized by innate immune cells. These molecules are pathogen-associated molecular patterns (PAMP) and they are identified by pattern-recognition receptors (PRR) expressed on antigen-presenting cells. Examples of PRR include toll-like receptors (TLR), receptors for advanced glycation endproducts (RAGE), nucleotide binding oligomerisation domain (NOD)-like receptors (NLR), c-type lectin receptors (CLR), RIG-I-like receptors (RLR), and intra-cytosolic DNA sensors. The reciprocal action between PAMP and PRR triggers the release of inflammatory mediators that regulate the elimination of invasive pathogens. Damage-associated molecular patterns (DAMP) are endogenous constituents released from damaged cells that also have the ability to activate the innate immune response. An increase of RAGE expression levels on neurons, astrocytes, microglia, and endothelial cells could be responsible for the accumulation of αβ-amyloid in dementia and related to the chronic inflammatory state that is found in neurodegenerative disorders.
Keywords: infection, virus, meningitis, sepsis, RAGE, αβ-amyloid, dementia
The central nervous system (CNS) is protected by a complex barrier system; however, a broad diversity of virus, bacteria, fungi, and protozoa can gain access and cause illness [1-2]. The blood-brain barrier (BBB) is an important structure consisting of microvascular endothelial cells, astrocytes, and pericytes. This barrier controls the passage of molecules into and out of the brain to preserve the microenvironment of the brain [3]. Pathogenic bacteria can cross the BBB through different mechanisms, including: transcellular traversal (Escherichia coli, Streptococcus pneumoniae, Neisseria meningitidis, Candida sp., and Cryptococcus neoformans); paracellular traversal (Trypanosoma spp. and Borrelia spp.); and Trojan-horse (Listeria monocytogenes and Mycobacterium tuberculosis) [2-3]. Transcellular traversal occurs when the pathogen crosses the BBB without any traces of intracellular tight-junction rupture. In paracellular traversal, the microorganism penetrates the BBB with or without evidence of tight-junction rupture. In the Trojan-horse mechanism the pathogen crosses endothelial cells by transmigrating in infected phagocytes [2]. Several virus species can directly infect endothelial cells to cross the BBB into the CNS [4-6].
Replication of the pathogen occurs concomitantly with the release of their component molecules. These components are all referred to as pathogen-associated molecular patterns (PAMP). PAMP are identified by pattern-recognition receptors (PRR), which are important mechanisms of the innate immune system [7-8]. The host immune response occurs when pathogens are identified by antigen-presenting cells. This identification occurs via the binding of PAMP to PRR on antigen-presenting cells. PPR examples include toll-like receptors (TLR), receptors for advanced glycation endproducts (RAGE), nucleotide binding oligomerisation domain (NOD)-like receptors (NLR), c-type lectin receptors (CLR), RIG-I-like receptors (RLR), and intra-cytosolic DNA sensors [8-9]. The reciprocal action between PAMP and PRR triggers the release of inflammatory mediators that regulate the response necessary to eliminate the invasive pathogens [10]. Innate immune cells are also activated by endogenous constituents that are released from damaged cells, such as high mobility group box-1 protein (HMGB-1), nucleic acids, amyloid β, cytochrome c, uric acid crystals, adenosine 5′-triphosphate (ATP), members of S100 protein family, histones, advanced glycation end products (AGE), and heat shock protein (HSP). These molecules are called damage-associated molecular patterns (DAMP) [11-12]. Thus, damage to CNS during infection commonly involves pathogenic mechanisms and the innate immune host response [13-14].
Recognition of Pathogen Infection by Innate Immune Sensors
TLR
The innate immune system identifies pathogens via numerous receptor families. Toll-like receptors (TLR) recognize molecular motifs that are expressed by pathogens or endogenous ligands released from damaged cells [15]. The TLR receptors can be separated into two categories: one group of receptors is expressed on the cell membrane for ligand recognition (TLR1, TLR2, TLR4, TLR5, TLR6, and TLR10) and the other group is localised in intracellular endosomal space for recognition of pathogen nucleic acids (TLR3, TLR7, TLR8, and TLR9) [16], Figure 1.
Figure 1.
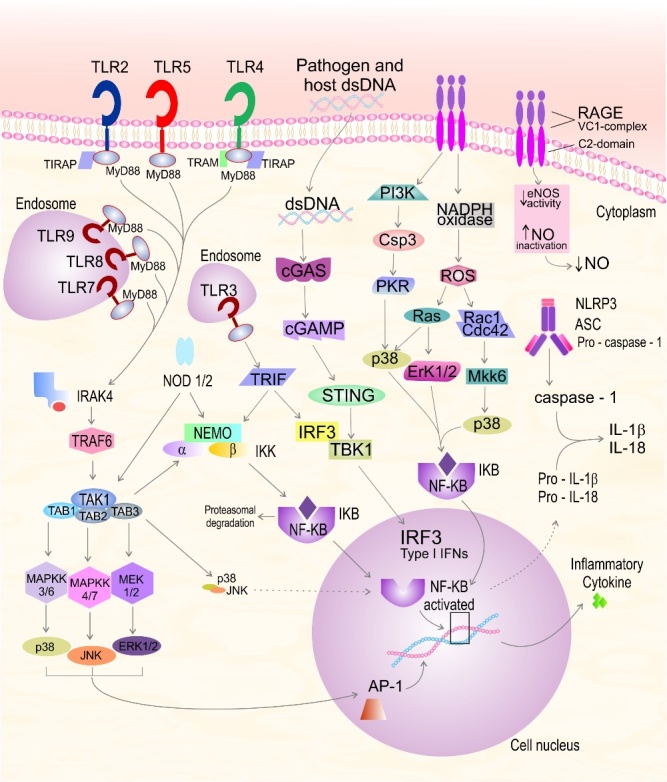
Recognition of pathogens by the innate immune system. Toll-like receptors (TLR) recognize molecular motifs that are expressed by pathogens or endogenous ligands released from damaged cells. AGE, advanced glycation end products; AP-1, activator protein-1; ASC, apoptosis-associated speck-like protein containing a caspase-recruitment domain; Cdc42, cell division control protein 42 homolog; Csp3, caspase-3; eNOS, endothelial nitric oxide synthase; ERK, extracellular signal regulated; IL, interleukin; IKK, NEMO/IKKα/IKKß complex; IRAK-4, interleukin-1 receptor-associated kinase 4; IκB, inhibitor of NF-κB; IKK, IκB kinase; INF, interferon; IRF3, interferon regulatory factor 3; JNK, c-jun N-terminal kinase; MAPK, mitogen-activated protein kinases; MyD88, myeloid differentiation factor 88; MEK, mitogen-activated protein kinase; MKK, MAPK kinase; NADPH, Nicotinamide adenine dinucleotide phosphate; NEMO, NF-κB essential modulator, IKKγ; NF-κB, nuclear transcription factor kappa B; NLRP3, nucleotide-binding domain and leucine-rich repeat protein 3; NO, nitric oxide; NODs, nucleotide binding oligomerisation domains; PI3K, phosphatidylinositide 3-kinase; PKB, protein kinase A; RAGE; receptor for advanced glycation endproducts; PKR, protein kinase RNA-activated; ROS, reactive oxygen species; TAB, TGF-β-activated kinase 1-binding protein 1; TAK1, TGFß activated kinase 1; TIRAP, TIR-containing protein; TLRs, toll-like receptors; TRAF, receptor-associated factor; TRAM, TRIF-related adaptor molecule; TRIF, TIR-domain-containing adapter-inducing interferon-β
The TLR activation signal is transmitted by a variety of intracellular protein adaptors. TLR5, TLR7, and TLR9 recruit the myeloid differentiation factor 88 adaptor (MyD88). TLR3 recruits the TRIF adaptor [TIR (Toll/interleukin-1 receptor) domain-containing adaptor protein inducing interferon-β)]. TLR1, TLR2, and TLR6 recruit (MyD88) or TIR-containing protein (TIRAP). In one example of signal transmission, TLR4 recruits TIRAP to bind MyD88 and TRIF-related adaptor molecule (TRAM) to bind TRIF leading to pro-inflammatory mediator release [17-18]. The MyD88 adaptor molecule connects with the serine/threonine kinase interleukin-1 receptor-associated kinase-4 (IRAK)-4, leading to phosphorylation of IRAK-1 and IRAK-2 [19]. Next, IRAK binds with the receptor-associated factor (TRAF) family and triggers a link to the TAK1/TAB1/TAB2/TAB3 complex [15]. TAK1 phosphorylates the NEMO/IKKα/IKKβ complex (IKK), which phosphorylates IKB, leading the nuclear translocation of the transcription factor nuclear factor-κB (NF-κB). NF-κB is a transcriptional activator of pro-inflammatory mediators [20-21]. TAK1 activation initiates the mitogen-activated protein kinase (MAPK) pathway. Subsequently, MAPKK-3/6, MAPKK-4/7, and MEK-1/2 trigger the activation of p38, c-jun N-terminal kinase (JNK), and extracellular signal regulated (ERK-1/2), respectively. These cascades trigger the translocation of protein-1 (AP-1) to the nucleus, thus inducing the transcription of inflammatory cytokines [15].
NOD1 and NOD2 Signalling
NLR are members of a family of cytosolic sensors that are responsible for the detection of different kinds of pathogens and the detection of DAMPs [16]. The NLR intracellular proteins have C-terminal leucine-rich repeat (LRR) domains and central NACHT nucleotide-binding domains (NBD) [22]. NOD1 (CARD4) and NOD2 (CARD15) activate numerous transcription factors to trigger the release of pro-inflammatory mediators [23]. Another group of NLR members activates the formation of a multiprotein complex known as the inflammasome. The inflammasome activates procaspase-1 and maturation of the IL-1 cytokines [17].
NOD1 and NOD2 recognize compounds from Gram-positive and Gram-negative bacteria and recruit the receptor-interacting protein-2 (RIP2), cellular inhibitors of apoptosis (cIAP2), and X-linked inhibitor of apoptosis protein (XIAP). This cascade provides a link to the TAK1/TAB1/TAB2/TAB3 complex and IKK complex. These complexes phosphorylate IKB, resulting in the release and nuclear translocation of the transcription factor, NF-κB and MAPK, to trigger the transcription of interleukin (IL)-1 cytokines [17, 24].
NLRP3 Inflammasome
The NLRP3 inflammasome is composed of nucleotide-binding oligomerisation domains (NOD)-like receptor 3 (NLRP3), pro-caspase-1, and the adaptor protein apoptosis-associated speck-like protein comprising a caspase recruitment domain (ASC) [17]. The NLRP3 is activated by several stimuli, such as bacterial toxins, bacterial compounds, and DAMPs [25]. Upon activation, NLRP3 oligomerises with ASC and pro-caspase 1. Procaspase-1 is converted to activated caspase-1, which subsequently matures and secretes the pro-inflammatory cytokines IL-1β and IL-18 in their mature forms [26]. In addition, NLRP3 contributes to amyloidosis and cognitive dysfunction in an experimental model of Alzheimer disease [27].
Cytosolic DNA Sensors
Intracellular DNA from virus and bacteria is important to initiate an efficient host immune response. The signaling adaptor protein Stimulator of Interferon Genes (STING) is a necessary innate immune pathway for cytosolic DNA sensing that drives type I IFN production [28]. STING is activated by cyclic dinucleotides generated by cyclic GMP-AMP synthase (cGAS). STING activation leads to activation of the serine/threonine-protein kinase tank-binding kinase 1 (TBK1). TBK1 phosphorylates interferon regulatory factor 3 (IRF3), thereby inducing type I IFN gene transcription. TBK1 also plays an important role in the regulation of the immune response expression of pro-inflammatory cytokines [29-30].
RAGE Receptor
The RAGE receptor is a central signalling molecule of the innate immune system. This receptor is involved in the inflammatory host response and is able to bind a variety of ligands with different properties and origin [31]. RAGE is an important receptor that recognizes AGE, members of S100 protein family, amyloid β and amyloid fibrils, HMGB-1, and β-integrin macrophage 1 antigen (Mac-1) [31-32]. Activated RAGE receptors trigger the Ras-extracellular signal-regulated kinase 1/2 (ERK1/2), Cdc42/Rac, stress-activated protein kinase/c-Jun-NH2-terminal kinase (SAPK/JNK), and p38 mitogen-activated protein (MAP) kinase pathways. These cascades result in the activation of the transcription factor NF-κB [33-34]. NF-κB then triggers gene transcription and the release of TNF-α, IL-1β, IL-6, M-CSF, and S100B. Secreted S100B and chronic RAGE activation contribute to microglial activation and increased oxidative stress. By these mechanisms, up-regulation NF-κB [35] leads to neurite degeneration and neuronal apoptosis that results in memory impairment [33], Figure 1.
Infectious Disease and Dementia
Chronic activation of microglia and astrocytes potentially cause injury to the BBB and neuronal impairment through the release of cytokines, chemokines, reactive oxygen species, DAMPs, and pro-inflammatory mediators. The exacerbated host immune response leads to perturbations in neuronal function and extensive brain degeneration. An important characteristic of dementia is its association with dysfunctions of cognitive memory such as calculation, space orientation, and speech impairment [36].
Systemic inflammation and neuroinflammation are emerging as important targets in cognitive decline in the aged and susceptible brain [37]. Sepsis-associated delirium is a clinical indication that there is CNS involvement in sepsis [38]. Severe sepsis can result in significant brain damage that is discernible when the patients present with delirium. A longer duration of delirium in the hospital is normally associated with worse global cognition and executive function scores of the patients [39]. The pathophysiology of sepsis-associated delirium involves hypoxemia, glucose dysregulation, systemic inflammatory host response, BBB disruption, abnormal neurotransmitter composition, impaired astrocyte function, and neuronal degeneration [38, 40]. Sepsis patients who present elevated levels of TNF-α, IL-1β, and IL-6 in the CSF may face long-term cognitive impairment [41-42]. In another study, septic patients presented higher expression of HMGB1 mRNA and protein in peripheral blood, and the levels of HMGB1 gradually decreased to normal after treatment [43]. Therefore, the endogenous proneurogenic response to sepsis could be triggered by, or involve signals in, pathway activation by amyloid β peptide, HMGB-1, and RAGE/NF-κB [44]. These potential proneurogenic response pathways also contribute to production of inflammatory mediators and neurotoxic compounds in the brain [44].
In an experimental sepsis model of cecal ligation and puncture (CLP) HMGB-1, IL-1β, and NADPH oxidase levels are responsible for long-term cognitive impairment [45-46]. These findings are interesting because these pro-inflammatory mediators are agonists of the RAGE receptor and trigger exacerbated pro-inflammatory cascades. In another sepsis model using lipopolysaccharide (a constituent of the cell wall of Gram-negative bacteria that activates TLR-4), induction of neuroinflammation and increased intracellular accumulation of amyloid precursor protein and amyloid β peptide was observed in transgenic mice [47-48]. Similarly, in the CLP sepsis model, brains from sepsis-survivor animals presented an increase in amyloid β content, a decrease in synaptophysin levels, and cognitive impairment [48].
Despite advances in medical care, severe bacterial meningitis often causes neurological deficits, including dementia, and infection is also associated with long-term cognitve impairment, principally in childhood [49-50]. Bacterial meningitis is accompanied by inflammatory mechanisms that can cause cell injury and release of DAMPs. An important DAMP in this process is HMGB1, a nuclear protein that has been detected in the CSF of paediatric patients with bacterial meningitis [51]. When released from cells, HMGB1 is recognized by TLR2, TLR4, and RAGE receptors [52-53]. Pro-inflammatory cytokines produced in the first hours post-infection include TNF-α, IL-1β, and IL-6 [54]. In one meningitis study, animals presented depressive-like-behavior and increased cytokines at ten days after infection. In this period the animals were free of infection, but the brain continued producing TNF-α 55. The increase of these cytokines has been demonstrated in both patients and in rodent meningitis models. The chronic production of cytokines could be related to long-term cognitive impairment [54, 56-57]. In another meningitis study, rodents receiving adjuvant treatment with antioxidant or immunomodulators had a decrese of TNF-α and a concurrent improvment in memory [58-60]. In clinical sepsis-associated delirium, patients with infectious meningitis, particularly mycotic or bacterial meningitis, present high levels of serum amyloid β, when compared to healthy controls [61]. Our hypothesis is that long-term cognitive impairment in sepsis-associated delirium or meningitis is associated with acumulation of amyloid peptide in the brain.
Conclusion
During infection, the severe inflammatory response may lead to neuronal damage and could potentially be detrimental for patients. Many stimuli including PAMPs, DAMPs and pro-inflammatory mediators are responsible for the activation of microglia cells that produce cytokines, chemokines, and reactive oxygen and nitrogen species to eliminate the invading pathogen. We propose that inhibition of the activity of HMGB-1 could prevent long-term cognitive impairment in patients with bacterial meningitis or sepsis.
Acknowledgments
Research from the Center for Translational Psychiatry (USA) is supported by grants from the Department of Psychiatry and Behavioral Sciences, The University of Texas Medical School at Houston. Research from Laboratorio de Microbiologia Experimental and Laboratório de Neurociências (Brazil) is supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Fundação de Amparo à Pesquisa e Inovação do Estado de Santa Catarina (FAPESC), and Universidade do Extremo Sul Catarinense (UNESC).
Footnotes
Conflict of interest
The authors declare that they have no conflict of interest.
References
- [1].Swanson PA 2nd,McGavern DB (2015). Viral diseases of the central nervous system. Curr Opin Virol, 11c:44-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kim KS (2008). Mechanisms of microbial traversal of the blood-brain barrier. Nat Rev Microbiol, 6(8):625-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kim KS (2006). Microbial translocation of the blood-brain barrier. Int J Parasitol, 36(5):607-14. [DOI] [PubMed] [Google Scholar]
- [4].Moses AV,Bloom FE,Pauza CD,Nelson JA (1993). Human immunodeficiency virus infection of human brain capillary endothelial cells occurs via a CD4/galactosylceramide-independent mechanism. Proc Natl Acad Sci U S A, 90(22):10474-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Coyne CB,Shen L,Turner JR,Bergelson JM (2007). Coxsackievirus entry across epithelial tight junctions requires occludin and the small GTPases Rab34 and Rab5. Cell Host Microbe, 2(3):181-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Coyne CB,Kim KS,Bergelson JM (2007). Poliovirus entry into human brain microvascular cells requires receptor-induced activation of SHP-2. Embo J, 26(17):4016-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sellner J,Täuber MG,Leib SL, (2010). Chapter 1 - Pathogenesis and pathophysiology of bacterial CNS infections, in:Karen LR andAllan RT, Editors. Handbook of Clinical Neurology. Elsevier, 1-16. [DOI] [PubMed] [Google Scholar]
- [8].Mook-Kanamori BB,Geldhoff M,van der Poll T,van de Beek D (2011). Pathogenesis and pathophysiology of pneumococcal meningitis. Clin Microbiol Rev, 24(3):557-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Savva A,Roger T (2013). Targeting Toll-Like receptors: promising therapeutic strategies for the management of sepsis-associated pathology and infectious Diseases. Front Immunol, 4:387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Iwasaki A,Medzhitov R (2010). Regulation of adaptive immunity by the innate immune system. Science, 327(5963):291-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wiersinga WJ,Leopold SJ,Cranendonk DR,van der Poll T (2014). Host innate immune responses to sepsis. Virulence, 5(1):36-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lu B,Wang C,Wang M,Li W,Chen F,Tracey KJ, et al. (2014). Molecular mechanism and therapeutic modulation of high mobility group box 1 release and action: an updated review. Expert Rev Clin Immunol, 10(6):713-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Barichello T,Generoso JS,Milioli G,Elias SG,Teixeira AL (2013). Pathophysiology of bacterial infection of the central nervous system and its putative role in the pathogenesis of behavioral changes. Rev Bras Psiquiatr, 35(1):81-7. [DOI] [PubMed] [Google Scholar]
- [14].Hu X,Liou AK,Leak RK,Xu M,An C,Suenaga J, et al. (2014). Neurobiology of microglial action in CNS injuries: Receptor-mediated signaling mechanisms and functional roles. Prog Neurobiol, 119-120:60-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Heiman A,Pallottie A,Heary RF,Elkabes S (2014). Toll-like receptors in central nervous system injury and disease: A focus on the spinal cord. Brain Behav Immun, 42:232-45. [DOI] [PubMed] [Google Scholar]
- [16].Kigerl KA,de Rivero Vaccari JP,Dietrich WD,Popovich PG,Keane RW (2014). Pattern recognition receptors and central nervous system repair. Exp Neurol, 258:5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kumar S,Ingle H,Prasad DV,Kumar H (2013). Recognition of bacterial infection by innate immune sensors. Crit Rev Microbiol, 39(3):229-46. [DOI] [PubMed] [Google Scholar]
- [18].Akira S (2009). Innate immunity to pathogens: diversity in receptors for microbial recognition. Immunol Rev, 227(1):5-8. [DOI] [PubMed] [Google Scholar]
- [19].Wang H,Zhang J,Wu H,Jiang C,Zheng Q,Li Z (2006). Inhibition of RAW264.7 macrophage inflammatory cytokines release by small haparin RNAi targeting TLR4. J Huazhong Univ Sci Technolog Med Sci, 26(5):500-3. [DOI] [PubMed] [Google Scholar]
- [20].Koedel U,Bayerlein I,Paul R,Sporer B,Pfister HW (2000). Pharmacologic interference with NF-kappaB activation attenuates central nervous system complications in experimental Pneumococcal meningitis. J Infect Dis, 182(5):1437-45. [DOI] [PubMed] [Google Scholar]
- [21].Kastenbauer S,Koedel U,Weih F,Ziegler-Heitbrock L,Pfister HW (2004). Protective role of NF-kappaB1 (p50) in experimental pneumococcal meningitis. Eur J Pharmacol, 498(1-3):315-8. [DOI] [PubMed] [Google Scholar]
- [22].Clarke TB,Weiser JN (2011). Intracellular sensors of extracellular bacteria. Immunol Rev, 243(1):9-25. [DOI] [PubMed] [Google Scholar]
- [23].Barbe F,Douglas T,Saleh M (2014). Advances in Nod-like receptors (NLR) biology. Cytokine Growth Factor Rev, 25(6):681-97. [DOI] [PubMed] [Google Scholar]
- [24].Damgaard RB,Gyrd-Hansen M (2011). Inhibitor of apoptosis (IAP) proteins in regulation of inflammation and innate immunity. Discov Med, 11(58):221-31. [PubMed] [Google Scholar]
- [25].Koppe U,Suttorp N,Opitz B (2012). Recognition of Streptococcus pneumoniae by the innate immune system. Cell Microbiol, 14(4):460-6. [DOI] [PubMed] [Google Scholar]
- [26].Schroder K,Zhou R,Tschopp J (2010). The NLRP3 inflammasome: a sensor for metabolic danger? Science, 327(5963):296-300. [DOI] [PubMed] [Google Scholar]
- [27].Heneka MT,Kummer MP,Stutz A,Delekate A,Schwartz S,Vieira-Saecker A, et al. (2013). NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature, 493(7434):674-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Dempsey A,Bowie AG (2015). Innate immune recognition of DNA: A recent history. Virology, 479-480c:146-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Cai X,Chiu YH,Chen ZJ (2014). The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol Cell, 54(2):289-96. [DOI] [PubMed] [Google Scholar]
- [30].Yu T,Yi YS,Yang Y,Oh J,Jeong D,Cho JY (2012). The pivotal role of TBK1 in inflammatory responses mediated by macrophages. Mediators Inflamm, 2012:979105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Fritz G (2011). RAGE: a single receptor fits multiple ligands. Trends Biochem Sci, 36(12):625-32. [DOI] [PubMed] [Google Scholar]
- [32].Harris HE,Andersson U,Pisetsky DS (2012). HMGB1: a multifunctional alarmin driving autoimmune and inflammatory disease. Nat Rev Rheumatol, 8(4):195-202. [DOI] [PubMed] [Google Scholar]
- [33].Kierdorf K,Fritz G (2013). RAGE regulation and signaling in inflammation and beyond. J Leukoc Biol, 94(1):55-68. [DOI] [PubMed] [Google Scholar]
- [34].Koch M,Chitayat S,Dattilo BM,Schiefner A,Diez J,Chazin WJ, et al. (2010). Structural basis for ligand recognition and activation of RAGE. Structure, 18(10):1342-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Tobon-Velasco JC,Cuevas E,Torres-Ramos MA,Santamaria A (2014). Receptor for AGEs (RAGE) as Mediator of NF-kB Pathway Activation in Neuroinflammation and Oxidative Stress. CNS Neurol Disord Drug Targets, 13(9):1615-26. [DOI] [PubMed] [Google Scholar]
- [36].Kubis AM,Janusz M (2008). [Alzheimer’s disease: new prospects in therapy and applied experimental models]. Postepy Hig Med Dosw (Online), 62:372-92. [PubMed] [Google Scholar]
- [37].Cunningham C,Hennessy E (2015). Co-morbidity and systemic inflammation as drivers of cognitive decline: new experimental models adopting a broader paradigm in dementia research. Alzheimers Res Ther, 7(1):33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Piva S,McCreadie VA,Latronico N (2015). Neuroinflammation in sepsis: sepsis associated delirium. Cardiovasc Hematol Disord Drug Targets, 15(1):10-8. [DOI] [PubMed] [Google Scholar]
- [39].Pandharipande PP,Girard TD,Jackson JC,Morandi A,Thompson JL,Pun BT, et al. (2013). Long-term cognitive impairment after critical illness. N Engl J Med, 369(14):1306-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zhang QH,Sheng ZY,Yao YM (2014). Septic encephalopathy: when cytokines interact with acetylcholine in the brain. Mil Med Res, 1:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Girard TD,Jackson JC,Pandharipande PP,Pun BT,Thompson JL,Shintani AK, et al. (2010). Delirium as a predictor of long-term cognitive impairment in survivors of critical illness. Crit Care Med, 38(7):1513-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].van Griensven M (2014). [Cytokines as biomarkers in polytraumatized patients]. Unfallchirurg, 117(8):699-702. [DOI] [PubMed] [Google Scholar]
- [43].Singh A,Feng Y,Mahato N,Li J,Wu C,Gong J (2015). Role of high-mobility group box 1 in patients with acute obstructive suppurative cholangitis-induced sepsis. J Inflamm Res, 8:71-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Meneghini V,Bortolotto V,Francese MT,Dellarole A,Carraro L,Terzieva S, et al. (2013). High-mobility group box-1 protein and beta-amyloid oligomers promote neuronal differentiation of adult hippocampal neural progenitors via receptor for advanced glycation end products/nuclear factor-kappaB axis: relevance for Alzheimer’s disease. J Neurosci, 33(14):6047-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Comim CM,Constantino LC,Barichello T,Streck EL,Quevedo J,Dal-Pizzol F (2009). Cognitive impairment in the septic brain. Curr Neurovasc Res, 6(3):194-203. [DOI] [PubMed] [Google Scholar]
- [46].Mina F,Comim CM,Dominguini D,Cassol-Jr OJ,Dall Igna DM,Ferreira GK, et al. (2014). Il1-beta involvement in cognitive impairment after sepsis. Mol Neurobiol, 49(2):1069-76. [DOI] [PubMed] [Google Scholar]
- [47].Sheng JG,Bora SH,Xu G,Borchelt DR,Price DL,Koliatsos VE (2003). Lipopolysaccharide-induced-neuroinflammation increases intracellular accumulation of amyloid precursor protein and amyloid beta peptide in APPswe transgenic mice. Neurobiol Dis, 14(1):133-45. [DOI] [PubMed] [Google Scholar]
- [48].Schwalm MT,Pasquali M,Miguel SP,Dos Santos JP,Vuolo F,Comim CM, et al. (2014). Acute brain inflammation and oxidative damage are related to long-term cognitive deficits and markers of neurodegeneration in sepsis-survivor rats. Mol Neurobiol, 49(1):380-5. [DOI] [PubMed] [Google Scholar]
- [49].Khandaker GM,Stochl J,Zammit S,Lewis G,Jones PB (2015). A population-based prospective birth cohort study of childhood neurocognitive and psychological functioning in healthy survivors of early life meningitis. Ann Epidemiol, 25(4):236-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Naito M,Johkura K,Momoo T,Nomiya T,Kudo Y,Kuroiwa Y (2010). Dementia and capsular genu ischemia in patients with severe bacterial meningitis. Neurol Sci, 31(2):133-6. [DOI] [PubMed] [Google Scholar]
- [51].Tang D,Kang R,Cao L,Zhang G,Yu Y,Xiao W, et al. (2008). A pilot study to detect high mobility group box 1 and heat shock protein 72 in cerebrospinal fluid of pediatric patients with meningitis. Crit Care Med, 36(1):291-5. [DOI] [PubMed] [Google Scholar]
- [52].Hohne C,Wenzel M,Angele B,Hammerschmidt S,Hacker H,Klein M, et al. (2013). High mobility group box 1 prolongs inflammation and worsens disease in pneumococcal meningitis. Brain, 136(Pt 6):1746-59. [DOI] [PubMed] [Google Scholar]
- [53].Saijo K,Glass CK (2011). Microglial cell origin and phenotypes in health and disease. Nat Rev Immunol, 11(11):775-87. [DOI] [PubMed] [Google Scholar]
- [54].Barichello T,dos Santos I,Savi GD,Simoes LR,Silvestre T,Comim CM, et al. (2010). TNF-alpha, IL-1beta, IL-6, and cinc-1 levels in rat brain after meningitis induced by Streptococcus pneumoniae. J Neuroimmunol, 221(1-2):42-5. [DOI] [PubMed] [Google Scholar]
- [55].Barichello T,Dos Santos I,Savi GD,Simoes LR,Generoso JS,Comim CM, et al. (2010). Depressive-like-behavior and proinflamatory interleukine levels in the brain of rats submitted to pneumococcal meningitis. Brain Res Bull, 82(5-6):243-6. [DOI] [PubMed] [Google Scholar]
- [56].Barichello T,Lemos JC,Generoso JS,Cipriano AL,Milioli GL,Marcelino DM, et al. (2011). Oxidative stress, cytokine/chemokine and disruption of blood-brain barrier in neonate rats after meningitis by Streptococcus agalactiae. Neurochem Res, 36(10):1922-30. [DOI] [PubMed] [Google Scholar]
- [57].Panato AP,Tomasi LT,Simon CS,Madeira K,Simoes LR,Medeiros LR, et al. (2014). Meta-analysis identifies Tumor Necrosis Factor-alpha and Interleukin-1 beta as Diagnostic Biomarkers for Bacterial and Aseptic Meningitis. Curr Neurovasc Res, 11(4):340-8. [DOI] [PubMed] [Google Scholar]
- [58].Barichello T,Santos AL,Savi GD,Generoso JS,Otaran P,Michelon CM, et al. (2012). Antioxidant treatment prevents cognitive impairment and oxidative damage in pneumococcal meningitis survivor rats. Metab Brain Dis, 27(4):587-93. [DOI] [PubMed] [Google Scholar]
- [59].Barichello T,Ceretta RA,Generoso JS,Moreira AP,Simoes LR,Comim CM, et al. (2012). Cannabidiol reduces host immune response and prevents cognitive impairments in Wistar rats submitted to pneumococcal meningitis. Eur J Pharmacol, 697(1-3):158-64. [DOI] [PubMed] [Google Scholar]
- [60].Barichello T,Simoes LR,Generoso JS,Sangiogo G,Danielski LG,Florentino D, et al. (2013). Erythropoietin prevents cognitive impairment and oxidative parameters in Wistar rats subjected to pneumococcal meningitis. Transl Res, 163(5):503-13. [DOI] [PubMed] [Google Scholar]
- [61].Kanoh Y,Ohara T,Akahoshi T (2011). Acute inflammatory biomarkers in cerebrospinal fluid as indicators of blood cerebrospinal fluid barrier damage in Japanese subjects with infectious meningitis. Clin Lab, 57(1-2):37-46. [PubMed] [Google Scholar]