

Abstract
Infection of some cell types by reovirus evokes a caspase-independent form of cell death resembling necrosis. While reovirus strain T3D induces necrosis much more efficiently than strain T1L, which viral components contribute to this difference is not known. In this study, we identified that the sialic acid binding property of the reovirus σ1 protein affects necrosis efficiency. We found that in addition to sialic acid engagement by the virus particles, viral gene expression, in the form of viral RNA or protein synthesis, is also required for necrosis induction. Our studies reveal that sialic acid does not directly participate in necrosis induction by initiating a signaling pathway. Instead, sialic acid engagement augments necrosis induction indirectly, by increasing reovirus gene expression in each infected cell. Comparison of our results with previous studies suggests that reovirus-induced apoptosis and necrosis are initiated by distinct stages of viral infection.
INTRODUCTION
Programmed cell death is an antiviral mechanism (Flint et al., 2009; Upton and Chan, 2014). To overcome cell death mediated restriction of virus replication, viruses encode strategies to prevent or delay cell death (Hay and Kannourakis, 2002; Lamkanfi and Dixit, 2010; Mocarski et al., 2012). Regardless of whether viruses are hampered by the cell death response or have evolved mechanisms to overcome it, death of the infected cells contributes to viral pathogenesis (Clarke and Tyler, 2009; Mocarski et al., 2014).
Mammalian reovirus, henceforth referred to as reovirus, is used as an experimental system to understand how the interplay between viral and cellular factors controls the induction and execution of death responses (Danthi, 2013). One form of programmed cell death, apoptosis, contributes to encephalitis and myocarditis following reovirus infection of cells within the central nervous system and heart, respectively (Beckham et al., 2010; Berens and Tyler, 2011; Danthi et al., 2008a; Danthi et al., 2008b; Danthi et al., 2010; DeBiasi et al., 2004; O'Donnell et al., 2005; Richardson-Burns et al., 2002). Apoptosis induction following reovirus infection involves activation of host transcription factor NF-κB by the upstream IκB kinase (IKK)(Connolly et al., 2000; Hansberger et al., 2007). NF-κB activation is required for caspase-8-mediated cleavage of Bid, a BH3-only member of the Bcl-2 family of proteins (Danthi et al., 2010; Kominsky et al., 2002a, b). Cleaved Bid, tBid, activates the intrinsic apoptotic cascade leading to the activation of effector caspases (Danthi et al., 2010). A role for other proteins such as transcription factor IRF-3, the protein kinase c-Jun N-terminal kinase (JNK), and the protease calpain in reovirus-induced cell death has also been suggested (Clarke et al., 2004; Debiasi et al., 1999; Holm et al., 2007; Knowlton et al., 2012). Recent studies indicate that reovirus infection of some cell types results in an alternative, necrotic form of cell death (Berger and Danthi, 2013). Necrosis following reovirus infection occurs in absence of NF-κB function or caspase activity, but instead is diminished by blocking the kinase activity of RIP1 (Berger and Danthi, 2013).
Events in reovirus replication that result in apoptosis induction have been extensively studied. The efficiency of apoptosis induction following reovirus infection is affected by receptor engagement (Barton et al., 2001b; Connolly et al., 2001). In addition, events that occur after penetration of host cell membranes by reovirus but prior to synthesis of viral RNA and proteins also are implicated in apoptosis induction (Connolly and Dermody, 2002; Danthi et al., 2006). Consistent with the effect of early events in virus infection on apoptosis induction, the efficiency of apoptosis following reovirus infection maps to the viral S1 and M2 gene segments, which respectively encode the σ1 attachment protein and the μ1 membrane penetration protein (Connolly et al., 2001; Danthi et al., 2006; Rodgers et al., 1997; Tyler et al., 1996; Tyler et al., 1995). The capacity of the σ1 protein to engage cell surface receptors affects NF-κB activation and caspase activation and therefore, is a major determinant of apoptotic potential (Barton et al., 2001b; Connolly et al., 2001). Whether σ1-receptor interactions contribute to cell death directly by activating a signaling pathway, or indirectly through effects on other aspects of viral replication, remains unknown.
While it is not precisely known how necrosis is initiated following reovirus infection, there is a key difference in the viral requirement for apoptosis and necrosis induction. Reovirus-induced apoptosis can be triggered by genome-deficient reovirus particles or UV-inactivated virus particles (Connolly and Dermody, 2002; Danthi et al., 2006). In contrast, reovirus-induced necrosis requires the presence of transcriptionally competent viral RNA (Berger and Danthi, 2013). Here, we demonstrate that necrosis induction following reovirus infection requires viral gene expression and that viral gene expression is enhanced by the capacity of the viral attachment protein σ1 to bind sialic acid. Both, the requirement for viral gene expression for necrosis induction and the means by which sialic acid engagement affects necrosis induction indicate that reovirus-induced apoptosis and necrosis are initiated via distinct mechanisms.
RESULTS
The reovirus S1 gene affects efficiency of necrosis induction
We previously showed that reovirus infection of L929 cells results in caspase-independent cell death (Berger and Danthi, 2013). Cell death in L929 cells is enhanced by RIP1 kinase activity and exhibits features of necrosis (Berger and Danthi, 2013). We also showed that among prototype reovirus strains, T3D induces necrosis much more efficiently than T1L (Berger and Danthi, 2013). Based on previous studies, which indicated that strain-specific differences in cell death induction are controlled by the viral S1 gene, we assessed the contribution of S1 to the necrosis-inducing capacity of reovirus (Connolly et al., 2001; Rodgers et al., 1997; Tyler et al., 1996; Tyler et al., 1995). For these experiments we utilized recombinant monoreassortant viruses T1L/T3DS1 and T3D/T1LS1 (Boehme et al., 2009; Kobayashi et al., 2010). The capacity of these viruses to induce necrosis in L929 cells was compared to the parental strains T1L and T3D. Consistent with our previous work, T3D induced significantly higher amounts of cell death than T1L (Berger and Danthi, 2013) (Figure 1). We observed that the presence of a T3D S1 gene in an otherwise T1L background (T1L/T3DS1) resulted in cell death equivalent to that observed following T3D infection. Conversely, the presence of a T1L S1 gene in an otherwise T3D background resulted in T1L-like levels of cell death. These data suggest that the viral S1 gene determines the efficiency of cell death induction following reovirus infection.
Fig. 1. The S1 gene determines efficiency with which reovirus induces necrosis.

ATCC L929 cells were adsorbed with 10 PFU/cell of T1L, T3D, T1L/T3DS1, or T3D/T1LS1. Following incubation at 37°C for 48 h, cells were stained with AOEB. The results are expressed as the mean percentages of cells undergoing cell death for three independent experiments. Error bars indicate SD. *, P < 0.05 by student's t test in comparison to T3D.
Sialic acid binding confers potency for necrosis
The viral S1 gene encodes the σ1 attachment protein and the σ1s non-structural protein (Dermody et al., 2013). In this study, we focused on the role of the σ1 protein. While T1L and T3D σ1 proteins both bind the JAM-A proteinaceous receptor, they bind different glycan receptors (Barton et al., 2001b; Campbell et al., 2005; Reiss et al., 2012; Reiter et al., 2011). Whereas T1L σ1 engages GM2 glycans, T3D σ1 engages GM3 glycans that terminate in sialic acid (Reiss et al., 2012; Reiter et al., 2011). To determine the contribution of sialic acid binding capacity of σ1 to the higher necrosis potential of T3D and T1L/T3DS1, we utilized reovirus strains T3SA+ and T3SA− (Barton et al., 2001a). While T3SA+ engages sialic acid, T3SA− contains a Leu-to-Pro change at residue 204 near the sialic acid binding site of σ1 and cannot bind sialic acid (Barton et al., 2001a; Reiter et al., 2011). All other gene segments in T3SA+ and T3SA− are derived from T1L and are identical. Comparison of cell death induction by T3SA+ and T3SA− indicate that T3SA+ induces significantly higher cell death than T3SA− at both 48 and 72 h following infection (Figure 2A). These data suggest that sialic acid binding controls efficiency of cell death induction in L929 cells. As an alternate strategy to evaluate the importance of sialic acid binding to cell death induction, we pretreated the cells with neuraminidase to remove cell surface sialic acid. Neuraminidase treatment diminished the necrotic potential of T3SA+ but had no effect on cell death induction by T3SA− after 48 hours of infection (Figure 2B). Together our data indicate that sialic acid binding is required for efficient induction of cell death.
Fig. 2. Sialic acid engagement is required for efficient induction of necrosis by reovirus.
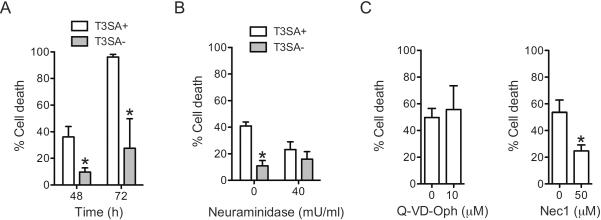
(A) ATCC L929 cells were adsorbed with 10 PFU/cell of T3SA+ or T3SA−. Following incubation at 37°C for 48 and 72 h, cells were stained with AOEB. The results are expressed as the mean percentages of cells undergoing cell death for three independent experiments. Error bars indicate SD. *, P < 0.05 by student's t test in comparison to T3SA+ at the comparable time point. (B) ATCC L929 cells were pretreated with PBS or 40mU/mL neuraminidase for 1 h at 37°C, washed with PBS and adsorbed with 10 PFU/cell of T3SA+ or T3SA−. Following incubation at 37°C for 48 h, cells were stained with AOEB. The results are expressed as the mean percentages of cells undergoing cell death for three independent experiments. Error bars indicate SD. *, P < 0.05 by student's t test in comparison to T3SA+ without neuraminidase. (C) ATCC L929 cells were adsorbed with 10 PFU/cell of T3SA+. Following incubation at 37°C for 48 in the presence of DMSO (0 μM) or the indicated concentration of Q-VD-OPh or Nec1 the, the cells were stained with AOEB. The results are expressed as the mean percentages of cells undergoing cell death for three independent experiments. Error bars indicate SD. *, P < 0.05 by student's t test in comparison to T3SA+ in the presence of DMSO.
Because both type 1 and type 3 reovirus strains kill L929 cells by necrosis (Berger and Danthi, 2013), we expected that T3SA+ would also induce necrotic cell death in this cell type. To confirm the mode of cell death induced by T3SA+, we measured the sensitivity of T3SA+ induced cell death to a pan-caspase inhibitor (Q-VD-OPh) and a RIP1 kinase inhibitor (Nec1)(Caserta et al., 2003; Degterev et al., 2008). Analogous to our previous observations with T1L and T3D (Berger and Danthi, 2013), cell death following infection was diminished by Nec1 but not Q-VD-OPh (Figure 2C). Thus, cell death following infection of L929 cells with T3SA+ occurs in a caspase independent manner and is sensitive to blockade of RIP1 kinase function.
Sialic acid binding alone is not sufficient for necrosis induction
To determine if sialic acid binding is sufficient for necrosis induction, we assessed the capacity of UV-treated T3SA+ to induce necrosis. To ensure that UV treatment did not adversely damage the integrity of reovirus particles, we exposed the virus to UV for only 1 min. To confirm that UV treatment does not affect virus attachment and entry, we compared disassembly kinetics of untreated and UV-treated virus over the first 6 h of infection. Based on the evidence that equivalent amount of virus was found attached to cells at 0 h post infection and that a comparable amount of δ was generated at 3 and 6 h following infection, we conclude that UV treatment did not influence virus attachment, internalization and disassembly (Figure 3A). Due to the short duration of UV treatment, we observed that virus infectivity was not abolished but was reduced by 2–3 orders of magnitude (data not shown). To ensure that any residual infectivity present in UV-treated particles does not confound the interpretation of our results, these experiments were terminated at 24 h following infection. We found that UV treatment diminished cell death following infection initiated both at an MOI of 10 and 100 PFU/cell (Figure 3B).
Fig. 3. Events subsequent to sialic acid engagement are required for reovirus-induced necrosis.

(A) ATCC L929 cells were absorbed with untreated or UV inactivated T3SA+ at an MOI of 100 PFU/cell. Cell lysates prepared following infection in the presence of CHX for various time intervals were immunoblotted for μ1 and PSTAIR loading control. μ1 resolves as μ1C on SDS-PAGE gels (Nibert et al., 2005). (B) ATCC L929 cells were adsorbed with 10 PFU/cell or 100 PFU/cell of T3SA+ or UV-treated T3SA+. Following incubation at 37°C for 24 h, cells were stained with AOEB. The results are expressed as the mean percentages of cells undergoing cell death for three independent experiments. Error bars indicate SD. *, P < 0.05 by student's t test in comparison to an equivalent MOI of untreated T3SA+. (C) ATCC L929 cells were pretreated with 0 or 200 μM ribavirin for 1 h at 37°C, then washed and adsorbed with 10 PFU/cell of T3SA+. Increase in viral RNA levels were determined following incubation with 0 or 200 μM ribavirin at 37°C for 6 h. The results are expressed as mean increase in RNA for three independent samples. Errors indicate SD. * P < 0.05 by student's t test in comparison to cell treated with 0 μM ribavirin at 0 h. (D) ATCC L929 cells were pretreated with 0 or 200 μM ribavirin for 1 h at 37°C, then washed and adsorbed with 10 PFU/cell of T3SA+. Following incubation at 37°C for 48 h in the presence of 0 or 200 μM ribavirin, cells were stained with AOEB. The results are expressed as the mean percentages of cells undergoing cell death for three independent experiments. Error bars indicate SD. *, P < 0.05 by student's t test in comparison to T3SA+ in the presence of 0 μM ribavirin. (E) ATCC L929 cells were pretreated with 0 or 200 μM ribavirin for 1 h at 37°C, then treated with 10 ng/ml TNFα and 25 μM Z-VAD-FMK for 12 h. The results are expressed as the mean percentages of cells undergoing cell death for three independent experiments. Error bars indicate SD.
The reduced capacity of UV-treated virus to induce necrosis suggests that necrosis requires steps in reovirus replication that occur following RNA synthesis. To directly examine this possibility, we measured necrosis induction in cells treated with ribavirin, an inhibitor of reovirus mRNA synthesis (Rankin et al., 1989). A concentration of 200 μM of ribavirin was used based on the previously demonstrated effect of this dose on blocking reovirus infection (Connolly and Dermody, 2002). While an ~ 600-fold increase in RNA was observed following infection for 6 h in the absence of ribavirin. In contrast, only ~ 6-fold increase was observed in the presence of ribavirin (Figure 3C). We observed that cell death inducing potential of T3SA+ was diminished following ribavirin treatment (Figure 3D). The potent induction of necrosis by treatment of cells with TNFα in the presence of a pancaspase inhibitor was not affected by ribavirin treatment indicating that ribavirin does not influence the function of the core necrosis machinery (Figure 3E) (Vercammen et al., 1998). Moreover, our data support the idea that either viral RNA synthesis or events that occur subsequent to viral RNA synthesis are required for efficient induction of necrosis.
Sialic acid binding enhances efficiency of infection
Because RNA synthesis or events that occur subsequent to it are required for necrosis induction, it was somewhat unexpected that the sialic acid binding capacity of reovirus would influence the efficiency of necrosis. We reasoned that the impact of sialic acid engagement on the difference in the capacity of T3SA+ and T3SA− to elicit necrosis are likely a consequence of disparity in their efficiency to synthesize RNA or complete a subsequent step in virus replication. Given the importance of events during and after RNA synthesis for necrosis induction by T3SA+, we first tested whether T3SA+ and T3SA− differ in their capacity to produce viral RNA. For this experiment, we measured the levels of viral RNA 6 h following infection. While infection with T3SA+ produced ~ 1000-fold increase in viral RNA, infection with T3SA− produced only about a ~ 300-fold increase in viral RNA (Figure 4A).
Fig. 4. Sialic acid attachment enhances efficiency of viral gene expression.
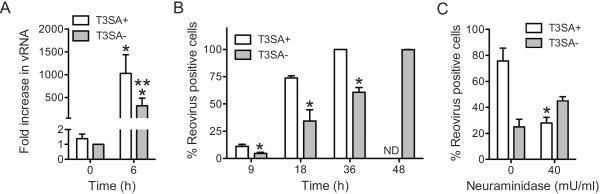
(A) ATCC L929 cells were adsorbed with 10 PFU/cell of T3SA+ or T3SA−. Increase in viral RNA levels was determined following incubation at 37°C for 6 h. The results are expressed as mean increase in RNA for three independent samples. Errors indicate SD. * P < 0.05 by student's t test in comparison to T3SA− at 0 h. ** P < 0.05 by student's t test in comparison to T3SA− at 6 h. (B) ATCC L929 cells were infected with T3SA+ or T3SA− at 10 PFU/cell. The results are expressed as the mean percentages of reovirus positive cells for three independent experiments. Error bars indicate SD. *, P < 0.05 by student's t test in comparison to T3SA+ at the comparable time point. ND, not determined (C) ATCC L929 cells pretreated with PBS or 40mU/mL neuraminidase at 37°C for 1 h, then washed and adsorbed with T3SA+ or T3SA− at 10 PFU/cell. Following incubation at 37°C for 18 h, reovirus positive cells were identified by indirect immunofluorescence. The results are expressed as the mean percentages of reovirus positive cells for three independent experiments. Error bars indicate SD. *, P < 0.05 by student's t test in comparison to T3SA+ in absence of neuraminidase.
To measure if this difference in RNA synthesis was a consequence of the fraction of cells that were infected by T3SA+ and T3SA−, we used indirect immunofluorescence to detect infected cells at 9, 18, 36 and 48 h following infection. At each time point until 36 h post infection, T3SA+ infected a greater proportion of cells in comparison to T3SA− (Figure 4B). Moreover, 100% of cells were infected in T3SA+ infected cells at 36 h following infection. A 100% of cells were found infected following infection with T3SA− only at 48 h following infection. Thus a lower level of RNA detected at 6 h following infection with T3SA− may be due to infection of a smaller fraction of reovirus positive cells or due to infection of these cells in a manner that produces a lower amount of viral proteins that are undetectable under the assay conditions used. Comparison of the infectivity data with those presented in Figure 2 indicate that despite infection of 100% of cells by 48 h, T3SA− fails to induce necrosis to levels equivalent to those observed by T3SA+. Thus, the differences in the necrosis-inducing potential of these two viral strains is not a direct consequence of the number of cells infected but likely due to the duration for which they are infected or the amount of gene expression taking place in each infected cell.
To confirm that the differences in infectivity of T3SA+ and T3SA− are due to differences in the capacity to engage sialic acid, we determined the effect of neuraminidase on the infectivity of T3SA+. We found that neuraminidase diminished the infectivity of T3SA+ but not T3SA− (Figure 4C). Thus, sialic acid binding confers reovirus a greater capacity to establish infection in L929 cells.
Virus attachment controls cell death by influencing events that occur after receptor engagement
L929 cells used for these experiment support infection by T1L, a virus that does not bind sialic acid (Berger and Danthi, 2013). Moreover, T3SA+ and T3SA− exhibit equivalent particle-to-PFU ratios in these cells (data not shown). Thus, why the numbers of infected cells are different following infection with T3SA+ and T3SA− is not understood. To determine if there are qualitative or quantitative differences in the capacity of T3SA+ and T3SA− to bind L929 cells, we measured virus attachment using flow cytometry. We found that the mean fluorescence intensity (MFI), a measure of the number of virus particles bound per cell was significantly higher for T3SA+ than T3SA− (Figure 5A). These results indicate that sialic acid binding allows a greater amount of virus to bind each cell. Our data are consistent with previous studies indicating that avidity of T3SA+ for cell binding is greater than that of T3SA− (Barton et al., 2001a; Barton et al., 2003). Moreover, these findings establish a correlation between sialic acid mediated enhancement in cell attachment and induction of necrotic cell death.
Fig. 5. T3SA− can overcome low cell death potential at higher doses.
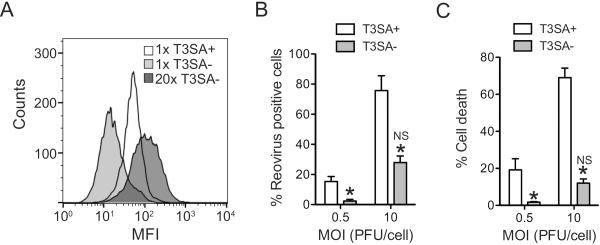
(A) ATCC L929 cells were adsorbed with 6 × 104 (1×) or 1.2 × 105 (20×) particles/cell of T3SA+ or T3SA−. Attached virions were detected by staining with anti-reovirus sera using a flow cytometer. (B) ATCC L929 cells were adsorbed with T3SA+ or T3SA− at the indicated MOI. Following incubation at 37°C for 18 h, reovirus positive cells were identified by indirect immunofluorescence. The results are expressed as the mean percentages of reovirus positive cells for three independent experiments. Error bars indicate SD. *, P < 0.05 by student's t test in comparison to T3SA+ at an equivalent MOI. NS, not significant in comparison to T3SA+ at an MOI of 0.5 PFU/cell. We note that 10PFU/cell samples are the same as those used in Figure 4. (C) ATCC L929 cells were adsorbed with T3SA+ or T3SA− at the indicated PFU/cell. Following incubation at 37°C for 48 h, cells were stained with AOEB. The results are expressed as the mean percentages of cells undergoing cell death for three independent experiments. Error bars indicate SD. *, P < 0.05 by student's t test in comparison to T3SA+ at the equivalent MOI. NS, not significant in comparison to T3SA+ at an MOI of 0.5 PFU/cell.
Our data demonstrate that sialic acid binding is required but not sufficient for necrosis induction (Figure 3). Moreover, our results indicate that viral RNA synthesis or a subsequent step in virus infection also is required for reovirus-induced necrosis. Therefore it can be hypothesized that necrotic cell death requires two signaling pathways - one initiated by sialic acid engagement and the other by virus replication - that function together to evoke necrosis. An alternate explanation for these observations could be that enhanced binding conferred by sialic acid attachment results in more efficient infection and thereby results in more cell death. If the latter possibility is true, then adding greater amounts of T3SA− at levels that recapitulate virus attachment efficiency of T3SA+ should produce infectivity and cell death to levels equivalent to T3SA+, even in absence of sialic acid engagement. When 20-fold more T3SA− was added, it displayed an MFI more closely resembling that of T3SA+ (Figure 5A). Under conditions where a 20-fold excess of T3SA− was used to initiate infection, infectivity (Figure 5B) and cell death induction (Figure 5C) by T3SA+ and T3SA− were also equivalent. The capacity of T3SA− to induce necrosis indicates that sialic acid binding per se is not required for necrosis induction. Thus, enhanced cell death efficiency conferred by sialic acid attachment is not related to signaling events initiated by sialic acid binding. Instead, the effect of sialic acid on cell death induction is a consequence of increasing the efficiency with which subsequent stages of reovirus infection occur.
The data presented thus far suggest that differences in the attachment efficiency of T3SA+ and T3SA− to cells determine viral infectivity and cell death. If the correlation between attachment and cell death is correct, we should expect that in a population of cells bound by T3SA+, those cells to which more T3SA+ particles are attached will exhibit a greater level of gene expression and will be more likely to succumb to cell death. To test this idea, we infected cells at an MOI of 20 or 200 PFU per cell of T3SA+. To quantify viral gene expression, viral RNA synthesis was measured 6 h following infection. We found that a greater amount of viral RNA accumulated in the cell as MOI increased (Figure 6A). Consistent with this difference in viral RNA synthesis, we found that the fraction of reovirus positive cells 9 h following infection also was higher as the MOI increased. Interestingly, at each MOI, nearly all cells displayed reovirus positivity within 18 h, a time frame roughly equivalent to one replication cycle (Figure 6B). Despite infection of an equivalent number of cells, we observed that 24 h following infection, a greater proportion of cells succumb to death as the MOI increased from 20 to 200 PFU/cell (Figure 6C). These data our similar to our findings presented in Figure 2 and 4 which indicate that under conditions where an equivalent fraction of cells is infected, necrotic potential of reovirus varies with the level to which viral gene products are expressed or accumulated. We showed above that when infection was initiated at an MOI of 10 PFU/cell, necrosis was sensitive to the viral RNA synthesis inhibitor, ribavirin (Figure 3). To ensure that this sensitivity was maintained at higher MOIs, we also measured necrosis in the presence of ribavirin (Figure 6D). We found that cell death at each MOI was significantly diminished when the cells were treated with ribavirin. This result highlights the importance of viral RNA synthesis or subsequent events in the induction of necrosis.
Fig. 6. Enhanced attachment promotes cell death by increasing viral gene expression.

ATCC L929 cells were adsorbed with T3SA+ at an MOI of 20 or 200 PFU/cell. (A) Increase in viral RNA levels was determined following incubation at 37°C for 6 h. The results are expressed as mean increase in RNA for three independent samples. Errors indicate SD. * P < 0.05 by student's t test in comparison to T3SA+ at MOI of 20 PFU/cell at 0 h. ** P < 0.05 by student's t test in comparison to T3SA+ at MOI of 20 PFU/cell at 6 h. (B) Following infection at 37°C for 9 h or 18 h, the fraction of infected cells was measured by indirect immunofluorescence and DAPI staining. The results are expressed as mean percentage of infected cells from three independent samples. Error bars indicate SD. * P < 0.05 by student's t test in comparison to T3SA+ at MOI of 20 PFU/cell at the comparable time point. ** P < 0.05 by student's t test in comparison to T3SA+ at MOI of 20 PFU/cell (C) Following infection at 37°C for 24 h in the presence of 0 or 200 μM ribavirin, the cells were stained with AOEB. The results are expressed as the mean percentages of cells undergoing cell death for three independent experiments. Error bars indicate SD. * P < 0.05 by student's t test in comparison to T3SA+ at an equivalent MOI. ** P < 0.05 by student's t test in comparison to T3SA+ at MOI of 20 PFU/cell in the absence of ribavirin.
DISCUSSION
Infection of some cell types with reovirus results in a RIP1 kinase dependent necrosis (Berger and Danthi, 2013). Toward gaining an understanding of how reovirus induces necrosis, we investigated the basis for the strain-specific differences in the capacity for necrosis induction. We identified that sialic acid binding capacity of the viral σs1 attachment protein controls necrosis. Though engagement of sialic acid enhances cell death, it is not sufficient for necrosis. Events dependent on production of viral RNA are also required for cell death induction. Our studies indicate that sialic acid binding enhances cell death by increasing the efficiency with which post-replicative events in the reovirus infectious cycle take place.
Unlike necrosis, apoptosis induction by reovirus does not require the presence of transcriptionally competent viral RNA (Connolly and Dermody, 2002). Thus, the incoming viral capsid is sufficient to activate proapoptotic signaling. Consistent with this, sialic acid binding reovirus strains induce greater levels of apoptosis than non-binding strains (Connolly et al., 2001). Evidence that even a 100-fold excess of T3SA−, a level that more than overcomes its lower binding efficiency (Barton et al., 2001a), fails to induce apoptosis to levels similar to T3SA+ suggest that differences in the apoptotic potential of T3SA+ and T3SA− are not merely due to increase in a binding to the cell but due to engaging sialic acid (Connolly et al., 2001). Because viral disassembly is also required for apoptosis induction, it is thought that two signals, one initiated by sialic acid engagement and other by disassembly cooperatively function to induce apoptosis (Connolly and Dermody, 2002). The contribution of sialic acid attachment for necrosis induction that we have shown in this study appears to be distinct. First, we show that when binding efficiency of T3SA− and T3SA+ are equalized (by using a 20-fold excess of T3SA+), the lower cell death efficiency of T3SA− can be overcome. Thus, unlike apoptosis efficiency, necrosis efficiency is dependent on levels of virus attached to cells, not the engagement of sialic acid itself. Additionally, we show here that T3SA+ induces higher necrosis because sialic acid engagement augments events in reovirus replication that are required for necrosis. Thus, sialic acid binding enhances apoptosis and necrosis following reovirus infection in a distinct manner.
We demonstrate here that even when all cells are infected, sialic acid binding engagement enhances necrosis. Following attachment to receptors, reovirus particles are endocytosed and disassembled (Dermody et al., 2013). The entry intermediates generated by disassembly deliver the viral inner core across the membrane (Dermody et al., 2013). Cores delivered into the cytoplasm initiate mRNA synthesis (Dermody et al., 2013). Thus, minimally, delivery of a single genetically and physically intact core would result in synthesis of viral RNA and produce a reovirus positive cell. We show in Figure 6 that an MOI of 20 PFU/cell, nearly all cells are reovirus positive within the time frame of a typical reovirus infectious cycle. Thus 20 PFU/cell is sufficient to allow delivery of at least one functional core into the cytoplasm of each cell. As the MOI increases to 200 PFU/cell, greater numbers of virions are attached to cells, and consequently a greater number of cores are delivered into the cytoplasm and produce more viral RNA. We think that the greater level of gene expression is also manifested in our infectivity assays where cells infected with MOI of 200 PFU/cell score reovirus-positive before those that are infected with MOI of 20 PFU/cell. Because necrotic cell death is dependent on RNA synthesis or a virus replication event dependent on viral RNA synthesis, cells that are attached by more virus, produce more viral RNA and therefore succumb to necrosis. Precisely how signaling pathways involving RIP1 kinase that lead to necrotic cell death are initiated by reovirus infection is under investigation.
Viruses used for our experiments were titered on L929 cells. Measurement of viral infectivity, viral RNA synthesis, and cell death were also performed on L929 cells. We observed that despite initiating infection with an MOI of 10 PFU/cell, a dose that should result in infection of all cells within the time frame of the first replication cycle (14–16 h) (Silverstein et al., 1976), we were unable to detect infection of all cells until multiple rounds of infection were completed. We also observed that even though infection of T3SA+ and T3SA− was initiated at equivalent MOIs, T3SA+ appears to propagate through the monolayer of L929 cells with faster kinetics than T3SA−. The basis for these peculiar observations is currently not known. One possibility is simply that our assay lacks the sensitivity to detect low-level viral gene expression. Another possibility could be that distinct signaling pathways are activated by different virus strains and that these signaling pathways affect the number of cells that are infected and the rate at which they are infected. These ideas remain to be formally tested.
Sialic acid binding controls the pathogenesis of reovirus disease. Sialic acid engagement has been demonstrated to be important for dissemination of reovirus, infection of cells of the bile duct and infection of cortical neurons (Barton et al., 2003; Frierson et al., 2012). As an attachment moiety on the cell surface, it is thought that sialic acid affects reovirus disease because it affects the tropism of the virus. However, based on our studies here, it is tempting to speculate that at least some of the effects of sialic acid engagement on reovirus pathogenesis is due to the enhancement of post attachment steps that lead to increased viral gene expression or cell killing. Additional experimental evidence is needed to support or refute this possibility.
MATERIALS AND METHODS
Cells
ATCC L929 cells were maintained in Eagle's MEM (EMEM) (Lonza) supplemented to contain 5% fetal bovine serum (FBS) (Invitrogen) and 2 mM L-glutamine (Invitrogen). Spinner-adapted murine L929 cells were maintained in Joklik's MEM (Lonza) supplemented to contain 5% FBS, 2 mM L-glutamine, 100 U/ml penicillin (Invitrogen), 100 μg/ml streptomycin (Invitrogen), and 25 ng/ml amphotericin B (Sigma-Aldrich). Virus titers and all experiments were performed in ATCC L929 cells. Spinner-adapted L929 cells were used for cultivating and purifying viruses.
Viruses
Prototype reovirus strains T1L and T3D and reassortant viruses T1L/T3DS1, T3D/T1LS1 were regenerated by plasmid-based reverse genetics (Kobayashi et al., 2007). T3SA+ and T3SA− are laboratory stocks that have been previously described (Barton et al., 2001a). Viral particles were Vertrel-XF (Dupont) extracted from infected cell lysates, layered onto 1.2 to 1.4 g/cm3 CsCl gradients, and centrifuged at 187,183 × g for 4 h. Bands corresponding to virions (1.36 g/cm3) were collected and dialyzed in virion-storage buffer (150 mM NaCl, 15 mM MgCl2, 10 mM Tris-HCl [pH 7.4]) (Berard and Coombs, 2009). The concentration of reovirus particles in purified preparations was determined from an equivalence of one OD unit at 260 nm equals 2.1 × 1012 virions per ml (Smith et al., 1969). Virus titers were determined as described (Berard and Coombs, 2009).
UV inactivation of virus
UV-inactivated virus was generated using a UV cross-linker (CL-1000 UV Crosslinker; UVP). T3SA+ virus (1.7 × 1011 PFU/ml) was irradiated with short-wave (254-nm) UV on ice at a distance of 10 cm for 1 min at 120,000 μJ/cm2 in a 60-mm tissue culture dish (Berger and Danthi, 2013).
Antibodies and reagents
Anti-reovirus polyclonal antibody was obtained from T. Dermody. Anti-μ1 4A3 mAb was purchased from Developmental Studies Hybridoma Bank. Alexa Fluorconjugated anti-rabbit IgG secondary antibody was purchased from Invitrogen. Ribavirin, Neuraminidase, Q-VD-OPh, Necrostatin (Nec1), Z-VAD-FMK were purchased from Sigma-Aldrich, MP biomedical, R&D systems, Santacruz, and ApexBio respectively and used at the indicated concentrations. None of the inhibitors displayed any cytotoxicity at the concentration used.
Assessment of ISVP formation in cells
ATCC L929 cells grown in 6-well plates were adsorbed with indicated doses of each virus at room temperature for 1 h. For analyzing ISVP formation, media added post adsorption also included 10 μg/ml cycloheximide. Cycloheximide was used to ensure that signal from only incoming viral proteins is detected. Cells were harvested at different time points, washed with PBS, and lysed using RIPA lysis buffer (50 mM Tris [pH 7.5], 50 mM NaCl, 1% TX-100, 1% DOC, 0.1% SDS, and 1 mM EDTA) containing a protease inhibitor cocktail (Roche), 500 μM DTT and 500 mM PMSF followed by centrifugation at 13,000 rpm at 4°C for 5 min to remove cell debris. The clarified lysates were resolved by electrophoresis in SDS-PAGE gels and transferred to nitrocellulose membranes. Membranes were blocked for at least 1 h in blocking buffer (TBS containing 5% milk) and incubated with anti-4A3 antibody (1:1000) and PSTAIR (1:10000) at 4°C overnight. Membranes were washed three times for 5 min each with washing buffer (TBS containing 0.1% Tween-20) and incubated with 1:20000 dilution of Alexa Fluor conjugated anti-mouse IgG (for PSTAIR) in blocking buffer. Following three washes, membranes were scanned using an Odyssey Infrared Imager (LICOR).
Assessment of infectivity by indirect immunofluorescence
ATCC L929 cells grown in 96-well plates were adsorbed with indicated doses of each virus at room temperature for 1 h Following infection for the necessary time interval, monolayers were fixed with chilled methanol at −20°C for a minimum of 30 min, washed twice with PBS, blocked with 2.5% Ig-free bovine serum albumin (Invitrogen). Cells were then incubated with polyclonal rabbit anti-reovirus serum at a 1:5000 dilution in PBS containing 0.25% Triton X-100 (TX-100) at room temperature for 30 min. Monolayers were washed twice with PBS and incubated with a 1:5000 dilution of Alexa Fluor 488-labeled anti-rabbit IgG (Invitrogen) in PBS containing 0.25% TX-100. DAPI was added to all samples to detect all cells and measure the fraction of infected cells. The infected cells were visualized by indirect immunofluorescence using a FITC filter set on an Olympus IX71 microscope. Infected cells were identified by the presence of intense cytoplasmic fluorescence that was excluded from the nuclei. No background staining of uninfected control monolayers was observed. Reovirus antigen-positive cells and total cells were quantified by counting fluorescent cells in random fields at a magnification of 20x.
Assessment of viral attachment by flow cytometry
L929 cells were detached using PBS supplemented with 20 mM EDTA and virus adsorption was performed by continuous rotation. After removal of the inoculum, the cells were harvested and resuspended in PBS supplemented with 5% BSA (PBS-BSA). Reovirus-specific rabbit polyclonal antiserum at 1:5000 was added and incubated at 4°C for 30 min with continuous rotation. The cells were washed twice with PBS-BSA followed by incubation at 4°C for 30 min with the Alexa Fluor 488-labeled goat anti-rabbit antibody at 1:1000 in PBS-BSA with continuous rotation. The cells were washed twice with PBS-BSA prior to being fixed with cold 1% paraformaldehyde in PBS. Viral attachment to cells was quantified using BD FACSCalibur Cell Analyzer and the CellQuestPro software.
RT-qPCR
RNA was extracted from infected cells using RNAeasy (Qiagen) 0.5 to 2 μg of RNA was reverse transcribed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) and random hexamers. The cDNA obtained was used for PCR using the SYBR Select Master Mix (Applied Biosystems) and primers specific for reovirus T1L S2 (AAGCGTTGGCAGATCAAACT and ATGGCTTCGTACGGAACATC) and murine GAPDH (ACCCAGAAGACTGTGGATGG, GGATGCAGGGATGATGTTCT). ΔCt values for each cDNA sample were calculated by subtracting Ct values of T1L S2 and Ct values for GAPDH. Fold change in gene expression with respect to the indicated sample was measured using the ΔΔCt method (Schmittgen and Livak, 2008).
Quantitation of cell death by Acridine orange and ethidium bromide (AOEB) staining
ATCC L929 cells grown in 24-well plates were adsorbed with T1L/T3SA+ or T1L/T3SA− at the indicated MOI at room temperature for 1 h. The percentage of dead cells after incubation in presence in untreated cells or cells treated with the indicated inhibitor was determined using AOEB staining as described (Tyler et al., 1995). For each experiment, >200 cells total cells were counted, and the percentage of isolated cells exhibiting orange staining (EB positivity) was determined by fluorescence microscopy using an Olympus IX71 microscope. For each experiment, cell death in control or inhibitor treated, but mock infected cells was also determined. Since cell death under these conditions was minimal (< 3%), these data are not included.
Highlights.
Differences in efficiency of necrosis induction by reovirus strains map to the viral S1 gene
The sialic acid binding capacity of the S1-encoded σ1 protein influences necrosis
Viral gene expression is also required for necrosis induction
Sialic acid engagement potentiates necrosis by increasing viral gene expression in each infected cell.
ACKNOWLEDGEMENTS
We thank members of our laboratory, Karl Boehme, and Tuli Mukhopadhyay for helpful suggestions and review of the manuscript. Flow cytometry was performed in the Indiana University Bloomington Flow Cytometry Core Facility with assistance from Christiane Hassel. This work was supported by funds from Public Health Service award 1R01AI110637, Walther Cancer Foundation, and Indiana University (P.D).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Barton ES, Connolly JL, Forrest JC, Chappell JD, Dermody TS. Utilization of sialic acid as a coreceptor enhances reovirus attachment by multistep adhesion strengthening. J Biol Chem. 2001a;276:2200–2211. doi: 10.1074/jbc.M004680200. [DOI] [PubMed] [Google Scholar]
- Barton ES, Forrest JC, Connolly JL, Chappell JD, Liu Y, Schnell FJ, Nusrat A, Parkos CA, Dermody TS. Junction adhesion molecule is a receptor for reovirus. Cell. 2001b;104:441–451. doi: 10.1016/s0092-8674(01)00231-8. [DOI] [PubMed] [Google Scholar]
- Barton ES, Youree BE, Ebert DH, Forrest JC, Connolly JL, Valyi-Nagy T, Washington K, Wetzel JD, Dermody TS. Utilization of sialic acid as a coreceptor is required for reovirus-induced biliary disease. J Clin Invest. 2003;111:1823–1833. doi: 10.1172/JCI16303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckham JD, Tuttle KD, Tyler KL. Caspase-3 activation is required for reovirus-induced encephalitis in vivo. J Neurovirol. 2010;16:306–317. doi: 10.3109/13550284.2010.499890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berard A, Coombs KM. Mammalian reoviruses: propagation, quantification, and storage. Current protocols in microbiology. 2009;Chapter 15(Unit15C):11. doi: 10.1002/9780471729259.mc15c01s14. [DOI] [PubMed] [Google Scholar]
- Berens HM, Tyler KL. The proapoptotic Bcl-2 protein Bax plays an important role in the pathogenesis of reovirus encephalitis. Journal of virology. 2011;85:3858–3871. doi: 10.1128/JVI.01958-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger AK, Danthi P. Reovirus activates a caspase-independent cell death pathway. mBio. 2013;4 doi: 10.1128/mBio.00178-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehme KW, Guglielmi KM, Dermody TS. Reovirus nonstructural protein sigma1s is required for establishment of viremia and systemic dissemination. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:19986–19991. doi: 10.1073/pnas.0907412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JA, Schelling P, Wetzel JD, Johnson EM, Forrest JC, Wilson GA, Aurrand-Lions M, Imhof BA, Stehle T, Dermody TS. Junctional adhesion molecule a serves as a receptor for prototype and field-isolate strains of mammalian reovirus. Journal of virology. 2005;79:7967–7978. doi: 10.1128/JVI.79.13.7967-7978.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caserta TM, Smith AN, Gultice AD, Reedy MA, Brown TL. Q-VD-OPh, a broad spectrum caspase inhibitor with potent antiapoptotic properties. Apoptosis. 2003;8:345–352. doi: 10.1023/a:1024116916932. [DOI] [PubMed] [Google Scholar]
- Clarke P, Meintzer SM, Wang Y, Moffitt LA, Richardson-Burns SM, Johnson GL, Tyler KL. JNK regulates the release of proapoptotic mitochondrial factors in reovirus-infected cells. Journal of virology. 2004;78:13132–13138. doi: 10.1128/JVI.78.23.13132-13138.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke P, Tyler KL. Apoptosis in animal models of virus-induced disease. Nat Rev Microbiol. 2009;7:144–155. doi: 10.1038/nrmicro2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly JL, Barton ES, Dermody TS. Reovirus binding to cell surface sialic acid potentiates virus-induced apoptosis. Journal of virology. 2001;75:4029–4039. doi: 10.1128/JVI.75.9.4029-4039.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly JL, Dermody TS. Virion disassembly is required for apoptosis induced by reovirus. Journal of virology. 2002;76:1632–1641. doi: 10.1128/JVI.76.4.1632-1641.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly JL, Rodgers SE, Clarke P, Ballard DW, Kerr LD, Tyler KL, Dermody TS. Reovirus-induced apoptosis requires activation of transcription factor NF-kappaB. Journal of virology. 2000;74:2981–2989. doi: 10.1128/jvi.74.7.2981-2989.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danthi P, Coffey CM, Parker JS, Abel TW, Dermody TS. Independent regulation of reovirus membrane penetration and apoptosis by the mu1 phi domain. PLoS Pathog. 2008a;4:e1000248. doi: 10.1371/journal.ppat.1000248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danthi P, Hansberger MW, Campbell JA, Forrest JC, Dermody TS. JAM-A-independent, antibody-mediated uptake of reovirus into cells leads to apoptosis. Journal of virology. 2006;80:1261–1270. doi: 10.1128/JVI.80.3.1261-1270.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danthi P, Holm GH, Stehle T, Dermody TS. Reovirus receptors, cell entry, and signaling. In: Pöhlmann S, Simmons G, editors. Viral Entry into Cells. Georgetown, TX: 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danthi P, Kobayashi T, Holm GH, Hansberger MW, Abel TW, Dermody TS. Reovirus apoptosis and virulence are regulated by host cell membrane penetration efficiency. Journal of virology. 2008b;82:161–172. doi: 10.1128/JVI.01739-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danthi P, Pruijssers AJ, Berger AK, Holm GH, Zinkel SS, Dermody TS. Bid regulates the pathogenesis of neurotropic reovirus. PLoS Pathog. 2010;6:e1000980. doi: 10.1371/journal.ppat.1000980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBiasi RL, Robinson BA, Sherry B, Bouchard R, Brown RD, Rizeq M, Long C, Tyler KL. Caspase inhibition protects against reovirus-induced myocardial injury in vitro and in vivo. Journal of virology. 2004;78:11040–11050. doi: 10.1128/JVI.78.20.11040-11050.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debiasi RL, Squier MK, Pike B, Wynes M, Dermody TS, Cohen JJ, Tyler KL. Reovirus-induced apoptosis is preceded by increased cellular calpain activity and is blocked by calpain inhibitors. Journal of virology. 1999;73:695–701. doi: 10.1128/jvi.73.1.695-701.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degterev A, Hitomi J, Germscheid M, Ch'en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nature chemical biology. 2008;4:313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dermody TS, Parker JC, Sherry B. Orthoreoviruses. In: Knipe DM, Howley PM, editors. Fields Virology. Sixth ed Lippincott Williams & Wilkins; Philadelphia: 2013. [Google Scholar]
- Flint SJ, Enquist LW, Racaniello VR, Skalka AM. Priciples of Virology. 3rd ed ASM press; 2009. [Google Scholar]
- Frierson JM, Pruijssers AJ, Konopka JL, Reiter DM, Abel TW, Stehle T, Dermody TS. Utilization of sialylated glycans as coreceptors enhances the neurovirulence of serotype 3 reovirus. Journal of virology. 2012;86:13164–13173. doi: 10.1128/JVI.01822-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansberger MW, Campbell JA, Danthi P, Arrate P, Pennington KN, Marcu KB, Ballard DW, Dermody TS. IkappaB kinase subunits alpha and gamma are required for activation of NF-kappaB and induction of apoptosis by mammalian reovirus. Journal of virology. 2007;81:1360–1371. doi: 10.1128/JVI.01860-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay S, Kannourakis G. A time to kill: viral manipulation of the cell death program. Journal of General Virology. 2002;83:1547–1564. doi: 10.1099/0022-1317-83-7-1547. [DOI] [PubMed] [Google Scholar]
- Holm GH, Zurney J, Tumilasci V, Leveille S, Danthi P, Hiscott J, Sherry B, Dermody TS. Retinoic acid-inducible gene-I and interferon-beta promoter stimulator-1 augment proapoptotic responses following mammalian reovirus infection via interferon regulatory factor 3. J Biol Chem. 2007;282:21953–21961. doi: 10.1074/jbc.M702112200. [DOI] [PubMed] [Google Scholar]
- Knowlton JJ, Dermody TS, Holm GH. Apoptosis induced by mammalian reovirus is beta interferon (IFN) independent and enhanced by IFN regulatory factor 3- and NF-kappaB-dependent expression of Noxa. Journal of virology. 2012;86:1650–1660. doi: 10.1128/JVI.05924-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Antar AAR, Boehme KW, Danthi P, Eby EA, Guglielmi KM, Holm GH, Johnson EM, Maginnis MS, Naik S, Skelton WB, Wetzel JD, Wilson GJ, Chappell JD, Dermody TS. A plasmid-based reverse genetics system for animal double-stranded RNA viruses. Cell Host Microbe. 2007;1:147–157. doi: 10.1016/j.chom.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Ooms LS, Ikizler M, Chappell JD, Dermody TS. An improved reverse genetics system for mammalian orthoreoviruses. Virology. 2010;398:194–200. doi: 10.1016/j.virol.2009.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kominsky DJ, Bickel RJ, Tyler KL. Reovirus-induced apoptosis requires both death receptor- and mitochondrial-mediated caspase-dependent pathways of cell death. Cell Death and Differentiation. 2002a;9:926–933. doi: 10.1038/sj.cdd.4401045. [DOI] [PubMed] [Google Scholar]
- Kominsky DJ, Bickel RJ, Tyler KL. Reovirus-induced apoptosis requires mitochondrial release of Smac/DIABLO and involves reduction of cellular inhibitor of apoptosis protein levels. Journal of virology. 2002b;76:11414–11424. doi: 10.1128/JVI.76.22.11414-11424.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamkanfi M, Dixit VM. Manipulation of host cell death pathways during microbial infections. Cell Host Microbe. 2010;8:44–54. doi: 10.1016/j.chom.2010.06.007. [DOI] [PubMed] [Google Scholar]
- Mocarski ES, Kaiser WJ, Livingston-Rosanoff D, Upton JW, Daley-Bauer LP. True grit: programmed necrosis in antiviral host defense, inflammation, and immunogenicity. J Immunol. 2014;192:2019–2026. doi: 10.4049/jimmunol.1302426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mocarski ES, Upton JW, Kaiser WJ. Viral infection and the evolution of caspase 8-regulated apoptotic and necrotic death pathways. Nat Rev Immunol. 2012;12:79–88. doi: 10.1038/nri3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nibert ML, Odegard AL, Agosto MA, Chandran K, Schiff LA. Putative autocleavage of reovirus m1 protein in concert with outer-capsid disassembly and activation for membrane permeabilization. Journal of Molecular Biology. 2005;345:461–474. doi: 10.1016/j.jmb.2004.10.026. [DOI] [PubMed] [Google Scholar]
- O'Donnell SM, Hansberger MW, Connolly JL, Chappell JD, Watson MJ, Pierce JM, Wetzel JD, Han W, Barton ES, Forrest JC, Valyi-Nagy T, Yull FE, Blackwell TS, Rottman JN, Sherry B, Dermody TS. Organ-specific roles for transcription factor NF-kappaB in reovirus-induced apoptosis and disease. J Clin Invest. 2005;115:2341–2350. doi: 10.1172/JCI22428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankin UT, Jr., Eppes SB, Antczak JB, Joklik WK. Studies on the mechanism of the antiviral activity of ribavirin against reovirus. Virology. 1989;168:147–158. doi: 10.1016/0042-6822(89)90413-3. [DOI] [PubMed] [Google Scholar]
- Reiss K, Stencel JE, Liu Y, Blaum BS, Reiter DM, Feizi T, Dermody TS, Stehle T. The GM2 glycan serves as a functional coreceptor for serotype 1 reovirus. PLoS Pathog. 2012;8:e1003078. doi: 10.1371/journal.ppat.1003078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter DM, Frierson JM, Halvorson EE, Kobayashi T, Dermody TS, Stehle T. Crystal structure of reovirus attachment protein sigma1 in complex with sialylated oligosaccharides. PLoS Pathog. 2011;7:e1002166. doi: 10.1371/journal.ppat.1002166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson-Burns SM, Kominsky DJ, Tyler KL. Reovirus-induced neuronal apoptosis is mediated by caspase 3 and is associated with the activation of death receptors. J Neurovirol. 2002;8:365–380. doi: 10.1080/13550280260422677. [DOI] [PubMed] [Google Scholar]
- Rodgers SE, Barton ES, Oberhaus SM, Pike B, Gibson CA, Tyler KL, Dermody TS. Reovirus-induced apoptosis of MDCK cells is not linked to viral yield and is blocked by Bcl-2. Journal of virology. 1997;71:2540–2546. doi: 10.1128/jvi.71.3.2540-2546.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Silverstein SC, Christman JK, Acs G. The reovirus replication cycle. Annual Review of Biochemistry. 1976;45:375–408. doi: 10.1146/annurev.bi.45.070176.002111. [DOI] [PubMed] [Google Scholar]
- Smith RE, Zweerink HJ, Joklik WK. Polypeptide components of virions, top component and cores of reovirus type 3. Virology. 1969;39:791–810. doi: 10.1016/0042-6822(69)90017-8. [DOI] [PubMed] [Google Scholar]
- Tyler KL, Squier MK, Brown AL, Pike B, Willis D, Oberhaus SM, Dermody TS, Cohen JJ. Linkage between reovirus-induced apoptosis and inhibition of cellular DNA synthesis: role of the S1 and M2 genes. Journal of virology. 1996;70:7984–7991. doi: 10.1128/jvi.70.11.7984-7991.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler KL, Squier MK, Rodgers SE, Schneider BE, Oberhaus SM, Grdina TA, Cohen JJ, Dermody TS. Differences in the capacity of reovirus strains to induce apoptosis are determined by the viral attachment protein sigma 1. Journal of virology. 1995;69:6972–6979. doi: 10.1128/jvi.69.11.6972-6979.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton JW, Chan FK. Staying alive: cell death in antiviral immunity. Mol Cell. 2014;54:273–280. doi: 10.1016/j.molcel.2014.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vercammen D, Beyaert R, Denecker G, Goossens V, Van Loo G, Declercq W, Grooten J, Fiers W, Vandenabeele P. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J Exp Med. 1998;187:1477–1485. doi: 10.1084/jem.187.9.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]