

Abstract
Ion transport across the mitochondrial inner and outer membranes is central to mitochondrial function, including regulation of oxidative phosphorylation and cell death. Although essential for ATP production by mitochondria, recent findings have confirmed that the c-subunit of the ATP synthase also houses a large conductance uncoupling channel, the mitochondrial permeability transition pore (mPTP), the persistent opening of which produces osmotic dysregulation of the inner mitochondrial membrane and cell death. This review will discuss recent advances in understanding the molecular components of mPTP, its regulatory mechanisms and how these contribute directly to its physiological as well as pathological roles.
Keywords: Mitochondria, calcium dysregulation, cell death, metabolism, ion channels
Graphical Abstract
Mitochondria at the center of cell metabolism and cell death
Mitochondria are complex organelles responsible for producing energy in the form of ATP for most eukaryotic cells. They regulate several other essential processes including calcium (Ca2+) homeostasis, heme and steroid biosynthesis. In addition, the mitochondrion lies at the center of the cellular response to stress and the control of cell death.
To produce energy in the form of ATP, mitochondria utilize substrates produced in the cytosol by carbohydrate, lipid and protein metabolic pathways. These products, particularly acetyl co-enzyme A, enter the tricarboxylic acid cycle. Turns of the TCA cycle synthesize NADH and FADH2 that donate their electrons to the electron transport chain. The energy of the bonds of NADH and FADH2 is used to pump H+ ions out of the matrix by the NADH dehydrogenase and other electron transport complexes, creating a proton motive force that in turn drives the F1FO ATP synthase [1]. Upon kinetic repositioning of the ATP synthase rotor, ATP is synthesized from ADP and Pi [2]. The machinery required for ADP/ATP exchange between the cytoplasm and the matrix including the outer membrane voltage dependent anion channel (VDAC) and the adenine nucleotide transporter (ANT) at the inner membrane are intimately linked to that of the ATP synthase [3].
Mitochondrial inner membrane leak-regulator of metabolic rate and uncoupling
There are two currents that complete the current loop of the proton pumping activity of the electron transport complexes. First, ATP is formed by hydrogen ion (H+) translocation through the ATP synthase in the opposite direction to that of the electron transport complexes. Second, an apparently wasteful leak in the inner mitochondrial membrane provides a pathway for uncoupling of oxidation from phosphorylation as H+ ions enter the matrix through channels independent of ATP production. Classically, uncoupling proteins carry out this role. Known physiological functions of uncoupling proteins are to generate heat for organisms with large surface to volume ratio, to depolarize mitochondria in order to temper oxidative damage and to regulate metabolic rate during hibernation and at other times [4–7]. In addition to uncoupling proteins, however, intrinsic uncoupling exists within other inner mitochondrial membrane channels and transporters and within the F1FO ATP synthase [8,9].
Mitochondrial inner membrane Ca2+ cycling regulates cellular Ca2+ dynamics: the example of neuronal short term synaptic plasticity
Mitochondrial inner membrane depolarization occurs not only through proton movement but also via the flux of other ions including Ca2+ across mitochondrial membranes. Ca2+ movement into the mitochondrial matrix is a physiological event that takes place in response to increased cytosolic Ca2+ levels. Ca2+ buffering is frequently employed by mitochondria in cells that experience rapidly changing cytosolic Ca2+ levels.
Mitochondria regulate cytosolic levels of Ca2+ and the release of Ca2+ and metabolites through an intricate system involving several ion channels. The discovery of the molecular structure for the Ca2+ uniporter ion channel (MCU) at the mitochondrial inner membrane has generated increasing interest in mechanisms of Ca2+ management within the cell body of many types of cells and also in the presynaptic terminals of neurons [10–13]. Additional isoforms of MCU and its helper MICU that confer tissue specificity and other behaviors have added to our understanding of the mechanisms of activity dependent energy production by mitochondria [14,15]. Mitochondrial Ca2+ release also appears to be highly regulated, involving both exchangers and channels, but, unlike the MCU, the molecular components of a Ca2+ release channel were only recently discovered and form the main focus of this review.
Ca2+ re-release from mitochondria determines short term synaptic plasticity in certain neuronal synapses [16]. During neuronal activity, Ca2+ influx across the plasma membrane occurs through glutamate receptors and voltage gated Ca2+ channels. After Ca2+ enters the cytosol, Ca2+ clearance is performed by the actions of Ca2+ ATPases at the plasma membrane and by buffering through uptake by intracellular stores including the endoplasmic reticulum (ER) and mitochondria [12,17]; these processes reset the normally low Ca2+ levels present in resting neurons or neuronal synapses. The Ca2+ that is buffered by intracellular stores is eventually re-released, providing, for example, for residual Ca2+ in presynaptic endings. Residual Ca2+ increases the amount of Ca2+ available for synaptic vesicle fusion, enhancing the amount of neurotransmitter released for a given stimulus [18,19].
Ca2+ -sensitive ligand gated mitochondrial channels, which are widely conserved and found in species from invertebrates to mammals, open in response to elevated Ca2+ within the mitochondrial matrix. In the squid presynaptic terminal, opening of a Ca2+ -activated mitochondrial channel is correlated with enhanced neurotransmitter release [20]. Electrophysiological recordings [21] demonstrate that within the resting presynaptic terminal, the conductance of mitochondrial membranes is low [20]. In contrast, during high frequency electrical stimulation of the presynaptic nerve, a large increase in mitochondrial membrane ion channel activity takes place [20]. The delay in onset of the mitochondrial activity and the persistence of the mitochondrial activity after stimulation are in keeping with the role of a channel and/or exchanger in re-releasing Ca2+ from mitochondria for short term plasticity [20,22,23]. Furthermore, mitochondrial activity and short term increases in post stimulation synaptic transmitter release are both abrogated by applying the uncoupler FCCP (carbonyl cyanide p-trifluoromethoxyphenylhydrazone), which depolarizes mitochondria, preventing Ca2+ handling [20].
Bcl-2 family proteins: regulators of mitochondrial outer membrane permeability and cell death
In contrast to the normal physiological role for Ca2+ release channels and exchangers in mitochondrial membranes, one of the main regulators of pathological mitochondrial permeability or leakiness are the proteins of the Bcl-2 family. Programmed cell death (apoptosis) in vertebrate cells may be initiated by signaling at the plasma membrane or by intracellular pathways that lead to changes in mitochondria [24]. The final common pathway for programmed cell death in many systems is mitochondrial outer membrane permeabilization (MOMP) [25–28]. Pro-apoptotic Bcl-2 family members such as Bax regulate MOMP by inducing the formation of large outer membrane pores comprised of activated oligomerized proteins, aided by other pro-apoptotic moieties [25,27,29]. In their canonical role, the anti-apoptotic Bcl-2 family proteins such as Bcl-xL protect cells against MOMP by interacting with, and inhibiting the pore forming properties of, the pro-apoptotic family members [28,30].
MOMP leads to the release of several inter-membrane space proteins such as cytochrome c [31,32]. The resultant decrease in cytochrome c levels compromises the ability of mitochondria to maintain the mitochondrial inner membrane potential and to produce ATP [33]. In addition, cytochrome c released into the cytoplasm activates downstream cytosolic enzyme pathways including effector caspases that execute cell death [34].
The mitochondrial permeability transition and pathophysiology
In some cases, an increase in mitochondrial outer membrane permeability may also be triggered by an acute inner membrane depolarization [35], particularly after cytosolic and mitochondrial Ca2+ overload. Although Ca2+ uptake and re-release from mitochondria is a normal physiological event in cells, accumulation of Ca2+ in the matrix can have detrimental effects, including diminution in energy production by the ATP synthase [36]. Ca2+ overload can produce an uncoupling process described historically as a rapid increase in permeability of the mitochondrial inner membrane to solutes and the halting of ATP production [37–39]. This phenomenon is termed permeability transition (PT).
PT can be reversible or irreversible [35,37,40–45]. If not reversed, PT leads to an even more extreme form of catastrophic PT associated with structural breakdown of the mitochondrial matrix accompanied by outer mitochondrial membrane rupture and cell death [35]. The interaction of this kind of mitochondrial cell death with apoptotic death produced by MOMP has been debated. Although the two types of cell death seem to be overlapping, it is safe to say that pathological PT is associated with necrotic cell death such as is found in ischemia or injury whereas MOMP occurring in the presence of sufficient amounts of ATP may have a more important role in developmental and genetically predetermined death [46,47]. Inter-membrane space pro-apoptotic factors such as cytochrome c and Smac/DIABLO are released during both forms of cell death. In MOMP, outer membrane permeabilization alone leads to release of these factors, whereas in prolonged PT, rupture of the outer membrane after inner membrane swelling releases pro-apoptotic factors into the cytosol [35].
Physiological functions of the permeability transition pore
PT has been extensively studied for its role in ischemic injury in brain, heart and other organs as well as in neurodegenerative conditions [48]. In the heart, data suggest that opening of the mPTP during early reperfusion after ischemia is a harmful event that precipitates further damage to the myocardium [49]. However additional data also suggest that transient mPTP opening during preconditioning can be protective, thus serving a physiological role even during injury [43].
Further evidence of physiological opening of the mPTP has emerged over the last few years [40–45]. Flickering of the mPTP was described in the late 1990s [50,51], and an association between transient mPTP opening and “superoxide flashes” has been described in striated muscle mitochondria [44]. It has been hypothesized that transient opening of the mPTP releases mitochondrial matrix Ca2+ to maintain mitochondrial homeostasis [52] although this function of the mPTP has recently be questioned [53], and as mentioned above, mitochondrial matrix Ca2+ re-release regulates synaptic transmission [20]. Finally, as discussed in more detail below, we have observed physiological, long-term opening of the mPTP in the early embryonic heart [54].
Regulation of the permeability transition pore
The mPTP is induced by elevated mitochondrial matrix Ca2+, ROS, inorganic phosphate, and intracellular acidification [55,56]. In contrast, it is inhibited by ATP/ADP and Mg2+ [40,57]. Recent reports have also confirmed increased activity of PT by polyphosphates, chains of 10s to 100s of repeating phosphates linked by ATP-like high energy bonds [58–61]. The actions of Ca2+ may also require polyhydroxybutyrate (PHB), which enters mitochondria and enhances the ability of Ca2+ to induce PT [62].
Many signaling cascades have been proposed to cause or protect against ischemia reperfusion injury by targeting the mPTP. The RISK (Reperfusion Injury Salvage Kinase) pathway, involving PI3K, Akt, and Erk 1/2; SAFE (Survivor Activating Factor Enhancement) pathway, involving TNFα and STAT-3; and PKA, PKC, and PKG may all converge on inactivation/phosphorylation of GSK3β, which is known to regulate the mPTP [63–65]. However, the importance of these signaling cascades, in comparison to simple oxidative stress, has been questioned [64,66].
Cellular metabolic pathways regulate the PT. First, electron transport chain activity regulates the mPTP; an increase in mitochondrial energization (membrane potential) inhibits the PT, while de-energization/depolarization (a fall in membrane potential) enhances it [37,38,64]. Components of the glycolytic pathway may also control opening of the mPTP, as the binding of hexokinase II to the OMM in cardiac myocytes inhibits MOMP and PT [67]. Furthermore, a complex of ANT, VDAC, hexokinase, and mitochondrial creatine kinase (mtCK) regulates PT [68,69]. Combined, these data suggest that mitochondrial function and metabolic channeling of substrates and products (high energy phosphate bonds in the form of creatine kinase) into and out of mitochondria play a large role in the regulation of the mPTP. Finally, the major pharmacologic agents used to manipulate the mPTP target these metabolic pathways. For example, inhibitors of ANT can either attenuate (bongkrekic acid) or enhance (atractyloside) mPTP opening [38,70,71]. In addition, the pharmacological agent most efficient in inhibiting PT is cyclosporine A (CsA), an immunosuppressant drug which binds to cyclophilin D (CypD) and inhibits the channel activity associated with PT. CyPD binds to ANT, F1FO ATP synthase, and the phosphate carrier (discussed below) further suggesting a role for metabolic pathways in the regulation of the PTP.
The quest for the PT pore
Electrophysiologic properties of the mPTP
Thus, PT is an important event that performs both physiologic and pathophysiologic functions. Most current studies make the assumption that PT begins as the opening of a Ca2+ sensitive ion channel in the inner mitochondrial membrane similar to the ion channel activity initiated by mitochondrial Ca2+ influx occurring during physiological mitochondrial Ca2+ cycling. Such a Ca2+ release channel is heavily regulated; therefore it is assumed that only after prolonged opening does pathological PT (with MOMP) occur [72]. The conversion of a physiological Ca2+ extrusion mechanism into a pathological channel opening is perhaps correlated with energy failure as a result of arrest of ATP synthesizing activity and slowing of energy dependent Ca2+ extrusion mechanisms. Despite these hypothetical models, the factors that regulate the transition from physiological to pathophysiological events are not completely understood. Therefore, identification of the molecular structure of the pore will provide the target for regulatory activities, allowing for a greater understanding of PT modulation during health and disease.
Description of the biophysical properties of the pore that opens in the inner membrane during PT (the mPTP) provided the earliest indication that PT was initiated by the opening of an ion channel. The first patch clamp recordings of mitochondrial inner membrane were published in 1987. This early report highlighted a ~100 pS channel recorded by patch-clamping giant mouse liver mitochondria produced by cuprizone application [73]. In the late 1980s, a putative mPTP was recorded by patch-clamping mitochondrial inner membrane or mitoplast preparations [74]. The activity occurred at positive potentials of the patch pipette and was found either in whole organelle mode or in single channel recordings in the organelle-attached configuration. The activity was slightly anion over cation selective with multiple sub-conductance states ranging from 30 pS to a peak single channel conductance of 1.3 nS. Lower conductances were attributed to substates of the larger channel openings because of long periods lacking activity followed by periods of multi-conductance behavior [74]. Conductances of 550 pS were frequently observed at positive potentials. Gating was less common at negative potentials but this observation was consistent with the presence of prolonged openings and fewer sub-conductance steps at negative patch potentials contrasted with increased flickering at positive potentials. The authors concluded that conductance levels were not sharply defined, consistent with the existence of many varied conductance levels of the channel.
Also in 1989, Kinnally et al. recorded a similar mitochondrial multiconductance channel (MMC) in mouse liver mitoplasts [75]. This channel changed over time, with low activity at the onset of the recording followed by progressively higher activity at later times during the recording. The channels were sometimes open more frequently at negative potentials but at times rectification occurred in the opposite direction (more frequently open at positive potentials). Channel activity displayed multiple conductances ranging from 10–1000 pS and was weakly cation-selective. These early studies began to establish expected criteria for activity of mPTP.
Shortly after the first recordings of the putative mPTP were performed, similar inner membrane activity was found to be inhibited by CsA. In patch clamp experiments performed in liver mitochondria, channel activity was rapidly inhibited by submicromolar concentrations of CsA in a manner consistent with the expression of the binding site on the matrix side of the inner membrane. Ca2+ -activated large conductance channel activity up to 1.3 nS was inhibitable, but a 107 pS inner membrane conductance similar to the first recorded inner mitochondrial membrane channel was also observed in the recordings. This smaller conductance was resistant to CsA, suggesting that this activity might be due to a separate ion channel [70]. The large conductance channel was sensitive to Mg2+, Mn2+, Ba2+ and Sr2+ in that order, which inhibited the activity in a competitive manner with Ca2+, the main activator of the channel [55].
Characterization of a molecular complex regulating the pore
The recent identification of the molecular structure matching the biophysical properties of mPTP was aided by seemingly unrelated sets of findings. One was that Bcl-xL enhances metabolic efficiency (decreases uncoupling) by binding to the β-subunit of the ATP synthase. The second finding was that CypD, which had been known for many years to regulate PT, binds to the stator arm of ATP synthase, specifically on the OSCP subunit. The third finding suggested that closure of the mPTP is related to the level of CypD activity in a developmentally regulated manner as activity falls at the onset of respiration in mammalian heart. The final project found that ATP synthase assembles into a very large complex with other proteins that may regulate the mPTP. These findings will now be discussed in greater detail.
Bcl-xL regulates metabolic efficiency by binding to the β-subunit of the ATP synthase
Inefficiency of metabolism is correlated with cell death under conditions of neurodegeneration or acute cellular injury such as occurs during PT [76–78]. In contrast, a highly efficient state of metabolism requires maximally decreased uncoupling of the inner membrane, and it has been found recently that Bcl-2 family proteins regulate this efficiency by binding directly to the ATP synthase [79–81]. It was known previously that Bcl-2 family proteins regulate mitochondrial outer membrane permeability to produce or inhibit synaptic degeneration and cell death [30,82–84]. Therefore, it seemed possible that Bcl-2 family proteins could form part of a large protein complex that regulates mPTP and cell death.
During the initiation of cell death an important function of Bcl-xL, in addition to protection from MOMP, is to increase the release of ATP through enhanced VDAC opening. This decreases the probability of MOMP in cancer cell lines by providing extra ATP to overcome cell death stimuli [33,85]. In the neuronal synapse, injection of either Bcl-xL or ATP enhances synaptic transmitter release [86], suggesting that Bcl-xL increases ATP levels in the synapse [87]. These findings raise the possibility that Bcl-xL might regulate not only the release but also the production of ATP.
In support of a role for Bcl-xL in the manufacture of ATP, hippocampal neurons overexpressing Bcl-xL show a large increase in cytoplasmic ATP levels. Surprisingly, this increase in ATP accompanies a decrease in neuronal oxygen uptake and aerobic glycolysis, consistent with the notion that Bcl-xL overexpression increases mitochondrial bioenergetic efficiency [80,81]. Bcl-xL depletion reverses these effects on metabolism, decreasing ATP production and increasing oxygen uptake by resting cells [80,88].
Direct interaction of Bcl-xL with the β-subunit of the ATP synthase maximizes the efficiency of ATP production [80,81] by decreasing a leak conductance in the mitochondrial inner membrane. This was first measured by patch clamp recordings of submitochondrial vesicles (SMVs) enriched in ATP synthase [80].
Closure of the leak within the inner membrane in the presence of Bcl-xL aids actively firing neurons to increase neurotransmitter release [88,89], consistent with a correlation between the increase in metabolic efficiency and the long-term higher efficacy of synaptic transmission found in Bcl-xL expressing neurons. In contrast, opening of the Bcl-xL-regulated leak decreases metabolic efficiency and predisposes neurons to death, providing yet another clue that the Bcl-xL-regulated inner membrane leak could be mPTP. Furthermore, neurons lacking Bcl-xL display a fluctuating mitochondrial inner membrane potential and a marked depolarization in the presence of the ATP synthase inhibitor oligomycin [81]. These data suggest that Bcl-xL may regulate inner membrane coupling and cell death via direct effects on F1FO ATP synthase.
Cyclophilin D binds to ATP synthase and regulates permeability transition
Another piece of the puzzle that helped determine the molecular components of mPTP was the discovery of the interaction between CypD and ATP synthase [90]. It has long been known that CypD, a chaperone protein and peptidyl-prolyl cis-trans isomerase that resides in the mitochondrial matrix, regulates the mPTP by enhancing its sensitivity to Ca2+. In keeping with this, once it was found that CsA inhibits PT, CsA rapidly became one of the major tools to study the mPTP. Later, it was found that CsA attenuates mPTP by inhibiting CypD [91–93].
In 2005, experiments using CypD null mice demonstrated that CypD was not itself the pore of the mPTP but that it played an important regulatory role, particularly in the modulation of the mPTP by Ca2+ [94–97]. Four groups showed that deletion of CypD decreased sensitivity to ischemia-reperfusion injury in the heart and brain [94–96] and the Molkentin group suggested that mPTP’s physiologic function is to maintain “homeostatic mitochondrial Ca2+ levels to match metabolism with alterations in myocardial workload” [52].
CypD expression varies widely among cell types. CypD is more highly expressed in aged hearts, and these changes in expression may regulate its association with a complex of proteins that increases mPTP opening during reperfusion [63]. Furthermore, CypD activity appears to be regulated by cell signaling and metabolic pathways [64,98–101] and by developmental cues in differentiating myocytes (see below).
Although the exact mechanism of CypD’s action in the mitochondrial matrix has not been clarified, recent data support the idea that this requires its binding to F1FO ATP synthase. First, the propensity toward PT is regulated by ATP hydrolysis and synthesis in as much as this regulates the membrane potential. Therefore, PT requires twice the Ca2+ load in mitochondria that are in the process of hydrolyzing ATP (making a membrane potential) versus synthesizing ATP (dissipating the membrane potential) [38,39,56]. Second, CypD binds to proteins on the stator of ATP synthase (OSCP, b and d-subunits, [71]) as well as to the F1FO ATP synthase binding partners ANT [102,103] and the phosphate carrier (PiC, [52,104,105]). Recent work on the binding of CypD to ATP synthase indicates that this occurs exclusively with OSCP, and depletion of OSCP using siRNA also depletes CypD [56] suggesting that the association regulates the expression of each protein. Interestingly, CsA decreases the propensity toward PT to the same degree (half) that decreases in OSCP expression increase the propensity toward PT (double), suggesting an inhibitory effect of OSCP on PT [56]. Third, purified ATP synthase dimers produce current consistent with mPTP in artificial lipid membranes [56]. The single channel activity has a maximal conductance of 1–1.3 nS with subconductance states. The current is not stimulated by Ca2+ or inhibited by CsA presumably because CypD is not present in the purified dimer preparation although the enzymatic activity of the F1FO ATP synthase is preserved. The ATPase dimer current is also not sensitive to bongkrekic acid or atractyloside, agents that primarily affect ANT activity, making it unlikely that ANT forms part of the pore of the channel [56]. These data emphasize the idea that the regulation of the mPTP may occur via the interaction of CypD and other molecules with F1FO ATP synthase and its binding partners.
PT activity regulates cardiac development
A third line of evidence that helped unravel the identity of the mPTP was a series of studies of mitochondrial function during cardiac development. Until recently, the general consensus was that opening of the mPTP was a devastating event that triggers cell death. However, as discussed above, over the last two decades, data have emerged suggesting that transient opening of the mPTP could serve a physiologic purpose.
In the heart, physiological variations in mPTP activity play a critical role in cardiac myocyte differentiation and cardiac development [106]. Interestingly, the mPTP is open in myocytes in the early embryonic mouse heart, and this opening is not associated with any form of cell death. However, by the mid-embryonic stage, the mPTP is closed [106]. This closure coincides with activation of complex I of the electron transport chain, assembly of electron transport chain supercomplexes called respirasomes, and activation of oxidative phosphorylation [54]. These changes cause a fall in mitochondrial-derived ROS that signals the myocyte to undergo further differentiation [106]. Furthermore, pharmacologically inhibiting or genetically deleting mPTP enhances myocyte differentiation, while opening mPTP inhibits differentiation [106]. These findings have been confirmed in cardiac stem cells [107,108], and various reports have stressed the importance of the mPTP during cardiac development and myocyte differentiation [109,110].
Regulatory molecules do not form the pore of mPTP
The F1FO ATP synthase interacts with a large number of proteins many of which have been candidates for mPTP. ANT was an early candidate to form the mPTP since atractyloside and bongkrekic acid, which inhibit ANT, affect the mPTP [38] and ANT was found to interact with CypD [92]. VDAC was also an early candidate to form the mPTP due to its high conductance and its association with ANT in immunoprecipitation experiments [102]. In addition, it was shown that a complex of ANT, VDAC, hexokinase, and mitochondrial creatine kinase (mtCK) could form high conductance pores when reconstituted into membranes [68,69]. Finally, the PiC is a more recent candidate to form the mPTP [105].
However, genetic deletion of ANT1 and 2 and of the PiC demonstrated that these proteins were not essential to mPTP formation, although these studies still supported their regulatory roles [104,111,112]. Furthermore, deletion of VDAC did not affect pore formation [113]. Additional data suggest that the conformation of ANT may be important for regulation of the mPTP [114]. Atractyloside induces mPTP opening and is known to stabilize the “c” conformation of ANT, such that the adenine nucleotide transport site faces the cytoplasmic, or intermembrane space, side of the IMM [114]. In contrast, bongkrekic acid prevents mPTP opening and stabilizes ANT in its “m,” or matrix facing conformation [114]. However, as both ATR and BKA inhibit ANT, it is unlikely that specific effects on ADP/ATP translocation regulate the mPTP and a more likely scenario is that the conformation of ANT itself can regulate the mPTP. This is supported by a report that Ca2+ stabilization of ANT’s “c” conformation is related to its proline isomerization and CypD is a peptidyl-prolyl, cis-trans isomerase [115].
It remains unclear how these candidate molecules regulate the mPTP, but evidence suggests that they form large macromolecular structures with F1FO ATP synthase in the inner mitochondrial membrane. ANT and PiC can form a complex with F1FO ATP synthase called the synthasome [3]. In addition, the large complex of ANT, VDAC, hexokinase and mtCK is likely also involved in the regulation of ATP synthesis [68,69]. Therefore, each of these molecules may regulate the structure and activity of F1FO ATP synthase, and, in doing so, modulate the opening of the mPTP.
The mPTP, a molecular definition
The c-subunit of F1FO ATP synthase comprises the PT pore
These various reports all agree that F1FO ATP synthase is a major factor in the formation of the mPTP, and recent evidence suggests that the FO or membrane portion of F1FO ATP synthase in fact forms the pore [48,116–120]. Mammalian F1FO ATP synthase is a ~600 kDa complex of 15 subunits. The membrane portion, or FO, contains a ring of 8 very hydrophobic c-subunits and subunits a, b, e, f, g, and A6L. A stalk composed of the δ, ε, and γ subunits connects the c-subunit ring to the catalytic F1 component made of a hexamer of alternating α and β subunits, where ATP synthesis and hydrolysis occur. Finally, a stator containing the b, d, F6, and OSCP subunits connects the lateral portion of FO the top of the F1. Movement of protons between the c-subunit and the a-subunit causes rotation of the c-subunit ring, the energy of which is transferred to F1 to synthesize ATP [121–125].
Age-dependent structural and functional alterations of F1FO ATP synthase in rat brain and heart mitochondria have been reported [126]. The Fo portion is found to be present at 3 months in the membranes of heart mitochondria in the absence of an equivalent complement of F1. During the period from 3 to 12 months, the level of F1 increases with an increase in ATPase activity and this accompanies a decrease in proton leak consequent to binding of F1 to FO. Decreases of F1 content with respect to that observed for FO are detected for aging heart in animals from 12 to 24 months suggesting the presence of lone FO leak channels in the membranes of these aging mitochondria [126].
To determine if the membrane portion of the F1FO ATP synthase comprises a cell death regulatory moiety that could produce PT under cell stress, the Pinton group performed different measures of cell death and PT after c-subunit expression levels were genetically manipulated either by depletion of all three isoforms of the c-subunit or by overexpression of a tagged version of the protein in HeLa cells [116]. Given that proliferating cell lines rely on glycolysis for ATP production in normal glucose-containing medium, depleting the c-subunit did not alter ATP levels in the cells. Depletion of the c-subunit, however, prevented both CsA-sensitive PT measured by the calcein-cobalt quench technique and by mitochondrial morphological analysis. Cell death brought on by H2O2 was also attenuated by c-subunit depletion as was cell death in neurons brought on by glutamate-induced excitotoxicity.
Although the above study indicated that the F1FO ATP synthase c-subunit was an important component of the mPTP [116], this work did not directly determine what portion of ATP synthase could form the pore of the mPTP. Subsequent to that publication, work from the Bernardi group suggested that the mPTP could only form from dimers of ATP synthase [56], demonstrating mPTP-like channel activity from purified dimers, and not monomers, of ATP synthase. However, the regulation of such a purified mPTP by traditional mechanisms has not been conclusively demonstrated, and the actual pore of the mPTP in this model remained to be determined. Furthermore, it was subsequently demonstrated by our group that purified monomers and c-subunits (discussed below) can form the pore of mPTP [117]. Finally, the ability of ATP synthase inhibitory factor 1 (IF1) to promote dimer formation yet prevent PT argues against the idea that ATP synthase dimers are necessary and sufficient for mPTP formation [127,128].
Membranous pores are usually formed by integral membrane proteins, and it is interesting that molecules that regulate PT are either not membrane proteins, or, if they are integral membrane proteins (see above), they have been shown not to form the pore. For example, CypD and Bcl-xL regulate ATP synthesis by interacting with the stator and β subunit of ATPase, respectively, and these proteins are not embedded in the inner mitochondrial membrane. However, ATP synthase contains integral membrane proteins in its FO segment. 9 polypeptides form the FO and the stator, but only three, a, b and c, are required for proton translocation and are evolutionarily highly conserved, like PT. In addition, ρ0 cells that lack mitochondrial DNA do not contain an a-subunit but do undergo PT. Attention focused on the c-subunit, and not the b-subunit, for a number of reasons. First, the structure of purified bacterial c-subunit rings suggests that the center of the mammalian c-subunit ring could form an ion conducting channel that would allow for uncoupling if the stalk partially or completely dissociated from it [129]. Furthermore, the c-subunit had been shown previously to express ion channel activity [130]. As a matter of fact, the mammalian c-subunit undergoes conformational changes from an α-helix to a β-sheet when in contact with water, encompassing spaces that form the walls of ion channels [131]. These ion channels have a diameter of 2.3 nm which allows molecules up to 1.5 kDa to pass, similar to PT.
Homologues of ATP synthase c-subunit are also present in the V0 subcomplex of various vacuolar H+-ATPases, which are located both on the plasma membrane and on intracellular membranes [132,133]. It has been shown that transmembrane domain 1 of V0V1-ATPase c-subunits faces the center of the V0 rotor structure and lines a water-accessible pore structure [134,135] V0 c-subunits have been shown to be involved in formation of mega-channels in gap junctions between cells [136]. Therefore pore-forming ability is an important feature of all homologous c-subunits which share similar amino acid sequence [118].
The c-subunit of ATP synthase creates the high conductance mPTP pore
More recent experiments have directly tested the hypothesis that the main membrane embedded portion of mammalian F1FO ATP synthase, i.e. the c-subunit ring, forms the pore of the mPTP [118,137]. Indeed, electrophysiologic recordings of the purified mitochondrial c-subunit yielded a multi-conductance, voltage dependent channel with prominent subconductance states [117]. Patches contained a ~100-pS conductance, which appeared to be a subconductance state of the multiconductance activity rather than a separate conductance. Peak single channel conductances of ~1.5–2 nS were similar to activity described previously for the mitochondrial multiple conductance channel (MCC) [75]. Also consistent with MCC, channel activity often but not always demonstrated negative rectification. At very positive patch pipette potentials of over 100 mV, single channel conductances of ~1.5 nS and ~2 nS were also consistently observed. There was a higher likelihood of observing single channel events or gating at positive potentials, most likely because of the negative rectification, in keeping with similar activity observed previously for the mPTP [74].
Regarding the voltage dependence of the c-subunit recordings, it should be pointed out that voltage dependence is an inherent property of the channel [138] and is not dependent on the mitochondrial inner membrane potential or on the solutions used to record the currents. It can be measured by varying the voltage across the membrane (the command or holding voltage). Early published current-voltage (I–V) curves of mPTP [74,75] reveal that the channel shows both non-rectifying conductances and a rectification in the I–V that can be either in the positive or negative voltage range. These data are consistent with I–V plots of recent recordings of the purified c-subunit [117] (Fig. 1).
Fig. 1.

The current voltage relationship (I–V) of proteoliposome recordings of the purified c-subunit mimics that of mitoplast recordings of mPTP. A, B, C. Graphs reproduced from Kinnally et al., 1989 (with kind permission of Springer Science+Business Media) showing examples of the different I–V relationships for the mPTP found in different mitoplast (isolated inner mitochondrial membrane) recordings. D, E. Graphs reproduced from Alavian et al., 2014 showing examples of the different I–V relationships found in different proteoliposome recordings of the purified c-subunit. Shown in E are a low and high conductance mode of the channel.
F1 regulates biophysical characteristics of the purified c-subunit
Purified c-subunit protein reconstituted into liposomes clearly lacks extrinsic regulatory moieties that are important for mPTP opening. The effects of Ca2+, a critical regulatory molecule, demonstrate this property. Ca2+ binding sites have not been detected in mitochondrial Fo, perhaps because mammalian c-subunits lack the formyl Met at the N-terminus to which Ca2+ binds to E. coli or chloroplast F1FO ATP synthase c-subunits [139]. In contrast, Ca2+ can bind to specific, low affinity and moderate capacity sites on the β-subunit of mitochondrial F1 ATP synthase [140]. Other sites in other ATP synthase-interacting molecules may also be important for regulation by Ca2+ and other agents [68,69]. Therefore, although the new models of the pore must account for all inducers and inhibitors, these molecules may not interact directly with the c-subunit pore itself, but may instead bind to sites in the F1 or to other molecules such as ANT that undergo structural re-arrangements to open and close the pore. Another example of this type of regulation is CypD.
In order to determine the location of the regulators, mitochondrial recordings were carried out using purified mitochondrial and F1FO ATP synthase preparations. In these studies, the absence of an effect of a modulator was taken as an indication that the ligand or binding site for that modulator had been removed by the purification process. These studies demonstrated that the c-subunit leak channel is regulated by the overlying F1 and peripheral regulatory proteins [117]. In mitochondria or inner membrane preparations lacking the outer membrane such as submitochondrial vesicles (SMVs)[3], Ca2+ activates the c-subunit leak channel while CsA and ATP/ADP inhibit it, suggesting that the Ca2+ and CsA sensitive sites are present in these preparations. In contrast, removal of the F1 and other peripheral membrane proteins by urea treatment of the inner membrane or removal of CypD by purification of ATP synthase monomers abrogates regulation of the c-subunit channel by CsA and Ca2+ and greatly diminishes sensitivity to ATP/ADP. These studies suggest that the CypD/Ca2+ binding site is contained within or associated with the F1 portion of the ATP synthase and that a second, low-affinity ATP binding site exists in the FO. These results are consistent with reports identifying the binding site of CypD and benzodiazepine 423, an ATP synthase-inhibitory and mPTP-sensitizing agent, on OSCP [141,142], and suggest that the assembly of F1FO ATP synthase into monomers, dimers, and higher order oligomers can regulate the formation of the mPTP.
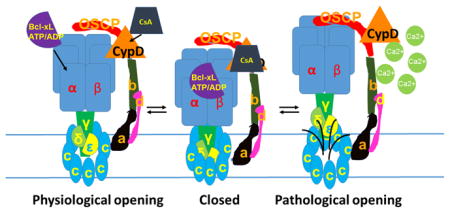
Channel activity of the purified c-subunit is inhibited by the purified β-subunit of F1, suggesting a structural rearrangement whereby the stalk and F1 of the ATP synthase inhibits opening of the c-subunit channel, aided by ADP/ATP/Bcl-xL binding to the β-subunit and opposed by CypD/Ca2+ interaction with OSCP (Fig. 2). In order to demonstrate this further, mitochondria were treated with Ca2+ following which the F1FO ATP synthase was immunoprecipitated using an antibody directed at the F1. These studies determined that Ca2+ exposure destabilizes the connection between the stalk and the c-subunit, disrupting protein/protein interaction between the c-subunit and F1 (Fig. 3). The model suggests that the channel of the mPTP forms within the c-subunit ring itself upon reversible CypD and Ca2+-dependent movement of the stalk away from the c-subunit [117].
Fig. 2.
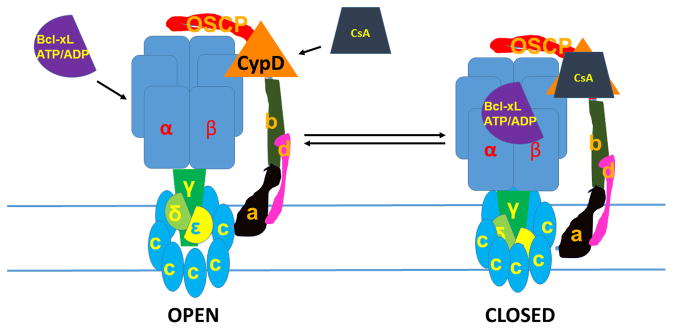
Model of physiological mPTP activity: β subunit, which binds Bcl-xL and ATP/ADP, blocks c-subunit conductance. CsA, inhibitor of CypD, blocks c-subunit conductance. Both Bcl-xL and CsA bind to F1 components during pore closure. The pore diameter also decreases during pore closure. The channel can switch reversibly between these open and closed states.
Fig. 3.
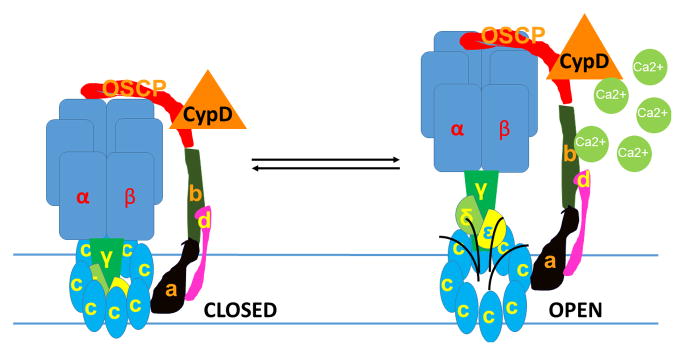
Model of pathophysiological mPTP activity: Ca2+ induces separation of F1 from FO during mPTP opening. The pore diameter also expands during Ca2+ -induced increases in mPTP conductance. The components of F1FO that separate from the c-subunit during this process are not yet completely known; the illustration here is schematic.
It should be emphasized that the loss of protein/protein interaction between F1 and FO does not require very drastic conditions; just 60 μM Ca2+ in the bath is enough to initiate what may indeed be reversible PT [117]. This concentration is well within the range of physiological Ca2+ concentrations found within the mitochondrial matrix [143–145] or concentrations measured adjacent to the mitochondria in Ca2+ microdomains at the plasma membrane or ER membrane [146,147]. That the loss of protein/protein interaction between F1 and FO is likely to be reversible has been shown upon chelation of Ca2+ in mitoplasts [68], intact mitochondria [148], intact neurons [20] as well as in reconstituted dimers of F-ATP synthase [149], suggesting that the F1 and the c-subunit can recombine to close the mPTP, reforming intact F1FO ATP synthase and re-initiating enzymatic function [150]. However, under certain conditions, this separation may become irreversible, forming pathophysiological PT (with MOMP).
Additional regulation of the mPTP may be due to the association of other molecules with F1FO ATP synthase. As discussed above, F1FO ATP synthase can complex with ANT and PiC. In the study of F1FO ATP synthase dimers, bongkrekic acid, which inhibits ANT, fails to attenuate the mPTP channel activity [56]; this is recapitulated in studies of SMVs [80]. Therefore, the regulation of the mPTP by ANT and PiC may occur through their association with the peripheral membrane components of F1FO ATP synthase. Furthermore, the association of F1FO ATP synthase and ANT with PiC, mtCK, VDAC and hexokinase may explain why mPTP regulation can occur via these molecules. Therefore, the layers of regulation that control formation of the c-subunit ring/mPTP are very complex and may involve many known and undiscovered proteins that associate directly or indirectly with F1FO ATP synthase, as suggested in previous reports [3,151,152].
Regulators of the mPTP may also work directly on the F1FO ATP synthase itself. F1 has binding sites that accommodate the effects of Ca2+, Mg2+, adenine nucleotides and Pi; and through CypD (un)binding those of H+, CsA and possibly of oxidants [153]. Therefore, in summary, the new model of mPTP describes either direct or indirect interaction with all known inducers, inhibitors and modulators of pore function.
Structural location of the pore within the c-subunit ring
The exact location of the pore within the c-subunit is becoming increasingly understood. Although it has been proposed that the pore of the ion channel sits between the two lateral stalks of a dimer of F1FO ATP synthases and not within the c-subunit ring [56], there is currently no electrophysiological evidence for the formation of such a channel, and regulation of the mPTP by components of F1 [56,117,154] argue against this. If the pore were located between dimeric membrane-associated subunits, then F1 would need to be positioned over the dimeric link, outside of the FO, directed to the side of the complex, in order to regulate channel activity. This seems unlikely to happen, and, thus, a model of the pore forming in the region between monomers of F1FO ATP synthase requires further hypothesis testing.
Rather, it is likely that the leak is located either within the central portion of the c-subunit ring, between the individual c-subunit monomers, or between the c-subunit and the other FO subunits, although the latter is less likely given the presence of PT in ρ0 cells that lack both mitochondrial DNA and the a-subunit [116]. In two separate experiments, it has been demonstrated that the c-subunit ring expands when it conducts ions, making it likely that the pore is formed by the c-subunit ring. The first experiment used fluorescent tetracysteine display with the placement of cysteine pairs on all c-subunit monomers within the ring. These studies showed that Ca2+ influx into cells causes a decrease in fluorescence consistent with expansion of the diameter of the c-subunit ring, while CsA increases fluorescence consistent with a decrease in ring diameter [117]. Mutagenesis to increase the diameter of the c-subunit ring also demonstrated that ring expansion is a means to increase conductance. Mutations targeted to four highly conserved glycines within the first (N terminus) alpha-helical region of the c-subunit [155] decrease the tight packing of the ring and increase average single-channel conductance compared with WT c-subunit rings [117].
These findings support the hypothesis that the c-subunit is necessary and sufficient to produce the pore of mPTP. When viewed from the inter-membrane space, the denuded c-subunit oligomer appears as a ring with a central pore-like structure that is normally obscured by the F1 stalk components gamma, delta and epsilon, suggesting that the pore may form within the center of the ring given the proper hydrophilic conformation [129]. Although it has been suggested that phosphlipids occupy the central cavity of the c-subunit ring in F1FO ATP synthases from different species [156–158] other evidence provides for formation of a proteolipid or proteophospholipid channel structure within the central lipid region [58,62,118,131,159]. Data suggest a working model whereby the c-subunit pore forms within the proteolipid milieu upon activation of mPTP (for example by elevated matrix Ca2+) whereupon the ring expands and F1 shifts; the pore is closed by a decrease in diameter of the ring and inactivated by binding of the F1 components to the ring (Figs. 2, 3). The details of these changes and their regulation remain a work in progress.
Conclusion
For many years investigators have sought the identity of the molecular structure underlying acute alterations in mitochondrial morphology and inner membrane conductance known collectively as mitochondrial PT. Early evidence asserted that PT was caused by opening of an inner membrane ion channel. More recent data have supported this idea and shown that the c-subunit of the F1FO ATP synthase forms a channel with similar biophysical characteristics to mPTP but lacking regulation by Ca2+ or CsA and with reduced sensitivity to adenine nucleotides. Depletion of c-subunit isoforms in cells blocks CsA-dependent PT and subsequent cell death. The traditional regulation of mPTP by Ca2+, Mg2+, Pi, adenine nucleotides, CsA, CypD and recently Bcl-xL has now been assigned to sites on the F1 including the stator complex and the enzymatic portion of ATP synthase. Inhibitors and activators may also work through peripheral regulatory moieties such as ANT, PiC and VDAC that exist in a large complex of proteins with the F1FO ATP synthase. Lipids and polyphosphates also may play an important role in pore gating or formation.
In this model, activators of the mPTP open the pore in a gating mechanism in which F1 moves away from the mouth of the c-subunit ring while the ring expands (Fig. 3). This process is reversible, perhaps due to binding of F1 components like the β-subunit to the ring or by the re-association of the entire F1 onto the ring (Fig. 2). Although an amazing amount of information has come to light recently regarding the molecular structure and regulation of mPTP, there is still much to do to understand the details. In addition, the role of mPTP during development of oxidative phosphorylation, in aging and in supercomplex formation comprise rapidly changing fields.
Supplementary Material
Acknowledgments
NIH NS045876; NS081746 to EAJ
American Heart Association Grant 12GRNT12060233 to GAP
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mitchell P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature. 1961;191:144–148. doi: 10.1038/191144a0. [DOI] [PubMed] [Google Scholar]
- 2.Watt IN, Montgomery MG, Runswick MJ, Leslie AG, Walker JE. Bioenergetic cost of making an adenosine triphosphate molecule in animal mitochondria. Proc Natl Acad Sci U S A. 2010;107:16823–16827. doi: 10.1073/pnas.1011099107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen C, Ko Y, Delannoy M, Ludtke SJ, Chiu W, Pedersen PL. Mitochondrial atp synthasome: Three-dimensional structure by electron microscopy of the atp synthase in complex formation with carriers for pi and adp/atp. J Biol Chem. 2004;279:31761–31768. doi: 10.1074/jbc.M401353200. [DOI] [PubMed] [Google Scholar]
- 4.Nicholls DG, Rial E. A history of the first uncoupling protein, ucp1. J Bioenerg Biomembr. 1999;31:399–406. doi: 10.1023/a:1005436121005. [DOI] [PubMed] [Google Scholar]
- 5.Andrews ZB, Diano S, Horvath TL. Mitochondrial uncoupling proteins in the cns: In support of function and survival. Nat Rev Neurosci. 2005;6:829–840. doi: 10.1038/nrn1767. [DOI] [PubMed] [Google Scholar]
- 6.Divakaruni AS, Brand MD. The regulation and physiology of mitochondrial proton leak. Physiology. 2011;26:192–205. doi: 10.1152/physiol.00046.2010. [DOI] [PubMed] [Google Scholar]
- 7.Fedorenko A, Lishko PV, Kirichok Y. Mechanism of fatty-acid-dependent ucp1 uncoupling in brown fat mitochondria. Cell. 2012;151:400–413. doi: 10.1016/j.cell.2012.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.D’Alessandro M, Turina P, Melandri BA. Intrinsic uncoupling in the atp synthase of escherichia coli. Biochim Biophys Acta. 2008;1777:1518–1527. doi: 10.1016/j.bbabio.2008.09.011. [DOI] [PubMed] [Google Scholar]
- 9.Caviston TL, Ketchum CJ, Sorgen PL, Nakamoto RK, Cain BD. Identification of an uncoupling mutation affecting the b subunit of f1f0 atp synthase in escherichia coli. FEBS Lett. 1998;429:201–206. doi: 10.1016/s0014-5793(98)00597-3. [DOI] [PubMed] [Google Scholar]
- 10.Billups B, Forsythe ID. Presynaptic mitochondrial calcium sequestration influences transmission at mammalian central synapses. J Neurosci. 2002;22:5840–5847. doi: 10.1523/JNEUROSCI.22-14-05840.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kang JS, Tian JH, Pan PY, Zald P, Li C, Deng C, Sheng ZH. Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell. 2008;132:137–148. doi: 10.1016/j.cell.2007.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol. 2012;13:566–578. doi: 10.1038/nrm3412. [DOI] [PubMed] [Google Scholar]
- 13.Chouhan AK, Ivannikov MV, Lu Z, Sugimori M, Llinas RR, Macleod GT. Cytosolic calcium coordinates mitochondrial energy metabolism with presynaptic activity. J Neurosci. 2012;32:1233–1243. doi: 10.1523/JNEUROSCI.1301-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Stefani D, Rizzuto R. Molecular control of mitochondrial calcium uptake. Biochemical and biophysical research communications. 2014;449:373–376. doi: 10.1016/j.bbrc.2014.04.142. [DOI] [PubMed] [Google Scholar]
- 15.Raffaello A, De Stefani D, Rizzuto R. The mitochondrial ca(2+) uniporter. Cell Calcium. 2012;52:16–21. doi: 10.1016/j.ceca.2012.04.006. [DOI] [PubMed] [Google Scholar]
- 16.Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]
- 17.Lopreiato R, Giacomello M, Carafoli E. The plasma membrane calcium pump: New ways to look at an old enzyme. J Biol Chem. 2014;289:10261–10268. doi: 10.1074/jbc.O114.555565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neher E, Sakaba T. Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron. 2008;59:861–872. doi: 10.1016/j.neuron.2008.08.019. [DOI] [PubMed] [Google Scholar]
- 19.Jonas E. Bcl-xl regulates synaptic plasticity. Molecular Interventions. 2006;6:208–222. doi: 10.1124/mi.6.4.7. [DOI] [PubMed] [Google Scholar]
- 20.Jonas EA, Buchanan J, Kaczmarek LK. Prolonged activation of mitochondrial conductances during synaptic transmission. Science. 1999;286:1347–1350. doi: 10.1126/science.286.5443.1347. [DOI] [PubMed] [Google Scholar]
- 21.Jonas EA, Knox RJ, Kaczmarek LK. Giga-ohm seals on intracellular membranes: A technique for studying intracellular ion channels in intact cells. Neuron. 1997;19:7–13. doi: 10.1016/s0896-6273(00)80343-8. [DOI] [PubMed] [Google Scholar]
- 22.Tang Y, Zucker RS. Mitochondrial involvement in post-tetanic potentiation of synaptic transmission. Neuron. 1997;18:483–491. doi: 10.1016/s0896-6273(00)81248-9. [DOI] [PubMed] [Google Scholar]
- 23.Friel DD, Tsien RW. An fccp-sensitive ca2+ store in bullfrog sympathetic neurons and its participation in stimulus-evoked changes in [ca2+]i. J Neurosci. 1994;14:4007–4024. doi: 10.1523/JNEUROSCI.14-07-04007.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kinnally KW, Martinez-Caballero S, Dejean LM. Detection of the mitochondrial apoptosis-induced channel (mac) and its regulation by bcl-2 family proteins. Current protoc toxicol. 2006;Chapter 2(Unit2):12. doi: 10.1002/0471140856.tx0212s30. [DOI] [PubMed] [Google Scholar]
- 25.Dejean LM, Martinez-Caballero S, Guo L, Hughes C, Teijido O, Ducret T, Ichas F, Korsmeyer SJ, Antonsson B, Jonas EA, Kinnally KW. Oligomeric bax is a component of the putative cytochrome c release channel mac, mitochondrial apoptosis-induced channel. Mol Biol Cell. 2005;16:2424–2432. doi: 10.1091/mbc.E04-12-1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dejean LM, Martinez-Caballero S, Manon S, Kinnally KW. Regulation of the mitochondrial apoptosis-induced channel, mac, by bcl-2 family proteins. Biochim Biophys Acta. 2006;1762:191–201. doi: 10.1016/j.bbadis.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 27.Antonsson B, Montessuit S, Lauper S, Eskes R, Martinou JC. Bax oligomerization is required for channel-forming activity in liposomes and to trigger cytochrome c release from mitochondria. Biochemical Journal. 2000;345(Pt 2):271–278. [PMC free article] [PubMed] [Google Scholar]
- 28.Adams JM, Cory S. The bcl-2 apoptotic switch in cancer development and therapy. Oncogene. 2007;26:1324–1337. doi: 10.1038/sj.onc.1210220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim H, Rafiuddin-Shah M, Tu HC, Jeffers JR, Zambetti GP, Hsieh JJ, Cheng EH. Hierarchical regulation of mitochondrion-dependent apoptosis by bcl-2 subfamilies [see comment] Nature cell biology. 2006;8:1348–1358. doi: 10.1038/ncb1499. [DOI] [PubMed] [Google Scholar]
- 30.Galonek HL, Hardwick JM. Upgrading the bcl-2 network [comment] Nat Cell Biol. 2006;8:1317–1319. doi: 10.1038/ncb1206-1317. [DOI] [PubMed] [Google Scholar]
- 31.Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 32.Martinez-Caballero S, Dejean LM, Jonas EA, Kinnally KW. The role of the mitochondrial apoptosis induced channel mac in cytochrome c release. J Bioenerg Biomembr. 2005;37:155–164. doi: 10.1007/s10863-005-6570-z. [DOI] [PubMed] [Google Scholar]
- 33.Gottlieb E, Armour SM, Thompson CB. Mitochondrial respiratory control is lost during growth factor deprivation. Proc Natl Acad Sci U S A. 2002;99:12801–12806. doi: 10.1073/pnas.202477599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Youle RJ, Strasser A. The bcl-2 protein family: Opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 35.Galluzzi L, Blomgren K, Kroemer G. Mitochondrial membrane permeabilization in neuronal injury. Nat Rev Neurosci. 2009;10:481–494. doi: 10.1038/nrn2665. [DOI] [PubMed] [Google Scholar]
- 36.Budd SL, Nicholls DG. Mitochondria, calcium regulation, and acute glutamate excitotoxicity in cultured cerebellar granule cells. J Neurochem. 1996;67:2282–2291. doi: 10.1046/j.1471-4159.1996.67062282.x. [DOI] [PubMed] [Google Scholar]
- 37.Haworth RA, Hunter DR. The ca2+-induced membrane transition in mitochondria. Ii Nature of the ca2+ trigger site. Archives of biochemistry and biophysics. 1979;195:460–467. doi: 10.1016/0003-9861(79)90372-2. [DOI] [PubMed] [Google Scholar]
- 38.Hunter DR, Haworth RA. The ca2+-induced membrane transition in mitochondria. I. The protective mechanisms. Archives of biochemistry and biophysics. 1979;195:453–459. doi: 10.1016/0003-9861(79)90371-0. [DOI] [PubMed] [Google Scholar]
- 39.Hunter DR, Haworth RA. The ca2+-induced membrane transition in mitochondria. Iii. Transitional ca2+ release. Archives of biochemistry and biophysics. 1979;195:468–477. doi: 10.1016/0003-9861(79)90373-4. [DOI] [PubMed] [Google Scholar]
- 40.Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochemical Journal. 1999;341:233–249. [PMC free article] [PubMed] [Google Scholar]
- 41.Huser J, Blatter LA. Fluctuations in mitochondrial membrane potential caused by repetitive gating of the permeability transition pore. Biochem J. 1999;343(Pt 2):311–317. [PMC free article] [PubMed] [Google Scholar]
- 42.Petronilli V, Miotto G, Canton M, Brini M, Colonna R, Bernardi P, Di Lisa F. Transient and long-lasting openings of the mitochondrial permeability transition pore can be monitored directly in intact cells by changes in mitochondrial calcein fluorescence. Biophys J. 1999;76:725–734. doi: 10.1016/S0006-3495(99)77239-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hausenloy D, Wynne A, Duchen M, Yellon D. Transient mitochondrial permeability transition pore opening mediates preconditioning-induced protection. Circulation. 2004;109:1714–1717. doi: 10.1161/01.CIR.0000126294.81407.7D. [DOI] [PubMed] [Google Scholar]
- 44.Wang W, Fang H, Groom L, Cheng A, Zhang W, Liu J, Wang X, Li K, Han P, Zheng M, Yin J, Mattson MP, Kao JP, Lakatta EG, Sheu SS, Ouyang K, Chen J, Dirksen RT, Cheng H. Superoxide flashes in single mitochondria. Cell. 2008;134:279–290. doi: 10.1016/j.cell.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Korge P, Yang L, Yang JH, Wang Y, Qu Z, Weiss JN. Protective role of transient pore openings in calcium handling by cardiac mitochondria. J Biol Chem. 2011;286:34851–34857. doi: 10.1074/jbc.M111.239921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bonora M, Pinton P. Shedding light on molecular mechanisms and identity of mptp. Mitochondrion. 2014 doi: 10.1016/j.mito.2014.10.001. [DOI] [PubMed] [Google Scholar]
- 47.Baines CP. The mitochondrial permeability transition pore and the cardiac necrotic program. Pediatric cardiology. 2011;32:258–262. doi: 10.1007/s00246-010-9880-9. [DOI] [PubMed] [Google Scholar]
- 48.Bonora M, Wieckowski MR, Chinopoulos C, Kepp O, Kroemer G, Galluzzi L, Pinton P. Molecular mechanisms of cell death: Central implication of atp synthase in mitochondrial permeability transition. Oncogene. 2014 doi: 10.1038/onc.2014.462. [DOI] [PubMed] [Google Scholar]
- 49.Griffiths EJ, Halestrap AP. Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem J. 1995;307 (Pt 1):93–98. doi: 10.1042/bj3070093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ichas F, Mazat JP. From calcium signaling to cell death: Two conformations for the mitochondrial permeability transition pore. Switching from low- to high-conductance state. Biochim Biophys Acta. 1998;1366:33–50. doi: 10.1016/s0005-2728(98)00119-4. [DOI] [PubMed] [Google Scholar]
- 51.Jouaville LS, Ichas F, Mazat JP. Modulation of cell calcium signals by mitochondria. Mol Cell Biochem. 1998;184:371–376. [PubMed] [Google Scholar]
- 52.Elrod JW, Wong R, Mishra S, Vagnozzi RJ, Sakthievel B, Goonasekera SA, Karch J, Gabel S, Farber J, Force T, Brown JH, Murphy E, Molkentin JD. Cyclophilin d controls mitochondrial pore-dependent ca(2+) exchange, metabolic flexibility, and propensity for heart failure in mice. J Clin Invest. 2010;120:3680–3687. doi: 10.1172/JCI43171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.De Marchi E, Bonora M, Giorgi C, Pinton P. The mitochondrial permeability transition pore is a dispensable element for mitochondrial calcium efflux. Cell Calcium. 2014;56:1–13. doi: 10.1016/j.ceca.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Beutner G, Eliseev RA, Porter GA., Jr Initiation of electron transport chain activity in the embryonic heart coincides with the activation of mitochondrial complex 1 and the formation of supercomplexes. PLoS One. 2014;9:e113330. doi: 10.1371/journal.pone.0113330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Szabo I, Bernardi P, Zoratti M. Modulation of the mitochondrial megachannel by divalent cations and protons. J Biol Chem. 1992;267:2940–2946. [PubMed] [Google Scholar]
- 56.Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, Glick GD, Petronilli V, Zoratti M, Szabo I, Lippe G, Bernardi P. Dimers of mitochondrial atp synthase form the permeability transition pore. Proc Natl Acad Sci U S A. 2013;110:5887–5892. doi: 10.1073/pnas.1217823110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kowaltowski AJ, Naia-da-Silva ES, Castilho RF, Vercesi AE. Ca2+-stimulated mitochondrial reactive oxygen species generation and permeability transition are inhibited by dibucaine or mg2+ Archives of biochemistry and biophysics. 1998;359:77–81. doi: 10.1006/abbi.1998.0870. [DOI] [PubMed] [Google Scholar]
- 58.Abramov AY, Fraley C, Diao CT, Winkfein R, Colicos MA, Duchen MR, French RJ, Pavlov E. Targeted polyphosphatase expression alters mitochondrial metabolism and inhibits calcium-dependent cell death. Proc Natl Acad Sci U S A. 2007;104:18091–18096. doi: 10.1073/pnas.0708959104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Seidlmayer LK, Blatter LA, Pavlov E, Dedkova EN. Inorganic polyphosphate--an unusual suspect of the mitochondrial permeability transition mystery. Channels. 2012;6:463–467. doi: 10.4161/chan.21939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Holmstrom KM, Marina N, Baev AY, Wood NW, Gourine AV, Abramov AY. Signalling properties of inorganic polyphosphate in the mammalian brain. Nature communications. 2013;4:1362. doi: 10.1038/ncomms2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stotz SC, Scott LO, Drummond-Main C, Avchalumov Y, Girotto F, Davidsen J, Gomez-Garcia MR, Rho JM, Pavlov EV, Colicos MA. Inorganic polyphosphate regulates neuronal excitability through modulation of voltage-gated channels. Molecular brain. 2014;7:42. doi: 10.1186/1756-6606-7-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Elustondo PA, Angelova PR, Kawalec M, Michalak M, Kurcok P, Abramov AY, Pavlov EV. Polyhydroxybutyrate targets mammalian mitochondria and increases permeability of plasmalemmal and mitochondrial membranes. PLoS One. 2013;8:e75812. doi: 10.1371/journal.pone.0075812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhu J, Rebecchi MJ, Glass PS, Brink PR, Liu L. Interactions of gsk-3beta with mitochondrial permeability transition pore modulators during preconditioning: Age-associated differences. The journals of gerontology Series A, Biological sciences and medical sciences. 2013;68:395–403. doi: 10.1093/gerona/gls205. [DOI] [PubMed] [Google Scholar]
- 64.Di Lisa F, Carpi A, Giorgio V, Bernardi P. The mitochondrial permeability transition pore and cyclophilin d in cardioprotection. Biochim Biophys Acta. 2011;1813:1316–1322. doi: 10.1016/j.bbamcr.2011.01.031. [DOI] [PubMed] [Google Scholar]
- 65.Xi J, Wang H, Mueller RA, Norfleet EA, Xu Z. Mechanism for resveratrol-induced cardioprotection against reperfusion injury involves glycogen synthase kinase 3beta and mitochondrial permeability transition pore. European journal of pharmacology. 2009;604:111–116. doi: 10.1016/j.ejphar.2008.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Clarke SJ, Khaliulin I, Das M, Parker JE, Heesom KJ, Halestrap AP. Inhibition of mitochondrial permeability transition pore opening by ischemic preconditioning is probably mediated by reduction of oxidative stress rather than mitochondrial protein phosphorylation. Circulation research. 2008;102:1082–1090. doi: 10.1161/CIRCRESAHA.107.167072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pasdois P, Parker JE, Halestrap AP. Extent of mitochondrial hexokinase ii dissociation during ischemia correlates with mitochondrial cytochrome c release, reactive oxygen species production, and infarct size on reperfusion. Journal of the American Heart Association. 2013;2:e005645. doi: 10.1161/JAHA.112.005645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Beutner G, Ruck A, Riede B, Brdiczka D. Complexes between porin, hexokinase, mitochondrial creatine kinase and adenylate translocator display properties of the permeability transition pore. Implication for regulation of permeability transition by the kinases. Biochim Biophys Acta. 1998;1368:7–18. doi: 10.1016/s0005-2736(97)00175-2. [DOI] [PubMed] [Google Scholar]
- 69.Beutner G, Ruck A, Riede B, Welte W, Brdiczka D. Complexes between kinases, mitochondrial porin and adenylate translocator in rat brain resemble the permeability transition pore. FEBS Lett. 1996;396:189–195. doi: 10.1016/0014-5793(96)01092-7. [DOI] [PubMed] [Google Scholar]
- 70.Szabo I, Zoratti M. The giant channel of the inner mitochondrial membrane is inhibited by cyclosporin a. J Biol Chem. 1991;266:3376–3379. [PubMed] [Google Scholar]
- 71.Giorgio V, Bisetto E, Soriano ME, Dabbeni-Sala F, Basso E, Petronilli V, Forte MA, Bernardi P, Lippe G. Cyclophilin d modulates mitochondrial f0f1-atp synthase by interacting with the lateral stalk of the complex. J Biol Chem. 2009;284:33982–33988. doi: 10.1074/jbc.M109.020115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bernardi P. Mitochondrial transport of cations: Channels, exchangers, and permeability transition. Physiol Rev. 1999;79:1127–1155. doi: 10.1152/physrev.1999.79.4.1127. [DOI] [PubMed] [Google Scholar]
- 73.Sorgato MC, Keller BU, Stuhmer W. Patch-clamping of the inner mitochondrial membrane reveals a voltage-dependent ion channel. Nature. 1987;330:498–500. doi: 10.1038/330498a0. [DOI] [PubMed] [Google Scholar]
- 74.Petronilli V, Szabo I, Zoratti M. The inner mitochondrial membrane contains ion-conducting channels similar to those found in bacteria. FEBS Lett. 1989;259:137–143. doi: 10.1016/0014-5793(89)81513-3. [DOI] [PubMed] [Google Scholar]
- 75.Kinnally KW, Campo ML, Tedeschi H. Mitochondrial channel activity studied by patch-clamping mitoplasts. J Bioenerg Biomembr. 1989;21:497–506. doi: 10.1007/BF00762521. [DOI] [PubMed] [Google Scholar]
- 76.Beal MF. Mitochondria and neurodegeneration. Novartis Found Symp. 2007;287:183–192. doi: 10.1002/9780470725207.ch13. discussion 192–186. [DOI] [PubMed] [Google Scholar]
- 77.Dodson MW, Guo M. Pink1, parkin, dj-1 and mitochondrial dysfunction in parkinson’s disease. Curr Opin Neurobiol. 2007;17:331–337. doi: 10.1016/j.conb.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 78.Brand MD. The efficiency and plasticity of mitochondrial energy transduction. Biochem Soc Trans. 2005;33:897–904. doi: 10.1042/BST0330897. [DOI] [PubMed] [Google Scholar]
- 79.Hockenbery D, Nunez G, Milliman C, Schreiber RD, Korsmeyer SJ. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature. 1990;348:334–336. doi: 10.1038/348334a0. [DOI] [PubMed] [Google Scholar]
- 80.Alavian KN, Li H, Collis L, Bonanni L, Zeng L, Sacchetti S, Lazrove E, Nabili P, Flaherty B, Graham M, Chen Y, Messerli SM, Mariggio MA, Rahner C, McNay E, Shore GC, Smith PJ, Hardwick JM, Jonas EA. Bcl-xl regulates metabolic efficiency of neurons through interaction with the mitochondrial f1fo atp synthase. Nat Cell Biol. 2011;13:1224–1233. doi: 10.1038/ncb2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chen YB, Aon MA, Hsu YT, Soane L, Teng X, McCaffery JM, Cheng WC, Qi B, Li H, Alavian KN, Dayhoff-Brannigan M, Zou S, Pineda FJ, O’Rourke B, Ko YH, Pedersen PL, Kaczmarek LK, Jonas EA, Hardwick JM. Bcl-xl regulates mitochondrial energetics by stabilizing the inner membrane potential. J Cell Biol. 2011;195:263–276. doi: 10.1083/jcb.201108059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jonas EA. Molecular participants in mitochondrial cell death channel formation during neuronal ischemia. Exp Neurol. 2009;218:203–212. doi: 10.1016/j.expneurol.2009.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Park HA, Licznerski P, Alavian KN, Shanabrough M, Jonas EA. Bcl-xl is necessary for neurite outgrowth in hippocampal neurons. Antioxid Redox Signal. 2014 doi: 10.1089/ars.2013.5570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jonas EA, Porter GA, Alavian KN. Bcl-xl in neuroprotection and plasticity. Frontiers in physiology. 2014;5:355. doi: 10.3389/fphys.2014.00355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vander Heiden MG, Li XX, Gottleib E, Hill RB, Thompson CB, Colombini M. Bcl-xl promotes the open configuration of the voltage-dependent anion channel and metabolite passage through the outer mitochondrial membrane. Journal of Biological Chemistry. 2001;276:19414–19419. doi: 10.1074/jbc.M101590200. [DOI] [PubMed] [Google Scholar]
- 86.Jonas EA, Hoit D, Hickman JA, Brandt TA, Polster BM, Fannjiang Y, McCarthy E, Montanez MK, Hardwick JM, Kaczmarek LK. Modulation of synaptic transmission by the bcl-2 family protein bcl-xl. J Neurosci. 2003;23:8423–8431. doi: 10.1523/JNEUROSCI.23-23-08423.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hickman JA, Hardwick JM, Kaczmarek LK, Jonas EA. Bcl-xl inhibitor abt-737 reveals a dual role for bcl-xl in synaptic transmission. J Neurophysiol. 2008;99:1515–1522. doi: 10.1152/jn.00598.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li H, Chen Y, Jones AF, Sanger RH, Collis LP, Flannery R, McNay EC, Yu T, Schwarzenbacher R, Bossy B, Bossy-Wetzel E, Bennett MV, Pypaert M, Hickman JA, Smith PJ, Hardwick JM, Jonas EA. Bcl-xl induces drp1-dependent synapse formation in cultured hippocampal neurons. Proc Natl Acad Sci U S A. 2008;105:2169–2174. doi: 10.1073/pnas.0711647105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li H, Alavian KN, Lazrove E, Mehta N, Jones A, Zhang P, Licznerski P, Graham M, Uo T, Guo J, Rahner C, Duman RS, Morrison RS, Jonas EA. A bcl-xl-drp1 complex regulates synaptic vesicle membrane dynamics during endocytosis. Nat Cell Biol. 2013;15:773–785. doi: 10.1038/ncb2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bernardi P. The mitochondrial permeability transition pore: A mystery solved? Frontiers in physiology. 2013;4:95. doi: 10.3389/fphys.2013.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Crompton M, Ellinger H, Costi A. Inhibition by cyclosporin a of a ca2+-dependent pore in heart mitochondria activated by inorganic phosphate and oxidative stress. Biochem J. 1988;255:357–360. [PMC free article] [PubMed] [Google Scholar]
- 92.Halestrap AP, Davidson AM. Inhibition of ca2(+)-induced large-amplitude swelling of liver and heart mitochondria by cyclosporin is probably caused by the inhibitor binding to mitochondrial-matrix peptidyl-prolyl cis-trans isomerase and preventing it interacting with the adenine nucleotide translocase. Biochem J. 1990;268:153–160. doi: 10.1042/bj2680153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.McGuinness O, Yafei N, Costi A, Crompton M. The presence of two classes of high-affinity cyclosporin a binding sites in mitochondria. Evidence that the minor component is involved in the opening of an inner-membrane ca(2+)-dependent pore. European journal of biochemistry/FEBS. 1990;194:671–679. doi: 10.1111/j.1432-1033.1990.tb15667.x. [DOI] [PubMed] [Google Scholar]
- 94.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin d reveals a critical role for mitochondrial permeability transition in cell death [see comment] Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 95.Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P. Properties of the permeability transition pore in mitochondria devoid of cyclophilin d. J Biol Chem. 2005;280:18558–18561. doi: 10.1074/jbc.C500089200. [DOI] [PubMed] [Google Scholar]
- 96.Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin d-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death [see comment] Nature. 2005;434:652–658. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 97.Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ. Cyclophilin d is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci U S A. 2005;102:12005–12010. doi: 10.1073/pnas.0505294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hafner AV, Dai J, Gomes AP, Xiao CY, Palmeira CM, Rosenzweig A, Sinclair DA. Regulation of the mptp by sirt3-mediated deacetylation of cypd at lysine 166 suppresses age-related cardiac hypertrophy. Aging. 2010;2:914–923. doi: 10.18632/aging.100252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nguyen TT, Stevens MV, Kohr M, Steenbergen C, Sack MN, Murphy E. Cysteine 203 of cyclophilin d is critical for cyclophilin d activation of the mitochondrial permeability transition pore. J Biol Chem. 2011;286:40184–40192. doi: 10.1074/jbc.M111.243469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Shulga N, Pastorino JG. Ethanol sensitizes mitochondria to the permeability transition by inhibiting deacetylation of cyclophilin-d mediated by sirtuin-3. J Cell Sci. 2010;123:4117–4127. doi: 10.1242/jcs.073502. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 101.Shulga N, Wilson-Smith R, Pastorino JG. Sirtuin-3 deacetylation of cyclophilin d induces dissociation of hexokinase ii from the mitochondria. J Cell Sci. 2010;123:894–902. doi: 10.1242/jcs.061846. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 102.Crompton M, Virji S, Ward JM. Cyclophilin-d binds strongly to complexes of the voltage-dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore. European Journal of Biochemistry. 1998;258:729–735. doi: 10.1046/j.1432-1327.1998.2580729.x. [DOI] [PubMed] [Google Scholar]
- 103.Woodfield K, Ruck A, Brdiczka D, Halestrap AP. Direct demonstration of a specific interaction between cyclophilin-d and the adenine nucleotide translocase confirms their role in the mitochondrial permeability transition. Biochem J. 1998;336 (Pt 2):287–290. doi: 10.1042/bj3360287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gutierrez-Aguilar M, Douglas DL, Gibson AK, Domeier TL, Molkentin JD, Baines CP. Genetic manipulation of the cardiac mitochondrial phosphate carrier does not affect permeability transition. Journal of molecular and cellular cardiology. 2014;72:316–325. doi: 10.1016/j.yjmcc.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Leung AW, Halestrap AP. Recent progress in elucidating the molecular mechanism of the mitochondrial permeability transition pore. Biochim Biophys Acta. 2008;1777:946–952. doi: 10.1016/j.bbabio.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 106.Hom JR, Quintanilla RA, Hoffman DL, de Mesy Bentley KL, Molkentin JD, Sheu SS, Porter GA., Jr The permeability transition pore controls cardiac mitochondrial maturation and myocyte differentiation. Developmental cell. 2011;21:469–478. doi: 10.1016/j.devcel.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cho SW, Park JS, Heo HJ, Park SW, Song S, Kim I, Han YM, Yamashita JK, Youm JB, Han J, Koh GY. Dual modulation of the mitochondrial permeability transition pore and redox signaling synergistically promotes cardiomyocyte differentiation from pluripotent stem cells. Journal of the American Heart Association. 2014;3:e000693. doi: 10.1161/JAHA.113.000693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fujiwara M, Yan P, Otsuji TG, Narazaki G, Uosaki H, Fukushima H, Kuwahara K, Harada M, Matsuda H, Matsuoka S, Okita K, Takahashi K, Nakagawa M, Ikeda T, Sakata R, Mummery CL, Nakatsuji N, Yamanaka S, Nakao K, Yamashita JK. Induction and enhancement of cardiac cell differentiation from mouse and human induced pluripotent stem cells with cyclosporin-a. PLoS One. 2011;6:e16734. doi: 10.1371/journal.pone.0016734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Drenckhahn JD. Heart development: Mitochondria in command of cardiomyocyte differentiation. Developmental cell. 2011;21:392–393. doi: 10.1016/j.devcel.2011.08.021. [DOI] [PubMed] [Google Scholar]
- 110.Folmes CD, Dzeja PP, Nelson TJ, Terzic A. Mitochondria in control of cell fate. Circulation research. 2012;110:526–529. doi: 10.1161/RES.0b013e31824ae5c1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallace DC. The adp/atp translocator is not essential for the mitochondrial permeability transition pore [see comment] Nature. 2004;427:461–465. doi: 10.1038/nature02229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kwong JQ, Davis J, Baines CP, Sargent MA, Karch J, Wang X, Huang T, Molkentin JD. Genetic deletion of the mitochondrial phosphate carrier desensitizes the mitochondrial permeability transition pore and causes cardiomyopathy. Cell Death Differ. 2014;21:1209–1217. doi: 10.1038/cdd.2014.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kinnally KW, Peixoto PM, Ryu SY, Dejean LM. Is mptp the gatekeeper for necrosis, apoptosis, or both? Biochim Biophys Acta. 2011;1813:616–622. doi: 10.1016/j.bbamcr.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gunter TE, Sheu SS. Characteristics and possible functions of mitochondrial ca(2+) transport mechanisms. Biochim Biophys Acta. 2009;1787:1291–1308. doi: 10.1016/j.bbabio.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pestana CR, Silva CH, Uyemura SA, Santos AC, Curti C. Impact of adenosine nucleotide translocase (ant) proline isomerization on ca2+-induced cysteine relative mobility/mitochondrial permeability transition pore. J Bioenerg Biomembr. 2010;42:329–335. doi: 10.1007/s10863-010-9297-4. [DOI] [PubMed] [Google Scholar]
- 116.Bonora M, Bononi A, De Marchi E, Giorgi C, Lebiedzinska M, Marchi S, Patergnani S, Rimessi A, Suski JM, Wojtala A, Wieckowski MR, Kroemer G, Galluzzi L, Pinton P. Role of the c subunit of the fo atp synthase in mitochondrial permeability transition. Cell Cycle. 2013;12:674–683. doi: 10.4161/cc.23599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Alavian KN, Beutner G, Lazrove E, Sacchetti S, Park HA, Licznerski P, Li H, Nabili P, Hockensmith K, Graham M, Porter GA, Jr, Jonas EA. An uncoupling channel within the c-subunit ring of the f1fo atp synthase is the mitochondrial permeability transition pore. Proc Natl Acad Sci U S A. 2014;111:10580–10585. doi: 10.1073/pnas.1401591111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Azarashvili T, Odinokova I, Bakunts A, Ternovsky V, Krestinina O, Tyynela J, Saris NE. Potential role of subunit c of f0f1-atpase and subunit c of storage body in the mitochondrial permeability transition Effect of the phosphorylation status of subunit c on pore opening. Cell Calcium. 2014;55:69–77. doi: 10.1016/j.ceca.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 119.Karch J, Molkentin JD. Identifying the components of the elusive mitochondrial permeability transition pore. Proc Natl Acad Sci U S A. 2014;111:10396–10397. doi: 10.1073/pnas.1410104111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chinopoulos C, Szabadkai G. What makes you can also break you, part iii: Mitochondrial permeability transition pore formation by an uncoupling channel within the c-subunit ring of the f1fo atp synthase? Frontiers in oncology. 2014;4:235. doi: 10.3389/fonc.2014.00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Carbajo RJ, Kellas FA, Runswick MJ, Montgomery MG, Walker JE, Neuhaus D. Structure of the f1-binding domain of the stator of bovine f1fo-atpase and how it binds an alpha-subunit. Journal of molecular biology. 2005;351:824–838. doi: 10.1016/j.jmb.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 122.Pedersen PL. Atp synthase. The machine that makes atp. Current biology: CB. 1994;4:1138–1141. doi: 10.1016/s0960-9822(00)00257-8. [DOI] [PubMed] [Google Scholar]
- 123.Jonckheere AI, Smeitink JA, Rodenburg RJ. Mitochondrial atp synthase: Architecture, function and pathology. Journal of inherited metabolic disease. 2012;35:211–225. doi: 10.1007/s10545-011-9382-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Walker JE. The atp synthase: The understood, the uncertain and the unknown. Biochem Soc Trans. 2013;41:1–16. doi: 10.1042/BST20110773. [DOI] [PubMed] [Google Scholar]
- 125.Wittig I, Schagger H. Supramolecular organization of atp synthase and respiratory chain in mitochondrial membranes. Biochim Biophys Acta. 2009;1787:672–680. doi: 10.1016/j.bbabio.2008.12.016. [DOI] [PubMed] [Google Scholar]
- 126.Guerrieri F, Capozza G, Kalous M, Papa S. Age-related changes of mitochondrial f0f1 atp synthase. Ann N Y Acad Sci. 1992;671:395–402. doi: 10.1111/j.1749-6632.1992.tb43813.x. [DOI] [PubMed] [Google Scholar]
- 127.Faccenda D, Tan CH, Seraphim A, Duchen MR, Campanella M. If1 limits the apoptotic-signalling cascade by preventing mitochondrial remodelling. Cell Death Differ. 2013;20:686–697. doi: 10.1038/cdd.2012.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Garcia JJ, Morales-Rios E, Cortes-Hernandez P, Rodriguez-Zavala JS. The inhibitor protein (if1) promotes dimerization of the mitochondrial f1f0-atp synthase. Biochemistry. 2006;45:12695–12703. doi: 10.1021/bi060339j. [DOI] [PubMed] [Google Scholar]
- 129.Pogoryelov D, Reichen C, Klyszejko AL, Brunisholz R, Muller DJ, Dimroth P, Meier T. The oligomeric state of c rings from cyanobacterial f-atp synthases varies from 13 to 15. Journal of bacteriology. 2007;189:5895–5902. doi: 10.1128/JB.00581-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.McGeoch JE, Guidotti G. A 0. 1–700 hz current through a voltage-clamped pore: Candidate protein for initiator of neural oscillations. Brain Res. 1997;766:188–194. doi: 10.1016/s0006-8993(97)00618-5. [DOI] [PubMed] [Google Scholar]
- 131.McGeoch JE, McGeoch MW. Entrapment of water by subunit c of atp synthase. Journal of the Royal Society, Interface/the Royal Society. 2008;5:311–318. doi: 10.1098/rsif.2007.1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mandel M, Moriyama Y, Hulmes JD, Pan YC, Nelson H, Nelson N. Cdna sequence encoding the 16-kda proteolipid of chromaffin granules implies gene duplication in the evolution of h+-atpases. Proc Natl Acad Sci U S A. 1988;85:5521–5524. doi: 10.1073/pnas.85.15.5521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Finbow ME, Harrison MA. The vacuolar h+-atpase: A universal proton pump of eukaryotes. Biochem J. 1997;324 (Pt 3):697–712. doi: 10.1042/bj3240697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Jones PC, Harrison MA, Kim YI, Finbow ME, Findlay JB. The first putative transmembrane helix of the 16 kda proteolipid lines a pore in the vo sector of the vacuolar h(+)-atpase. Biochem J. 1995;312 (Pt 3):739–747. doi: 10.1042/bj3120739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Harrison M, Durose L, Song CF, Barratt E, Trinick J, Jones R, Findlay JB. Structure and function of the vacuolar h+-atpase: Moving from low-resolution models to high-resolution structures. J Bioenerg Biomembr. 2003;35:337–345. doi: 10.1023/a:1025728915565. [DOI] [PubMed] [Google Scholar]
- 136.Peters C, Bayer MJ, Buhler S, Andersen JS, Mann M, Mayer A. Trans-complex formation by proteolipid channels in the terminal phase of membrane fusion. Nature. 2001;409:581–588. doi: 10.1038/35054500. [DOI] [PubMed] [Google Scholar]
- 137.Alavian KN, Beutner G, Lazrove E, Sacchetti S, Park HA, Licznerski P, Li H, Nabili P, Hockensmith K, Graham M, Porter GA, Jr, Jonas EA. An uncoupling channel within the c-subunit ring of the f1fo atp synthase is the mitochondrial permeability transition pore. Proc Natl Acad Sci U S A. 2014 doi: 10.1073/pnas.1401591111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Borjesson SI, Elinder F. Structure, function, and modification of the voltage sensor in voltage-gated ion channels. Cell biochemistry and biophysics. 2008;52:149–174. doi: 10.1007/s12013-008-9032-5. [DOI] [PubMed] [Google Scholar]
- 139.Zakharov SD, Li X, Red’ko TP, Dilley RA. Calcium binding to the subunit c of e. Coli atp-synthase and possible functional implications in energy coupling. J Bioenerg Biomembr. 1996;28:483–494. doi: 10.1007/BF02110438. [DOI] [PubMed] [Google Scholar]
- 140.Hubbard MJ, McHugh NJ. Mitochondrial atp synthase f1-beta-subunit is a calcium-binding protein. FEBS Lett. 1996;391:323–329. doi: 10.1016/0014-5793(96)00767-3. [DOI] [PubMed] [Google Scholar]
- 141.Giorgio V, Bisetto E, Soriano ME, Dabbeni-Sala F, Basso E, Petronilli V, Forte MA, Bernardi P, Lippe G. Cyclophilin d modulates mitochondrial f0f1-atp synthase by interacting with the lateral stalk of the complex. J of Biological Chemistry. 2009;284:33982–33988. doi: 10.1074/jbc.M109.020115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, Glick GD, Petronilli V, Zoratti M, Szabo I, Lippe G, Bernardi P. Dimers of mitochondrial atp synthase form the permeability transition pore. PNAS. 2013;110:5887–5892. doi: 10.1073/pnas.1217823110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Csordas G, Thomas AP, Hajnoczky G. Calcium signal transmission between ryanodine receptors and mitochondria in cardiac muscle. Trends in cardiovascular medicine. 2001;11:269–275. doi: 10.1016/s1050-1738(01)00123-2. [DOI] [PubMed] [Google Scholar]
- 144.Rizzuto R, Bernardi P, Pozzan T. Mitochondria as all-round players of the calcium game. J Physiol. 2000;529(Pt 1):37–47. doi: 10.1111/j.1469-7793.2000.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Rizzuto R, Pozzan T. Microdomains of intracellular ca2+: Molecular determinants and functional consequences. Physiol Rev. 2006;86:369–408. doi: 10.1152/physrev.00004.2005. [DOI] [PubMed] [Google Scholar]
- 146.Rizzuto R, Marchi S, Bonora M, Aguiari P, Bononi A, De Stefani D, Giorgi C, Leo S, Rimessi A, Siviero R, Zecchini E, Pinton P. Ca(2+) transfer from the er to mitochondria: When, how and why. Biochim Biophys Acta. 2009;1787:1342–1351. doi: 10.1016/j.bbabio.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Schneggenburger R, Neher E. Presynaptic calcium and control of vesicle fusion. Curr Opin Neurobiol. 2005;15:266–274. doi: 10.1016/j.conb.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 148.Roestenberg P, Manjeri GR, Valsecchi F, Smeitink JA, Willems PH, Koopman WJ. Pharmacological targeting of mitochondrial complex i deficiency: The cellular level and beyond. Mitochondrion. 2012;12:57–65. doi: 10.1016/j.mito.2011.06.011. [DOI] [PubMed] [Google Scholar]
- 149.Gomez L, Thibault H, Gharib A, Dumont JM, Vuagniaux G, Scalfaro P, Derumeaux G, Ovize M. Inhibition of mitochondrial permeability transition improves functional recovery and reduces mortality following acute myocardial infarction in mice. Am J Physiol Heart Circ Physiol. 2007;293:H1654–1661. doi: 10.1152/ajpheart.01378.2006. [DOI] [PubMed] [Google Scholar]
- 150.Pedersen PL, Hullihen J. Adenosine triphosphatase of rat liver mitochondria. Capacity of the homogeneous f1 component of the enzyme to restore atp synthesis in urea-treated membranes. J Biol Chem. 1978;253:2176–2183. [PubMed] [Google Scholar]
- 151.Bernardi P, Di Lisa F. The mitochondrial permeability transition pore: Molecular nature and role as a target in cardioprotection. Journal of molecular and cellular cardiology. 2014 doi: 10.1016/j.yjmcc.2014.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Halestrap AP, Richardson AP. The mitochondrial permeability transition: A current perspective on its identity and role in ischaemia/reperfusion injury. Journal of molecular and cellular cardiology. 2014 doi: 10.1016/j.yjmcc.2014.08.018. [DOI] [PubMed] [Google Scholar]
- 153.Kruse SE, Watt WC, Marcinek DJ, Kapur RP, Schenkman KA, Palmiter RD. Mice with mitochondrial complex i deficiency develop a fatal encephalomyopathy. Cell metabolism. 2008;7:312–320. doi: 10.1016/j.cmet.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Alavian KN, Dworetzky SI, Bonanni L, Zhang P, Sacchetti S, Li H, Signore AP, Smith PJ, Gribkoff VK, Jonas EA. The mitochondrial complex v-associated large-conductance inner membrane current is regulated by cyclosporine and dexpramipexole. Molecular pharmacology. 2015;87:1–8. doi: 10.1124/mol.114.095661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Norris U, Karp PE, Fimmel AL. Mutational analysis of the glycine-rich region of the c subunit of the escherichia coli f0f1 atpase. Journal of bacteriology. 1992;174:4496–4499. doi: 10.1128/jb.174.13.4496-4499.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Oberfeld B, Brunner J, Dimroth P. Phospholipids occupy the internal lumen of the c ring of the atp synthase of escherichia coli. Biochemistry. 2006;45:1841–1851. doi: 10.1021/bi052304+. [DOI] [PubMed] [Google Scholar]
- 157.Meier T, Matthey U, Henzen F, Dimroth P, Muller DJ. The central plug in the reconstituted undecameric c cylinder of a bacterial atp synthase consists of phospholipids. FEBS Lett. 2001;505:353–356. doi: 10.1016/s0014-5793(01)02837-x. [DOI] [PubMed] [Google Scholar]
- 158.Matthies D, Preiss L, Klyszejko AL, Muller DJ, Cook GM, Vonck J, Meier T. The c13 ring from a thermoalkaliphilic atp synthase reveals an extended diameter due to a special structural region. J Mol Biol. 2009;388:611–618. doi: 10.1016/j.jmb.2009.03.052. [DOI] [PubMed] [Google Scholar]
- 159.Pavlov E, Zakharian E, Bladen C, Diao CT, Grimbly C, Reusch RN, French RJ. A large, voltage-dependent channel, isolated from mitochondria by water-free chloroform extraction. Biophys J. 2005;88:2614–2625. doi: 10.1529/biophysj.104.057281. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.