

Summary
The importance and complexity associated with the totality of glycan structures, i.e. the glycome, has garnered significant attention from chemists and biologists alike. However, what is lacking from this biochemical picture is how cells, tissues, and organisms interpret glycan patterns and translate this information into appropriate responses. Lectins, glycan-binding proteins, are thought to bridge this gap by decoding the glycome and dictating cell fate based on the underlying chemical identities and properties of the glycome. Yet, our understanding of the in vivo ligands and function for most lectins is still incomplete. This review focuses on recent advances in chemical tools to study the specificity and dynamics of mammalian lectins in live cells. A picture emerges of lectin function that is highly sensitive to its organization, which in turn drastically shapes immunity and cancer progression. We hope this review will inspire biologists to make use of these new techniques and stimulate chemists to continue developing innovative approaches to probe lectin biology in vivo.
Introduction
The glycome is an information-rich dataset that influences almost every facet of molecular, cellular and organismal biology (Varki et al., 2008). The potential complexity of the glycome exceeds that of other biomolecules, e.g., proteins and nucleic acids (Ohtsubo and Marth, 2006). In mammals, this molecular diversity arises from the action of ~250 glycosyltransferases that catalyze the addition of different sugar building blocks to their substrates with various regio- and stereochemical specificities. The primary decoders of the glycome are lectins, glycan-binding proteins that are abundant in all domains of life (Lee and Lee, 1995; Sharon, 2007). Lectin interactions with specific glycan structures are responsible for protein folding and sorting, cell adhesion and trafficking, pathogen recognition and mammalian fertilization, among other processes (Varki et al., 2008).
Modern genome sequencing efforts have led to prediction of over 160 putative lectins in humans alone, rich starting point for new discoveries in glycobiology. However, identifying the authentic biological ligands of each lectin remains challenging, as does elucidation of the lectin’s role in normal and pathological processes. In earlier decades, lectins were often characterized based on their ability to aggregate cells – the so-called agglutination assay (Sharon and Lis, 2004). Their ligand specificities were assigned by screening available saccharides as agglutination inhibitors. The results of such crude assays are still evident in the stated specificities of commercial lectins that are widely used as research tools. Yet, we now know that these ligand assignments are at best incomplete and in many cases misleading. In modern times, more accurate ligand assignments can be made by glycan microarray screens (Rillahan and Paulson, 2011). The most advanced of these platforms comprise over 600 vertebrate glycan structures (Wang et al., 2014) or 300 microbially-derived structures, immobilized on slides that can be screened using fluorophore-conjugated lectins. The results of such screens have revised our understanding of lectin specificity and improved our appreciation of their biological roles (Geissner et al., 2014).
Still, present-day glycan microarrays do not capture critical elements of the biological situation. In nature, glycans are displayed on particular protein or lipid scaffolds, which in many cases contribute to lectin specificity (Pouyani and Seed, 1995; Sako et al., 1995; Wilkins et al., 1995). As well, the spatial arrangement and density of glycan ligands, in concert with the mutivalency and distribution of cognate lectins, factor into the biological response these interactions mediate. These facets of lectin biology are difficult to emulate in artificial experimental platforms and therefore have been hard to study.
As more lectins are discovered and characterized, it has also become clear that individual lectin families have their own unique properties and peculiarities. Many membrane-associated lectins exist in different oligomeric states that have been proposed to regulate ligand binding, signaling and organization of higher order complexes (Brewer et al., 2002). Some lectins interact with cognate ligands on opposing cells in a manner dependent on force (Marshall et al., 2003), while others can interact with ligands in the same cell membrane (Han et al., 2005). Further, certain lectins have been found to possess activating or inhibiting motifs, essential elements for proper signal transduction. Since most lectins are thought to lack regulatory domains or allosteric control, lectin trafficking and sub-cellular localization, and their respective dynamics, control how and when lectins exert their function in many cases (Gabius et al., 2011).
The complexities inherent to lectin biology have motivated the development of new experimental approaches, many of which are rooted in chemistry. In this review, the focus will be on recent contributions aimed at identifying a lectin’s endogenous biological ligands or on probing the dynamic aspects of lectin biology using live cells. We concentrate here on three chemical strategies: (A) metabolic labeling with cross-linking sugars for discovery of endogenous ligands, (B) use of synthetic glycoconjugate mimics to study lectin function, and (C) imaging strategies to probe lectin organization and dynamics (Figure 1). While this is by no means an exhaustive list of techniques for interrogating lectin binding and dynamics, these tools have transformed our understanding of lectin-glycan interactions in vivo and will continue to reveal unexpected features of mammalian lectins in the future.
Figure 1.
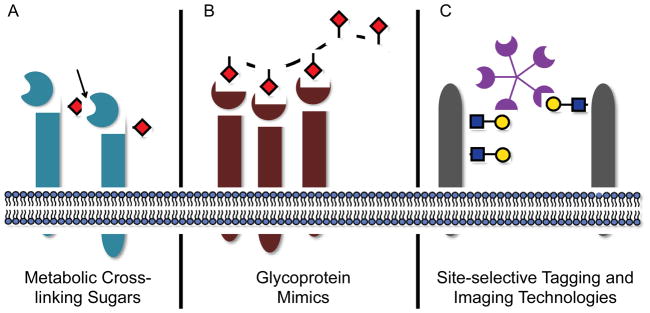
Recent chemical methods have begun to elucidate the binding partners and dynamic behavior of mammalian lectins in vivo. (A) Metabolic labeling with cross-linking sugars can report on the in situ interactions between lectins and glycans. (B) Glycoprotein mimics are synthetic, multimeric structures designed to emulate the spatial distribution of glycan epitopes on natural glycoconjugate ligands. (C) New tagging and imaging technologies for monitoring lectin trafficking and organization in vivo.
Metabolic labeling with cross-linking sugars
A major goal in lectin research is the identification of endogenous biological ligands – the true binding partners whose engagement triggers a biological response. Some lectins interact with glycan structures that can be found on a variety of underlying scaffolds, such as N- and O-glycoproteins as well as glycolipids. Figuring out which subset of these are the predominant ligands in a biological setting is a daunting task. An emerging approach to identifying native lectin ligands is to trap them in a covalent complex. The challenge of performing such an experiment in the context of live cells lies in arming potential glycoconjugate ligands with a cross-linkable functional group. This has been accomplished using the technique of metabolic glycan engineering.
First demonstrated by Reutter and co-workers (Kayser et al., 1992a, 1992b), metabolic glycan engineering relies on the native cellular biomachinery to incorporate non-natural monosaccharides into native glycoconjugates. In their classic work, N-acetylmannosamine (ManNAc) analogs bearing unnatural N-acyl chains were shown to be converted to the corresponding sialic acids, which were incorporated into glycoproteins in cultured cells and laboratory animals (Keppler et al., 2001). We and others expanded upon this idea using chemically functionalized ManNAc analogs bearing bioorthogonal ketone, thiol, azide and alkyne groups (Bussink et al., 2007; Hsu et al., 2007; Mahal et al., 1997; Sampathkumar et al., 2006; Saxon and Bertozzi, 2000). Similarly, these bioorthogonal functional groups have been incorporated into glycans containing N-acetylgalactosamine (GalNAc) (Hang et al., 2003), N-acetylglucosamine (GlcNAc) (Vocadlo et al., 2003; Zaro et al., 2011), fucose (Dehnert et al., 2011; Hsu et al., 2007; Rabuka et al., 2006; Al-Shareffi et al., 2013), and xylose residues (Beahm et al., 2014) in vertebrate systems. Incorporation of a bioorthogonal group into cellular glycans enabled subsequent reaction with fluorescent probes for imaging or affinity reagents for enrichment and proteomic analyses. These pioneering efforts provided evidence that similar glycan structures on a variety of different protein and lipid scaffolds could be targeted using a metabolism-based approach. Recently, a number of reports have detailed the combination of metabolic labeling and Förster Resonance Energy Transfer (FRET) for imaging proteins bearing particular glycan structures (Belardi et al., 2013; Haga et al., 2012; Lin et al., 2014).
Paulson and coworkers were the first to adapt metabolic glycan engineering for the purpose of capturing native lectin-ligand complexes. Their focus was Siglec-2, or CD22, a member of the sialic acid-binding immunoglobin-like lectin (siglec) family known to modulate immune cell activation (Walker and Smith, 2008). CD22 comprises an extracellular sialic acid binding domain, a single transmembrane domain, and a cytosolic immunoreceptor tyrosine-based inhibitory motif (ITIM) that when phosphorylated recruits SH2 domain-containing phosphatase 1 (Tedder et al., 2005). Engagement of CD22 at the immune synapse inhibits downstream signaling associated with B-cell receptor (BCR) triggering, thereby limiting autoimmunity. On resting B cells, however, it was hypothesized that cis interactions with other B cell surface glycoproteins masked CD22’s lectin domain, preventing CD22 and BCR co-localization.
To identify the cis ligands of CD22, Han et al. metabolically labeled B cells with a C-9 azidoaryl sialic acid analog (9-AAz-NeuAc) (Han et al., 2005). Upon UV exposure, azidoarenes form reactive nitrenes can capture proximal proteins by C-H bond insertion. Immunoprecipitation of CD22 from 9-AAz-NeuAc-treated and irradiated B cells revealed a single cross-linked glycoprotein ligand – CD22 itself. That is, CD22 appears to bind in cis to other CD22 molecules through interaction of its lectin domain with CD22-associated glycans. Glycoproteins that bound soluble CD22 fusion constructs in vitro, for example CD45 and CD19, were not cross-linked to CD22 in vivo, emphasizing the potential specificity of this metabolism-based approach. In addition, the cross-linked products had apparent molecular weights > 400 kDa, suggesting that CD22 forms multimers on the cell surface. This last finding also reflects the unique organization of CD22 in vivo. CD22’s cytosolic domain has a motif that is recognized by a component of clathrin-coated pits and therefore may reinforce CD22 self-association and microdomain formation on resting B cells.
Paulson and co-workers sought to apply this same photocross-linking strategy to identify trans ligands of CD22 on opposing target cells (Ramya et al., 2010). During B cell engagement, glycoproteins on apposing cells unmask CD22, changing its organization on the cell surface. Subsequent localization to cell-cell synapses enriched in BCR downregulate immune signaling. The authors generated a list of potential glycoprotein candidates that interact during B cell-B cell contact by first incubating K20 cells, which are deficient in endogenous sialic acid production, with 9-AAz-NeuAc. Intact cells were then treated with a soluble CD22-Fc chimera, a surrogate for B cells, and exposed to UV light. After lysis, cross-linked species were characterized via various proteomics methods, revealing 27 potential trans ligands of CD22, of which IgM (a component of the BCR), CD45, and Basigin were validated in cell-cell binding assays. IgM was the most robustly cross-linked, and thus subjected to further studies. CD22 and IgM were found to redistribute and colocalize preferentially to the sites of B cell-B cell contact in a lectin- and glycan-dependent manner.
There are limitations associated with functionalizing C-9 of sialic acid. The hydroxyl group at this position can undergo further modifications, such as 9-O-acetylation, 9-O-lactylation and 9-phosphorylation (Yu and Chen, 2007), which precludes 9-AAz-NeuAc’s use in situations where elaborated versions of sialic acid are under investigation. Kohler and coworkers circumvented this problem by replacing the N-acetyl group of ManNAc with an N-acyl diazirine photolabile functionality (Tanaka and Kohler, 2008). Diazirines are known to generate carbenes after UV illumination and can crosslink binding partners that are in close association (MacKinnon and Taunton, 2009). Another liability with 9-AAz-NeuAc is its low incorporation levels in normal mammalian cell lines, while the smaller structural perturbation associated with the diazirine group enhances competition with endogenous sialic acids, leading to greater levels of metabolic incorporation. Both K20 cells and wildtype K88 cells fed the fully protected diazirine-ManNAc analogue, Ac4ManNDAz, incorporated SiaDAz into cell surfaces glycans. Once there, SiaDAz crosslinked CD22 upon UV irradiation. This result provided further evidence that CD22 forms cis multimers on resting B cells. Kohler and coworkers went on to show that the chain length of N-acyl diazirines was crucial for efficient metabolic incorporation (Bond et al., 2011). ManNDAz derivatives containing two (2me), three (3me), or four (4me) methylene groups in the N-acyl chain were synthesized and evaluated (Figure 2). In Daudi B cells, only Ac4ManNDAz (2me) was found to crosslink CD22, which defines the maximal structural perturbation permissible by the metabolic machinery.
Figure 2.

Structures of metabolic crosslinking sugars that generate nitrenes or carbenes (red) upon UV exposure. These intermediates then react with lectins in vivo to form stable covalent adducts.
In less than a decade, metabolic crosslinking sugars have unearthed surprising features of CD22 on B cells. Many glycoproteins are modified with an N-acetylneuraminic acid α(2,6) galactose structure (Neu5Ac-α(2,6)-Gal), but among the plethora of possible binding partners of the lectin CD22, only CD22 and IgM appear to govern cis and trans interactions in vivo, respectively. Kohler and coworkers have recently expanded the library of crosslinking sugars to other monosaccharide structures (Yu et al., 2012), and we anticipate that many of these derivatives will be used to detect additional lectin-glycan interactions in the future.
Moving forward, we envision research towards inverting the current placement of the crosslinking functional group. If the crosslinking moiety is placed within the lectin, then this may provide a means to efficiently capture in vivo glycoprotein ligands, particularly in cases where the carbohydrate specificity of a lectin is not well understood. Incorporation of photoreactive benzophenones into proteins has already been accomplished in mammalian cells through unnatural amino acid mutagenesis (Hino et al., 2005). This technology could be expanded to the recognition domain of most lectins in vivo. Mass spectrometry (MS) will certainly play a major role in sequencing the captured ligands post-crosslinking. MS/MS has become in indispensible tool and in many cases routine for glycoproteomics (Mariño et al., 2010; Pan et al., 2011; Kailemia et al., 2014). Yet, for years, MS-based techniques to profile protein glycosylation have relied on liberating the glycan moiety from the protein or peptide backbone, which would obscure many of the fine details of lectin ligands. Fortunately, new unbiased MS methods have emerged that profile intact glycopeptides (Yin et al., 2013; Wu et al., 2014; Woo et al., 2015). We imagine these approaches and new developments in MS will eventually provide unprecedented information on the precise glycoprotein structures involved in lectin adhesion in vivo.
Glycoprotein Mimics
Many glycoproteins, especially mucin-type O-glycoproteins, are heavily elaborated biomolecules with multiple glycosylation sites. The glycan structures on a single protein backbone can vary dramatically from site to site, making analysis of lectin-glycoprotein interactions difficult. The protein backbone is known to modulate glycan presentation, sometimes restricting glycan conformation and subcellular localization. Further still, many possess weak affinity for their putative monomeric glycan ligands (Kd’s of 100 μM to 1 mM). Multivalency, the presence of numerous glycan ligands on a single scaffold, can dramatically increase the avidity of lectins for their oligomeric targets.
As a post-translational modification, glycosylation is not amenable to direct genetic manipulations, and as such, encoding a specific glycan structure along a single polypeptide backbone with genetic engineering is not possible. Chemists have harnessed biomimicry in order to construct molecular surrogates that emulate the essential features of glycoconjugates but differ in tunable composition and properties (Poupon and Nay, 2011). Three general classes of well-defined glycoprotein mimics have been synthesized and used to study mammalian lectins both in vitro and in vivo: glycopolymers (Ladmiral et al., 2004), linear polymers with pendant glycans; glycodendrimers (Turnbull and Stoddart, 2002), highly branched architectures with terminal glycan residues; and glycoparticles (de la Fuente and Penadés, 2006), organic or inorganic micro/nanoscale materials, or liposomal compositions that are functionalized with glycans through various surface chemistries. As opposed to the native heterogeneous glycoforms of proteins, these synthetic glycoconjugates offer the ability to incorporate homogeneous glycans structures onto a spatially defined platform. This section will focus on glycopolymers as mimics of glycoproteins, since these materials have yielded profound insights into lectin biology.
The original synthesis of glycopolymers dates back to the 1970’s (Schnaar and Lee, 1975). Since then, glycopolymers have found widespread use as lectin inhibitors, for instance during bacterial and viral infection (Sigal et al., 1996), and as tools for describing the biophysics of multivalency in vitro (Becer, 2012; Deniaud et al., 2011; Spain et al., 2007). In the past decade, chemical biologists have continued to push glycopolymers into the biological milieu (Kiessling and Grim, 2013). Free glycopolymers resemble soluble, secreted glycoproteins, and owing to improved synthetic methodology, it is now possible to prepare glycopolymers that mimic membrane-bound glycoproteins. One caveat to the vast majority of these materials is that they eliminate an important binding surface for the lectin, i.e. the protein itself. Still, with these tools in hand, a number of questions relating to lectin dynamics are now accessible and have been addressed for three large families of lectins, namely C-type lectins, siglecs, and galectins.
Selectins, calcium-dependent lectins (C-type lectins), are critical mediators of cell adhesion, especially during immune cell homing (Rosen, 2004). The three selectins, P, L, and E-, are expressed on different cell types, platelets, leukocytes, and endothelial cells, respectively (P-selectin is also expressed on activated endothelium). During the inflammatory response, leukocytes roll along endothelial cells, adhere to the vascular substrate and extravasate into the damaged tissue. It is the early rolling stage that involves selectin-ligand interactions. Likewise, the upregulation of L-selectin on leukocytes can lead to enhanced leukocyte migration and inflammatory damage, and therefore L-selectin density is tightly regulated on leukocyte surfaces. The glycan ligands of L-selectin possess sulfated versions of the sialyl Lewis x epitope (sLex, Neu5Acα2-3Galβ1-4(Fucα1-3)GlcNAc).
Kiessling and coworkers were interested in how the differential sulfation of this epitope affected L-selectin binding and leukocyte rolling (Gordon et al., 1998a, 1998b; Mowery et al., 2004; Sanders et al., 1999). They employed a type of living polymerization, ring opening metathesis polymerization (ROMP), to produce well-defined glycopolymers with pendant sLex derivatives. Starting from functionalized norbornenes, different glycopolymers bearing unsulfated, mono- or disulfated sLex were prepared. Once their relative binding was ascertained, the glycopolymers were evaluated for their abilities to induce shedding of cell-surface L-selectin, a phenomenon thought to naturally regulate its inflammatory contribution. A 3′,6-disulfated Lewis x glycopolymer caused the most significant decrease in surface L-selectin levels after binding, whereas monovalent analogs were inactive. The data provided evidence that clustering of L-selectin by the multivalent ligand induced proteolysis from the cell surface (Figure 3). Importantly, these findings suggest a mode of therapeutic intervention wherein L-selectin shedding is induced, and inflammatory leukocyte recruitment muted, by synthetic glycopolymers.
Figure 3.
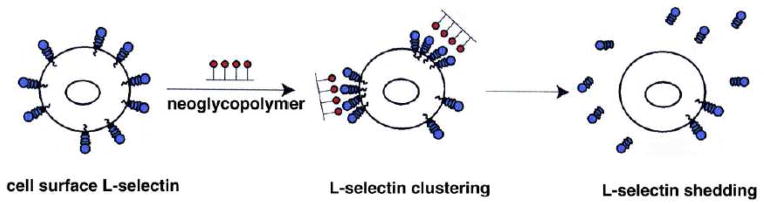
A 3′,6-disulfated Lewis x glycopolymer promotes L-selectin shedding from leukocytes by forming membrane clusters. From [56]. Reprinted with permission from Elsevier.
To address how spacing of these glycan epitopes along a scaffold might modulate the consequences of L-selectin clustering, Kiick and coworkers prepared artificial glycoproteins based on alanine-rich recombinant polypeptides (Liu and Kiick, 2011). Glycan conjugation to glutamic acid residues at defined positions yielded spatially-defined, α-helical glycoprotein mimics. Specifically, disulfated galactosyl amines were coupled to the polypeptide backbone with HBTU and arranged at two different distances, 1.7–3.5 nm and 3.5–5.0 nm. Jurkat T cells were incubated with these glycopolypeptides, and the amount of L-selectin shedding was determined by an enzyme-linked immunosorbent assay (ELISA) for the protein. As anticipated, monovalent glycans and an unstructured glycopolypeptide did not induce L-selectin shedding. However, while the 1.7–3.5 nm construct resulted in L-selectin cleavage, the 3.5–5.0 nm glycopolypeptide, unexpectedly, did not. This differential shedding activity highlights that the dynamics of selectin organization on cell surfaces has functional consequences. Clustering is thought to induce calmodulin (CaM) release from the cytoplasmic tail of L-selectin, which is then followed by a conformational change that enhances susceptibility to sheddase activity (Grailer et al., 2009). This work suggests another parameter that influences signaling and shedding of L-selectin: the spatial arrangement of L-selectin binding epitopes.
Another C-type lectin, Dendritic cell-specific ICAM-3-grabbing nonintegrin (DC-SIGN), carries out a multitude of functions on DCs through interactions with high mannose glycans and fucosylated Lewis-type antigens on ICAM-3 and ICAM-2 (Švajger et al., 2010). DC-SIGN stabilizes immunological synapses during T-cell engagement and can mediate trans-endothelial migration. The lectin was also thought to be involved in antigen uptake and processing, a proposal confirmed by Kiessling and coworkers using synthetic glycoconjugates (Prost et al., 2012). Bovine serum albumin (BSA), serving as a polymeric backbone, was decorated at its lysine residues with mannosyl or fucosyl glycans. Fluorophore-functionalized BSA glycoconjugates were administered to Raji cells, a B cell line, stably transfected with the gene for DC-SIGN (Raji/DC-SIGN). The Raji/DC-SIGN cells, but not the wildtype Raji cell line, internalized all three glycoconjugates. The glycoprotein surrogates also stimulated JNK signaling in response to interaction with DC-SIGN. Phosphorylation of JNK was not seen in other studies of DC-SIGN binding, leading to the proposal that DC-SIGN reorganization and perhaps clustering is necessary for signaling to take place. C-type lectin-dependent uptake may be a general feature in antigen processing, as polyacrylamide (PAA)-GalNAc glycopolymers were found by Irimura and coworkers to be readily internalized by DCs through Macrophage Galactose-type C-type Lectin 2 (MGL2) (Denda-Nagai et al., 2010).
As discussed above, CD22 belongs to the siglec family of lectins. An important siglec on B cells, CD22 downregulates immune cell activation via recruitment of SHP-1 phosphatase to the site of receptor signaling. Cis α2,6-linked sialic acid-CD22 interactions were shown to mask CD22 on resting B cells. But one question remained: what are the properties of trans ligands that promote escape of CD22 from cis ligand-dependent multimers? Paulson and coworkers examined this question using a series of sialoside glycopolymers (Collins et al., 2006). Biotinylated PAA polymers of either 30 or 1000 kDa were used as scaffolds for the attachment of sialosides, including NeuAc and its C-9-biphenyl derivative (BPC-NeuAc) as well as the native N-glycolylneuraminic acid (NeuGc) and its C-9-biphenyl derivative (BPA-NeuGc). Aromatic groups at the C-9 position of sialic acid had been previously shown to increase affinity toward CD22 (Zaccai et al., 2003). The prepared glycopolymers were added to either BJAB or Daudi B cells, and flow cytometry was used to detect glycopolymer binding. Only the 1000 kDa BPC-NeuAc-PAA and BPA-NeuGc-PAA polymers adhered to the B cell surface. The native sialoside polymers, NeuAc-PAA and NeuGc-PAA, as well as the 30 kDa glycopolymers did not exhibit cell surface binding. A simple interpretation of these findings is that CD22 exists in a dynamic equilibrium on the surface of B cells. If presented with exogenous ligands of sufficient avidity, the CD22 binding equilibrium favors unmasking from cis ligands, whereas weak exogenous ligands cannot compete with self-association in cell-surface multimers (Figure 3). Only in the case when CD22 is presented with highly multivalent glycopolymers, 1000 kDa, containing high affinity glycan derivatives, i.e. BPC-NeuAc and BPA-NeuGc, does unmasking occur. The authors also provide evidence that the CD22-bound PAA glycopolymers undergo clathrin-mediated endocytosis into early endosomes that is dynamin dependent.(Tateno et al., 2007)
In an elegant follow-up report, Kiessling and coworkers employed glycopolymers to study the consequences of engaging both CD22 and BCR on B cell activation (Courtney et al., 2009). A well-defined, ROMP-derived N-hydroxysuccinimidyl (NHS) ester polymer was elaborated either with BCR ligands (2,4-dinitrophenyl, DNP), CD22 ligands (CD22L) or both (DNP/CD22L). Polymers with BCR and CD22 ligands on the same polymer backbone induced clustering of the two proteins (Figure 5). As well, cells treated with DNP polymer displayed an increase in cytosolic calcium concentration. The DNP/CD22L copolymer, on the other hand, had no such effect. No calcium influx was observed for the copolymer, and downstream BCR signaling targets, such as Syk and PLC γ2, had reduced phosphorylation compared with DNP polymer-treated cells. Interestingly, incubation with the DNP polymer or the DNP/CD22L copolymer produced similar early signaling patterns in B cells; both Lyn and CD22 became phosphorylated upon polymer engagement. The authors proposed that after BCR attachment, the increase in local concentration of polymer on the membrane facilitates CD22 trans association. Collectively, the data also indicate that co-clustering of BCR and CD22 on the cell surface elicits early B cell activation but attenuates downstream signaling effectors. The copolymer data strongly suggest that CD22 dynamics can be modulated by the sialylation state of an antigen, which could be an innate form of self-recognition, an insight difficult to obtain without the aid of homogeneous glycoprotein mimics.
Figure 5.

A soluble copolymer engages both BCR and CD22 simultaneously. Glycopolymer binding reorganizes CD22 to sites of BCR activation and blocks downstream signaling. The CD22 ligands are composed of sialic acid residues, suggesting that sialylation may be a molecular form of ‘self’ in mammals.
To further explore the influence of B cell siglecs on immune tolerance, polyacrylamide (PA) copolymers were synthesized bearing both a BCR ligand (NP) and either a native CD22 ligand (N-glycolyl neuraminic acid (NeuGc)) or a high-affinity unnatural CD22 ligand (9-biphenyl NeuGc, bNeuGc) (Duong et al., 2010). Mice immunized with NP-PA produced a strong antibody response as expected. In contrast, mice immunized either with the NP-PA-NeuGc copolymer or the NP-PA-bNeuGc copolymer failed to elicit high anti-NP antibody titers. These results agree with the above findings that B cell signaling is attenuated by co-engagement of CD22 and BCR. To test whether glycopolymer antigen challenge shaped the organism’s subsequent immune response, mice that had been treated with sialylated antigen were then immunized with unsialylated NP-PA. Drastically reduced antibody titers were observed, a hallmark of antibody suppression and immune tolerance. Tolerance induction was associated with suboptimal B cell proliferation and arrest of plasma B cell differentiation. Additionally, long-term tolerance was correlated with the strength of siglec binding, as NP-PA-bNeuGc copolymers produced the highest immune tolerance. CD22 can thus confer a graded output when presented with different glycosylated antigens. This work points to an underexplored area of immunotherapy, namely targeting siglecs, and the potential of harnessing single glycoconjugates simultaneously presenting BCR and inhibitory siglec ligands as immunoregulators.
Yet, what mechanisms are responsible for producing a spectrum of B cell activation? Kiessling and coworkers explored how the dynamics of lectin trafficking relate to B cell signaling (Courtney et al., 2014). B cells were incubated with DNP polymer or DNP/CD22L copolymers, and BCR endocytosis was tracked by fluorescence microscopy. The cell surface half-life of BCR was significantly longer for cells treated with DNP polymer compared to cells treated with DNP/CD22L copolymer. Co-clustering of BCR and CD22 on B cell surfaces triggered endocytosis, which resulted in accumulation of BCR in early endosomes. Glycosylation-dependent internalization was mediated by dynamin and initiated by early phosphorylation of BCR and CD22 through Lyn. This mechanism of BCR regulation provides a means for B cells to tailor their BCR levels based on the sialylation state of an antigen and, in turn, increase the threshold of B cell activation to subsequent antigen encounter.
Immune cells other than B cells also possess unique repertoires of siglecs. One example is natural killer (NK) cells that act as early responders to pathogens and cancerous cells, leading to cell death and clearance (Kumar and McNerney, 2005). Human NK cells prominently express Siglec-7 and to a lesser extant Siglec-9 (Crocker et al., 2007). Early work by Nicoll et al. used soluble glycopolymers to establish that, similar to CD22, Siglec-7 is indeed masked on NK cells by the disialylated ganglioside GD3 (Nicoll et al., 2003). The similar domain organization of Siglecs-7 and -9 to CD22 suggests roles in downregulating NK cell activation, but the precise roles of these siglecs in NK cell biology was unclear. As an integral part of the innate immune response, NK cells act to contain the growth and metastasis of tumor cells. Aggressive cancers though are able to evade NK cells through the release of activating receptors or expression of inhibitory motifs. Interestingly, there is a body of literature associating hypersialylation of cells with malignancy, prompting speculation that this glycomic signature might protect cancers cells from immune surveillance by downregulating NK cells.
To test this hypothesis, our lab designed sialylated glycopolymers functionalized with a lipid at one terminus, enabling insertion into live cell membranes (Rabuka et al., 2008). Hudak et al. employed these reagents to modify cancer cells and thereby test the effects of engineered hypersialylation on susceptibility to NK cell killing (Hudak et al., 2014). Jurkat cells, a hyposialylated cell line that are known to be susceptible to NK cell killing, were decorated with polymers and then incubated with either purified NK cells or peripheral blood mononuclear cells (PBMC). NK cytotoxicity of Jurkat cells was strongly inhibited by sialic acid-containing glycopolymers but not polymers lacking the terminal sugar or polymers possessing similar charge, e.g. acetic acid, or polarity, e.g. glycerol. Function-blocking antibodies against Siglec-7, but not Siglec-9, abolished the protection afforded by sialic acid polymers. Fluorescence microscopy revealed that Siglec-7 on NK cells had reorganized to concentrate at the synapse with sialopolymer-coated Jurkat cells. A role for immune cell siglecs in sialic acid-dependent cancer immune surveillance was later confirmed in mouse models (Läubli et al., 2014) and implicated from human clinical samples (Jandus et al., 2014).
Galectins, another family of carbohydrate binding proteins, differ considerably in structure from selectins and siglecs (Di Lella et al., 2011). These proteins are soluble, and whereas siglecs and selectins bind sialylated structures, galectins recognize galactose- and N-acetyllactosamine-based motifs. Moreover, many galectins oligomerize noncovalently in the presence of ligand, whereas others possess two carbohydrate recognition domains (CRDs) in a single polypeptide, illustrating two distinct mechanisms for achieving multivalency. Stemming from their oligomeric nature, it was proposed that galectins form a lattice-like structure with glycoprotein ligands on cell surfaces (Rabinovich et al., 2007). Dennis and coworkers found that limiting the number of LacNAc residues on cell surface glycoproteins altered the internalization rates of a number of growth factor receptors, indicating that galectin lattices may extend the cell surface residence time of certain glycoproteins (Partridge et al., 2004).
Unfortunately, direct experimental validation of galectin-glycoprotein lattices had eluded researchers. Without a means of detection, physical parameters, such as size and lifetime, were largely undefined for galectin-mediated aggregation. Our lab leveraged membrane-anchored glycopolymers and fluorescence spectroscopy to verify the existence of galectin lattices (Belardi et al., 2012). Reversible addition-fragmentation chain transfer (RAFT) polymerization, another type of living polymerization, offers the opportunity to individually manipulate both ends of a polymer chain while also affording predictable degree of polymerization.(Keddie, 2014; Lowe and McCormick, 2007) Lactosyl and cellobiosyl (negative control) acrylamide monomers were therefore subjected to RAFT polymerization conditions in the presence of a lipid-functionalized chain transfer agent. Subsequent end group modification with a fluorophore yielded glycopolymers containing either a single FRET donor or acceptor dye. After embedding donor and acceptor glycopolymers (1:1) into live cell membranes, a decrease in fluorescence lifetime, which was indicative of FRET, of the donor-functionalized lactosyl polymer was observed in the presence of galectin-1. Moreover, the diffusion time, a parameter measured by Fluorescence Correlation Spectroscopy and related to clustering, for the lactosyl glycopolymer increased only in the presence of galectin-1. By examining the data in real time, it appears that galectin-1 mediates the dynamic cross-linking of membrane glycoprotein mimics on the minutes timescale and results in extended galectin networks, which are themselves stable for tens of minutes.
Now considered invaluable tools in the chemical biologist’s toolkit, glycoprotein mimics have been utilized in vivo to understand the flow of information from a glycosylated biomolecule to a lectin and the resulting cellular decision-making that is altered by carbohydrate binding. What has emerged is a picture of lectins whose functions are highly dependent on changes in organization that occur upon ligand binding. Following glycoprotein mimic association, lectins redistribute, form signaling clusters, and process signals, all of which are regulated by the nature of the glycan partners themselves. Glycoprotein mimics have been instrumental in defining the parameters that affect lectin-mediated signaling and membrane organization both in cell culture and in organismal studies. Present mimics, however, lack the signaling domains associated with many glycoprotein ligands. In the future, it would be interesting to combine mucin-type polymeric structures with functional domains through semi-synthesis. Using this approach, questions regarding the feedback between lectin dynamics and glycoprotein function can start to be assessed.
Site-selective Tagging and Imaging Technologies
Some problems in lectin biology require direct observation of the lectins themselves, with fast temporal and high spatial resolution. Chemical tools, both reagents and instrumentation, are becoming ever more important for meeting these challenges. For example, biologists have long debated how DC-SIGN efficiently recognizes glycan epitopes and captures large viral particles and pathogens. Given the dimensions of viral particles (80–100 nm), researchers were interested in whether DC-SIGN formed small clusters on the surface of immature dendritic cells. To examine DC-SIGN’s organization below optical resolution (~ 200 nm), Blink Microscopy, a super-resolution method, was creatively applied by Jacobson and coworkers (Itano et al., 2012). Blink Microscopy relies on driving the majority of fluorophores in a densely-labeled sample into a dark state through a combination of redox reagents, leaving a few dyes fluorescing for short on times (~ms). DC-SIGN was imaged and found to reside in nanodomains with a diameter of 76 nm. By analyzing the number of blink localizations per cluster, the occupancy was calculated to be ~12 molecules, or three tetramers of DC-SIGN, indicating that the cluster was sparsely occupied. This finding was consistent with the ability of lipid molecules to easily diffuse through DC-SIGN membrane domains. The multivalency associated with DC-SIGN and its limited density in clusters suggests an effective, yet flexible mechanism to ensure entry of weakly-binding, large antigens, e.g. HIV.
For years, another important question in lectin biology, namely how leukocyte attachment mediated by L-selection interactions is enhanced under flow, was particularly vexing. This finding was partly rationalized by changes in cell surface area or number of adhesions under shear force. But to examine L-selection dynamics under force in detail, Yago et al. immobilized the L-selectin ligand, P-selectin glycoprotein ligand-1 (PSGL-1), on a substrate and measured biophysical parameters that govern adhesion under flow with fast microscopy detectors (250–500 frames per second) (Yago et al., 2004). The authors monitored off-rates (koff), among other parameters, for the L-selectin-PSGL-1 interaction in neutrophils under various tether forces. They observed decreases in koff as shear force was increased. This behavior is known as a catch bond, where bond lifetime increases under force. In this case, instrumentation and chemical modification were pivotal for defining the catch-bond behavior of L-selectin. This work also supports the notion that lectin dynamics, e.g. force-induced conformation changes, are important for biological phenomena, like leukocyte rolling and extravasation.
Galectin-3 (Gal-3) is the only member of the galectin family that belongs to the chimera subtype. This protein has been reported to oligomerize in vitro upon binding glycan ligands, forming multimers in solution (Ahmad et al., 2004; Lepur et al., 2012). Nieminen et al. were interested in observing Gal-3 oligomerization using FRET in the context of neutrophil-endothelial cell interfaces (Nieminen et al., 2006). Yet, large fluorescent protein fusions (~25 kDa) were problematic for Gal-3 since both the N- and C-terminus were considered indispensible for assembly and glycan binding, respectively. To address this, a small donor or acceptor fluorophore, Alexa Fluor 488- or Alexa Fluor 555-cadaverine, was conjugated to the C-terminus of Gal-3 in a site-specific manner by the enzyme, transglutaminase. Taking advantage of Gal-3 outfitted with either a donor or acceptor dye, robust FRET signals were observed in unprimed neutrophils alone as well as in the context of neutrophil-endothelial cell adhesions. On the other hand, Gal-3 monomers modified non-specifically at lysine residues showed limited FRET. Together, these results suggest that Gal-3 is oligomerized within the glycocalyx on cell surfaces. The study also emphasizes the unmet need and demand for developing methods capable of site-selectively modifying lectins without perturbing activity or quaternary lectin structure. For instance, in recent work, Gal-3 has been expressed with a SNAP-tag that can be modified by a myriad of benzylguanine derivatives, opening the door for a range of markers useful for observing lectin dynamics/organization (Kupper et al., 2013).
Finally, fluorescent amino acids are beneficial since they are small in size and can be placed at many points throughout the protein backbone. Fluorescence is extremely sensitive to the local environment and has been used extensively to detect binding events in vivo. Based on these advantages, Hohsaka and coworkers recently pioneered the incorporation of fluorescent amino acids into lectins (Iijima and Hohsaka, 2009; Ito and Hohsaka, 2013). Unnatural BODIPY fluorescent amino acids were efficiently installed in maltose-binding protein as well as in a sialic acid-binding protein (Figure 6). In both examples, placement of the fluorophore near the glycan-binding site created a fluorogenic lectin, i.e. fluorescence of the amino acid is quenched by a nearby tryptophan residue in the absence of ligand, but when maltose or sialyllactose was administered fluorescence turns “on.” These examples are encouraging since they suggest that fluorescent amino acids may be well suited to visualize lectin binding events in vivo, obfuscating addition of large protein fusions or peptide tags to a lectin’s sequence. They also illustrate a path to differentiate between lectins that are engaged with their glycoprotein ligands and the free protein.
Figure 6.

Fluorescent amino acids that have been incorporated into maltose-binding protein and a sialic acid-binding protein.
Chemical tools have revolutionized the way molecules are imaged and tracked inside complex biological milieu. Many of these techniques are just now being applied to lectins. Lectins assemble, disassemble, form cross-linked ‘bridges’ between glycoproteins and adjacent cells, and generally act in complex, multimeric forms. Non-traditional methods are needed to observe the debated details of these processes in vivo. Recently, new hardware and chemical reagents have revealed, for instance, the nature of lectin binding under force and the spatial organization of lectin domains in membranes. We believe that by leveraging new methods in protein modification and better imaging hardware/techniques our present understanding of lectin dynamics and binding behavior will give way to a more refined description. As well, we hope that in the future researchers will take advantage of single molecule techniques, such as single particle tracking, coupled with site-specific fluorophore-lectin conjugates to resolve lectin dynamics in different subcellular compartments, an unexplored frontier.
Conclusion
Chemists have transformed the field of glycobiology by contributing a number of methods to interrogate mammalian lectin dynamics in vivo. These tools have come in a variety of molecular forms, ranging from small molecule metabolites to large macromolecular agents. Cross-linking sugars have revealed extraordinary features of CD22 that were wholly inaccessible using typical molecular biology techniques. The fact that CD22 is segregated into homomultimers in resting B cells has shed light on the in vivo consequences of lectin reorganization for tuning immune cell activation. Glycoprotein mimics have, for instance, clarified a novel glycan-based mechanism of NK cytotoxicity evasion by cancer cells. Escaping immunosurveillance was, not surprisingly, predicated on glycan-siglec interactions. And new imaging methods, such as super-resolution microscopy, have settled long-standing questions concerning DC-SIGN membrane domains. By precisely locating individual DC-SIGN molecules, DC-SIGN clusters were found to be composed of only 12 molecules, which explains their ability to efficiently mobilize and capture viral particles. However, in vivo specificity is still undefined for the vast majority of lectins and only recently have researchers begun to appreciate that assembly and subcellular dynamics of lectins profoundly dictate function and cell behavior (Croci et al., 2014). We are in desperate need of techniques to probe the glycoprotein ligands of lectins in full detail, i.e. sequencing the intact glycoconjugate, and methods that define the assembly state and trafficking history of lectins are severely limited. We believe that chemical tools will be indispensible for elucidating how lectins interpret glycomic changes in the future. Our hope is that this review will stimulate further interest and work in this new, exciting area at the interface of chemistry and biology.
Figure 4.
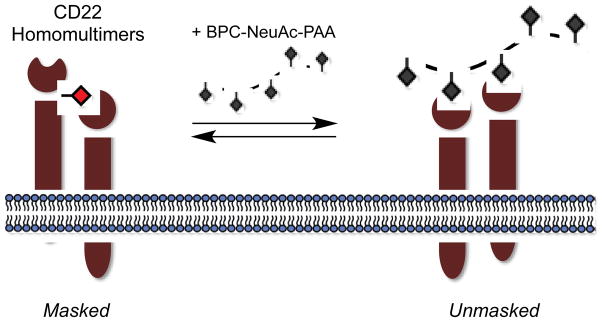
The siglec, CD22, exists in homomultimers on resting B cells but is unmasked in the presence of a multivalent glycopolymer, BPC-NeuAc-PAA, of sufficient avidity.
Acknowledgments
We would like to thank Elliot Woods for critical reading of the manuscript. This work was funded by a grant from the NIH to C.R.B. (GM59907). B.B. was supported by an NSF predoctoral fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmad N, Gabius HJ, André S, Kaltner H, Sabesan S, Roy R, Liu B, Macaluso F, Brewer CF. Galectin-3 Precipitates as a Pentamer with Synthetic Multivalent Carbohydrates and Forms Heterogeneous Cross-linked Complexes. J Biol Chem. 2004;279:10841–10847. doi: 10.1074/jbc.M312834200. [DOI] [PubMed] [Google Scholar]
- Beahm BJ, Dehnert KW, Derr NL, Kuhn J, Eberhart JK, Spillmann D, Amacher SL, Bertozzi CR. A visualizable chain-terminating inhibitor of glycosaminoglycan biosynthesis in developing zebrafish. Angew Chem Int Ed Engl. 2014;53:3347–3352. doi: 10.1002/anie.201310569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becer CR. The Glycopolymer Code: Synthesis of Glycopolymers and Multivalent Carbohydrate–Lectin Interactions. Macromol Rapid Commun. 2012;33:742–752. doi: 10.1002/marc.201200055. [DOI] [PubMed] [Google Scholar]
- Belardi B, O’Donoghue GP, Smith AW, Groves JT, Bertozzi CR. Investigating Cell Surface Galectin-Mediated Cross-Linking on Glycoengineered Cells. J Am Chem Soc. 2012;134:9549–9552. doi: 10.1021/ja301694s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belardi B, de la Zerda A, Spiciarich DR, Maund SL, Peehl DM, Bertozzi CR. Imaging the glycosylation state of cell surface glycoproteins by two-photon fluorescence lifetime imaging microscopy. Angew Chem Int Ed. 2013;52:14045–14049. doi: 10.1002/anie.201307512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond MR, Zhang H, Kim J, Yu SH, Yang F, Patrie SM, Kohler JJ. Metabolism of diazirine-modified N-acetylmannosamine analogues to photo-cross-linking sialosides. Bioconjug Chem. 2011;22:1811–1823. doi: 10.1021/bc2002117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer CF, Miceli MC, Baum LG. Clusters, bundles, arrays and lattices: novel mechanisms for lectin-saccharide-mediated cellular interactions. Curr Opin Struct Biol. 2002;12:616–623. doi: 10.1016/s0959-440x(02)00364-0. [DOI] [PubMed] [Google Scholar]
- Bussink AP, van Swieten PF, Ghauharali K, Scheij S, van Eijk M, Wennekes T, van der Marel GA, Boot RG, Aerts JMFG, Overkleeft HS. N-azidoacetylmannosamine-mediated chemical tagging of gangliosides. J Lipid Res. 2007;48:1417–1421. doi: 10.1194/jlr.C700006-JLR200. [DOI] [PubMed] [Google Scholar]
- Collins BE, Blixt O, Han S, Duong B, Li H, Nathan JK, Bovin N, Paulson JC. High-affinity ligand probes of CD22 overcome the threshold set by cis ligands to allow for binding, endocytosis, and killing of B cells. J Immunol. 2006;177:2994–3003. doi: 10.4049/jimmunol.177.5.2994. [DOI] [PubMed] [Google Scholar]
- Courtney AH, Puffer EB, Pontrello JK, Yang ZQ, Kiessling LL. Sialylated multivalent antigens engage CD22 in trans and inhibit B cell activation. Proc Natl Acad Sci. 2009;106:2500. doi: 10.1073/pnas.0807207106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtney AH, Bennett NR, Zwick DB, Hudon J, Kiessling LL. Synthetic Antigens Reveal Dynamics of BCR Endocytosis during Inhibitory Signaling. ACS Chem Biol. 2014;9:202–210. doi: 10.1021/cb400532y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croci DO, Cerliani JP, Dalotto-Moreno T, Méndez-Huergo SP, Mascanfroni ID, Dergan-Dylon S, Toscano MA, Caramelo JJ, García-Vallejo JJ, Ouyang J, Mesri EA, Junttila MR, Bais C, Shipp MA, Salatino M, Rabinovich GA. Glycosylation-dependent lectin-receptor interactions preserve angiogenesis in anti-VEGF refractory tumors. Cell. 2014;156:744–758. doi: 10.1016/j.cell.2014.01.043. [DOI] [PubMed] [Google Scholar]
- Crocker PR, Paulson JC, Varki A. Siglecs and their roles in the immune system. Nat Rev Immunol. 2007;7:255–266. doi: 10.1038/nri2056. [DOI] [PubMed] [Google Scholar]
- Dehnert KW, Beahm BJ, Huynh TT, Baskin JM, Laughlin ST, Wang W, Wu P, Amacher SL, Bertozzi CR. Metabolic labeling of fucosylated glycans in developing zebrafish. ACS Chem Biol. 2011;6:547–552. doi: 10.1021/cb100284d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denda-Nagai K, Aida S, Saba K, Suzuki K, Moriyama S, Oo-Puthinan S, Tsuiji M, Morikawa A, Kumamoto Y, Sugiura D, et al. Distribution and function of macrophage galactose-type C-type lectin 2 (MGL2/CD301b): efficient uptake and presentation of glycosylated antigens by dendritic cells. J Biol Chem. 2010;285:19193–19204. doi: 10.1074/jbc.M110.113613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deniaud D, Julienne K, Gouin SG. Insights in the rational design of synthetic multivalent glycoconjugates as lectin ligands. Org Biomol Chem. 2011;9:966–979. doi: 10.1039/c0ob00389a. [DOI] [PubMed] [Google Scholar]
- Duong BH, Tian H, Ota T, Completo G, Han S, Vela JL, Ota M, Kubitz M, Bovin N, Paulson JC, et al. Decoration of T-independent antigen with ligands for CD22 and Siglec-G can suppress immunity and induce B cell tolerance in vivo. J Exp Med. 2010;207:173–187. doi: 10.1084/jem.20091873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De la Fuente JM, Penadés S. Glyconanoparticles: types, synthesis and applications in glycoscience, biomedicine and material science. Biochim Biophys Acta. 2006;1760:636–651. doi: 10.1016/j.bbagen.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Gabius HJ, André S, Jiménez-Barbero J, Romero A, Solís D. From lectin structure to functional glycomics: principles of the sugar code. Trends Biochem Sci. 2011;36:298–313. doi: 10.1016/j.tibs.2011.01.005. [DOI] [PubMed] [Google Scholar]
- Geissner A, Anish C, Seeberger PH. Glycan arrays as tools for infectious disease research. Curr Opin Chem Biol. 2014;18:38–45. doi: 10.1016/j.cbpa.2013.11.013. [DOI] [PubMed] [Google Scholar]
- Gordon EJ, Sanders WJ, Kiessling LL. Synthetic ligands point to cell surface strategies. Nature. 1998a;392:30–31. doi: 10.1038/32073. [DOI] [PubMed] [Google Scholar]
- Gordon EJ, Strong LE, Kiessling LL. Glycoprotein-inspired materials promote the proteolytic release of cell surface l-Selectin. Bioorg Med Chem. 1998b;6:1293–1299. doi: 10.1016/s0968-0896(98)00122-9. [DOI] [PubMed] [Google Scholar]
- Grailer JJ, Kodera M, Steeber DA. L-selectin: Role in regulating homeostasis and cutaneous inflammation. Journal of Dermatological Science. 2009;56:141–147. doi: 10.1016/j.jdermsci.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haga Y, Ishii K, Hibino K, Sako Y, Ito Y, Taniguchi N, Suzuki T. Visualizing specific protein glycoforms by transmembrane fluorescence resonance energy transfer. Nat Commun. 2012;3:907. doi: 10.1038/ncomms1906. [DOI] [PubMed] [Google Scholar]
- Han S, Collins BE, Bengtson P, Paulson JC. Homomultimeric complexes of CD22 in B cells revealed by protein-glycan cross-linking. Nat Chem Biol. 2005;1:93–97. doi: 10.1038/nchembio713. [DOI] [PubMed] [Google Scholar]
- Hang HC, Yu C, Kato DL, Bertozzi CR. A metabolic labeling approach toward proteomic analysis of mucin-type O-linked glycosylation. Proc Natl Acad Sci USA. 2003;100:14846–14851. doi: 10.1073/pnas.2335201100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hino N, Okazaki Y, Kobayashi T, Hayashi A, Sakamoto K, Yokoyama S. Protein photo-cross-linking in mammalian cells by site-specific incorporation of a photoreactive amino acid. Nat Methods. 2005;2:201–206. doi: 10.1038/nmeth739. [DOI] [PubMed] [Google Scholar]
- Hsu TL, Hanson SR, Kishikawa K, Wang SK, Sawa M, Wong CH. Alkynyl sugar analogs for the labeling and visualization of glycoconjugates in cells. Proc Natl Acad Sci USA. 2007;104:2614–2619. doi: 10.1073/pnas.0611307104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudak JE, Canham SM, Bertozzi CR. Glycocalyx engineering reveals a Siglec-based mechanism for NK cell immunoevasion. Nat Chem Biol. 2014;10:69–75. doi: 10.1038/nchembio.1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima I, Hohsaka T. Position-specific incorporation of fluorescent non-natural amino acids into maltose-binding protein for detection of ligand binding by FRET and fluorescence quenching. ChemBioChem. 2009;10:999–1006. doi: 10.1002/cbic.200800703. [DOI] [PubMed] [Google Scholar]
- Itano MS, Steinhauer C, Schmied JJ, Forthmann C, Liu P, Neumann AK, Thompson NL, Tinnefeld P, Jacobson K. Super-Resolution Imaging of C-Type Lectin and Influenza Hemagglutinin Nanodomains on Plasma Membranes Using Blink Microscopy. Biophys J. 2012;102:1534–1542. doi: 10.1016/j.bpj.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito Y, Hohsaka T. Incorporation of Fluorescent Nonnatural Amino Acid into Sialic Acid-Binding Lectin for Fluorescence Detection of Ligand-Binding. Bull Chem Soc Jpn. 2013;86:729–735. [Google Scholar]
- Jandus C, Boligan KF, Chijioke O, Liu H, Dahlhaus M, Démoulins T, Schneider C, Wehrli M, Hunger RE, Baerlocher GM, et al. Interactions between Siglec-7/9 receptors and ligands influence NK cell–dependent tumor immunosurveillance. J Clin Invest. 2014;124:1810–1820. doi: 10.1172/JCI65899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kailemia MJ, Ruhaak LR, Lebrilla CB, Amster IJ. Oligosaccharide analysis by mass spectrometry: a review of recent developments. Anal Chem. 2014;86:196–212. doi: 10.1021/ac403969n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayser H, Zeitler R, Kannicht C, Grunow D, Nuck R, Reutter W. Biosynthesis of a nonphysiological sialic acid in different rat organs, using N-propanoyl-D-hexosamines as precursors. J Biol Chem. 1992a;267:16934–16938. [PubMed] [Google Scholar]
- Kayser H, Geilen CC, Paul C, Zeitler R, Reutter W. Incorporation of N-acyl-2-amino-2-deoxy-hexoses into glycosphingolipids of the pheochromocytoma cell line PC 12. FEBS Lett. 1992b;301:137–140. doi: 10.1016/0014-5793(92)81233-c. [DOI] [PubMed] [Google Scholar]
- Keddie DJ. A guide to the synthesis of block copolymers using reversible-addition fragmentation chain transfer (RAFT) polymerization. Chem Soc Rev. 2014;43:496. doi: 10.1039/c3cs60290g. [DOI] [PubMed] [Google Scholar]
- Keppler OT, Horstkorte R, Pawlita M, Schmidt C, Reutter W. Biochemical engineering of the N-acyl side chain of sialic acid: biological implications. Glycobiology. 2001;11:11R–18R. doi: 10.1093/glycob/11.2.11r. [DOI] [PubMed] [Google Scholar]
- Kiessling LL, Grim JC. Glycopolymer probes of signal transduction. Chem Soc Rev. 2013;42:4476–4491. doi: 10.1039/c3cs60097a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V, McNerney ME. A new self: MHC-class-I-independent Natural-killer-cell self-tolerance. Nat Rev Immunol. 2005;5:363–374. doi: 10.1038/nri1603. [DOI] [PubMed] [Google Scholar]
- Kupper CE, Böcker S, Liu H, Adamzyk C, van de Kamp J, Recker T, Lethaus B, Jahnen-Dechent W, Neuss S, Müller-Newen G, et al. Fluorescent SNAP-tag galectin fusion proteins as novel tools in glycobiology. Curr Pharm Des. 2013;19:5457–5467. doi: 10.2174/1381612811319300017. [DOI] [PubMed] [Google Scholar]
- Ladmiral V, Melia E, Haddleton DM. Synthetic glycopolymers: an overview. Eur Polym J. 2004;40:431–449. [Google Scholar]
- Läubli H, Pearce OMT, Schwarz F, Siddiqui SS, Deng L, Stanczak MA, Deng L, Verhagen A, Secrest P, Lusk C, et al. Engagement of myelomonocytic Siglecs by tumor-associated ligands modulates the innate immune response to cancer. Proc Natl Acad Sci USA. 2014;111:14211–14216. doi: 10.1073/pnas.1409580111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YC, Lee RT. Carbohydrate-Protein Interactions: Basis of Glycobiology. Acc Chem Res. 1995;28:321–327. [Google Scholar]
- Lepur A, Salomonsson E, Nilsson UJ, Leffler H. Ligand Induced Galectin-3 Protein Self-Association. J Biol Chem. 2012;287:21751–21756. doi: 10.1074/jbc.C112.358002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Lella S, Sundblad V, Cerliani JP, Guardia CMA, Estrin DA, Vasta GR, Rabinovich GA. When Galectins Recognize Glycans: From Biochemistry to Physiology and Back Again. Biochemistry. 2011;50:7842–7857. doi: 10.1021/bi201121m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Du Y, Zhu Y, Chen X. A Cis-Membrane FRET-Based Method for Protein-Specific Imaging of Cell-Surface Glycans. J Am Chem Soc. 2014;136:679–687. doi: 10.1021/ja410086d. [DOI] [PubMed] [Google Scholar]
- Liu S, Kiick K. Architecture effects on L-selectin shedding induced by polypeptide-based multivalent ligands. Polym Chem. 2011;2:1513–1522. doi: 10.1039/C1PY00063B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe AB, McCormick CL. Reversible addition–fragmentation chain transfer (RAFT) radical polymerization and the synthesis of water-soluble (co)polymers under homogeneous conditions in organic and aqueous media. Prog Polym Sci. 2007;32:283–351. [Google Scholar]
- MacKinnon AL, Taunton J. Target Identification by Diazirine Photo-Cross-linking and Click Chemistry. Curr Protoc Chem Biol. 2009;1:55–73. doi: 10.1002/9780470559277.ch090167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahal LK, Yarema KJ, Bertozzi CR. Engineering Chemical Reactivity on Cell Surfaces Through Oligosaccharide Biosynthesis. Science. 1997;276:1125–1128. doi: 10.1126/science.276.5315.1125. [DOI] [PubMed] [Google Scholar]
- Mariño K, Bones J, Kattla JJ, Rudd PM. A systematic approach to protein glycosylation analysis: a path through the maze. Nat Chem Biol. 2010;6:713–723. doi: 10.1038/nchembio.437. [DOI] [PubMed] [Google Scholar]
- Marshall BT, Long M, Piper JW, Yago T, McEver RP, Zhu C. Direct observation of catch bonds involving cell-adhesion molecules. Nature. 2003;423:190–193. doi: 10.1038/nature01605. [DOI] [PubMed] [Google Scholar]
- Mowery P, Yang ZQ, Gordon EJ, Dwir O, Spencer AG, Alon R, Kiessling LL. Synthetic glycoprotein mimics inhibit L-selectin-mediated rolling and promote L-selectin shedding. Chem Biol. 2004;11:725–732. doi: 10.1016/j.chembiol.2004.03.027. [DOI] [PubMed] [Google Scholar]
- Nicoll G, Avril T, Lock K, Furukawa K, Bovin N, Crocker PR. Ganglioside GD3 expression on target cells can modulate NK cell cytotoxicity via siglec-7-dependent and -independent mechanisms. Eur J Immunol. 2003;33:1642–1648. doi: 10.1002/eji.200323693. [DOI] [PubMed] [Google Scholar]
- Nieminen J, Kuno A, Hirabayashi J, Sato S. Visualization of Galectin-3 Oligomerization on the Surface of Neutrophils and Endothelial Cells Using Fluorescence Resonance Energy Transfer. J Biol Chem. 2006;282:1374–1383. doi: 10.1074/jbc.M604506200. [DOI] [PubMed] [Google Scholar]
- Ohtsubo K, Marth JD. Glycosylation in Cellular Mechanisms of Health and Disease. Cell. 2006;126:855–867. doi: 10.1016/j.cell.2006.08.019. [DOI] [PubMed] [Google Scholar]
- Pan S, Chen R, Aebersold R, Brentnall TA. Mass Spectrometry Based Glycoproteomics - From a Proteomics Perspective. Mol Cell Proteomics. 2011;10:R110. doi: 10.1074/mcp.R110.003251. 003251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partridge EA, Le Roy C, Di Guglielmo GM, Pawling J, Cheung P, Granovsky M, Nabi IR, Wrana JL, Dennis JW. Regulation of Cytokine Receptors by Golgi N-Glycan Processing and Endocytosis. Science. 2004;306:120–124. doi: 10.1126/science.1102109. [DOI] [PubMed] [Google Scholar]
- Poupon E, Nay B. Biomimetic Organic Synthesis. John Wiley & Sons; 2011. [Google Scholar]
- Pouyani T, Seed B. PSGL-1 recognition of P-selectin is controlled by a tyrosine sulfation consensus at the PSGL-1 amino terminus. Cell. 1995;83:333–343. doi: 10.1016/0092-8674(95)90174-4. [DOI] [PubMed] [Google Scholar]
- Prost LR, Grim JC, Tonelli M, Kiessling LL. Noncarbohydrate Glycomimetics and Glycoprotein Surrogates as DC-SIGN Antagonists and Agonists. ACS Chem Biol. 2012;7:1603–1608. doi: 10.1021/cb300260p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovich GA, Toscano MA, Jackson SS, Vasta GR. Functions of cell surface galectin-glycoprotein lattices. Curr Opin Struct Biol. 2007;17:513–520. doi: 10.1016/j.sbi.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabuka D, Hubbard SC, Laughlin ST, Argade SP, Bertozzi CR. A Chemical Reporter Strategy to Probe Glycoprotein Fucosylation. J Am Chem Soc. 2006;128:12078–12079. doi: 10.1021/ja064619y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabuka D, Forstner MB, Groves JT, Bertozzi CR. Noncovalent Cell Surface Engineering: Incorporation of Bioactive Synthetic Glycopolymers into Cellular Membranes. J Am Chem Soc. 2008;130:5947–5953. doi: 10.1021/ja710644g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramya TNC, Weerapana E, Liao L, Zeng Y, Tateno H, Liao L, Yates JR, Cravatt BF, Paulson JC. In Situ trans Ligands of CD22 Identified by Glycan-Protein Photocross-linking-enabled Proteomics. Mol Cell Proteomics. 2010;9:1339–1351. doi: 10.1074/mcp.M900461-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rillahan CD, Paulson JC. Glycan microarrays for decoding the glycome. Annu Rev Biochem. 2011;80:797–823. doi: 10.1146/annurev-biochem-061809-152236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen SD. Ligands for L-Selectin: Homing, Inflammation, and Beyond. Annu Rev Immunol. 2004;22:129–156. doi: 10.1146/annurev.immunol.21.090501.080131. [DOI] [PubMed] [Google Scholar]
- Sako D, Comess KM, Barone KM, Camphausen RT, Cumming DA, Shaw GD. A sulfated peptide segment at the amino terminus of PSGL-1 is critical for P-selectin binding. Cell. 1995;83:323–331. doi: 10.1016/0092-8674(95)90173-6. [DOI] [PubMed] [Google Scholar]
- Sampathkumar SG, Li AV, Jones MB, Sun Z, Yarema KJ. Metabolic installation of thiols into sialic acid modulates adhesion and stem cell biology. Nat Chem Biol. 2006;2:149–152. doi: 10.1038/nchembio770. [DOI] [PubMed] [Google Scholar]
- Sanders WJ, Gordon EJ, Dwir O, Beck PJ, Alon R, Kiessling LL. Inhibition of L-selectin-mediated Leukocyte Rolling by Synthetic Glycoprotein Mimics. J Biol Chem. 1999;274:5271–5278. doi: 10.1074/jbc.274.9.5271. [DOI] [PubMed] [Google Scholar]
- Saxon E, Bertozzi CR. Cell Surface Engineering by a Modified Staudinger Reaction. Science. 2000;287:2007–2010. doi: 10.1126/science.287.5460.2007. [DOI] [PubMed] [Google Scholar]
- Schnaar RL, Lee YC. Polyacrylamide gels copolymerized with active esters. New medium for affinity systems. Biochemistry. 1975;14:1535–1541. doi: 10.1021/bi00678a030. [DOI] [PubMed] [Google Scholar]
- Al-Shareffi E, Chaubard JL, Leonhard-Melief C, Wang SK, Wong CH, Haltiwanger RS. 6-Alkynyl fucose is a bioorthogonal analog for O-fucosylation of epidermal growth factor-like repeats and thrombospondin Type-1 repeats by protein O-fucosyltransferases 1 and 2. Glycobiology. 2013;23:188–198. doi: 10.1093/glycob/cws140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon N. Lectins: Carbohydrate-specific Reagents and Biological Recognition Molecules. J Biol Chem. 2007;282:2753–2764. doi: 10.1074/jbc.X600004200. [DOI] [PubMed] [Google Scholar]
- Sharon N, Lis H. History of lectins: from hemagglutinins to biological recognition molecules. Glycobiology. 2004;14:53R–62R. doi: 10.1093/glycob/cwh122. [DOI] [PubMed] [Google Scholar]
- Sigal GB, Mammen M, Dahmann G, Whitesides GM. Polyacrylamides Bearing Pendant α-Sialoside Groups Strongly Inhibit Agglutination of Erythrocytes by Influenza Virus:3 The Strong Inhibition Reflects Enhanced Binding through Cooperative Polyvalent Interactions. J Am Chem Soc. 1996;118:3789–3800. [Google Scholar]
- Spain SG, Gibson MI, Cameron NR. Recent advances in the synthesis of well-defined glycopolymers. J Polym Sci A Polym Chem. 2007;45:2059–2072. [Google Scholar]
- Švajger U, Anderluh M, Jeras M, Obermajer N. C-type lectin DC-SIGN: An adhesion, signalling and antigen-uptake molecule that guides dendritic cells in immunity. Cellular Signalling. 2010;22:1397–1405. doi: 10.1016/j.cellsig.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Kohler JJ. Photoactivatable crosslinking sugars for capturing glycoprotein interactions. J Am Chem Soc. 2008;130:3278–3279. doi: 10.1021/ja7109772. [DOI] [PubMed] [Google Scholar]
- Tateno H, Li H, Schur MJ, Bovin N, Crocker PR, Wakarchuk WW, Paulson JC. Distinct endocytic mechanisms of CD22 (Siglec-2) and Siglec-F reflect roles in cell signaling and innate immunity. Mol Cell Biol. 2007;27:5699–5710. doi: 10.1128/MCB.00383-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedder TF, Poe JC, Haas KM. CD22: a multifunctional receptor that regulates B lymphocyte survival and signal transduction. Adv Immunol. 2005;88:1–50. doi: 10.1016/S0065-2776(05)88001-0. [DOI] [PubMed] [Google Scholar]
- Turnbull WB, Stoddart JF. Design and synthesis of glycodendrimers. J Biotechnol. 2002;90:231–255. doi: 10.1016/s1389-0352(01)00062-9. [DOI] [PubMed] [Google Scholar]
- Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME. Essentials of Glycobiology. Cold Spring Harbor N.Y: Cold Spring Harbor Laboratory Press; 2008. [PubMed] [Google Scholar]
- Vocadlo DJ, Hang HC, Kim EJ, Hanover JA, Bertozzi CR. A chemical approach for identifying O-GlcNAc-modified proteins in cells. Proc Natl Acad Sci USA. 2003;100:9116–9121. doi: 10.1073/pnas.1632821100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JA, Smith KGC. CD22: an inhibitory enigma. Immunology. 2008;123:314–325. doi: 10.1111/j.1365-2567.2007.02752.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Cummings RD, Smith DF, Huflejt M, Campbell CT, Gildersleeve JC, Gerlach JQ, Kilcoyne M, Joshi L, Serna S, et al. Cross-platform comparison of glycan microarray formats. Glycobiology. 2014;24:507–517. doi: 10.1093/glycob/cwu019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins PP, Moore KL, McEver RP, Cummings RD. Tyrosine Sulfation of P-selectin Glycoprotein Ligand-1 Is Required for High Affinity Binding to P-selectin. J Biol Chem. 1995;270:22677–22680. doi: 10.1074/jbc.270.39.22677. [DOI] [PubMed] [Google Scholar]
- Woo CM, Iavarone AT, Spiciarich DR, Palaniappan KK, Bertozzi CR. Isotope-targeted glycoproteomics (IsoTaG): a mass-independent platform for intact N- and O-glycopeptide discovery and analysis. Nat Methods. 2015;12:561–567. doi: 10.1038/nmeth.3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SW, Pu TH, Viner R, Khoo KH. Novel LC-MS2 product dependent parallel data acquisition function and data analysis workflow for sequencing and identification of intact glycopeptides. Anal Chem. 2014;86:5478–5486. doi: 10.1021/ac500945m. [DOI] [PubMed] [Google Scholar]
- Yago T, Wu J, Wey CD, Klopocki AG, Zhu C, McEver RP. Catch bonds govern adhesion through L-selectin at threshold shear. J Cell Biol. 2004;166:913–923. doi: 10.1083/jcb.200403144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X, Bern M, Xing Q, Ho J, Viner R, Mayr M. Glycoproteomic analysis of the secretome of human endothelial cells. Mol Cell Proteomics. 2013;12:956–978. doi: 10.1074/mcp.M112.024018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Chen X. Carbohydrate post-glycosylational modifications. Org Biomol Chem. 2007;5:865–872. doi: 10.1039/b700034k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu SH, Boyce M, Wands AM, Bond MR, Bertozzi CR, Kohler JJ. Metabolic labeling enables selective photocrosslinking of O-GlcNAc-modified proteins to their binding partners. Proc Natl Acad Sci USA. 2012;109:4834–4839. doi: 10.1073/pnas.1114356109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccai NR, Maenaka K, Maenaka T, Crocker PR, Brossmer R, Kelm S, Jones EY. Structure-Guided Design of Sialic Acid-Based Siglec Inhibitors and Crystallographic Analysis in Complex with Sialoadhesin. Structure. 2003;11:557–567. doi: 10.1016/s0969-2126(03)00073-x. [DOI] [PubMed] [Google Scholar]
- Zaro BW, Yang YY, Hang HC, Pratt MR. Chemical reporters for fluorescent detection and identification of O-GlcNAc-modified proteins reveal glycosylation of the ubiquitin ligase NEDD4-1. Proc Natl Acad Sci USA. 2011;108:8146–8151. doi: 10.1073/pnas.1102458108. [DOI] [PMC free article] [PubMed] [Google Scholar]