

Abstract
Somatic mutations of isocitrate dehydrogenase 1 (IDH1) at R132 are frequently found in certain cancers such as glioma. With losing the activity of wild-type IDH1, the R132H and R132C mutant proteins can reduce α-ketoglutaric acid (α-KG) to D-2-hydroxyglutaric acid (D2HG). The resulting high concentration of D2HG inhibits many α-KG-dependent dioxygenases, including histone demethylases, to cause broad histone hypermethylation. These aberrant epigenetic changes are responsible for initiation of these cancers. We report the synthesis, structure activity relationships, enzyme kinetics and binding thermodynamics of a novel series of 2-thiohydantoin and related compounds, among which several compounds are potent inhibitors of mutant IDH1 with Ki as low as 420 nM. X-ray crystal structures of IDH1(R132H) in complex with two inhibitors are reported, showing their inhibitor-protein interactions. These compounds can decrease the cellular concentration of D2HG, reduce the levels of histone methylation, and suppress proliferation of stem-like cancer cells in BT142 glioma with IDH1 R132H mutation.
INTRODUCTION
Isocitrate dehydrogenase (IDH) catalyzes oxidative decarboxylation of isocitric acid (ICT) to α-ketoglutaric acid (α-KG) (Figure 1A), which is one of the key reactions in the tricarboxylic acid cycle.1 Among the three isoforms of IDH in humans, frequent somatic mutations of IDH1 have recently been found in certain cancers.2-4 Particularly noted is that ~75% of lower grade (grade II and III) gliomas, the most common form of brain tumors, contain IDH1 mutations.5,6 Although growing relatively slowly, these gliomas are invasive and eventually develop to become secondary glioblastoma multiforme (GBM, grade IV glioma), a highly malignant tumor with 5-year survival rates of <10%. Distinct from many other mutations, the mutation site in IDH1 is always located in the R132 residue, with R132H being predominant (~93%). IDH mutations (including IDH1 and IDH2) have also been found in ~20% acute myeloid leukemia (AML) and ~50% of certain sarcomas.4,7 Moreover, clinical data suggest the IDH mutation is an early event in cancer initiation and development.5,6
Figure 1.

Reactions catalyzed by (A) Wild-type IDH1, (B) Mutant IDH1, and (C) α-KG-dependent histone demethylases. D2HG, the product of mutant IDH1, inhibits α-KG-dependent histone demethylases, resulting in histones with more methylated lysine residues; (D) The structures of two known inhibitors of mutant IDH1.
Biochemically, all of the characterized mutant IDH1 proteins in cancer almost lose the wild-type (WT) enzyme activity. However, these mutant enzymes can reduce α-KG to D-2-hydroxyglutaric acid (D2HG), using NADPH as the cofactor (Figure 1B).4,8,9 This new function causes a high concentration of D2HG to be accumulated in cancer cells (as well as in patient plasma). D2HG was found to be an inhibitor of many α-KG-dependent dioxygenases, including the JmjC family of histone demethylases (Figure 1C).3 This inhibitory activity leads to genome-wide hypermethylation of histones in IDH mutated cancers.10,11 Given the crucial roles of histone lysine methylation in gene regulation as well as cancer initiation, the observed aberrant epigenetic changes were hypothesized to be responsible for the initiation and/or maintenance of the IDH mutated cancers.12,13
As representatively shown in Figure 1D, three chemo-types of inhibitors (1 – 3) of mutant IDH1 have been reported.14-18 These compounds can reduce the cellular levels of D2HG, showing on-target activity in cells. Compounds 1 and 2 can also inhibit proliferation of IDH1 mutated cancer cells,15,17 demonstrating that mutant IDH1 is a drug target for these cancers. Here, we report the synthesis and structure activity relationships (SAR) of a series of 2-thiohydantoin-containing compounds as a new chemo-type of inhibitors of mutant IDH1. X-ray crystallographic, enzyme kinetics as well as binding thermodynamics studies of selected compounds showed the binding structures and the mode of action. Biological activity testing demonstrated these compounds can reduce cellular concentration of D2HG, inhibit histone lysine methylation, and impair self-renewal ability of IDH1 mutated glioma cells.
RESULTS AND DISCUSSION
Synthesis and structure activity relationship studies
Recombinant human IDH1 R132H mutant protein was expressed in E. coli and purified with a series of column chromatography, using our previously reported methods.16,17 The activity and inhibition of IDH1(R132H) were determined based on initial linear consumption of NADPH (Figure 1B) monitored at 340 nm, where NADPH has the maximal optical absorption. Through compound screening using our published biochemical method,16,17 3-benzyl-5-(3,4-dihydroxybenzylidene)-2-thiohydantoin (4)
 was identified to be an inhibitor of IDH1(R132H) with a Ki value of 4.7 μM against the substrate α-KG. This compound is of interest since it possesses good inhibitory activity and is amenable for medicinal chemistry studies.
was identified to be an inhibitor of IDH1(R132H) with a Ki value of 4.7 μM against the substrate α-KG. This compound is of interest since it possesses good inhibitory activity and is amenable for medicinal chemistry studies.
Medicinal chemistry was performed with modifications on the 3-substituent (−R1, Table 1), 5-substituent (−R2, Table 1) and the 2-thiohydantoin core (Chart 1) of compound 4. These compounds were efficiently synthesized using a general method as shown in Scheme 1. Glycine was reacted with R1 isothiocyanate and the resulting thiourea product was cyclized in the presence of sulfuric acid to give 3-R1-substituted 2-thiohydantoin. The condensation reaction of a 2-thiohydantoin (or related heterocyclic) compound with an aldehyde in the presence of sodium acetate in refluxing acetic acid to give a R2-substituted 5-methylene-2-thiohydantoin compound in 60-90% yield. The synthesis and characterization of compounds 4-41 are described in the Supporting Information Experimental Section.
Table 1.
Structures and Ki values of 5- or 6-substituted compounds against IDH1(R132H).
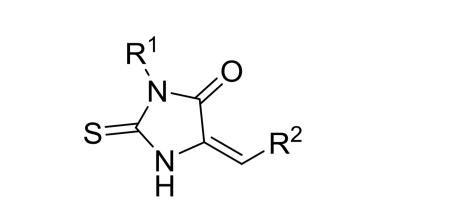
|
|||
|---|---|---|---|
|
| |||
| R1 | R2 | Ki (μM) | |
| 5 | Bn | 3,4-OMe-Ph | >50 |
| 6 | Ph | 3,4-OMe-Ph | >50 |
| 7 | 4-OMe-Bn | 3,4-OMe-Ph | >50 |
| 8 | Ph | 3,4-OH-Ph | 16.7 |
| 9 | 4-OH-Bn | 3,4-OH-Ph | 13.4 |
| 10 | Bn | Pyridin-3-yl | >50 |
| 11 | Bn | 2-OMe-pyridin-5-yl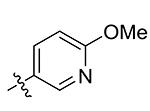
|
>50 |
| 12 | Bn | 2-Pyridinone-5-yl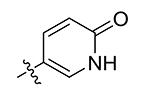
|
4.8 |
| 13 | Et | 2-OMe-pyridin-5-yl | 9.2 |
| 14 | Et | 2-Pyridinone-5-yl | 3.6 |
| 15 | Me | 2-OMe-pyridin-5-yl | 25.0 |
| 16 | Me | 2-Pyridinone-5-yl | 0.75 |
| 17 | H | 2-OMe-pyridin-5-yl | 7.8 |
| 18 | H | 2-Pyridinone-5-yl | 0.42 |
| 19 | −CH2COOH | 2-OMe-pyridin-5-yl | >50 |
| 20 | −CH2COOH | 2-Pyridinone-5-yl | >50 |
| 21 | Me | 1-Me-2-pyridinone-5-yl | 0.47 |
| 22 | Me | 1-Bn-2-Pyridinone-5-yl | 4.6 |
| 23 | Me | 1-Oxo-2-OMe-pyridin-5-yl
|
4.3 |
| 24 | Me | 1-OH-2-pyridinone-5-yl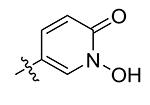
|
5.9 |
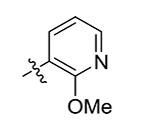
| 25 | Me |
|
25.3 |
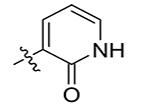
| 26 | Me |
|
>50 |
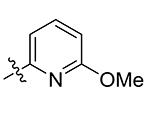
| 27 | Me |
|
34.2 |
| 28 | Me |
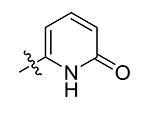
|
11.3 |
| 29 | Me | Pyridin-4-yl | >50 |
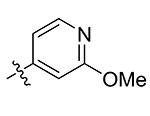
| 30 | Me |
|
2.6 |
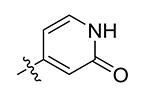
| 31 | Me |
|
9.2 |
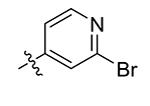
| 32 | H |
|
>50 |
| 33 | H |
|
5.6 |
| 34 | Me |
|
3.2 |
Chart 1.

Structures and activities of compounds 35-41.
Scheme 1.

General synthetic method.a
aReagents and conditions: (i) R1NCS, KOH, H2O-EtOH; (ii) H2SO4, Acetone; (iii) an aldehyde, e.g., 6-methoxynicotinaldehyde, NaOAc, HOAc, reflux; (iv) HCl, reflux.
Compound 5 (Table 1) having a 3,4-dimethoxyphenyl group was found to be inactive (Ki > 50 μM) against IDH1(R132H). Similarly, compounds 6 and 7, which contain the same R2 group of 3,4-dimethoxyphenyl, are also inactive. This observation suggests that at least one of the two –OH groups in compound 4 is important for the activity. Indeed, after removal of the methyl groups of 6 and 7, the corresponding hydroxyl compounds 8 and 9 can inhibit the mutant enzyme with Ki values of 16.7 and 13.4 μM, respectively, although they are ~3-fold less active than compound 4 (R1 = benzyl), which should be due to a different R1 group (i.e., phenyl and 4-OH-benzyl for 8 and 9 versus benzyl in 4).
Because undesirable properties associated with the 3,4-dihydroxyphenyl (catechol-containing) group, which is a strong metal chelator and is easily oxidized, compounds 10 – 12 were synthesized in an effort to find another R2 group that has comparable or improved activity. Compound 10 with a 3-pyridinyl and compound 11 with a 2-methoxy-5-pyridinyl group have one and two H-bond acceptors. Both compounds are inactive. However, compound 12 with a 2-pyridinone-5-yl group possesses a good inhibitory activity with a Ki value of 4.8 μM against IDH1(R132H), showing a comparable activity to compound 4.
Next, compounds 13 – 20 (Table 1) were synthesized to optimize the R1 group. Although compounds 13, 15 and 17 containing a 2-methoxy-5-pyridinyl R2 group are still moderate or weak inhibitors of IDH1(R132H) with Ki values of 9.2, 25 and 7.8 μM, their activities are superior to that of compound 11 (Ki > 50 μM), suggesting a smaller R1 group (i.e., −Et in 13, −Me in 15 and −H in 17) is more favorable. The demethylated compounds 14, 16 and 18 (with a 2-pyridinone-5-yl R2 group) were found to possess strong activities. While compound 14 with an −Et R1 group has a Ki of 3.6 μM, compounds 16 and 18 with a −Me and −H R1 group, respectively, showed potent inhibitory activities with Ki values of 750 and 420 nM. The increasing activities of compounds 12, 14, 16 and 18 suggest a SAR trend for the R1 group of Bn < Et < Me < H, with small substituents Me and H being most favorable. Compounds 19 and 20 were synthesized to find if a carboxyl group or its ester is a good R1. However, none of these compounds are active.
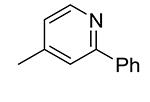
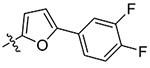
Compounds 21 – 34 (Table 1) were synthesized to investigate the SAR for the R2 group. Compound 21 with an N-methyl substituent on the 2-pyridinone ring was found to have a slight improved activity with a Ki of 470 nM, as compared with 16. However, an N-benzyl substituent considerably reduces the inhibitory activity of compound 22 (Ki = 4.6 μM). Compound 23 and its demethylated compound 24, which have an O atom (or –OH) attached to N1 of the pyridine-2-one ring also show decreased activities (Ki = 4.3 and 5.9 μM, respectively). Compounds with other pyridinyl R2 substituents were synthesized for SAR studies. Compounds 25 and 26 containing 2-methoxy-pyridin-3-yl and pyridine-2-one-3-yl group, respectively, were found to have very weak or no inhibitory activity (Ki ≥ 25 μM) against IDH1(R132H). Compound 27 with 6-methoxypyridin-2-yl R2 group is inactive, while the demethylated compound 28 has a modest activity (Ki = 11.3 μM). Compounds bearing a 4-pyridinyl R2 group are of interest. Although compound 29 with a 4-pyridinyl group is inactive (Ki > 50 μM), compounds 30 and 31 having 2-methoxy-4-pyridinyl and pyridin-2-one-4-yl groups are good inhibitors with Ki values of 2.6 and 9.2 μM, respectively. In addition, compound 30 is the only −OMe containing compound that is considerably more active than its −OH analog 31, suggesting a more hydrophobic group might be favorable at this position and several compounds were therefore synthesized. Loss of activity for compound 32 indicates the 3-bromo-4-pyridinyl R2 group is disfavored, while compound 33 bearing 3-phenyl-4-pyridinyl group showed good inhibitory activity (Ki = 5.6 μM). Similarly, compound 34 having a 2,5-substituted furan R2 group also exhibited a good Ki of 3.2 μM.
The SAR studies described above show that a good R2 group requires a strong H-bond acceptor (e.g., the 4-O atom in 4 and 2-O in 12, 14, 16, 18, 21, 22 and 24) at the para-position. The potent activity of compound 21 (as well as that of 22) indicates that an H-bond donor (e.g., 3-OH in 4 and 1-NH in pyridin-2-one containing compounds) is not essential.
SARs for the 2-thiohydantoin ring were investigated and the results are shown in Chart 1. We synthesized compounds 35 and 36 with a 2-thioxothiazolidin-4-one core structure, 37 and 38 with a hydantoin, and compound 39 with a 2-amino-imidazol-4-one core. The activities of compounds 35 and 36 (Ki = >50 and 3.1 μM, respectively) are significantly lower than those of the corresponding 2-thiohydantoin compounds 17 and 18 (Ki = 7.8 and 0.42 μM), showing −NH− at the 1-position of the ring is more favorable than −S−. Complete loss of activity for compounds 37 and 38 was unexpected, indicating the 2-carbonyl O atom of the hydantoin ring is highly disfavored, as compared to the corresponding S atom of 2-thiohydantoin ring in 17 and 18. Similarly, compound 39 having an –NH2 at this position is also inactive. In compound 40, the methyl group attached to the S atom causes the compound to completely lose the inhibitory activity against IDH1(R132H). Moreover, adding a methyl group to the N3 atom of the 2-thiohydantoin ring in compound 41 (Ki = 16.5 μM) resulted in a 22-fold activity reduction, as compared to compound 16 (Ki = 0.75 μM).
Activity against R132C and WT IDH1
Compounds 16, 18 and 22 were selected to be tested for their abilities to inhibit R132C (a frequent mutation in leukemia and sarcoma) mutant as well as WT IDH1. These two recombinant IDH1 proteins were also expressed in E. coli and purified with our previously reported methods.16,17 The activity and inhibition of IDH1(R132C) were similarly determined as those of the R132H mutant protein. For WT IDH1, the reaction rate was calculated based on the production of NADPH (Figure 1B), resulting in an increase of optical density at 340 nm.
As can be seen in Table 2, these three compounds were found to be also inhibitors of IDH1(R132C). While compound 18 is the most potent inhibitor of IDH1(R132H) (KiR132H = 0.42 μM), it is less inhibitory against the R132C mutant protein with a Ki of 2.3 μM. Compound 16 exhibited a Ki value of 1.2 μM against IDH1(R132C), which is, however, comparable to that against IDH1(R132H) (KiR132H = 0.75 μM). In addition, compound 22 showed a less inhibitory active against IDH1(R132C) with a Ki of 12.5 μM. These three compounds have moderate to good selectivity for the mutant IDH1 enzymes. Compounds 16, 18 and 22 were found to be relatively weak inhibitors of WT IDH1, with Ki values of 8.8, 10.3 and 32.9 μM, respectively. Thus, compound 16 shows selectivity indices of 11.7 and 7.3 (Table 2) for the R132H and R132C mutant protein, respectively. Compound 18 was found to be 24.5- and 4.5-fold more active for the R132H and R132C mutants, as compared to the WT enzyme. Similarly, compound 22 possesses selectivity indices of 7.2 and 2.6-fold for inhibition of R132H and R132C, respectively.
Table 2.
Activity (Ki, μM) and selectivity of compounds 16, 18 and 22.
| Selectivity Index | |||||
|---|---|---|---|---|---|
| R132H | R132C | WT | KiWT/KiR132H | KiWT/KiR132C | |
| 16 | 0.75 ± 0.39 | 1.2 ± 0.3 | 8.8 ± 0.4 | 11.7 | 7.3 |
| 18 | 0.42 ± 0.21 | 2.3 ± 0.7 | 10.3 ± 0.5 | 24.5 | 4.5 |
| 22 | 4.6 ± 0.7 | 12.5 ± 0.7 | 32.9 ± 1.2 | 7.2 | 2.6 |
X-ray crystallography
X-ray crystallography was used to find how these novel 2-thiohydantoin inhibitors bind to IDH1(R132H). We determined the tertiary structures of IDH1(R132H) in complex with NADPH and compounds 16 and 22 to a resolution of 3.2 Å, similar to those reported previously.16,19 Statistics for diffraction data and structural refinement are in Supporting Information Table S1. As shown in Figure 2A and Supporting Information Figure S1, the protein IDH1(R132H) was found to crystallize as a homodimer with one NADPH molecule co-crystallized in each protein subunit. Only one molecule of 2-thiohydantoin inhibitor 16 (or 22) can be found in the protein homodimer, based on the electron density and omit maps (Figures 2B, C and Figure S2). These observations were also found in our previous structures of IDH1(R132H) in complex with 1-hydroxypyridinone inhibitors (e.g., compound 2).16
Figure 2.

X-ray structures of IDH1(R132H):inhibitor complexes. (A) The overall structure of IDH1(R132H) in complex with compound 16 (ball & stick model) and NADPH (tube model); (B) The 2Fo-Fc electron density map of IDH1(R132H):16 at the inhibitor-binding site, contoured at 1σ; (C) The Fo-Fc omit map of IDH1(R132H):16, contoured at 3σ; (D) The superimposed structures of IDH1(R132H) (electrostatic surface model) in complex with 16 (with C atoms in green) and 22 (in pink); and (E) The close-up views of compound 16 and (F) compound 22 in IDH1(R132H). The protein backbones are shown as blue lines and H-bonds as purple dotted lines.
Compounds 16 and 22 are located deeply inside a cleft between the two protein homodimers (Figure 2D), sitting in a pocket surrounded by the residues Thr77, Ser94, Asn96, Gly97, Arg100, Asn101, Arg109 and NADPH. Figures 2E and S3A show the close-up view of the binding site and the ligand-protein interactions of compound 16. The almost flat inhibitor molecule lies on a “bed surface” composed of Gly97, Asn96 and Ser94, with the distance between the two almost parallel planes being ~3.3-3.6 Å. The two partially negatively charged O atoms of 16 form three H-bonds as well as electrostatic interactions with the positively charged side chains of Arg100 and Asn101. The S atom of compound 16 was found to have favorable, mostly hydrophobic interactions with Thr77 and NADPH. Figures 2F and S3B show the binding site and the ligand-protein interactions of compound 22. The conjugated rings of 2-thiohydantoin and pyridinone adopt a similar binding pose as observed for compound 16, having favorable interactions with Gly97, Asn96 and Ser94. In addition, the two O atoms of 22 also have three H-bond and electrostatic interactions with Arg100 and Asn101. Therefore, it appears that these two types of common interactions between the protein and compounds 16 and 22 provide the majority of binding affinity for these inhibitors. The 2-thiohydantoin ring of compound 22 was found to orientate slightly differently from that of compound 16 (Figure 2D), with, for example, the distance between the two S atoms being ~2.2 Å. While such a movement for 22 causes loss of the interactions between the S atom and NADPH and Thr77, it renders the 3-methyl group of 22 to have van der Waals interactions with the side chain of Arg109. The benzyl group of compound 22 bends and inserts into a deep side-pocket, having mostly hydrophobic interactions with the side chains of Lys93, Gly97 and Thr98 (Figure 2D and F).
Previous crystallographic studies showed that unlike the WT protein, the R132H IDH1 mutant has two ligand binding sites.16,19 Site I is where ICT as well as 1-hydroxypyridinone inhibitors (e.g., compound 2) bind to IDH1(R132H). The binding site II is the catalytically active site where the substrate α-KG is located.8 It is also noted that the two binding sites in IDH1(R132H) do not co-exist. A large protein conformational change occurs to switch the two ligand binding sites in IDH1(R132H) that recognize a different ligand (e.g., ICT versus α-KG). However, in the WT IDH1, there is only one ligand binding site that corresponds to site II in IDH1(R132H).
Superposition of the crystal structures of IDH1(R132H) in complex with ICT, α-KG and compounds 16 and 22, shown in Figure 3A, demonstrated that the two inhibitors occupy the ligand binding site I, the same location where ICT and 1-hydroxypyridinone inhibitors (e.g., 2) are located in IDH1(R132H). α-KG is ~6 Å away in the binding site II. Figure 3B shows the large protein conformational differences between the two binding states of IDH1(R132H). In addition, the binding poses of NADPH in the two structures also differ significantly (Figure 3B).
Figure 3.

(A) Superimposed structures of IDH1(R132H):16 (protein as blue lines) and IDH1(R132H):α-KG (protein as brown lines), showing the two ligand binding sites. Also aligned are the crystal structures of IDH1(R132H):ICT (with C atoms in gray) and :22 (in purple), showing both ligands are also located in the binding site I; (B) Superposition of the structures of IDH1(R132H):16 (protein as blue ribbons) and IDH1(R132H):α-KG (as yellow ribbons), showing a large protein conformational difference. Compound 16 (C atoms in green) and α-KG (in brown) are shown as ball and stick models. In addition, the conformation as well as the binding site of NADPH of the two structures (tube models in green and brown, respectively) are also significantly different.
Enzyme kinetic studies
We also performed a steady-state enzyme kinetic study for the most potent compound 18, with which an X-ray crystal structure of IDH1(R132H) cannot be obtained. Initial velocities of IDH1(R132H) were determined in the presence of increasing concentrations of compound 18, the substrate α-KG and cofactor NADPH. These data were imported into the program SigmaPlot and fitted to competitive, uncompetitive and noncompetitive kinetic models, using a Lineweaver-Burk or Michaelis-Menten plot. The best model, as judged by the R2 and AICc values, showed the mode of action for the inhibitor. Thus, compound 18 was found to exert a competitive mode of action against the substrate α-KG, as shown in Figures 4A (competitive fitting) and S4A (uncompetitive and noncompetitive fitting). In addition, compound 18 was determined to be a noncompetitive inhibitor against the cofactor NADPH (Figures 4B and S4B). Moreover, enzyme kinetic results for compound 18 are in line with the crystal structure of compound 16 described above, which also suggests that it is competitive to α-KG and noncompetitive to NADPH.
Figure 4.

The best fitting models for enzyme kinetic studies of compound 18. (A) Competitive inhibition model with variable concentrations of α-KG using a Lineweaver-Burk plot; and (B) Noncompetitive inhibition model with variable concentrations of NADPH using a Michaelis-Menten plot.
Binding thermodynamics studies
We performed isothermal titration calorimetry (ITC) experiments to further characterize the binding as well as mode of action for compound 18. First, binding thermodynamics of compound 18 to IDH1(R132H) (100 μM) without NADPH was investigated in 30 mM Tris buffer (pH 7.4) with 4 mM MgCl2 and 150 mM NaCl at 298K. Use of a relatively high concentration of the protein was because of the small amount of heat generated for the binding. As shown representatively in Figure 5A, the binding of 18 to IDH1(R132H) alone was slightly exothermic (ΔH = −1.5 kJ/mol) with a Ka (affinity constant) value of 1.68 × 105 M−1, or a Kd (dissociate constant) value of 5.9 μM. This equals a ΔG value of −29.8 kJ/mol, showing the binding is mostly entropy driven (with a calculated ΔS of 95.0 J K−1 mol−1). Next, binding of compound 18 to IDH1(R132H) (133 μM) in the presence of NADPH (300 μM) was found to be slightly more exothermic (ΔH = −2.5 kJ/mol) with a Kd value of 8.9 μM (Figure 5B). Comparable Kd values as well as similar thermodynamics for these two experiments suggest that the binding of compound 18 to IDH1(R132H) is not significantly affected by NADPH. These two molecules could bind to different binding sites in the protein. Third, compound 18 was titrated into IDH1(R132H) (100 μM) with NADPH (300 μM) and α-KG (300 μM). As shown representatively in Figure 5C, the observed binding affinity of 18 to the protein (Kd = 46.0 μM) was found to be reduced by ~5-folds in the presence of α-KG, suggesting inhibitor 18 and α-KG are competitive to each other. These results are consistent with the enzyme kinetics data as well as the X-ray crystallographic studies of compounds 16 and 22.
Figure 5.

Representative ITC results and fitting curves for (A) compound 18 binding to IDH1(R132H) (100 μM); (B) 18 binding to IDH1(R132H) (100 μM) in the presence of NADPH (300 μM); and (C) 18 binding to IDH1(R132H) (100 μM) in the presence of NADPH (300 μM) and α-KG (300 μM).
Biological activity evaluation
First we tested if the most potent inhibitors 16 and 18 inhibit mutant IDH1 enzymes in vivo (cells), using human fibrosarcoma cell line HT1080, which harbors R132C mutation of IDH1, as a cell model. Thus, HT1080 cells in an exponential growth phase were treated with increasing concentrations of compounds 16 and 18 for 2 days, after which the concentrations of D2HG were determined by HPLC-MS using our previous method.17 As can be seen in Figure 6A, treatment with compounds 16 and 18 significantly reduced the production of D2HG, showing these inhibitors are cell membrane permeable and on-target in cells.
Figure 6.

Cellular activity of mutant IDH1 inhibitors. (A) Treatment of compounds 16 and 18 caused significantly reduced amount of D2HG in HT1080 cells; (B) Treatment of 18 significantly reduced methylation levels on histone lysine residues in HT1080 cells, while inactive compound 20 did not significantly affect histone methylation levels; (C) Treatment of compounds 18, 16 and 1 inhibited neurosphere formation of BT142 glioma cells with IDH1 R132H mutation, but (D) did not affect neurosphere formation of BXD-3752 glioma cells without an IDH1 mutation.
Upon confirming their ability to inhibit cellular mutant IDH1, we next investigated whether the activity can alter levels of histone methylation. Since D2HG broadly inhibits α-KG-dependent histone lysine demethylases,3 it is expected that treatment with inhibitors of mutant IDH1 can restore the activity of these histone demethylases and therefore reduce the levels of histone lysine methylation. HT1080 cells were treated with compound 18 for 4 days. Histone proteins from both control and treated cells were extracted and subjected to Western blot analysis, probed by specific antibodies of human histone H3, trimethylated and dimethylated histone H3 lysine 4 (H3K4me3 and H3K4me2), dimethylated histone H3 lysine 27 (H3K27me2) and dimethylated histone H4 lysine 20 (H4K20me2). As shown in Figure 6B, compound 18 was found to significantly reduce the methylation levels of these histone lysine residues in a dose dependent manner. To further confirm the effects were due to inhibition of mutant IDH1, inactive compound 20 (Table 1) was used in the experiments and it did not significantly reduce D2HG (data not shown) as well as histone lysine methylation levels in HT1080 (Figure 6B). These results show that by reducing the cellular concentration of D2HG, the inhibitors of mutant IDH1 can maintain the functions of histone demethylases and reverse the histone hypermethylation phenotype in HT1080 with IDH1 R132C mutation.
Compounds 16 and 18 were next tested for their in vitro antitumor activity, together with potent known inhibitor 1 as a comparison. These three compounds were found to have no or negligible activity (EC50 > 50 μM) against proliferation of MCF-7 (breast), A549 (lung) cancer cells as well as WI-38 normal fibroblast cells. In addition, compounds 1, 16 and 18 did not significantly inhibit the growth of HT1080 cells (EC50 > 50 μM). These results showed these potent inhibitors of mutant IDH1 do not have general cytotoxicity. Lack of growth inhibition against these tumor and normal cells also suggests that compounds 16 and 18 do not appear to affect critical cellular metabolism and signaling pathways, showing possibly good selectivity, although an enzyme selectivity profiling needs to be done for future studies.
We further tested whether these compounds inhibit cancer stem cells (CSC) containing an IDH1 mutation. CSCs represent a small fraction of cancer cells that possess certain stem cell traits including ability to self-renew and differentiate. Studies have found that only CSCs can generate new tumors when transplanted into a new host.20 In the perspective of cancer treatment, CSCs are more resistant to chemotherapies and also believed to be responsible for tumor relapse and metastasis. It is therefore of importance to find compounds have selective activity against CSCs. To this end, patient sample derived BT142 glioma cells21 bearing IDH1 R132H mutation were cultured in serum-free media and grew as “neurospheres”, which are colonies of cells with a diameter of 30 - 300 μm. Stem-like glioma cells are found to be enriched in these neurospheres.22-24 BXD-3752 glioma cells25 without an IDH1 mutation were included as a control cell model. As shown in Figure 6C, compounds 16 and 18 were able to significantly inhibit proliferation of BT142 glioma stem-like cells, although these compounds are non-cytotoxic to normal and malignant cancer cells. However, compounds 16 and 18 did not have activity against proliferation of BXD-3752 glioma stem-like cells (Figure 6D), showing their anti-CSC activity is selective for IDH1 mutated cancer cells. Moreover, 2-thiohydantoin inhibitors 16 and 18 exerted comparable anti-CSC activity to that of known inhibitor 1 (Figures 6C and D). Compound 1 can inhibit growth of IDH1 mutated BT142 glioma stem-like cells, but did not affect BXD-3752 cells without IDH1 mutation. These results from the two chemo-types of inhibitors (i.e., compounds 1, 16 and 18) show that the function of mutant IDH1 seems of importance for maintenance of glioma stem cells bearing an IDH1 mutation. However, presumably due to the presence of other gene mutations, inhibition of mutant IDH1 alone does not significantly affect proliferation of the bulk of non-stem like cancer cells, such as those in HT1080. This probably explains the reason why these mutant IDH1 inhibitors did not affect the overall proliferation rate of HT1080, but impaired self-renewal ability of the HT1080 stem-like cells.
Although 2-thiohydantoin and related compounds have been reported to be associated with a number of other biological activities and implicated as promiscuous inhibitors,30 our SAR studies showed this is unlikely to be the case, because our compounds showed a wide range of activity (Ki: 0.4 - >50 μM) as well as several SARs for inhibition of IDH1(R132H). Enzyme kinetics and ITC studies also suggested that compound 18 is a competitive inhibitor of IDH1(R132H) against the substrate α-KG. Our X-ray crystallographic investigation further revealed the exact binding structures of compounds 16 and 22 in IDH1(R132H), which could serve as a useful platform for more rational inhibitor design and development.
Conclusion
Genetic studies have shown frequent mutations of IDH are found in certain cancers, especially in gliomas. Biochemical studies have revealed that the mutant IDH enzymes can reduce α-KG, an important cellular metabolite, to D2HG, which inhibits histone lysine demethylation. Further biological and pharmacological studies demonstrated that mutant IDH1 is a viable drug target for these cancers. However, there have been a limited chemotypes of inhibitors of mutant IDH1.
Several series of 5-substituted 2-thiohydantoin and related compounds were synthesized, among which several compounds were found to be potent and selective inhibitors of mutant IDH1 with Ki values as low as 420 nM. SAR studies of these compounds showed that 1) 2-thiohydantoin ring is the most favorable core, 2) a small substituent, such as –H or –Me, is more favored at the 3-position, and 3) 2-pyridinone-5-yl group at the 5-postion is the most potent for inhibition of mutant IDH1. X-ray crystallographic studies revealed the binding structures of compounds 16 and 22 in IDH1(R132H), providing a platform for further structure-based inhibitor design. Enzyme kinetics as well as binding thermodynamics studies indicated that the most potent inhibitor 18 is a competitive inhibitor with respect to the substrate α-KG and exerts a noncompetitive mode of action to the cofactor NADPH. Cell biology studies showed that mutant IDH1 inhibitors 16 and 18 can lower down the concentrations of D2HG in HT1080 cells. These compounds can also reverse the hypermethyled phenotype of histones, and selectively suppress self-renewal ability of glioma stem-like cells with IDH1 R132H mutation. These compounds are therefore novel chemical probes for studies of mutant IDH1 in cancer and represent a new scaffold for drug discovery targeting IDH1 mutated cancers.
Experimental Section
All reagents were purchased from Alfa Aesar (Ward Hill, MA) or Aldrich (Milwaukee, WI). Compounds were characterized by 1H NMR on a Varian (Palo Alto, CA) 400-MR spectrometer and identities of selected compounds 16, 18 and 22 were confirmed with high resolution mass spectra (HRMS) using a ThermoFisher LTQ-Orbitrap mass spectrometer. Compound purity was monitored by a Shimadzu Prominence HPLC with a Phenomenex C18 column (4.6 × 250 mm, Methonal:H2O 60:40, monitored at 254 and 280 nm). The purities of all compounds were found to be >95%.
Synthesis and characterization of compounds 4 – 41 can be found in Supporting Information Experimental Section.
Expression, purification and inhibition of human WT and mutant IDH1
Expression, purification and inhibition of the WT, R132H and R132C IDH1 enzymes followed our previous published methods.16,17 Enzyme inhibition assay for IDH1(R132H) and IDH1(R132C) was performed in a 96-well microplate in 50 mM HEPES buffer (pH = 7.5) containing the enzyme (100 nM), 4 mM MgCl2, 100 μM NADPH and increasing concentrations an inhibitor. Upon incubation for 5 min, α-KG (1 mM) was added to initiate the reaction. The optical absorbance of each well was monitored every 30s at 340 nm with a Beckman DTX-880 microplate reader. The inhibition assay of WT IDH1 was carried out in 50 mM HEPES buffer containing the enzyme (30 nM), 4 mM MgCl2, 1 mM NADP+, an inhibitor as well as 200 μM sodium (D)-isocitrate. The reaction was monitored by an increase in optical absorbance at 340 nm. The data were imported into Prism (version 5.0, GraphPad) and IC50s were calculated by using a standard dose response curve fitting. For compounds with IC50s >> [enzyme], Ki was calculated using the Cheng-Prusoff equation Ki = IC50/(1+[S]/Km), where [S] is the concentration of α-KG (1 mM) and Km is the literature value of 0.965 mM for R132H and 0.295 mM for R132C. For compounds with IC50s < 1 μM, Ki values were determined with the Morrison tight inhibition modeling in Prism. The reported results are the average from at least three independent experiments.
Crystallization and structure determination
IDH1(R132H) (10 mg/mL) was crystallized together with 2 mM of compounds 2 or 3, 10 mM NADPH and 10 mM CaCl2 in a solution of 1.75 M (NH4)2SO4 and 0.1 M NaOAc (pH 5.6). Prism-like single crystals appeared in ~5 days. Diffraction data were collected using a Rigaku FR-E+ SuperBright X-ray source at Baylor College of Medicine and processed with the program the program HKL2000.26 The initial structure was obtained by molecular replacement using the program Phaser,27 with the coordinates of 3MAP as a target. Structural refinement was performed using the program Refmac28 or PHENIX29. The coordinates of IDH1(R132H) in complex with 16 and 22 were deposited into Protein Data Bank as entries 4XRX and 4XS3, respectively.
Steady-state kinetic study
Steady-state kinetic inhibition experiment was conducted by measuring the initial velocities of reactions catalyzed by IDH1(R132H) while varying the concentrations of compound 18, α-KG and NADPH. Data were imported into SigmaPlot (version 13, Systat Software, Inc.) and fitted to competitive, noncompetitive and uncompetitive inhibition models. The best kinetic models were determined by the highest R2 and lowest AICc values. Lineweaver-Burk or Michaelis-Menten plots were generated by Sigmaplot.
Isothermal titration calorimetry study
ITC experiments were performed using a Nano ITC LV (190 μL) instrument from TA Instruments (New Castle, DE) at 298 K. A 190 μL solution containing specified concentrations of IDH1(R132H), NADPH and α-KG in 30 mM Tris-HCl buffer (pH = 7.4) with 4 mM MgCl2 and 150 mM NaCl was added into the sample cell. Compound 18 (1 mM) in the same buffer solution (50 μL) was transferred into the syringe of the instrument. Upon reaching temperature equilibrium, 25 injections (2 μL each) of the inhibitor were used for the titration. ITC data were then imported into NanoAnalyze software (TA Instruments, New Castle, DE). The titration baseline was corrected and the ΔH and Ka values for the binding of the inhibitor were obtained by fitting into the independent model in the software. All experiments were repeated for at least one time to ensure the reported data are reliable.
Cell culture and growth inhibition for cells that attach to a plate
5,000 - 105 cells/well were inoculated into each well of a 96-well plate and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) at 37 °C in a 5% CO2 atmosphere with 100% humidity. Upon culturing overnight for cell attachment, increasing concentrations of a compound were added and incubated for 48-96 h. Cell viability was then assessed with an MTT assay.
D2HG production inhibition in HT1080 cells
Inhibition of cellular mutant IDH1 was determined following our previous protocol.17 105 HT1080 cells/well were cultured in DMEM containing10% dialyzed FBS and treated with increasing concentrations of an inhibitor for 48h. Medium was collected, diluted with MeOH (8 mL) and centrifuged (20,000 rpm for 3 min). D2HG in the supernatant was determined by HPLC-MS using our previous published methods.17
Inhibition of the proliferation of glioma stem-like cells
Two glioma cells BT142 and BXD-3752 were cultured as neurospheres in serum-free cell growth media at 37 °C in a 5% CO2 atmosphere with 100% humidity as described previously.17 2000 cells/well were added into 96-well plates and treated with increasing concentrations of a compound for 14 days. Cell viability was determined by Cell Counting Kit-8 (Dojindo Molecular Technologies, Rockville, MD) according to the manufacturer’s instructions.
Supplementary Material
Acknowledgment
This work was supported by a grant (RP140469) from Cancer Prevention and Research Institute of Texas (CPRIT) and a grant (R01NS080963) from National Institute of Neurological Disorders and Stroke (NINDS/NIH) to Y.S.
ABBREVATIONS
- α-KG
α-ketoglutaric acid
- D2HG
D-2-hydroxyglutaric acid
- ICT
isocitric acid
- IDH
isocitrate hydrogenase
- ITC
isothermal titration calorimetry
- R132H
Arg132 mutation to His
- R132C
Arg132 mutation to Cys
- SAR
structure activity relationship
- WT
wild-type
Footnotes
Supporting Information Available. Table S1, Figure S1-S4 and Experimental Section showing compound synthesis and characterization. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- (1).Xu X, Zhao J, Xu Z, Peng B, Huang Q, Arnold E, Ding J. Structures of human cytosolic NADP-dependent isocitrate dehydrogenase reveal a novel self-regulatory mechanism of activity. J. Biol. Chem. 2004;279:33946–33957. doi: 10.1074/jbc.M404298200. [DOI] [PubMed] [Google Scholar]
- (2).Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, Keir S, Nikolskaya T, Nikolsky Y, Busam DA, Tekleab H, Diaz LA, Jr., Hartigan J, Smith DR, Strausberg RL, Marie SK, Shinjo SM, Yan H, Riggins GJ, Bigner DD, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Xiao MT, Liu LX, Jiang WQ, Liu J, Zhang JY, Wang B, Frye S, Zhang Y, Xu YH, Lei QY, Guan KL, Zhao SM, Xiong Y. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Gross S, Cairns RA, Minden MD, Driggers EM, Bittinger MA, Jang HG, Sasaki M, Jin S, Schenkein DP, Su SM, Dang L, Fantin VR, Mak TW. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J. Exp. Med. 2010;207:339–344. doi: 10.1084/jem.20092506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D, Herndon J, Kinzler KW, Velculescu VE, Vogelstein B, Bigner DD. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Hartmann C, Meyer J, Balss J, Capper D, Mueller W, Christians A, Felsberg J, Wolter M, Mawrin C, Wick W, Weller M, Herold-Mende C, Unterberg A, Jeuken JW, Wesseling P, Reifenberger G, von Deimling A. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol. 2009;118:469–474. doi: 10.1007/s00401-009-0561-9. [DOI] [PubMed] [Google Scholar]
- (7).Amary MF, Bacsi K, Maggiani F, Damato S, Halai D, Berisha F, Pollock R, O’Donnell P, Grigoriadis A, Diss T, Eskandarpour M, Presneau N, Hogendoorn PC, Futreal A, Tirabosco R, Flanagan AM. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J. Pathol. 2011;224:334–343. doi: 10.1002/path.2913. [DOI] [PubMed] [Google Scholar]
- (8).Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, Marks KM, Prins RM, Ward PS, Yen KE, Liau LM, Rabinowitz JD, Cantley LC, Thompson CB, van der Heiden MG, Su SM. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE, Rabinowitz JD, Carroll M, Su SM, Sharp KA, Levine RL, Thompson CB. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225–234. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, Wellen KE, O’Rourke DM, Berger SL, Chan TA, Levine RL, Mellinghoff IK, Thompson CB. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, Campos C, Fabius AW, Lu C, Ward PS, Thompson CB, Kaufman A, Guryanova O, Levine R, Heguy A, Viale A, Morris LG, Huse JT, Mellinghoff IK, Chan TA. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483:479–483. doi: 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Prensner JR, Chinnaiyan AM. Metabolism unhinged: IDH mutations in cancer. Nat. Med. 2011;17:291–293. doi: 10.1038/nm0311-291. [DOI] [PubMed] [Google Scholar]
- (13).Garber K. Oncometabolite? IDH1 discoveries raise possibility of new metabolism targets in brain cancers and leukemia. J. Natl. Cancer Inst. 2010;102:926–928. doi: 10.1093/jnci/djq262. [DOI] [PubMed] [Google Scholar]
- (14).Popovici-Muller J, Saunders JO, Salituro FG, Travins JM, Yan S, Zhao F, Gross S, Dang L, Yen KE, Yang H, Straley KS, Jin S, Kunii K, Fantin VR, Zhang S, Pan Q, Shi D, Biller SA, Su SM. Discovery of the first potent inhibitors of mutant IDH1 that lower tumor 2-HG in vivo. ACS Med. Chem. Lett. 2012;3:850–855. doi: 10.1021/ml300225h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, Tsoi J, Clark O, Oldrini B, Komisopoulou E, Kunii K, Pedraza A, Schalm S, Silverman L, Miller A, Wang F, Yang H, Chen Y, Kernytsky A, Rosenblum MK, Liu W, Biller SA, Su SM, Brennan CW, Chan TA, Graeber TG, Yen KE, Mellinghoff IK. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340:626–630. doi: 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Zheng B, Yao Y, Liu Z, Deng L, Anglin JL, Jiang H, Prasad BV, Song Y. Crystallographic Investigation and Selective Inhibition of Mutant Isocitrate Dehydrogenase. ACS Med. Chem. Lett. 2013;4:542–546. doi: 10.1021/ml400036z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Liu Z, Yao Y, Kogiso M, Zheng B, Deng L, Qiu JJ, Dong S, Lv H, Gallo JM, Li X-N, Song Y. Inhibition of Cancer-Associated Mutant Isocitrate Dehydrogenases: Synthesis, SAR and Selective Antitumor Activity. J. Med. Chem. 2014;57:8307–8318. doi: 10.1021/jm500660f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Deng G, Shen J, Yin M, McManus J, Mathieu M, Gee P, He T, Shi C, Bedel O, McLean LR, Le-Strat F, Zhang Y, Marquette JP, Gao Q, Zhang B, Rak A, Hoffmann D, Rooney E, Vassort A, Englaro W, Li Y, Patel V, Adrian F, Gross S, Wiederschain D, Cheng H, Licht S. Selective inhibition of mutant isocitrate dehydrogenase 1 (IDH1) via disruption of a metal binding network by an allosteric small molecule. J. Biol. Chem. 2015;290:762–774. doi: 10.1074/jbc.M114.608497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Yang B, Zhong C, Peng Y, Lai Z, Ding J. Molecular mechanisms of “off-on switch” of activities of human IDH1 by tumor-associated mutation R132H. Cell Res. 2010;20:1188–1200. doi: 10.1038/cr.2010.145. [DOI] [PubMed] [Google Scholar]
- (20).Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA, Dick JE. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–648. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- (21).Luchman HA, Stechishin OD, Dang NH, Blough MD, Chesnelong C, Kelly JJ, Nguyen SA, Chan JA, Weljie AM, Cairncross JG, Weiss S. An in vivo patient-derived model of endogenous IDH1-mutant glioma. Neuro. Oncol. 2012;14:184–191. doi: 10.1093/neuonc/nor207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Wan F, Zhang S, Xie R, Gao B, Campos B, Herold-Mende C, Lei T. The utility and limitations of neurosphere assay, CD133 immunophenotyping and side population assay in glioma stem cell research. Brain Pathol. 2010;20:877–889. doi: 10.1111/j.1750-3639.2010.00379.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Laks DR, Masterman-Smith M, Visnyei K, Angenieux B, Orozco NM, Foran I, Yong WH, Vinters HV, Liau LM, Lazareff JA, Mischel PS, Cloughesy TF, Horvath S, Kornblum HI. Neurosphere formation is an independent predictor of clinical outcome in malignant glioma. Stem Cells. 2009;27:980–987. doi: 10.1002/stem.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Chaichana K, Zamora-Berridi G, Camara-Quintana J, Quinones-Hinojosa A. Neurosphere assays: growth factors and hormone differences in tumor and nontumor studies. Stem Cells. 2006;24:2851–2857. doi: 10.1634/stemcells.2006-0399. [DOI] [PubMed] [Google Scholar]
- (25).Liu Z, Zhao X, Mao H, Baxter PA, Huang Y, Yu L, Wadhwa L, Su JM, Adesina A, Perlaky L, Hurwitz M, Idamakanti N, Police SR, Hallenbeck PL, Hurwitz RL, Lau CC, Chintagumpala M, Blaney SM, Li XN. Intravenous injection of oncolytic picornavirus SVV-001 prolongs animal survival in a panel of primary tumor-based orthotopic xenograft mouse models of pediatric glioma. Neuro. Oncol. 2013;15:1173–1185. doi: 10.1093/neuonc/not065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Otwinowski Z, Minor W. In: Macromolecular Crystallography, Part A, Method Enzymology. Carter CW Jr., Sweet RM, editors. Vol. 276. Academic Press; New York: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- (27).McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J. Appl. Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta. Crystallogr. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- (29).Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta. Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Tomašić T, Mašič LP. Rhodanine as a scaffold in drug discovery: a critical review of its biological activities and mechanisms of target modulation. Expert Opin. Drug Discov. 2012;7:549–560. doi: 10.1517/17460441.2012.688743. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.