

Abstract
Pseudomonas aeruginosa is a Gram-negative bacterium known to cause opportunistic infections in immune-compromised or immunosuppressed individuals that often prove fatal. New drugs to combat this organism are therefore sought after. To this end, we subjected the gene products of predicted perturbative genes to structure-based druggability predictions using DrugPred. Making this approach suitable for large-scale predictions required the introduction of new methods for calculation of descriptors, development of a workflow to identify suitable pockets in homologous proteins and establishment of criteria to obtain valid druggability predictions based on homologs. We were able to identify 29 perturbative proteins of P. aeruginosa that may contain druggable pockets, including some of them with no or no drug-like inhibitors deposited in ChEMBL. These proteins form promising novel targets for drug discovery against P. aeruginosa.
Introduction
Attrition rates in the drug discovery process are high. Studies have revealed that 80% of drug discovery projects fail to produce clinical candidates and only 2% actually produce marketed drugs. Poor target selection was found to be a major factor for the failures [1]. Hence, target selection is an important consideration for drug discovery. The advent of large-scale genomics projects has introduced a plethora of plausible targets. Target selection has traditionally been guided by biological or technical aspects such as assay feasibility. However, high-throughput screening against targets where chemical tractability has not been established may lead to unsatisfying hit rates [2]. This raises the need for low-cost methodologies to assist with target selection. Recently, computational methods have been introduced for this purpose. One option is to prioritize targets based on homology with other targets already possessing high-affinity ligands [3–7]. Alternatively, one can assess the binding sites of potential targets to estimate their suitability as drug targets.
Targets likely to bind orally bioavailable drugs with high affinity possess binding sites that complement the nature of these molecules These binding sites are commonly referred to as being druggable [8]. This distinguishes them from other binding sites which are referred to as “less-druggable” or “non-druggable”. The probability of deriving a drug-like ligand with high binding affinity for a protein possessing a “druggable” pocket is therefore higher than for others. The term “druggability” itself is hotly debated and several alternative terms such as “bindability”, “ligandability”, “tractability” or “chemical tractability” have been proposed. We will use the term “druggability” throughout this manuscript because it is the prevalent term used in literature.
Over the last few years, several methods have been reported that are able to segregate druggable pockets from less-druggable ones [9–20]. DrugPred is one such druggability prediction method [21]. DrugPred describes the size and shape of the binding site using a “superligand”, which is obtained by merging predicted binding modes of drug molecules that were docked into the pocket using only steric constraints. Descriptors encoding polarity and size of the pocket are subsequently calculated based on the superligand and used to predict the druggability of the binding site. We had previously found DrugPred to perform uniformly on the NRDLD dataset containing proteins with druggable and less druggable binding sites as well as other datasets, with ~90% prediction accuracy, which was superior to other methods we had tested [21]. Desaphy et al. found that our linear model was approximately as accurate as their Support Vector Machine-based method, both of which were better than two other methods [11]. Even though the variability in metrics used to determine robustness of druggability prediction methods makes it difficult to meaningfully compare and contrast them, it is clear that DrugPred is one of the more accurate and reliable methods reported thus far [8,11]. Here, we test the use of this method in identifying druggable proteins on a genome-wide scale. For this study, we chose Pseudomonas aeruginosa as a model organism.
P. aeruginosa is a Gram-gram negative bacterium that has proven to be difficult to treat with antibiotics. It often causes opportunistic infections in hospitalized patients of cystic fibrosis [22] and burn victim who are immunosuppressed or immunocompromised [23]. Chemotherapeutic intervention is therefore required, which is made difficult when infection is caused by resistant strains of bacteria. Studies with transposon mutant libraries have identified perturbative proteins in P. aeruginosa, i.e. proteins that are either essential, potentially essential or else virulence factors [24,25]. A comprehensive database of P. aeruginosa genes and related information is available in the AEROPATH database (aeropath.lifesci.dundee.ac.uk), including essentiality labels as described by the above studies [24,25]. There are 5677 genes reported in the AEROPATH database, of which 992 are predicted to be perturbative. Crystal structures are available in the public domain (RCSB Protein Data Bank) for 77 of the perturbative gene products. Crystal structures are also available for homologs of 565 of the remaining perturbative proteins.
Structures of perturbative genes in the AEROPATH database were analysed using DrugPred in order to evaluate the use of such methods for genome-wide druggability predictions and to prioritize proteins for drug discovery. While it was straightforward to assess pockets of available crystal structures of P. aeruginosa proteins, the real challenge was to make predictions for pockets in proteins where no solved structure was available. To this end, we established a work flow for homology-based druggability assessment. We also compared the predictions to chemogenomics-based predictions and discuss similarities between the two systems, along with the advantage of using both systems simultaneously in order to prioritize targets. Finally, we suggest potential new drug targets for P. aeruginosa.
Results
Constructing DrugPred 2.0
Analysis of protein druggability on a large scale required the use of a high-throughput druggability prediction algorithm. In order to scale up the original DrugPred’s performance, we re-engineered the method for smooth execution on a compute cluster and introduced some changes in the descriptor calculation methodology. Previously, the relative polar surface area (psa_r), total hydrophobic surface area (hsa_t) and total contact surface area (csa) of a binding site were calculated using the MOLCAD module in SYBYL-X (Tripos, St. Louis, Missouri, USA) [21]. In the current version, these descriptors were calculated with Openeye’s OEChem TK and Spicoli TK (OpenEye Scientific Software, Santa Fe, NM) [26,27]. This decision was made due to license restrictions and easier integration in the Python code. All other descriptors were calculated as reported previously [21]. Retraining and revalidation of the original DrugPred statistical models was carried out to identify any changes in performance. The NRDLD dataset of 115 structures was modified as per previous recommendations [21]. Thiamine purophosphokinase (TPK) was eliminated because there are no clinically used drugs for this target, apart from thiamine, which is actually a substrate, making it unclear if TPK is druggable. Urokinase Plasminogen activator (uPA) and human thymidine phosphorylase (HTP) were eliminated, because all known modulators are either highly charged or else require administration as prodrugs, against the very definition of druggability [21]. The binding sites of hydroxynitrile lyase (HNL) and angiotensin-converting enzyme 1 (ACE-1) vary significantly from other members of the dataset, making them unsuitable for model development [21]. Thereafter, the new dataset containing 110 proteins was divided into a training set of 75 structures and a validation set of 35 structures (Table A in S1 File). Descriptors were calculated for the training set and a new Partial Least Squares-Discriminant Analysis (PLS-DA) model was built as done previously [21]. The new model, which we named DrugPred 2.0, was used to obtain scores for all training and test set structures, following which accuracy, precision and recall values were calculated as done previously [21]. Briefly, accuracy describes the success rate of categorizing pockets as druggable or less-druggable, precision describes the rate of correct calls by the model (e.g. how many data points predicted to be druggable were truly druggable), and recall describes the ability of the model to correctly identify members of a category (e.g., how many of the druggable proteins were correctly categorized). Comparing the two models, we found that all three statistical values were similar for both models. In our original report, we had also employed an ambiguous zone. This is a region of scores where it is difficult to identify targets as belonging to either the druggable or less-druggable categories [21]. When applying such a zone with DrugPred 2.0, similar results were obtained (Table 1). For the sake of simplification, during the rest of this manuscript, we will highlight only those results where an ambiguous zone was employed and any data points that lay within this zone were discarded for calculation of final results.
Table 1. Accuracy, recall and precision values for training and validation sets for DrugPred 1 and 2.0.
| DrugPred version | Data set | With/ without ambiguous zone | Accuracy | Recall (Druggable/ Less druggable) | Precision (Druggable/Less druggable) |
|---|---|---|---|---|---|
| 1 Ω | Training | Without | 0.91 | 0.96/0.83 | 0.90/0.93 |
| 2.0 | 0.91 | 0.94/0.86 | 0.92/0.89 | ||
| 1 Ω | With | 0.92 | 0.95/0.86 | 0.91/0.93 | |
| 2.0 | 0.95 | 0.98/0.9 | 0.95/0.95 | ||
| 1 Ω | Validation | Without | 0.89 | 0.91/0.85 | 0.91/0.85 |
| 2.0 | 0.94 | 0.95/0.93 | 0.95/0.93 | ||
| 1 Ω | With | 0.91 | 0.91/0.92 | 0.95/0.86 | |
| 2.0 | 0.97 | 1.00/0.92 | 0.95/1.00 |
Ω Values taken from Krasowski et al [21]
Homolog-based druggability predictions
For the majority of essential or potentially essential proteins from P. aeruginosa in the AEROPATH database no crystal structure was deposited in the PDB. However, structures of homologous proteins were available for 565 of them. It is common practice to assume that homologs of a target already known to be modulated by small molecules are druggable as well, particularly if the sequence homology is high [3–7,17]. It was therefore interesting to test whether DrugPred predictions could be transferred between homologous pockets as well. We also wanted to establish a sequence identity cut-off at which such transfers could be made and a minimum number of structures required for reliable transfers.
With this aim in mind, we embarked on a study to identify structural homologs of the modified NRDLD dataset and to score their pockets using DrugPred 2.0. The predictions were then compared to the classification of the parent structures. Homologous structures were found for all but three proteins in the dataset. For 19 proteins, none of the homologous structures contained a ligand to mark the binding site and they were therefore not considered further. The druggability of the homologous binding sites in the remaining 88 proteins was predicted. The predictions for all homologs of six of these proteins were outside the model as judged by high distance-to-model in X-plane (DModX) values. DModX represents the distance of a data point from a hyperplane that represents the model. Smaller values demonstrate a higher likelihood that data points are within the predictive domain of the model, while higher values demonstrate that predictions for the data points may be unreliable. Predictions with a high DModX value were therefore not analysed further. Thus, the final dataset consisted of 3186 homologous pockets for 82 proteins. The total number of homologous pockets per dataset pocket ranged from 1 to 208 and the sequence identity between the homologs and parent proteins from 22.3 to 89.9% (Table B in S1 File). The percentage of homologous pockets whose classification correctly reflected the druggability of the parent pocket ranged from 0% to 100% (Fig 1A). In the majority of the cases the druggability prediction of the homologous pockets was correctly transferable to the parent pocket, e.g. for 57 out of 82 proteins at least 90% of the homologous pockets provided the correct prediction. Of these 57, 51 showed 100% correct predictions. However, there were 12 instances where more than 50% of predictions for homologs did not match the druggability of the dataset pocket. Therefore, we attempted to identify filter criteria to obtain more reliable predictions.
Fig 1. The basis for homolog-based druggability predictions.

(A) Homologous pockets whose classification correctly reflected the druggability of the parent pocket. The data was binned according to percent correct predictions among the scored pockets for each parent homolog. The number of NRDLD proteins that fitted into each category was then plotted (frequency and percentages are both shown). (B) Correct predictions in relationship to sequence identity. The percent identity between NRDLD dataset structures and homologous chains was noted. The homologs were then binned according to their percent sequence identity. The percent of homologs whose predictions matched that of the NRDLD dataset pocket was plotted for each bin. (C) Correct predictions in relationship to sequence identity of binding site residues only. Plotted as described for (B), but instead of the sequence identity only the identity of the binding site residues was used. (D) Percent correct predictions in relationship to number of assessed homologs. (E) Percent consensus in relationship to percentage of correctly predicted NRDLD dataset pockets. The NRDLD dataset pockets were binned into two categories, where either <80% or ≥80% consensus (see methods) in druggability predictions for their respective homologs was observed. The percentage of NRDLD pockets whose druggability was correctly reflected by consensus amongst their homologs was then plotted for each of these bins.
It would be useful if we could establish (A) a sequence-cut off, (B) a requirement of minimum number of homologs, and/or (C) a minimum percent consensus in predictions for homologs, as suitable criteria to make the predictions more reliable. Hence, we first plotted the percent homologs that reflected the parent structure’s druggability (% correct predictions) for various bins of percent sequence identity to the parent structure (Fig 1B). There was no relationship between sequence identity of the homologous proteins and the percentage of correct predictions observed. The same was observed when instead of the sequence identity only the identity of the binding site residues was considered (Fig 1C). Similarly, percent correct predictions did not increase with the number of homologs used for assessment (Fig 1D). We went on to investigate if the parent protein pocket’s druggability could be predicted by a consensus between its homologs. For this purpose, percent consensus between different homologous pockets belonging to the same NRDLD parent entry was calculated using the formula: (100*|#druggable—#less-druggable|)/(total number of predictions). This measure enabled us to identify how many homologous pockets gave us the same prediction regardless of the prediction itself. We then binned the NRDLD pockets into two categories, with either ≥80% consensus or <80% consensus amongst all homologs (Fig 1E). With <80% consensus in their druggability prediction, only ~62% of the parent NRDLD pocket’s druggabilities was predicted correctly. However, with at least 80% consensus, ~93% of the parent NRDLD pockets were correctly predicted. The latter value increased to 100%, when additionally at least 5 homologs were assessed. Hence, we identified a dual-filter approach to obtain reliable homolog-based druggability predictions using DrugPred 2.0. This formed the basis for the subsequent evaluation of P aeruginosa proteins where no crystal structure has yet been solved.
Structure-based druggability analysis of P. aeruginosa proteins
Information for 5677 genes is stored in the AEROPATH database (Table 2). Of those, 992 are annotated to be perturbative. Crystal structures for 77 of those gene products have been determined. Homologous structures for a further 565 perturbative gene products were available. The available crystal structures were subjected to structure-based druggability predictions using DrugPred 2.0.
Table 2. Overview of triaging the genes stored in the AEROPATH database to enable structure-based druggability predictions for P. aeruginosa proteins.
| Number of entities | |
|---|---|
| 1) Genes stored in AEROPATH database | 5677 |
| 2) Annotated to be perturbative | 992 |
| 3) Perturbative gene products originating from P. aeruginosa for which crystal structures are available | 77 |
| 3a) Proteins for which valid druggability predictions were obtained | 24 |
| 3b) Proteins predicted to be druggable (Table 3) | 13 |
| 4) Proteins encoded by perturbative genes for which crystal structures of homologous proteins were available | 565 |
| 4a) Proteins for which valid druggability predictions were obtained | 241 |
| 4b) Proteins predicted to be druggable (Table 4) | 16 |
The analysis of available Pseudomonas protein structures was straightforward. Pockets were identified by bound ligands. Subsequently, the druggability was assessed using DrugPred 2.0 and pockets not fitting to the model were removed (as judged by DModX values). A gene product was considered druggable if it possessed at least one pocket with a druggable score. Following this procedure druggability scores could be obtained for 103 pockets from 34 Pseudomonas proteins (out of 77 perturbative genes for which crystal structures were available, Table 2). Of those 13 were found to possess pockets likely to bind drug-like molecules with high affinity (Table 3).
Table 3. List of P. aeruginosa protein crystal structures containing pockets that are predicted to be druggable.
| Pseudomonas gene code | Product name | Gene name | PDB code | Ligand | Chemogenomics-based druggability rank a |
|---|---|---|---|---|---|
| PA0019 | Polypeptide deformylase | def | 1lry | BB2 | 3 (2) |
| 1ix1 | BB2 | ||||
| PA0395 | Twitching motility protein | pilT | 3jvv | ACP | - |
| PA1148 | Exotoxin A precursor | toxA | 1aer | AMP | - |
| 1dma | AMP | ||||
| NCA | |||||
| 1aer | TIA | ||||
| 1xk9 | P34 | ||||
| 1zm9 | P34 | ||||
| PA1430 | Transcriptional regulator | lasR | 2uv0 | OHN | 8 (6) |
| 3ix3 | OHN | ||||
| 3ix4 | TX1 | ||||
| 3jpu | TY4 | ||||
| 3ix8 | TX3 | ||||
| PA1900 | Probable phenazine biosynthesis protein | phzB2 | 3ff0 | UNL | - |
| PA2386 | L-ornithine-N5-oxygenase | pvdA | 3s61 | ORN | - |
| PA3155 | UDP-2-acetamido-2-dideoxy-D-ribohex-3-uluronic acid transaminase | wbpE | 3nyu | LLP | - |
| PA3540 | GDP-mannose-6-dehydrogenase | algD | 1muu | GDX | - |
| 1mv8 | GDX | ||||
| PA3724 | Elastase | lasB | 3dbk | RDF | 4 (3) |
| PA4279 | Hypothetical protein (probable pantothenate kinase) | 2f9w | PAU | - | |
| PA4406 | UDP-3-O-acyl-N-acetylglucosamine deacetylase (LpxC) | lpxC | 2ves | GVR | 1 |
| PA4407 | Cell division protein | ftsZ | 1ofu | GDP | 115 (29) |
| PA5163 | Glucose-1-phosphate thymidylyltransferase (RmlA) | rmlA | 1g3l | TRH | - |
| 1fxo | TMP |
aRanks obtained when only perturbative proteins are considered are given in brackets.
The analysis of gene products lacking crystal structures, but where homologous structures were available, was more convoluted. In the AEROPATH database crystal structures of homologous proteins are linked to P. aeruginosa gene products. However, there is no annotation if these crystal structures cover the same sequence segment of the gene, which is particularly relevant for multi-domain and multi-pocket proteins (Fig 2A). Therefore, it had to be ensured that only corresponding pockets of the homologs were compared when assessing the druggability of a parent protein. This was achieved using the following workflow (Fig 2B): First, DrugPred 2.0 was used to score all pockets marked by a ligand in the structures of homologous proteins for a particular parent sequence. In the next step any pockets with high DModX values or scores within the ambiguous zone were discarded. Subsequently, the sequence of residues forming each homologous pocket was determined. Next, the domains represented by this sequence was determined by pairwise BLAST [28] searches with an E-value cut-off of 1e-5. For each domain identified, the homologous structures were aligned and the number of pockets represented was then determined by pairwise comparison of ligand positions. For final assessment, only corresponding pockets represented in at least five different crystal structures were retained. A protein pocket was considered to be druggable, if at least 80% consensus was reached among the homologs and the majority of the pockets were predicted druggable. Following this workflow, starting from 565 gene products for which homologous structures were available, valid (low DModX) predictions were obtained for 241 (Table 3). A total of 16 P. aeruginosa gene products were found to possess pockets likely to bind drug-like ligands (Table 4).
Fig 2. Complexity of homolog-based identification of pertinent pockets in proteins.
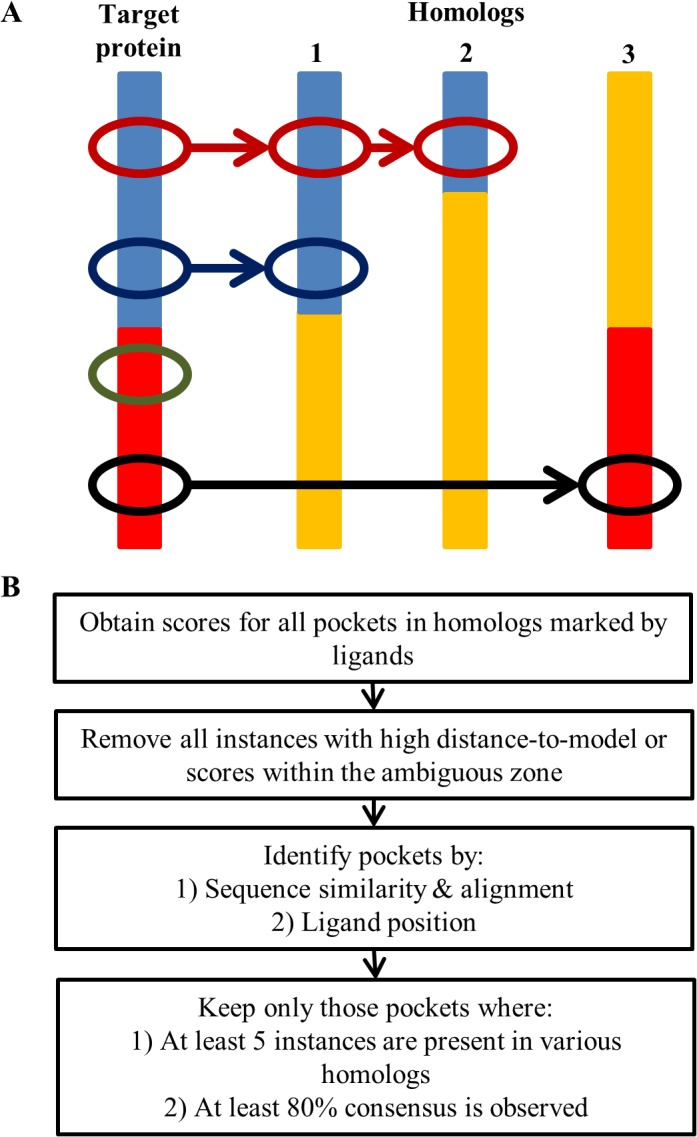
A) A hypothetical target protein is depicted with three homologous proteins. The target protein consists of two domains, one shown in blue and the other in red. These domains may be represented by complete or partial sequences in homologs. For example, homologs 1 and 2 possess short domains homologous to the target protein's blue domain. On the other hand, homolog 3 possesses a sequence match for the red domain. Each target protein domain possesses pockets (denoted by black, green, red and blue ovoids), which may or may not be identified by the presence of ligands in homologs. Here, the black pocket is also represented in homolog 3, but the green pocket is not. The red pocket is observed in both, homolog 1 and 2, but the blue pocket is only represented in homolog 1. B) Workflow to identify druggable pockets in homologs proteins.
Table 4. List of P. aeruginosa proteins predicted to possess a druggable pocket.
| PA Code | Representative homolog (PDB code / ligand) | # of HA α | % Sequence ID range | # of predictions druggable / less druggable / ambiguous | Product Name | Gene Name | Rank Ω |
|---|---|---|---|---|---|---|---|
| PA0350 | 2qk8 / MTX | 11 | 39.76–48.78 | 11 / 0 / 0 | Dihydrofolate reductase (DHFR) | folA | 85 (22) |
| PA0363 | 1b6t / COD | 7 | 40.51–60.00 | 6 / 1 / 0 | Phosphopante-theine adenylyltrans-ferase (PPAT) | coaD | - |
| PA1648 | 2dm6 / IMN | 6 | 38.07–39.66 | 6 / 0 / 0 | Probable oxidoreductase | 685 (126) | |
| PA1671 | 1ig1 / ANP | 9 | 26.85–28.66 | 9 / 0 / 0 | Serine-threonine kinase Stk1 | Stk1 | 195 (42) |
| PA1778 | 1cbf / SAH | 9 | 28.65–46.15 | 9 / 0 / 0 | Uroporphyrin-III C-methyltrans-ferase | cobA | - |
| PA2086 | 1vj5 / CIU | 7 | 29.82–29.82 | 7 / 0 / 0 | Probable epoxide hydrolase | 336 (62) | |
| PA2344 | 1tz3 / AIS | 6 | 25.10–30.35 | 5 / 0 / 1 | Fructokinase | mtlZ | - |
| PA2965 | 1b3n / CER | 9 | 39.49–66.50 | 9 / 0 / 0 | Beta-ketoacyl carrier protein synthase II | fabF1 | 119 (30) |
| PA2967 | 1doh / NID | 8 | 34.23–37.70 | 7 / 0 / 1 | 3-oxoacyl-[acyl carrier protein] reductase | fabG | 80 (21) |
| PA3883 | 1a27 / EST | 9 | 34.81–35.19 | 9 / 0 / 0 | Probable short-chain dehydrogenase | 32 (11) | |
| PA4068 | 1lrl / UPG | 6 | 30.97–33.55 | 6 / 0 / 0 | Probable epimerase | 544 (98) | |
| PA4385 | 1a6e / ADP | 17 | 21.93–80.69 | 17 / 0 / 0 | GroEL protein | groEL | - |
| PA4386 | 1aon / ADP | 7 | 57.45–61.46 | 7 / 0 / 0 | GroES protein | groES | - |
| PA4439 | 1i6k / TYM | 11 | 31.66–33.73 | 9 / 0 / 2 | Tryptophanyl-tRNA synthetase | trpS | - |
| PA5174 | 1b3n / CER | 8 | 27.88–28.36 | 8 / 0 / 0 | Probable beta-ketoacyl synthase | 488 (83) | |
| PA5288 | 1v3s / ATP | 12 | 41.07–77.68 | 10 / 1 / 1 | Nitrogen regulatory protein P-II 2 | glnK | - |
α HA: distinct homologous pockets assessed
Ω Rank: Chemogenomics-based druggability rank. Ranks obtained when only perturbative proteins are considered are given in brackets.
Experimental support for DrugPred-based findings
It is already known that some proteins, out of those predicted to be druggable using DrugPred 2.0, are capable of strongly binding drug-like molecules; this is strong support for the predictions. For example, UDP-3-O-acyl-N-acetylglucosamine deacetylase (LpxC, Table 3) is a well-studied target for antibacterial compounds and a number of drug-like ligands has been discovered [29]. Similar, dihydrofolate reductase (DHFR, Table 4) is an established target for antibiotics which are routinely used in man [30]. Recently, drug-like inhibitors of phosphopantetheine adenylyltransferase (PPAT) were discovered and their activity against a range of bacterial strains was demonstrated [31]. An interesting case is also glucose-1-phosphate thymidylyltransferase (RmlA, Table 3). The crystal structure of RmlA was reported earlier [32] (RCSB PDB code: 1fxo), with TMP bound at the active and allosteric sites. Both these sites were assessed using DrugPred 2.0 and it was found that the allosteric site scored druggable while the active site scored less druggable. Interestingly, a high-throughput screen, followed by rational drug design resulted in nanomolar inhibitors of RmlA [33]. These inhibitors were characterised crystallographically and it was observed that all these compounds bound at the allosteric regulatory site that scored druggable.
Comparison with a chemogenomics-based druggability rankings
Bickerton et al. have introduced a chemogenomics-based druggability prediction method for P. aeruginosa proteins, which is available through the AEROPATH database. This method utilizes ChEMBL [34] data to identify compounds possessing high affinity or ligand efficiency for specific targets, which are further quantified by Quantitative Estimate of Druggability (QED) measurements [35]. Proteins with a larger number of QED-positive compounds are judged to be more druggable. Furthermore, homologs of druggable protein domains are also considered to be druggable. The P. aeruginosa proteins were then ranked according to the scores obtained (http://aeropath.lifesci.dundee.ac.uk/pages/background).
We compared DrugPred 2.0 predictions with this chemogenomics-based ranking system. Predictions could be obtained for 158 perturbative gene products using the latter system, and for 265 gene products using DrugPred 2.0 (Table 2). There was an overlap of 38 gene products using both methods, 17 of which were predicted to possess druggable pockets by DrugPred. A good agreement was found between DrugPred 2.0 predictions and top scoring chemogenomics-based predictions. All gene products for which crystal structures from P. aeruginosa proteins were available and which ranked among the top 10 perturbative gene products using the chemogenomics-based system were predicted to be druggable by DrugPred 2.0 (Table 3). Some gene products that ranked low in the chemogenomics-based method were still classified to be druggable by DrugPred 2.0 (Table 4).
Discussion
The DrugPred methodology was redevised for high-throughput operation, involving the introduction of robust methods for calculation of descriptors. The new version of this druggability prediction method showed accuracy, precision and recall parameters similar to the older version (Table 1) and it is therefore suitable for large-scale predictions.
Criteria for homolog-based druggability predictions were established. Coverage of perturbative P. aeruginosa gene products by crystal structures is low (Table 2). Crystal structures for only 77 out of 992 perturbable gene products were available. Homolog-based predictions can be used in order to enhance coverage of the genome. It is commonplace to assume that homologs of druggable proteins will be druggable as well [3–7]. Yet, our analysis of a data set containing crystal structures of homologous proteins for the NRDLD dataset does not support such assumptions (Fig 1 and Table B in S1 File). Similar successful predictions were observed for homologs with sequence identities ranging between 20–90%. However, when at least five instances of homologous pockets were present with at least 80% consensus in druggability predictions, 100% correct predictions were obtained (Fig 1E). While the exact numbers will be data set dependent, this provides guidelines on which criteria to apply when transferring structure-based druggability predictions between homologous proteins.
Full coverage of all perturbative P. aeruginosa proteins could not be achieved despite including homolog-based druggability predictions (Table 2). The main reason is the lack of structural data. While there are 992 perturbative proteins in the P. aeruginosa proteome, only 77 of them have solved structures. This number increases to 642 when structures of homologs are also considered. However, as more than one crystal structure of a homologous protein is needed to obtain reliable predictions using DrugPred, this number is actually smaller. Another reason lies in the difficulty of identifying pertinent pockets for computational analysis. DrugPred accepts as input to mark a binding site also spheres generated by a pocket prediction program such as FPOCKET [21,36]. However, for the current study we opted to only assess binding sites that contained a ligand. This was done for the following two reasons. 1) Many groups have attempted to devise new pocket prediction methods, however, unambiguous identification of binding sites is not yet possible [37–48]. Consequently, including predicted pocked in a large scale study is likely to introduce errors. 2) The apo-structure binding site conformation does not necessarily represent the bound conformation [49,50]. This makes such pockets less useful for predictions. Even so restricting the input structures to holo-structures further limited the number of proteins we could address we believe that the obtained predictions are more meaningful. It is evident from this discussion that there is a clear need for more structural data to enable better coverage, especially in the presence of ligands. Previous endeavours have been highly successful in this regard and must continue in the future [51].
Druggable proteins were identified by DrugPred 2.0 using crystal structures from P. aeruginosa proteins itself, or else through its homologs (Table 3 and Table 4). It must be noted that proteins are not necessarily less-druggable per se just because a pocket with druggable score was not observed. It’s possible that the crystal structure of the relevant domain or containing a pocket with the required binding site conformation has not yet been crystallized. We therefore consider the druggability status of these proteins as unknown, rather than less-druggable. Some of the predicted druggable proteins like LpxC and DHFR are well known drug targets for antibiotics while others like RmlA and PPAT were only recently pursued and drug-like inhibitors were identified [29,30]. These findings validate our predictions and add confidence that the predictions reported here are reliable. Other proteins were also found to possess druggable binding pockets but have not yet been investigated using small molecules. For 15 out of the 29 druggable proteins, no inhibitors were submitted in ChEMBL to allow a chemogenomics-based ranking. These proteins form promising novel targets for drug discovery against P. aeruginosa.
The druggability predictions based on chemogenomics-based scoring and made by DrugPred complemented each other. Predictions for 385 perturbative proteins are obtained by combining both methods. The overlap between the methods was rather small with only 38 gene products. The reasons for this are either the lack of crystal structures suitable for druggability predictions (resulting in ranking by only the chemogenomics-based scoring system) or no precedence of drug-like ligands (resulting in predictions only by DrugPred). This clearly demonstrates that a larger coverage can be reached when complementary methods are used. The chemogenomics-based method provides a rank-ordered list but does not provide a direct classification of druggable or less-druggable. Therefore, a direct comparison between both methods is difficult. Still, when looking at the predicted druggable proteins for which crystal structures from P. aeruginosa were available, a good agreement was observed (Table 3). Low chemogenomics-based ranks for predicted druggable proteins were also observed (Table 4) and can have several reasons: it is possible that the compounds used to derive the chemogenomics-based score target a different binding site than those used for the structure-based predictions. It might also be that the structures used for scoring do not contain the binding site in a conformation relevant for binding a drug-like ligand. Further, there is also possible that drug-like ligands have not yet been described or were not present in the ChEMBL database. Finally, both computational methods have got short-comings and any predictions obtained should always be viewed critically. Nevertheless, assessment by two such independent methods provides confidence in those targets found to be druggable by both and tremendously extends the coverage of the genome. Together, these tools can direct drug discovery efforts.
Conclusions
The DrugPred methodology was redevised for high-throughput operation, involving the introduction of new methods for calculation of descriptors. The old and new versions of DrugPred showed similar accuracy, precision and recall parameters. Thus, DrugPred 2.0 is suitable for large scale predictions.
A robust workflow and criteria to score homologous protein structures was established to extend genome coverage. This procedure can readily be applied to other organisms for which drug targets are sought.
This work was limited by the availability of solved structures, but with important advances being made all the time in the field of structural biology, such bottlenecks may seize to exist in the not-too-distant future. In the meantime, identification of 29 perturbative proteins of P. aeruginosa that may contain druggable pockets, including some of them with no or no drug-like inhibitors deposited in ChEMBL, is a remarkable achievement that might drive drug discovery efforts in the right direction.
Methods
Construction of DrugPred 2.0
For speed and scale-up reasons, minor changes were introduced to the previous DrugPred version. In particular, Openeye’s OEChem TK (OEChem, version 1.7.4, OpenEye Scientific Software, Inc., Santa Fe, NM, USA) and Spicoli TK (Spicoli, version 1.1.2, OpenEye Scientific Software, Inc., Santa Fe, NM, USA) were used to calculate the relative polar surface area (psa_r), total hydrophobic surface area (hsa_t) and total contact surface area (csa), instead of using Sybyl-X (Tripos, St. Louis, Missouri, USA) as done previously. [26,27] For that purpose, any atoms whose solvent accessibility changed in the presence of the superligand were identified, and classified as polar and hydrophobic, respectively. Their solvent accessible surface in the unbound state (SASAi) was then determined and used to calculate the relevant descriptors using the following formulae:
For building and validating a Partial Least Squares-Discriminant Analysis (PLS-DA) model, the NRDLD dataset was modified as suggested previously and detailed in the result section [21]. The new dataset of 110 structures was thus formulated accordingly. This modified dataset was divided into a training set of 75 structures and a validation set of 35 structures. Descriptors were calculated for the training set and a Partial Least Squares-Discriminant Analysis (PLS-DA) model was built as done previously [21]. The new model was called DrugPred 2.0.
An ambiguous zone for uncertain predictions had previously been defined mathematically using a one-sided 90% cut-off for both categories; 1.28 times the standard deviation of scores for the less druggable structures was added to their mean score. Likewise, the same was subtracted from the mean score for all druggable structures. This exercise generated a region of statistical uncertainty, aptly named the ambiguous zone. DrugPred performed reasonably well outside this zone of uncertainty, but a drop in performance was observed when data points within the ambiguous zone were included. Accordingly, we attempted to establish the ambiguous zone during the construction of DrugPred 2.0 as well. Using the same definition, we found the ambiguous zone to be unreasonably small; suggesting this method for setting boundaries for the ambiguous zone could no longer be used. However, it is reasonable to expect a region of uncertainty with such models, so we arbitrarily built an ambiguous zone with the magnitude of two score units, from 0.4–0.6, which is the middle of the scale and of similar size than the ambiguous zone established with DrugPred 1.0.
Accuracy, precision and recall values were calculated as before [21]. Statistical analysis, including DModX value determination, was carried out in the SIMCA-P+ package (www.umetrics.com/products/simca).
Preparation of structures for druggability analysis
Ligands were identified in each PDB file and unless a particular ligand was part of the list of cofactors or common additives during the crystallography process (Table C in S1 File), it was treated as a marker for its binding site. Any protein chains, cofactors and metals presenting at least one atom within 3 Å of the ligand were retained as components of the binding site, while the rest were deleted. These reduced PDB files were processed using DrugPred.
Identification of homologous structures of the NRDLD dataset
The NRDLD dataset, along with ligands marking the binding sites has been reported previously [21] and was modified as described above. The ligand marking the binding site for each dataset structure was identified and the surrounding chain that forms maximum contacts with it was determined. Homologous structures for this chain were identified by sequence abstraction and BLAST searches using a database containing all sequences for structures reported in the RCSB PDB using an E-value cut-off of 1e-5. They were downloaded, followed by structure preparation as detailed above. These structures of homologous proteins were then subjected to structural alignment using PyMOL (The PyMOL Molecular Graphics System, Version 1.2r3pre, Schrödinger, LLC.) and the Openeye OEChem toolkit was used to confirm vicinity of ligands in the homologous chain, confirming that only pockets that are analogous to the one reported in the modified NRDLD dataset were included.
The binding site identity between the parent pocket and the homologous pockets was calculated as follows: 1) A consensus pocket was generated. The consensus pocket consisted of all residues that were either part of the parent pocket or the pocket found in the homolog. For that purpose, the same pocket definition as for the descriptor calculation was used (e.g. change of solvent accessibility in the presence of the superligand). 2) The sequences of the parent protein and the homolog were aligned using ClustalW.[52] 3) For each residue in the consensus pocket it was noted if a sequence match in the alignment was found. The sequence identity of the two binding sites was then calculated as 100*(number of sequence matches)/(number of binding site residues) whereas residues that occurred at the same position in the alignment were only counted once.
Percent consensus between different homologous pockets for the same NRDLD dataset pocket was calculated using the formula: (100*|#druggable—#less-druggable|)/(total number of predictions). This measure enabled us to identify how many homologous pockets gave us the same prediction regardless of the prediction itself.
Identification of crystal structures for P. aeruginosa proteins and their homologs
The AEROPATH database (aeropath.lifesci.dundee.ac.uk, current release from 2010) contains lists of crystal structures for P. aeruginosa proteins and their homologs. These structures were downloaded from the in-house RCSB PDB mirror and subjected to structure preparation as detailed above. Only those structures were retained where at least one ligand was obtained. Repeated occurrences of the same ligand within the same PDB chain were not retained as it is rare that the same ligand occupies different pockets in the same protein structure. An exception to this rule was RmlA, where prior knowledge about a second pocket binding the cocrystallized ligand was available; hence, a separate analysis was conducted in order to include both, the active site and the allosteric pocket.
Druggability predictions for gene products for which only crystal structure from homologous proteins were available
For each parent gene product it was ensured that the binding sites of the crystal structures of the homologous proteins covered the same sequence segment (Fig 2B). For final assessment, only corresponding pockets represented in at least five different crystal structures were retained. A protein pocket was considered to be druggable, if at least 80% consensus was reached among the homologs and the majority of the pockets were predicted druggable. For this purpose, percent consensus between different homologous pockets belonging to the same parent entry was calculated using the formula: (100*|#druggable—#less-druggable|)/(total number of predictions).
Supporting Information
Results from homolog-based druggability predictions for the NRDLD dataset structures when using an ambiguous zone (Table B). List of PDB 3-letter codes for cofactors or common additives during the crystallography process. These compounds were not considered to mark a binding pocket (Table C). Scripts are available on https://github.com/ruthbrenk/DrugPred2.0.git.
(DOCX)
Acknowledgments
We would like to acknowledge Richard Bickerton’s assistance in understanding the complexity and scale of genome-scale analysis projects, Thomas Wehler and Raphael Klein for critical reading of the manuscript, and Openeye for providing free software licenses. Marko Hyvonen helped us accumulate a list of common additives often found in crystal structures. Jonathan Downward’s and Tom Walsh’s efforts in providing system administration and maintenance of the high-performance computing cluster respectively are also thankfully acknowledged.
Data Availability
All relevant data are in the AEROPATH database (http://aeropath.lifesci.dundee.ac.uk/). Scripts are available on Github (https://github.com/ruthbrenk/DrugPred2.0.git). Scripts are open source but rely on the Python package, which is free for academic researchers. Python license can be obtained from Openeye.
Funding Statement
This work was funded by the European Commission Seventh Framework Programme (FP7/2007–2013; Aeropath, http://ec.europa.eu/research/fp7/index_en.cfm). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Brown D, Superti-Furga G. Rediscovering the sweet spot in drug discovery. Drug Discov Today. 2003;8: 1067–77. [DOI] [PubMed] [Google Scholar]
- 2. Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov. 2007;6: 29–40. 10.1038/nrd2201 [DOI] [PubMed] [Google Scholar]
- 3. Hopkins AL, Bickerton GR, Carruthers IM, Boyer SK, Rubin H, Overington JP. Rapid analysis of pharmacology for infectious diseases. Curr Top Med Chem. 2011;11: 1292–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Agüero F, Al-Lazikani B, Aslett M, Berriman M, Buckner FS, Campbell RK, et al. Genomic-scale prioritization of drug targets: the TDR Targets database. Nat Rev Drug Discov. 2008;7: 900–7. 10.1038/nrd2684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Patel MN, Halling-Brown MD, Tym JE, Workman P, Al-Lazikani B. Objective assessment of cancer genes for drug discovery. Nat Rev Drug Discov. 2013;12: 35–50. 10.1038/nrd3913 [DOI] [PubMed] [Google Scholar]
- 6. Shanmugham B, Pan A. Identification and characterization of potential therapeutic candidates in emerging human pathogen Mycobacterium abscessus: a novel hierarchical in silico approach. PLoS One. 2013;8: e59126 10.1371/journal.pone.0059126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Uddin R, Saeed K. Identification and characterization of potential drug targets by subtractive genome analyses of methicillin resistant Staphylococcus aureus. Comput Biol Chem. Elsevier Ltd; 2014;48: 55–63. 10.1016/j.compbiolchem.2013.11.005 [DOI] [PubMed] [Google Scholar]
- 8. Volkamer A, Rarey M. Exploiting structural information for drug-target assessment. Future Med Chem. 2014;6: 319–31. 10.4155/fmc.14.3 [DOI] [PubMed] [Google Scholar]
- 9. Seco J, Luque FJ, Barril X. Binding site detection and druggability index from first principles. J Med Chem. 2009;52: 2363–71. 10.1021/jm801385d [DOI] [PubMed] [Google Scholar]
- 10. Cheng AC, Coleman RG, Smyth KT, Cao Q, Soulard P, Caffrey DR, et al. Structure-based maximal affinity model predicts small-molecule druggability. Nat Biotech. 2007;25: 71–5. 10.1038/nbt1273 [DOI] [PubMed] [Google Scholar]
- 11. Desaphy J, Azdimousa K, Kellenberger E, Rognan D. Comparison and druggability prediction of protein-ligand binding sites from pharmacophore-annotated cavity shapes. J Chem Inf Model. 2012;52: 2287–99. 10.1021/ci300184x [DOI] [PubMed] [Google Scholar]
- 12. Hajduk PJ, Huth JR, Fesik SW. Druggability indices for protein targets derived from NMR-based screening data. J Med Chem. 2005;48: 2518–25. 10.1021/jm049131r [DOI] [PubMed] [Google Scholar]
- 13. Halgren TA. Identifying and characterizing binding sites and assessing druggability. J Chem Inf Model. 2009;49: 377–89. 10.1021/ci800324m [DOI] [PubMed] [Google Scholar]
- 14. Nisius B, Sha F, Gohlke H. Structure-based computational analysis of protein binding sites for function and druggability prediction. J Biotechnol. Elsevier B.V.; 2012;159: 123–34. 10.1016/j.jbiotec.2011.12.005 [DOI] [PubMed] [Google Scholar]
- 15. Perola E, Herman L, Weiss J. Development of a rule-based method for the assessment of protein druggability. J Chem Inf Model. 2012;52: 1027–38. 10.1021/ci200613b [DOI] [PubMed] [Google Scholar]
- 16. Schmidtke P, Barril X. Understanding and predicting druggability. A high-throughput method for detection of drug binding sites. J Med Chem. 2010;53: 5858–67. 10.1021/jm100574m [DOI] [PubMed] [Google Scholar]
- 17. Sheridan RP, Maiorov VN, Holloway MK, Cornell WD, Gao Y-D. Drug-like density: a method of quantifying the “bindability” of a protein target based on a very large set of pockets and drug-like ligands from the Protein Data Bank. J Chem Inf Model. 2010;50: 2029–40. 10.1021/ci100312t [DOI] [PubMed] [Google Scholar]
- 18. Volkamer A, Kuhn D, Grombacher T, Rippmann F, Rarey M. Combining global and local measures for structure-based druggability predictions. J Chem Inf Model. 2012;52: 360–72. 10.1021/ci200454v [DOI] [PubMed] [Google Scholar]
- 19. Nayal M, Honig B. On the nature of cavities on protein surfaces: application to the identification of drug-binding sites. Proteins. 2006;63: 892–906. 10.1002/prot.20897 [DOI] [PubMed] [Google Scholar]
- 20. Huang N, Jacobson MP. Binding-site assessment by virtual fragment screening. PLoS One. 2010;5: e10109 10.1371/journal.pone.0010109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Krasowski A, Muthas D, Sarkar A, Schmitt S, Brenk R. DrugPred: a structure-based approach to predict protein druggability developed using an extensive nonredundant data set. J Chem Inf Model. 2011;51: 2829–42. 10.1021/ci200266d [DOI] [PubMed] [Google Scholar]
- 22. Oliver A. High Frequency of Hypermutable Pseudomonas aeruginosa in Cystic Fibrosis Lung Infection. Science. 2000;288: 1251–1253. 10.1126/science.288.5469.1251 [DOI] [PubMed] [Google Scholar]
- 23. Wang CY, Jerng JS, Chen KY, Cheng KY, Lee LN, Yu CJ, et al. Pandrug-resistant Pseudomonas aeruginosa among hospitalised patients: clinical features, risk-factors and outcomes. Clin Microbiol Infect. 2006;12: 63–8. 10.1111/j.1469-0691.2005.01305.x [DOI] [PubMed] [Google Scholar]
- 24. Liberati NT, Urbach JM, Miyata S, Lee DG, Drenkard E, Wu G, et al. An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants. Proc Nat Acad Sci USA. 2006;103: 2833–8. 10.1073/pnas.0511100103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jacobs MA, Alwood A, Thaipisuttikul I, Spencer D, Haugen E, Ernst S, et al. Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc Nat Acad Sci USA. 2003;100: 14339–44. 10.1073/pnas.2036282100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee B, Richards FM. The interpretation of protein structures: estimation of static accessibility. J Mol Biol. 1971;55: 379–400. 10.1016/0022-2836(71)90324-X [DOI] [PubMed] [Google Scholar]
- 27. Sharp KA, Nicholls A, Fine RF, Honig B. Reconciling the magnitude of the microscopic and macroscopic hydrophobic effects. Science. 1991;252: 106–109. 10.1126/science.2011744 [DOI] [PubMed] [Google Scholar]
- 28. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215: 403–10. 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- 29. Zhang J, Zhang L, Li X, Xu W. UDP-3-O-(R-3-hydroxymyristoyl)-N-Acetylglucosamine Deacetylase (LpxC) Inhibitors: A New Class of Antibacterial Agents. Curr Med Chem. 2012;19: 2038–2050. [DOI] [PubMed] [Google Scholar]
- 30. Schweitzer BI, Dicker AP. Dihydrofolate reductase as a therapeutic target. FASEB J. 1990;4: 2441–2452. [DOI] [PubMed] [Google Scholar]
- 31. De Jonge BLM, Walkup GK, Lahiri SD, Huynh H, Neckermann G, Utley L, et al. Discovery of inhibitors of 4’-phosphopantetheine adenylyltransferase (PPAT) to validate PPAT as a target for antibacterial therapy. Antimicrob Ag Chemother. 2013;57: 6005–15. 10.1128/AAC.01661-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Blankenfeldt W, Asuncion M, Lam JS, Naismith JH. The structural basis of the catalytic mechanism and regulation of glucose-1-phosphate thymidylyltransferase (RmlA). EMBO J. 2000;19: 6652–63. 10.1093/emboj/19.24.6652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Alphey MS, Pirrie L, Torrie LS, Boulkeroua WA, Gardiner M, Sarkar A, et al. Allosteric Competitive Inhibitors of the Glucose-1-phosphate Thymidylyltransferase (RmlA) from Pseudomonas aeruginosa. ACS Chem Biol. 2012; 10.1021/cb300426u [DOI] [PubMed] [Google Scholar]
- 34. Gaulton A, Bellis LJ, Bento a P, Chambers J, Davies M, Hersey A, et al. ChEMBL: a large-scale bioactivity database for drug discovery. Nucl Acids Res. 2012;40: D1100–7. 10.1093/nar/gkr777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bickerton GR, Paolini G V, Besnard J, Muresan S, Hopkins AL. Quantifying the chemical beauty of drugs. Nat Chem. 2012;4: 90–8. 10.1038/nchem.1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Le Guilloux V, Schmidtke P, Tuffery P. Fpocket: an open source platform for ligand pocket detection. BMC Bioinformatics. 2009;10: 168 10.1186/1471-2105-10-168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tripathi A, Kellogg GE. A novel and efficient tool for locating and characterizing protein cavities and binding sites. Proteins. 2010;78: 825–42. 10.1002/prot.22608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. An J, Totrov M, Abagyan R. Pocketome via comprehensive identification and classification of ligand binding envelopes. Mol Cell Proteom. 2005;4: 752–61. 10.1074/mcp.M400159-MCP200 [DOI] [PubMed] [Google Scholar]
- 39. Brady GP, Stouten PF. Fast prediction and visualization of protein binding pockets with PASS. J Comput Mol Des. 2000;14: 383–401. [DOI] [PubMed] [Google Scholar]
- 40. Brylinski M, Skolnick J. A threading-based method (FINDSITE) for ligand-binding site prediction and functional annotation. Proc Nat Acad Sci USA. 2008;105: 129–34. 10.1073/pnas.0707684105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Weisel M, Proschak E, Schneider G. PocketPicker: analysis of ligand binding-sites with shape descriptors. Chem Cent J. 2007;1: 7 10.1186/1752-153X-1-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Huang B, Schroeder M. LIGSITEcsc: predicting ligand binding sites using the Connolly surface and degree of conservation. BMC Struct Biol. 2006;6: 19 10.1186/1472-6807-6-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Laskowski RA. SURFNET: a program for visualizing molecular surfaces, cavities, and intermolecular interactions. J Mol Graph. 1995;13: 323–30, 307–8. [DOI] [PubMed] [Google Scholar]
- 44. Laurie ATR, Jackson RM. Q-SiteFinder: an energy-based method for the prediction of protein-ligand binding sites. Bioinformatics. 2005;21: 1908–16. 10.1093/bioinformatics/bti315 [DOI] [PubMed] [Google Scholar]
- 45. Liang J, Edelsbrunner H, Woodward C. Anatomy of protein pockets and cavities: measurement of binding site geometry and implications for ligand design. Prot Sci. 1998;7: 1884–97. 10.1002/pro.5560070905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Peters KP, Fauck J, Frömmel C. The automatic search for ligand binding sites in proteins of known three-dimensional structure using only geometric criteria. J Mol Biol. 1996;256: 201–13. 10.1006/jmbi.1996.0077 [DOI] [PubMed] [Google Scholar]
- 47. Fauman EB, Rai BK, Huang ES. Structure-based druggability assessment—identifying suitable targets for small molecule therapeutics. Curr Opin Chem Biol. Elsevier Ltd; 2011;15: 463–8. 10.1016/j.cbpa.2011.05.020 [DOI] [PubMed] [Google Scholar]
- 48. Henrich S, Salo-Ahen OMH, Huang B, Rippmann FF, Cruciani G, Wade RC. Computational approaches to identifying and characterizing protein binding sites for ligand design. J Mol Recognit. 2010;23: 209–19. 10.1002/jmr.984 [DOI] [PubMed] [Google Scholar]
- 49. McGovern SL, Shoichet BK. Information decay in molecular docking screens against holo, apo, and modeled conformations of enzymes. J Med Chem. 2003;46: 2895–907. 10.1021/jm0300330 [DOI] [PubMed] [Google Scholar]
- 50. Schmidt T, Bergner A, Schwede T. Modelling three-dimensional protein structures for applications in drug design. Drug Discov Today. Elsevier Ltd; 2014;19: 890–7. 10.1016/j.drudis.2013.10.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Moynie L, Schnell R, McMahon SA, Sandalova T, Boulkerou WA, Schmidberger JW, et al. The AEROPATH project targeting Pseudomonas aeruginosa: crystallographic studies for assessment of potential targets in early-stage drug discovery. Acta Crystallogr Sect F Struct Biol Cryst Commun. International Union of Crystallography; 2013;69: 25–34. 10.1107/S1744309112044739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Larkin MA, Blackshields G, Brown NP, Chenna R, Mcgettigan PA, McWilliam H, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23: 2947–2948. 10.1093/bioinformatics/btm404 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Results from homolog-based druggability predictions for the NRDLD dataset structures when using an ambiguous zone (Table B). List of PDB 3-letter codes for cofactors or common additives during the crystallography process. These compounds were not considered to mark a binding pocket (Table C). Scripts are available on https://github.com/ruthbrenk/DrugPred2.0.git.
(DOCX)
Data Availability Statement
All relevant data are in the AEROPATH database (http://aeropath.lifesci.dundee.ac.uk/). Scripts are available on Github (https://github.com/ruthbrenk/DrugPred2.0.git). Scripts are open source but rely on the Python package, which is free for academic researchers. Python license can be obtained from Openeye.