

Abstract
Background
Activation of the immune system affects the circadian clock. Tumor necrosis factor (TNF) and Interleukin (IL)-1β inhibit the expression of clock genes including Period (Per) genes and the PAR-bZip clock-controlled gene D-site albumin promoter-binding protein (Dbp). These effects are due to cytokine-induced interference of E-box mediated transcription of clock genes. In the present study we have assessed the two E-box binding transcriptional regulators Twist1 and Twist2 for their role in cytokine induced inhibition of clock genes.
Methods
The expression of the clock genes Per1, Per2, Per3 and of Dbp was assessed in NIH-3T3 mouse fibroblasts and the mouse hippocampal neuronal cell line HT22. Cells were treated for 4h with TNF and IL-1β. The functional role of Twist1 and Twist2 was assessed by siRNAs against the Twist genes and by overexpression of TWIST proteins. In luciferase (luc) assays NIH-3T3 cells were transfected with reporter gene constructs, which contain a 3xPer1 E-box or a Dbp E-box. Quantitative chromatin immunoprecipitation (ChIP) was performed using antibodies to TWIST1 and CLOCK, and the E-box consensus sequences of Dbp (CATGTG) and Per1 E-box (CACGTG).
Results
We report here that siRNA against Twist1 protects NIH-3T3 cells and HT22 cells from down-regulation of Period and Dbp by TNF and IL-1β. Overexpression of Twist1, but not of Twist2, mimics the effect of the cytokines. TNF down-regulates the activation of Per1-3xE-box-luc, the effect being prevented by siRNA against Twist1. Overexpression of Twist1, but not of Twist2, inhibits Per1-3xE-box-luc or Dbp-E-Box-luc activity. ChIP experiments show TWIST1 induction by TNF to compete with CLOCK binding to the E-box of Period genes and Dbp.
Conclusion
Twist1 plays a pivotal role in the TNF mediated suppression of E-box dependent transactivation of Period genes and Dbp. Thereby Twist1 may provide a link between the immune system and the circadian timing system.
Introduction
Clock genes mediate circadian rhythmicity and thereby control behavior, sleep-wake cycles, metabolism and inflammation [1–3]. Synchronization of the cellular clocks in the body is coordinated by the suprachiasmatic nuclei (SCN) in the brain. At the molecular level the clock consists of a network of transcription factors that functions in self regulated transcription-translation feedback loops. The heterodimerized transcription factors CLOCK-BMAL1 (brain and muscle ARNT-like protein) activate transcription by binding to the cis-regulatory element E-box in the promoters of Period (Per), Cryptochrome (Cry), and of the members of the PAR-bZip family of transcription factor genes including the D-site albumin promoter-binding protein (Dbp) [4, 5]. PER and CRY are transported into the cell nucleus where they inhibit the transcriptional activity of CLOCK-BMAL1 complexes, thereby inhibiting their own gene expression. The same positive and negative regulatory components also govern the rhythmic expression of the nuclear orphan receptor Rev-Erb-alpha, which in turn represses the transcription of Bmal1. These pathways drive the 24-h rhythms in physiology.
Sickness behavior syndrome (SBS) with fatigue and depression is observed in states of immune activation in the course of infections and autoimmune diseases [6]. SBS can be induced by peripheral or central administration of proinflammatory cytokines. The effect of the cytokines is associated with altered expression of clock genes. TNF and IL-1β have been shown to lead to inhibition of expression of Per1, Per2, Per3, Cry1 and Cry2 and of the PAR-bZip clock- controlled genes Dbp, tyrotroph embryonic factor (Tef), and hepatic leukemia factor (Hlf) [7]. In NIH-3T3 fibroblasts TNF exerts its effect by inhibiting CLOCK-BMAL1 induced activation of E-box regulatory elements in Dbp and Per promoters [7]. Interference with clock gene expression is also mediated by transforming growth factor beta (TGFβ) [8, 9]. TGFβ is overexpressed in neurons in Alzheimer`s disease and has been proposed to alter the circadian rhythm in aged patients [9].
The mechanisms involved in dysregulation of clock genes by immune activation have just recently been addressed. Because the addition of TNF to NIH-3T3 cells was found to increase Per and Cry genes in the first three hours before exerting its inhibitory effects in the following hours, it was thought that TNF triggers the well described negative feedback loop by which PER-CRY inhibit their own transcription. Experiments using Per1,Per2 -/- and Cry1,Cry2 -/- mouse embryonic fibroblasts did not confirm this hypothesis [10]. Another approach made use of recent studies, which indicate that the cold-inducible RNA-binding protein (CIRBP) is required for high amplitude circadian gene expression [11]. Among the transcripts interacting with CIRBP are the mRNAs encoding Per2, Per3, and Dbp, which were decreased in Cirbp-depleted cells [11, 12]. TNF was found to modulate clock gene expression by impairing the production of CIRBP [13]. However lower Cirbp expression was only responsible for the effect of TNF when using suboptimal TNF concentrations in the NIH-3T3 cell assay. Taken collectively the pathways involved in TNF mediated abnormal clock gene expression are still unknown. This contrasts the inhibitory effect of TGFβ on clock genes, which results from the induction of Dec1 [8]. This E-box binding protein competes with CLOCK:BMAL1 for the DNA binding in clock genes.
In the search for factors, which upon induction by TNF interfere with E-box mediated transcription we have put our focus on the transcriptional regulators Twist 1 and Twist 2. The two members of the basic helix-loop-helix (bHLH) family of proteins have been characterized as transcriptional regulators, which bind to E-boxes [14]. TWIST2 is 66% identical to TWIST1 and the identity increases to 98% in the basic and HLH regions of the proteins. At the C-terminus a repressor domain termed “Twist box” is located, which is identical in TWIST1 and TWIST2. However, the two DNA binding proteins differ in their individual roles as activators and repressors of gene transcription. Twist1 is expressed in embryonic stages and is important for early development and osteoblast differentiation [15, 16]. Recent work also shows that Twist1 is overexpressed in many human cancers and cancer cell lines, thereby driving tumor invasion and metastasis [17–19]. TWIST1 and -2 are known to repress cytokine gene expression and epithelial–mesenchymal transition through interaction with NFκB mediated transcription [20]. Both proteins form dimers and depending on their phosphorylation state may interact with other bHLH family members including the E-proteins E12/E47 [14].
The property of TNF to suppress the expression of clock genes may involve TWIST1 and TWIST2. This hypothesis is based on (i) the effect of TNF to interact with the Clock / Bmal1 induced activation of E-boxes of Period genes and Dbp [7], (ii) the property of TWIST1 and TWIST2 to bind to E-boxes, and (iii) on the induction of Twist1 and Twist2 by TNF, the effect being mediated by NF-κB [20, 21]. The hypothesis of Twist genes being involved in TNF mediated clock gene inhibition is supported by the recent finding of Twist1 and Twist2 to inhibit the expression of TNF, which may indicate bidirectional regulatory control of their expression [20]. Here, we describe the novel function of Twist1 to serve as a transcriptional repressor mediating TNF effects on Period and Dbp genes by interfering with CLOCK induced E-box binding and functions thereof.
Materials and Methods
Cell culture
The murine fibroblast cell line NIH-3T3 (CRL-1658) was obtained from the American Type Culture Collection. The mouse hippocampal neuronal cell line HT22 was obtained from David Schubert at the Salk Institute (La Jolla, CA). [22]. NIH-3T3 cells and HT22 cells were grown in DMEM (Gibco) with 10% FBS (Sigma) at 37°C and 5% CO2. For assessing the effect of Twist on clock genes cells were synchronized for 1h with serum deprivation and then treated with TNF (0.1, 1.0 or 10ng/ml), IL-1β (0.1, 1 or 10ng/ml) or TGFβ2 (2ng/ml) (Peprotech) for 4h as described recently [7]. Time kinetic analysis was performed by treating the cells with TNF (10ng/ml) for 24hr, cells being harvested every three hours.
SiRNA experiments and overexpression
Knockdown experiments were performed in NIH-3T3 and HT22 using ON-TARGETplus siRNA (Dharmacon) against Twist1 and a DharmaFECT transfection reagent. Twenty-four hours before transfection, the cells were seeded in 6-well plates (150,000 cells/well). The transfections used siRNA solutions (5 μM) in 1× siRNA buffer (Dharmacon). The transfection reactions were performed according to the manufacturer's instructions. After 48 h, the cells were harvested for mRNA analyses.
Overexpression of Twist1 and Twist2 was established by stable transfection of TrueClone pCMV6::Twist1, pCMV6::Twist2 and pCMV6::empty plasmid (Origene) into NIH-3T3 cells with FugeneHD transfection reagent (Promega) according to the manufacturer's instructions. G418 (Promega) was used as selection antibiotics at a concentration of 0.5mg/ml.
RNA extraction, cDNA synthesis and qRT-PCR analyses
Total RNA from NIH-3T3 and HT22 cells were obtained with the NucleoSpin RNAII Kit (Macherey-Nagel), including DNase digestion. Total RNA (1 μg), anchored oligo(dT)18 primer (2.5 μM) and reverse transcriptase (10 U) from the nanoScript 2 reverse transcription kit (PrimerDesign) were used for the first strand synthesis according to the manufacturer's suggestions. The cDNA was stored at −20°C.
The qRT-PCR primers for Twist1, Twist2, Dbp, Per1, Per2 and Per3 were commercially available from PrimerDesign Each of the samples was examined in triplicate and subjected to qRT-PCR using PrecisionPLUS qPCR Mastermix (PrimerDesign). HPRT and eEF1a1 probes were used as internal controls. Relative RNA expression levels were determined by normalizing to the internal controls, and the values were calculated using the comparative Ct method [23].
Western blot analysis
Immunoblots were performed with samples containing total protein (40 μg) and 12% NuPage Bis-Tris or 7% Tris-Acetat polyacrylamide gels (LifeTechnologies). The membranes were probed with rabbit polyclonal anti-PER1 (1:500; Abcam ab136451), rabbit polyclonal anti PER2 (1:250; ab180655), rabbit monoclonal anti-PER3 (1:2500; ab177482), rabbit polyclonal anti-DBP (1:400; ab22824), rabbit polyclonal anti-Twist1 (1:400; ab49254) and rabbit polyclonal anti-TWIST2 antibodies (1μg/ml; ab66031). The secondary antibody was a horseradish peroxidase-linked goat anti-rabbit IgG (various concentrations; ab97051), and it was detected by the chemiluminescence technique using the Immobilon Western ECL system (Millipore). For a loading control, we used a mouse monoclonal anti-p84 antibody (nuclear matrix protein 84; 1:2000; Abcam ab487). The secondary antibody was a horseradish peroxidase-linked goat anti-mouse IgG (1:5000; Abcam; ab20043). Densitometric analysis was performed using the ImageJ software and the intensity of the control lanes were set as 1.
Cotransfection and dual luciferase assay
NIH-3T3 cells were grown as described above. The transfections were performed with Effectene transfection reagent (Qiagen) according to the manufacturer's protocol. Briefly, 24 h before transfection, cells were seeded in 12-well plates (200,000 cells/well). The transfections required DNA (450 ng), the pGL3 promoter vector (firefly) with our inserts, the pRL SV40 vector (renilla) as an internal control, and 240.8 μl of transfection reagent for triplicates (212 μl of EC Buffer, 10.8 μl of Enhancer and 18 μl Effectene). At 48 h after transfection, cells were washed with PBS and lysed with passive lysis buffer (Promega) to perform a conventional dual luciferase assay (DLR). Lysate (20 μl) was placed into each well of a white 96-well plate and measured with an Infinite M200 pro Luminometer (Tecan). The DLR was performed according to the manufacturer's instructions, except that the amount of substrate was reduced to 50 μl per aliquot. In this assay we tested first a construct containing 3 E-boxes from the Period1 promoter (TTTAGCCACGTGACAGTGTAAGCACACGTGGGCCCTCAAGTCCACGTGCAGGGA), as well as the mutated counterpart (TTTAGCACCGGTACAGTGTAAGCAACCGGTGGCCCTCAAGTCAC CG GTCAGGGA). From the Dbp promoter one E-box, wild type (CACGTCCCATGTGGCC) and mutated (CACGTC G CA GTTGGCC) was cloned into the reporter plasmid according to: [24, 25]. The Period1 E-Box, as well as the mutated sequence were inserted into the pGL3 promoter vector via the Nhe1/Xho1 site. The cloned sequence consists of three E-boxes within 2.0 kb of the 5’-flanking region of the mouse Per1 gene. The Dbp E-Box and the mutated counterpart were amplified out of a shuttle plasmid and ligated into the BglII restriction site of the pGL3 promoter vector.
Chromatin immunoprecipitation (ChIP)
NIH-3T3 cells were grown in 15 cm petri dishes under normal conditions up to 80% confluency and fixed with formaldehyde. Fixation, enzymatic shearing and immunoprecipitation were performed with the ChIP-IT Express Enzymatic Kit (Active Motif) according to the manufacturer’s instructions. Chromatin was precipitated with either a monoclonal ChIP grade antibody against TWIST1 (Abcam, ab50887) or anti CLOCK antibody (ab 3517). As positive control a monoclonal RNA Pol II antibody, as negative control a IgG antibody was used 10μg of sheared chromatin and 2μg of antibody was used in 100μl reaction volume. Eluted and reverse cross-linked chromatin was analyzed via qPCR of region of interest. Primers for E-box regions:
Per1 E-box:
fw: 5’-GGGTAGTTTCCCTCCCTCAC-3’
rv: 5’- TGGCATCTGATTGGCTACTG-3’.
Dbp E-box:
fw: 5’-TCAACATGGTACAGCCCAGA-3’
rv: 5’-TGTGGGAGCTGAGCACATAG-3’.
The analysis method entailed solving for the DNA quantity of the ChIP and IgG samples, then calculating the fold enrichment of the ChIP sample relative to the IgG sample.
Statistics
All experiments were performed in triplets and analyzed with GraphPad Prism (GraphPad Software). To assess the statistical significance between single series of measurements (i.e. untreated vs. treated) we used the two-tailed Student’s t-test. For grouped measurements with one variable for the selected genes, an one-way ANOVA was used, followed by a Bonferroni post-hoc test to decrease the likelihood of a rare event in multiple comparisons. In the case of two variables (i.e. cytokine treatment and siRNA/overexpression) a two-way ANOVA was used to measure the statistical significance, also followed by a Bonferroni post-hoc test to decrease the α-error margin. P values less than 0.05 were considered significant, 0.01 in ANOVAs with post-hoc tests. +/- SEM is shown by error bars.
Results
Recent studies showed TNF and IL-1β to increase the expression of Twist genes in various types of cells [21, 26]. To test whether NIH-3T3 fibroblasts and HT22 hippocampal neuronal cells similarly respond to cytokines with upregulation of Twist1 cells were treated for 4 h with TNF and IL-1β at various concentrations. Both TNF and IL-1β induced the expression of Twist1 in a dose dependent fashion (Fig 1A and 1B). In contrast to TNF and IL-1β, TGFβ2 failed to augment the expression of Twist1 mRNA in NIH-3T3 cells (Fig 1C). Upregulation of TWIST1 by TNF and IL-1β, but not by TGFβ2 was also seen at the protein level (Fig 1D). Densitometric analysis of the Western blot showed TNF and IL-1β to increase TWIST1 and TGFβ2 to have only a minor inhibitory effect. Compared to untreated cells TWIST1 expression in cells treated with the aforementioned cytokines was 3.3, 2.8 and 0.9-fold respectively.
Fig 1. Twist1 is induced by TNF and IL-1β, but not by TGFβ2.

Dose dependent expression of Twist1 mRNA in NIH-3T3 (A) and HT22 (B) cells after a 4 hr treatment with different concentrations of TNF (0.1, 1 and 10ng/ml), and IL-1β (0.1, 1 and 10ng/ml). (C) Expression of Twist1 mRNA in NIH-3T3 after a 4 hr treatment with TNF (10ng/ml) and IL-1β (10ng/ml). The dose of TGFβ2 added (2ng/ml) was chosen from previous studies on effects of this cytokine on the expression of clock genes [8, 9]. (D) Western blot showing upregulation of TWIST1 in cell lysates of NIH-3T3 cells treated for 4 hr with TNF (10ng/ml), IL-1β (10ng/ml), but not after TGFβ2 treatment (2ng/ml). Nuclear matrix protein p84 was used as loading control. Data of RT-qPCR assays show the mean +/- SEM (error bars) of triplicates from three independent experiments. Significance of grouped results were calculated with one way-ANOVA and Bonferroni post-hoc test;*** p<0.001. Western blot shows one representative experiment of three.
Cytokines have been shown to interfere with the expression of clock genes [7–9]. Consistent with these reports we find a 4h treatment of NIH-3T3 cells with TNF and IL-1β to inhibit the expression of Dbp, Per1, Per2, and Per3 (Fig 2A). While having no effect on Twist1 expression, TGFβ2 shared with TNF and IL-1β its effects on clock genes. Next we determined whether Twist1 induction is responsible for the down-regulation of clock genes by cytokines. The hypothesis was tested by suppressing Twist1 expression by siRNA. NIH-3T3 were transfected for 36 h with siRNA against Twist1 and siNOtarget control. Depletion of Twist1 resulted in an almost complete prevention of the TNF and IL-1β mediated suppression of Dbp, Per1, Per2 and Per3 (Fig 2B). However, siRNA against Twist1 did not alter the inhibitory effect of TGFβ2 on the expression of clock genes. Whereas siRNA against Twist1 had no effect on basal expression of Dbp, Per1 and Per2, it upregulated the expression of Per3 irrespective of whether the cells were exposed to TNF or not (Fig 2B). Moreover in NIH-3T3 cells treated with siRNA against Twist1 the upregulation of Per3 was not seen in the presence of IL-1β or TGFβ2. To assess whether the prevention of cytokine mediated inhibition of Period genes and of Dbp by siRNA against Twist1 is unique to NIH-3T3 cells we studied another type of cell. As observed in NIH-3T3 cells also HT22 neuronal cells respond to TNF with up-regulation of Twist1 (Fig 1B); prevention of this effect by siRNA against Twist1 abolished the inhibitory effect of TNF on Period genes and on Dbp (Fig 2B). siRNA against Twist1 up-regulated the expression of Per3 only in NIH-3T3 cells, but not in HT22 cells (Fig 1B). The latter type of cell, however, responded to siRNA against Twist1 with an up-regulation of Per1 when exposed to IL-1β (Fig 2B). Taken collectively siRNA against Twist1 prevents from TNF and IL-1β induced suppression of Period genes and Dbp in both NIH-3T3 and HT22 cells. The mechanisms, which lead to different effects on expression of Per1 and Per3 in the two types of cells treated with siRNA against Twist1 are not yet clear.
Fig 2. siRNA against Twist1 prevent from inhibition of clock gene expression by TNF and IL-1β.

(A) Expression of clock genes in NIH-3T3 cells treated with TNF (10 ng/ml), IL-1β (10 ng/ml) and TGFβ2 (2 ng/ml) for 4 hr. Data of RT-qPCR assays of Dbp, Per1, Per2, Per3, and Twist1 mRNA expression show the mean +/- SEM (error bars) of triplicates from three independent experiments. (B) Effect of cytokines on clock genes in NIH-3T3 cells (upper panel) and HT22 cells (lower panel), the cells being pretreated with siRNA against Twist1. (siTwist1 ). (C) Western blot of lysates from NIH-3T3 cells, which were pretreated with siRNA against Twist1 and thereafter with TNF for 4 hr. Matrix protein p84 was used as loading control. (D) Intensity of bands, which are shown in Fig 2C by Western blot. The data are given as ratio of untreated versus treated cells. Untreated lanes were set as 1. The concentrations of the cytokines used in the experiments are given in legend to Fig 1C. Significance of RT-qPCR data was calculated with two way-ANOVA and Bonferroni post-hoc test;* p<0.01, ** p<0.01, *** p<0.001. Western blot shows one representative experiment of three.
When assessing the effect of TNF on clock genes at the protein level, treatment of NIH-3T3 cells with TNF was found to reduce the amount of DBP, PER1, PER2 and PER3 in NIH-3T3 cells (Fig 2C and 2D). The inhibitory effect of TNF on clock proteins was prevented by siRNA against Twist1. The increase of expression of Per3 mRNA in cells treated with siRNA agaist Twist1 was not observed at the protein level (Fig 2C and 2D).
Next we performed time kinetic experiments using NIH-3T3 cells. As reported the NIH-3T3 cell line used in our study responds to serum deprivation with a rhythmic expression of Dbp mRNA; peak expression is observed at 24 hr (Fig 3A) [7]. Inhibition of Dbp mRNA by TNF was significant at all time points assessed. The induction of Twist1 by TNF was maximal at 3hr and declined thereafter. Decreased expression of Twist1 was paralleled by an increase of Dbp mRNA (Fig 3A and 3B). To determine the potential of Twist1 to act as a repressor of clock genes we overexpressed Twist1 and assessed the expression of clock genes in the absence of exogenously added cytokines. Overexpression of Twist1 interfered profoundly with Dbp expression during the 24 hr study period (Fig 3A). The effectiveness of Twist1 transfection using pCMV6 vectors was controlled and found to result in an increase of Twist1 mRNA between 3.5 and 4.5 fold above the empty vector control (Fig 3B). As shown in Fig 3C NIH-3T3 cells, which overexpress Twist1 for 4 hr show not only a reduction of Dbp, but also of Per1, Per2 and Per3 expression when compared to cells transfected with empty vector control. Overexpression of Twist1 did not alter the expression of Twist2 mRNA (Fig 3B). Taken collectively our data characterize Twist1 as a repressor of clock genes.
Fig 3. Time kinetic of TNF effects on Twist1 and Dbp expression, and suppression of Dbp by overexpressiom of Twist1.

(A) Expression of Dbp and (B) of Twist1 mRNA in NIH-3T3, which were treated with TNF (10 ng/ml) for 3 to 24 hr or transfected with the pCMV6::Twist1 expression vector. The control consisted of transfection with the empty vector (pCMV6). (C) Expression of clock gene mRNA after transfection with pCMV6::Twist1 expression vector or empty vector control in NIH-3T3 cells. Overexpression of Twist1 was controlled by RT-qPCR and the effect on Twist2 assessed at 4hr. Data of RT-qPCR assays of Dbp, Per1, Per2, Per3, and Twist1 mRNA expression show the mean +/- SEM (error bars) of triplicates from three independent experiments. Data are analyzed with one way-ANOVA and Bonferroni post-hoc test; * p<0.01, ** p<0.01, *** p<0.001
In the light of the high degree of homology of Twist1 with Twist2 we studied the effect of Twist2 on clock genes. As observed with Twist1, also Twist2 mRNA and protein are upregulated by treatment of NIH-3T3 cells with TNF and IL-1β, but not when adding TGFβ2 to the cells (Fig 4A and 4B). The overexpression of TWIST2 failed, however, to interfere with the expression of clock genes (Fig 4C). Overexpression of TWIST2 was verified by Western blot (Fig 4D). Taken collectively we find that Twist2 does not share Twist1‘s inhibitory effects on clock genes.
Fig 4. Twist2 does not impair the expression of clock genes.

(A) Expression of Twist2 mRNA in NIH-3T3 cells after a 4 hr treatment with TNF, IL-1β and TGFβ2. The concentrations of the cytokines used are given in the legend to Fig 1. Data of RT-qPCR assays show the mean +/- SEM of three independent experiments. (B) Western blot showing upregulation of TWIST2 in cell lysates of NIH-3T3 cells treated with TNF for 4 hr. Nuclear matrix protein p84 was used as loading control. (C) Transfection of NIH-3T3 cells with pCMV6::Twist2 show no significant effect on expression of Dbp, Per1, Per2 and Per3 compared to the empty vector control. (D) Data show TWIST2 expression in untreated (empty vector control) and TNF treated NIH-3T3 cells and in cells transfected with pCMV6::Twist2. Data of RT-qPCR assays show the mean +/- SEM (error bars) of triplicates from three independent experiments. Significance was calculated with two-way ANOVA and Bonferroni post-hoc test; *** p<0.001. Western blots show one representative experiment of three.
E-boxes are functionally important components of DNA promoters that guide the expression of clock genes. Rhythmic binding of CLOCK and BMAL1 depends on E-boxes and is a prerequisite for robust waves of gene expression characteristic of circadian transcription [27, 28]. Previous data show that only clock genes with E-boxes in their promoter, the PAR bZip genes Dbp, Tef, and Hlf and the Per genes, are affected by TNF, whereas clock genes devoid of E-boxes such as Clock and Bmal1 are not affected by TNF [7]. To explore the predication that Twist1 operates at the site of E-elements in clock promoters we tested the effect of cytokines on Per1-3xE-box-luciferase (luc) reporter in the presence or absence of siRNA against Twist1. TNF down-regulates the activation of Per1-3xE-box-luc (Fig 5A). This effect of TNF was not observed when cells were transfected with siRNA against Twist1. On the other hand inhibition of TNF mediated Per1-3xE-box-luc activity was mimicked by overexpression of Twist1, but not of Twist2. An analogous picture emerged when testing IL-1β. The IL-1β induced suppression of Per1-3xE-box-luc was found to depend on the presence of Twist1 (Fig 5B). However, depletion of Twist1 by siRNA failed to prevent the inhibitory effect of TGFβ2 on Per1-3xE-box-luc activity (Fig 5C). The experiments using cytokines and siRNA against Twist1 were also performed using Dbp-E-Box-luc. Confirming the experiments with the Per1-3xE-box-luc construct, the TNF and IL-1β induced inhibition of Dbp-E-box-luc activity was dependent on Twist1 (Fig 5D and 5E). Again the effect of TGFβ2 was independent of the presence or absence of Twist1 (Fig 5F). This finding is consistent with the failure of TGFβ2 to enhance the expression of Twist1.
Fig 5. Dual luciferase assay for different E-box containing constructs.

NIH-3T3 cells were transfected with different reporter gene constructs, which contain a 3xPer1 E-box (A-C) or a Dbp E-box (D-F). The concentrations of the cytokines used are provided in the legend to Fig 1. Results are given for wild type and mutated E-box, as well as cytokine treated cells (4 hr) and co-transfection with Twist1 and Twist2 overexpression plasmids respectively. pGL3 basic was used as negative, pGL3 promoter as positive control. White bars represent the constructs containing the corresponding E-box. Data of RT-qPCR assays show the mean +/- SEM (error bars) of triplicates from three independent experiments. ** p<0.01, *** p<0.001; two-way ANOVA followed by the Bonferroni post-hoc test (n = 3)
To test whether the E-box of Dbp and Per1 are transcriptional targets of TWIST1, we performed quantitative ChIP experiments using anti-TWIST1 and anti-CLOCK antibodies. We found that TWIST1 is highly enriched at E-box elements of Dbp in the presence of TNF (120- fold). In the absence of TNF, CLOCK is 243-fold enriched at the Dbp E-box. The knock-down of Twist1 by siRNA treatment abrogates the enrichment of TWIST1 to the Dbp E-box (22-fold) and despite of the presence of TNF enhanced the binding of CLOCK (221-fold) to the E-box (Fig 6A). The overall enrichment at the Per1 E-box is less pronounced. However, as observed with the Dbp E-box treatment with TNF led to an enrichment of TWIST1 compared to CLOCK (63-fold vs. 8-fold) (Fig 6B). In the absence of TNF the ratio is inverted to TWIST1 4-fold, CLOCK 113-fold. Silencing of Twist1 expression by siRNA increased CLOCK binding at the Per1 E-box despite of the presence of TNF.
Fig 6. Quantitative ChIP for TWIST1 and CLOCK showed a preference of TWIST1 binding to E-box consensus sequence in the presence of TNF.
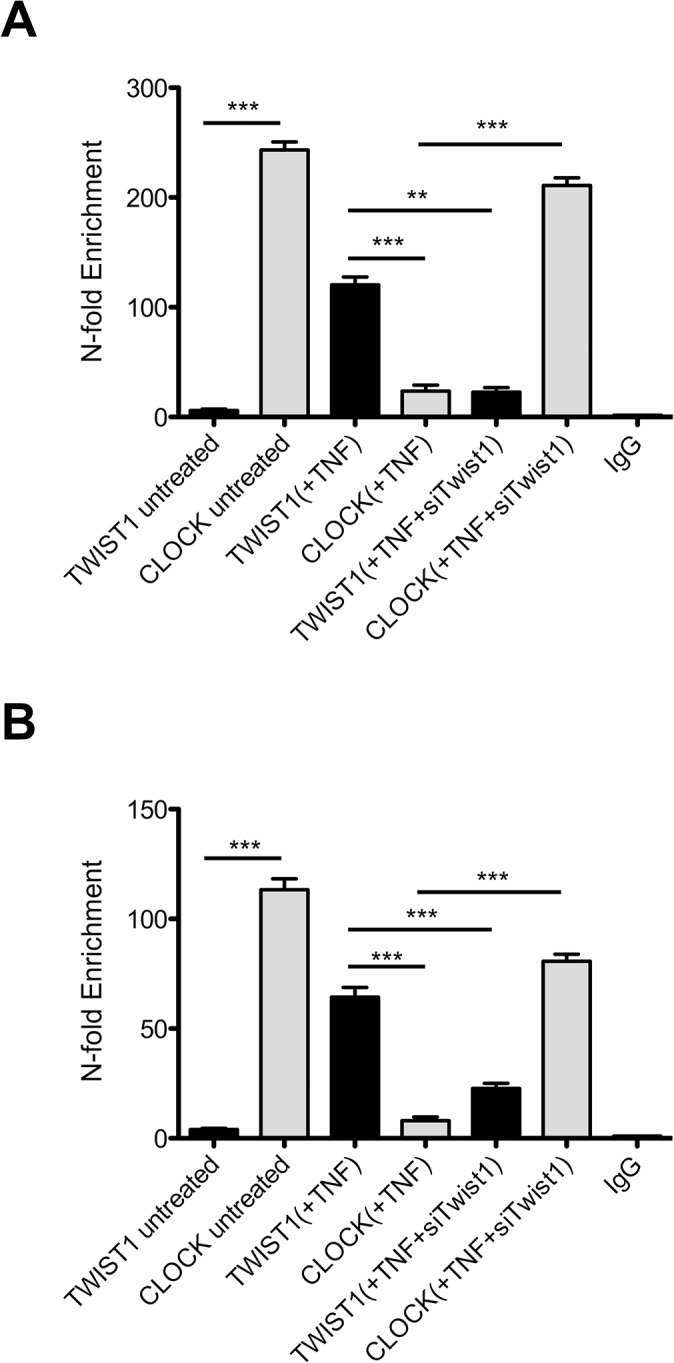
Binding of TWIST1 and CLOCK on the Dbp (CATGTG) E-box (A) and Per1 E-box (CACGTG) (B) in NIH-3T3 cells. The correspondent treatment is given in brackets. TNF (10ng/ml) treatment was performed for 4 hr. IgG antibody was used as negative control. Data of RT-qPCR assays show the mean +/- SEM (error bars) of triplicates from three independent experiments. The significance was determined by student’s t-test; ** p<0.01, *** p<0.001.
Taken together these results support the hypothesis that TWIST1 induction by TNF competes with CLOCK binding to the E-box.
Discussion
Dysregulation of clock gene expression by proinflammatory cytokines has been described in fibroblasts, human pancreatic cancer cells, and in leukocytes in vitro, and in the liver and SCN of mice with CD40 mediated inflammatory syndrome or upon treatment with lipopolysaccharide (LPS) from gram-negative bacteria, interferon alpha (IFN) or TNF [29–34]. The intracellular pathway responsible for the inhibition of clock genes by cytokines has not yet been identified. Our data identify Twist1 as a TNF induced repressor of the expression of Per1, Per2, Per3 and Dbp. Upon treatment with TNF and IL-1β NIH-3T3 fibroblasts and HT22 hippocampal neuronal cells show a striking upregulation of Twist1 and Twist2. These data are in agreement with recent reports showing TNF and IL-1β to induce both DNA binding proteins in different types of cells including fibroblasts, macrophages, breast epithelial cells, and various tumor cells [21, 26]. Besides the aforementioned cytokines, also IFNα stimulates Twist1 expression [26]. However, its induction in macrophages requires treatment of the cells for two days. The delayed response was found to be due to the dependence of Twist1 induction on IFN induced production of Axl. This member of the Tyro 3 kinase family is preferentially expressed in myeloid, neuronal and reproductive system cells. Both the delayed kinetics of Twist1 induction by IFNα and the cell specificity of Axl expression may explain absence of inhibition of clock gene expression when treating NIH-3T3 cells for 4 hr with IFNα.
Twist1 is involved in development and osteoblast differentiation and has been characterized as an essential player in cancer stem cell generation, tumor invasion and metastasis. The data provided here show Twist1 to act as a repressor of the transcriptional activity of CLOCK on E-box mediated induction of Per1 and Dbp expression. siRNA against Twist1 leads to an almost complete prevention from TNF and IL-1β induced inhibition of clock genes. This effect is not shared with effects of siRNA against Twist2. Likewise the overexpression of Twist1, but not of Twist2 inhibits clock gene expression in NIH-3T3 cells. Thus Twist1 is characterized here as the main TNF-induced repressor of clock genes in NIH-3T3 cells. Recently the effect of TNF was shown to be dependent on down-regulation of Cirbp [13], which is required for high amplitude expression of clock genes [11, 13]. However, experiments using siRNA against Cirbp show that downregulation of Cirbp plays only a role in inhibition of clock gene expression at low doses (1 ng/ml) of TNF. At high doses (10 ng/ml) TNF operates in a Cirbp independent way [13], the effect of TNF being dependent on induction of Twist1.
Many of the target genes of TWIST1 and TWIST2 have multiple E-boxes in their promoters. Induction of transcriptional inhibition by TWIST1 includes blocking of DNA binding and/or the action of other transcriptional activators, binding E-boxes as heterodimers with E-proteins, and recruitment of histone deacetylases [14]. In TNF treated NIH-3T3 cells we find TWIST1 binding to E-box elements in the Per1 and Dbp promoter to be associated with absence of CLOCK protein at the respective sites. These data suggest that by occupying the E-box elements in the Per1 and Dbp promoters TWIST1 inhibits binding of CLOCK. It remains to be seen whether Twist1 functions as a homodimer or, as reported previously, the effects of Twist1 may depend on formation of heterodimers with e.g. other class B bHLH proteins [14]. Moreover it can not be excluded that TWIST1 interacts with the master clock genes CLOCK, BMAL1 and/or NPAS2 and thereby hinders their access to the E-box or alters the intrinsic histone acetyltransferase activity of CLOCK, which has been well described to contribute to chromatin-remodeling events implicated in circadian control of gene expression [35]. But generally TWIST binds direct to the E-box to induce or repress transcription [36, 37].
While TGFβ2 shares with TNF and IL-1β its effect to inhibit the expression of Period and of the PAR bZip genes Dbp, Tef, and Hlf, but not of Bmal1 and Clock [9], the interference of TGFβ2 with the clock system is found here to be independent of Twist (see above). In contrast to TNF and IL-1β, TGFβ2 does not activate Twist1 expression. In line with absence of Twist1 activation we find siRNA against Twist1 not to be protective for the effect of TGFβ2 to inhibit clock gene expression. Previous work showed TGFβ to act on the circadian system by induction of Dec1 [8]. Dec1, another bHLH factor, is induced by CLOCK:BMAL1 heterodimer via the CACGTG E-box in the promoter and, thereafter, suppresses transcription by competing with CLOCK-BMAL1 for the DNA binding. DEC1 upregulation by TGFβ resets the circadian phase of peripheral clocks [38]. The different pathways involved in the negative control of clock gene expression by TNF/IL-1β and TGFβ is interesting from an immunological point of view. While TNF/IL-1β are mainly increased in the acute phase of the immune response, TGFβ is a cytokine, which dampens immune effector mechanisms and mediates repair processes at later stages of inflammatory diseases.
Being induced by proinflammatory cytokines, Twist1 may be increased in the periphery and / or in the brain in states of immune activation in infectious–and autoimmune diseases. Upregulated Twist1 may lead to low levels of E-box dependent transcription induced by CLOCK-BMAL1. Since E-boxes are very frequent in the mammalian genome, CLOCK-BMAL1 heterodimers regulate the transcription of many genes besides the clock genes Per and Cry. Thus the TNF mediated increase of Twist1 may modulate a multitude of physiological processes. Experimental in-vivo evidence in support of the hypothesis would require mice with a deletion of the Twist1 gene. However, Twist1 -/- mice die at E10.5 with increased apoptosis in multiple tissues [15]. Mutations in human Twist1 lead to the Saethre-Chotzen syndrome, which is associated with severe abnormalities in fusion of the cranial bones and digit defects [39]. Thus cell-type specific inducible deletions of the Twist1 gene are required to substantiate the hypothesis that Twist1 transmits signals of the immune system to the clock gene network at the transcriptional level.
The ability of TWIST1 to bind to E-box elements and to compete with CLOCK-BMAL1 mediated activation of Period genes and Dbp points to a previously unrecognized function of Twist1 and suggests that Twist1 plays a critical role linking the immune and clock systems.
Supporting Information
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
The work was supported to A.F. by the Swiss National Science Foundation (Project 141055), the Baugarten Stiftung and the Lotex Foundation. Adriano Fontana is Hertie Senior Research Professor for Neuroscience of the Gemeinnützige Hertie-Stiftung.
References
- 1. Eckel-Mahan K, Sassone-Corsi P. Metabolism and the circadian clock converge. Physiol Rev. 2013;93(1):107–35. 10.1152/physrev.00016.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Franken P. A role for clock genes in sleep homeostasis. Current opinion in neurobiology. 2013;23(5):864–72. 10.1016/j.conb.2013.05.002 . [DOI] [PubMed] [Google Scholar]
- 3. Curtis AM, Bellet MM, Sassone-Corsi P, O'Neill LA. Circadian clock proteins and immunity. Immunity. 2014;40(2):178–86. 10.1016/j.immuni.2014.02.002 . [DOI] [PubMed] [Google Scholar]
- 4. Dibner C, Schibler U, Albrecht U. The mammalian circadian timing system: organization and coordination of central and peripheral clocks. Annu Rev Physiol. 2010;72:517–49. 10.1146/annurev-physiol-021909-135821 . [DOI] [PubMed] [Google Scholar]
- 5. Ko CH, Takahashi JS. Molecular components of the mammalian circadian clock. Hum Mol Genet. 2006;15 Spec No 2:R271–7. 10.1093/hmg/ddl207 . [DOI] [PubMed] [Google Scholar]
- 6. Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9(1):46–56. 10.1038/nrn2297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cavadini G, Petrzilka S, Kohler P, Jud C, Tobler I, Birchler T, et al. TNF-alpha suppresses the expression of clock genes by interfering with E-box-mediated transcription. Proc Natl Acad Sci U S A. 2007;104(31):12843–8. Epub 2007/07/25. 10.1073/pnas.0701466104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kon N, Hirota T, Kawamoto T, Kato Y, Tsubota T, Fukada Y. Activation of TGF-beta/activin signalling resets the circadian clock through rapid induction of Dec1 transcripts. Nat Cell Biol. 2008;10(12):1463–9. Epub 2008/11/26. 10.1038/ncb1806 . [DOI] [PubMed] [Google Scholar]
- 9. Gast H, Gordic S, Petrzilka S, Lopez M, Muller A, Gietl A, et al. Transforming growth factor-beta inhibits the expression of clock genes. Ann N Y Acad Sci. 2012;1261:79–87. Epub 2012/07/25. 10.1111/j.1749-6632.2012.06640.x . [DOI] [PubMed] [Google Scholar]
- 10. Petrzilka S, Taraborrelli C, Cavadini G, Fontana A, Birchler T. Clock gene modulation by TNF-alpha depends on calcium and p38 MAP kinase signaling. J Biol Rhythms. 2009;24(4):283–94. Epub 2009/07/25. 10.1177/0748730409336579 . [DOI] [PubMed] [Google Scholar]
- 11. Morf J, Rey G, Schneider K, Stratmann M, Fujita J, Naef F, et al. Cold-inducible RNA-binding protein modulates circadian gene expression posttranscriptionally. Science. 2012;338(6105):379–83. Epub 2012/08/28. 10.1126/science.1217726 . [DOI] [PubMed] [Google Scholar]
- 12. Liu Y, Hu W, Murakawa Y, Yin J, Wang G, Landthaler M, et al. Cold-induced RNA-binding proteins regulate circadian gene expression by controlling alternative polyadenylation. Sci Rep. 2013;3:2054 Epub 2013/06/26. 10.1038/srep02054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lopez M, Meier D, Muller A, Franken P, Fujita J, Fontana A. Tumor necrosis factor and transforming growth factor beta regulate clock genes by controlling the expression of the cold inducible RNA-binding protein (CIRBP). J Biol Chem. 2014;289(5):2736–44. 10.1074/jbc.M113.508200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Franco HL, Casasnovas J, Rodriguez-Medina JR, Cadilla CL. Redundant or separate entities?—roles of Twist1 and Twist2 as molecular switches during gene transcription. Nucleic acids research. 2011;39(4):1177–86. 10.1093/nar/gkq890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen ZF, Behringer RR. twist is required in head mesenchyme for cranial neural tube morphogenesis. Genes Dev. 1995;9(6):686–99. . [DOI] [PubMed] [Google Scholar]
- 16. Bialek P, Kern B, Yang X, Schrock M, Sosic D, Hong N, et al. A twist code determines the onset of osteoblast differentiation. Developmental cell. 2004;6(3):423–35. . [DOI] [PubMed] [Google Scholar]
- 17. Ansieau S, Bastid J, Doreau A, Morel AP, Bouchet BP, Thomas C, et al. Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer cell. 2008;14(1):79–89. 10.1016/j.ccr.2008.06.005 . [DOI] [PubMed] [Google Scholar]
- 18. Ma JL, Han SX, Zhu Q, Zhao J, Zhang D, Wang L, et al. Role of Twist in vasculogenic mimicry formation in hypoxic hepatocellular carcinoma cells in vitro. Biochem Biophys Res Commun. 2011;408(4):686–91. 10.1016/j.bbrc.2011.04.089 . [DOI] [PubMed] [Google Scholar]
- 19. Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117(7):927–39. 10.1016/j.cell.2004.06.006 . [DOI] [PubMed] [Google Scholar]
- 20. Sosic D, Richardson JA, Yu K, Ornitz DM, Olson EN. Twist regulates cytokine gene expression through a negative feedback loop that represses NF-kappaB activity. Cell. 2003;112(2):169–80. . [DOI] [PubMed] [Google Scholar]
- 21. Li CW, Xia W, Huo L, Lim SO, Wu Y, Hsu JL, et al. Epithelial-mesenchymal transition induced by TNF-alpha requires NF-kappaB-mediated transcriptional upregulation of Twist1. Cancer research. 2012;72(5):1290–300. 10.1158/0008-5472.CAN-11-3123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li Y, Maher P, Schubert D. A role for 12-lipoxygenase in nerve cell death caused by glutathione depletion. Neuron. 1997;19(2):453–63. . [DOI] [PubMed] [Google Scholar]
- 23. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–8. 10.1006/meth.2001.1262 . [DOI] [PubMed] [Google Scholar]
- 24. Honma S, Kawamoto T, Takagi Y, Fujimoto K, Sato F, Noshiro M, et al. Dec1 and Dec2 are regulators of the mammalian molecular clock. Nature. 2002;419(6909):841–4. 10.1038/nature01123 . [DOI] [PubMed] [Google Scholar]
- 25. Ripperger JA, Shearman LP, Reppert SM, Schibler U. CLOCK, an essential pacemaker component, controls expression of the circadian transcription factor DBP. Genes Dev. 2000;14(6):679–89. [PMC free article] [PubMed] [Google Scholar]
- 26. Sharif MN, Sosic D, Rothlin CV, Kelly E, Lemke G, Olson EN, et al. Twist mediates suppression of inflammation by type I IFNs and Axl. The Journal of experimental medicine. 2006;203(8):1891–901. 10.1084/jem.20051725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Munoz E, Brewer M, Baler R. Circadian Transcription. Thinking outside the E-Box. J Biol Chem. 2002;277(39):36009–17. 10.1074/jbc.M203909200 . [DOI] [PubMed] [Google Scholar]
- 28. Ripperger JA, Schibler U. Rhythmic CLOCK-BMAL1 binding to multiple E-box motifs drives circadian Dbp transcription and chromatin transitions. Nat Genet. 2006;38(3):369–74. 10.1038/ng1738 . [DOI] [PubMed] [Google Scholar]
- 29. Haimovich B, Calvano J, Haimovich AD, Calvano SE, Coyle SM, Lowry SF. In vivo endotoxin synchronizes and suppresses clock gene expression in human peripheral blood leukocytes. Crit Care Med. 2010;38(3):751–8. Epub 2010/01/19. 10.1097/CCM.0b013e3181cd131c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ohdo S, Koyanagi S, Suyama H, Higuchi S, Aramaki H. Changing the dosing schedule minimizes the disruptive effects of interferon on clock function. Nat Med. 2001;7(3):356–60. Epub 2001/03/07. 10.1038/85507 . [DOI] [PubMed] [Google Scholar]
- 31. Okada K, Yano M, Doki Y, Azama T, Iwanaga H, Miki H, et al. Injection of LPS causes transient suppression of biological clock genes in rats. J Surg Res. 2008;145(1):5–12. Epub 2008/02/19. 10.1016/j.jss.2007.01.010 . [DOI] [PubMed] [Google Scholar]
- 32. Suzuki T, Sato F, Kondo J, Liu Y, Kusumi T, Fujimoto K, et al. Period is involved in the proliferation of human pancreatic MIA-PaCa2 cancer cells by TNF-alpha. Biomed Res. 2008;29(2):99–103. Epub 2008/05/16. . [DOI] [PubMed] [Google Scholar]
- 33. Naidoo N, Davis JG, Zhu J, Yabumoto M, Singletary K, Brown M, et al. Aging and sleep deprivation induce the unfolded protein response in the pancreas: implications for metabolism. Aging cell. 2014;13(1):131–41. 10.1111/acel.12158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Taraborrelli C, Palchykova S, Tobler I, Gast H, Birchler T, Fontana A. TNFR1 is essential for CD40, but not for lipopolysaccharide-induced sickness behavior and clock gene dysregulation. Brain Behav Immun. 2011;25(3):434–42. 10.1016/j.bbi.2010.11.001 . [DOI] [PubMed] [Google Scholar]
- 35. Hirayama J, Sahar S, Grimaldi B, Tamaru T, Takamatsu K, Nakahata Y, et al. CLOCK-mediated acetylation of BMAL1 controls circadian function. Nature. 2007;450(7172):1086–90. 10.1038/nature06394 . [DOI] [PubMed] [Google Scholar]
- 36. Casas E, Kim J, Bendesky A, Ohno-Machado L, Wolfe CJ, Yang J. Snail2 is an essential mediator of Twist1-induced epithelial mesenchymal transition and metastasis. Cancer research. 2011;71(1):245–54. 10.1158/0008-5472.CAN-10-2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chang AT, Liu Y, Ayyanathan K, Benner C, Jiang Y, Prokop JW, et al. An evolutionarily conserved DNA architecture determines target specificity of the TWIST family bHLH transcription factors. Genes Dev. 2015;29(6):603–16. 10.1101/gad.242842.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kato Y, Kawamoto T, Fujimoto K, Noshiro M. DEC1/STRA13/SHARP2 and DEC2/SHARP1 coordinate physiological processes, including circadian rhythms in response to environmental stimuli. Current topics in developmental biology. 2014;110:339–72. 10.1016/B978-0-12-405943-6.00010-5 . [DOI] [PubMed] [Google Scholar]
- 39. Howard TD, Paznekas WA, Green ED, Chiang LC, Ma N, Ortiz de Luna RI, et al. Mutations in TWIST, a basic helix-loop-helix transcription factor, in Saethre-Chotzen syndrome. Nat Genet. 1997;15(1):36–41. 10.1038/ng0197-36 . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.