

Abstract
Background
Prolonged air leak (PAL) is an important cause of morbidity and mortality after lung resection, but its pathogenesis has not been elucidated. Migration of alveolar type II epithelial cells is essential for lung wound repair. Here we determined the role of C-X-C motif chemokine 12 (CXCL12) on alveolar epithelial cell migration and lung wound healing.
Methods
CXCL12 in the pleural fluid of patients was analyzed using enzyme-linked immunosorbent assay. Human A549 and murine MLE12 alveolar epithelial cell lines were used for wound closure, cell migration, and proliferation assays. Western blot was used to analyze Rac1 and cofilin.
Results
Pleural CXCL12 was decreased in patients with PAL (1,389 ± 192 vs 3,270 ± 247 pg/mL; P < .0001). CXCL12 enhanced scratch wound closure in both A549 (77.9 ± 0.7% vs 71.5 ± 0.4%; P = .0016) and MLE12 (92.9 ± 4.9% vs 66.0 ± 4.8%; P = .017). CXCL12 enhanced migration by 57% in A549 (P = .0008) and by 86% in MLE12 (P < .0001). AMD3100, a selective CXCR4 antagonist, prevented the effects of CXCL12. CXCL12 increased Rac1 and cofilin activation but did not change bromodeoxyuridine incorporation or cell counts.
Conclusion
Reduced pleural CXCL12 is associated with PAL. CXCL12 promotes alveolar epithelial cell migration by binding to its receptor CXCR4 and may have a role in lung healing. CXCL12- mediated alveolar epithelial cell migration is associated with Rac1 and cofilin activation.
Prolonged lung parenchymal air leak (PAL) is a frequent complication after pulmonary resection.1 Defined as a leak persisting for >5 days, the incidence of PAL has been reported to be as high as 58%.2 PAL increases the risk of morbidity by 4-fold and remains one of the most important contributors of mortality after lung surgery.3,4 Risk factors shown to be associated with PAL include reduced pulmonary function, use of steroids, upper lobectomy, pleural adhesions,1 incomplete fissures,5 emphysema,6 and low diffusion capacity.7 However, the underlying pathogenesis of PAL is not understood fully.8-11
Alveolar type II pneumocytes are important in the repair after lung injury.12,13 The alveolar type II pneumocytes proliferate in response to lung injury and migrate over the injured surface to repair lung injury, differentiating into type I alveolar pneumocytes that are responsible for gas exchange.12-15 Although proliferation has been studied extensively, the factors promoting alveolar type II pneumocyte cell migration remain unknown. Stromal cell-derived factor 1, also known as C-X-C motif chemokine 12 (CXCL12), is known to promote cell migration in a variety of cancer cells. Further, it is responsible for cell migration during tissue development in fetal life.16 Although many growth factors are known to affect cell migration, the effects of CXCL12 do not appear to be tissue restricted. Therefore, we hypothesized that CXCL12 might play a role in lung tissue repair after surgery by promoting alveolar epithelial cell migration upon binding to its receptor, CXCR4.
Materials and Methods
Human subjects
Patients undergoing lobectomy through video-assisted thoracoscopic surgery were included. PAL was defined as leak persisting for >5 days. For pleural CXCL12 analysis, the first 10 patients with PAL and the first 10 patients without PAL were included during the study. For analysis, 5 mL of fluid was collected on postoperative day 1 and analyzed using standardized enzyme-linked immunosorbent assay ELISA; (R&D Systems Inc, Minneapolis, MN). The study was approved by the Institutional Review Board of Northwestern University.
Reagents
Recombinant human and murine CXCL12 were purchased from Peprotech (Rocky Hill, NJ). Human CXCL12 was used in experiments involving A549 cells, and murine CXCL12 was used in experiments involving MLE12 cells. Concentration response curves were performed and in accordance with prior published reports14 a dose of 100 ng/mL was used for both cell proliferation and cell migration assays. The CXCR4 antagonist AMD3100 was obtained from Sigma (St. Louis, MO). Cell proliferation assays were performed by analyzing the incorporation of bromodeoxyuridine (BrdU), which was obtained from BD Pharmingen (San Diego, CA). Anti-phosphorylated cofilin antibody and anti-cofilin antibody were purchased from Abcam (Cambridge, MA). Anti-beta-tubulin antibody was obtained from Santa Cruz Biotechnology (Dallas, TX).
Cell lines and culture
Human (A549) and murine (MLE12) alveolar epithelial cell lines were obtained from the American Type Culture Collection (Manassas, VA). Cells were grown in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, 100 μg/mL streptomycin, and 20 mmol/L N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES) buffer at 37°C in a humidified atmosphere containing 5% CO2/95% air.
Wound healing assay
Cells were seeded into 30-mm wells in 6-well plates. After the cells grew to confluence, they were washed with phosphate-buffered saline (PBS), 1 mL of fresh DMEM (2% FBS) was placed in each well, and a scratch wound was made by sterile pipette tip. Photographs were taken, and wound sizes were measured for 4 fields in each well, using a Nikon TE-2000-U microscope ( × 100) and Metamorph image analysis software (Molecular Devices, Sunnyvale, CA). The medium was refreshed, CXCL12 (100 ng/mL) was added, and AMD3100 (6 μg/mL) was added 15 minutes earlier when required. Photographs of the same fields were taken, and cells were incubated at 37°C. Wounds were measured at 24, 48, and 72 hours, with the medium refreshed at each interval, along with CXCL12 and AMD3100 treatments.
Transwell migration assay
Migration was analyzed in Boyden chambers (6.5-mm Transwell plates with 8.0-μm pore polyester membrane inserts, Corning, Corning, NY). Cells were trypsinized and counted. Then, 5 × 104 cells were added in 0.1 mL of serum-free medium to the upper chamber, and 0.6 mL of medium with 10% FBS was added to the lower chamber. When required, CXCL12 (100 ng/mL) was added to both upper and lower chambers, and AMD3100 (6 μg/mL) was added 15 minutes earlier when required. Transwells were incubated at 37°C for 4 hours. The polyester membrane inserts were removed from the Boyden chambers, placed upside down on microscope slides, fixed with formaldehyde (3.7% in PBS) and stained with Hoechst 33342, (Life Technologies, Grand Island, NY). Photographs of 5 fields were taken using a Zeiss Axio-plan 2 Imaging microscope ( × 400), and the cells were counted.
BrdU proliferation assay
Cells were plated on coverslips in 35-mm plates at either 5 × 104 or 1 × 105 cells per plate in complete medium and were incubated at 37°C. At 24 hours, cells were treated with CXCL12 (100 ng/mL) when necessary, and with BrdU (10 ng/mL) at the time of the CXCL12 treatment. After another 24 hours, BrdU incorporation was analyzed using an immunofluo-rescence kit (eBioscience, San Diego, CA). Briefly, cells were fixed and permeabilized. After antigen retrieval, cells were treated with a BrdU antibody followed by a streptavidin-conjugated fluorophore. Photographs of 5 fields were taken using a Zeiss Axioplan 2 Imaging microscope ( × 400), and the cells were counted.
Cell count assay
Cells were plated on 35-mm plates at either 5 × 104 or 1 × 105 cells per plate in complete medium and were incubated at 37°C. CXCL12 was added to the medium 8 hours later and then every 24 hours thereafter. At 24, 48, and 72 hours, cells were trypsinized and counted using an automated Scepter cell counter (Millipore, Billerica, MA).
Rac1 activation pull-down assay
Cells were plated on 60-mm plates and incubated at 37°C. Once cells reached ∼90% confluence, they were treated with CXCL12 (100 ng/mL) for 5 minutes. A Rac1 activation assay was then run according to manufacturers' instructions (Millipore, Temecula, CA). Briefly, cells were lysed, scraped, transferred to an Eppendorf tube, passed through a syringe, precleared, and centrifuged. Ten microliters of supernatant was set aside for total Rac1 analysis, and the manufacturer's Rac1 agarose reagent was added to the rest of the supernatant, followed by a 1 hour rotation at 4°C. The beads were then centrifuged, washed, reconstituted with loading dye, and loaded into a polyacrylamide gel. After sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), the membrane was incubated with the kit's anti-Rac1 antibody overnight, followed by a goat anti-mouse secondary antibody.
Cofilin activation assay
Cells were plated on 60-mm plates and incubated at 37°C. Once cells reached ∼90% confluence, they were treated with CXCL12 (100 ng/mL) for 5 minutes. Plates were then washed twice with ice-cold PBS, and lysis buffer was added. Cells were scraped, transferred to an Eppendorf tube, and centrifuged for 5 minutes. The supernatant was removed, and a Bradford assay was run to load 15 μg of protein into a polyacrylamide gel. After SDS-PAGE, the membrane was divided. The lower portion was incubated with anti-phosphorylated cofilin antibody (∼20 kDa) overnight followed by a goat anti-rabbit secondary antibody, and the upper portion was incubated with anti–beta-tubulin (∼50 kDa) overnight followed by a goat anti-rabbit secondary antibody. The lower portion was then stripped and incubated with anti-cofilin antibody followed by a goat anti-mouse secondary antibody.
Results
PALs are associated with decreased pleural CXCL12 levels
Patients undergoing surgical lobectomy were classified into PAL-positive or PAL-negative groups (n = 10 each). PAL was diagnosed if the air leak persisted for >5 days after surgery. There were no differences in the clinical or pathologic variables between PAL-positive and PAL-negative groups (age 56 ± 7.5 vs 55 ± 6.8 years; males 60% vs 50%; Caucasians 60% in both; all P = .9). There was no difference in smoking status between PAL-positive and PAL-negative groups. All patients underwent video-assisted thoracoscopic lobectomy. The mean post-resection predictive forced expiratory volume in 1 second (FEV1) as well as diffusion capacity of carbon monoxide were >40% for all patients. The mean duration of air leaks was 8.9 ± 2.3 days in the PAL-positive group and 2.3 ± 0.9 in PAL-negative (P = .001). On postoperative day 1, pleural fluid (5 mL) was collected from each of the patients and analyzed using standardized ELISA. As shown in Fig 1, the pleural fluid of patients in the PAL-positive group contained significantly lower CXCL12 levels (1389 ± 192 vs 3270 ± 247 pg/mL; P < .0001).
Fig 1.

Patients with prolonged air leaks (PAL) after lung resection have decreased pleural C-X-C motif chemokine 12 (CXCL12). Controls included patients undergoing lung resection but without PAL. Difference in the CXCL12 levels was significant (P = .009).
CXCL12 enhances wound closure by alveolar epithelial cells
We next investigated whether CXCL12 could play a role in lung wound repair using in vitro scratch wound assays. Human (A549) as well as murine (MLE12) cells were grown to confluence, at which point a standardized scratch wound was created. Wound closure was determined using percentage of the wound area remaining at various time points compared with the starting area. Wound closure was conducted in medium containing 2% FBS to limit cell proliferation. As shown in Fig 2, CXCL12 promoted wound closure in both A549 and MLE cells. At 48 hours, 77.9 ± 0.7% of the wound remained open in A549 cells, whereas in the presence of CXCL12, 71.5 ± 0.4% of the wound remained (P = .0016). In MLE12 cells, 7.1 ± 4.9% of the wound remained open at 48 hours in the presence of CXCL12, compared with 34.0 ± 4.8% without (P = .017).
Fig 2.

C-X-C motif chemokine 12 (CXCL12) promotes wound closure of alveolar epithelial cells in vitro. A, Scratch wound assays demonstrating increased wound closure in the presence of recombinant CXCL12 (100 ng/mL) in both MLE and A549 cells. B, Blocking CXCR4 by AMD3100 (6 μg/mL) 15 minutes before the addition of CXCL12 reversed the effects in both cell types. A549 cells demonstrated increased wound closure at 48 hours in the presence of CXCL12 (28.5 ± 0.4% vs 22.1 ± 0.7%; P = .0016), which was reversed by AMD3100. Similarly, MLE12 cells showed much greater wound closure at 48 hours in the presence of CXCL12 (92.9 ± 4.9% vs 66.0 ± 4.8%; P = .017), which was inhibited by AMD3100. Red line, control; Green line, CXCL12; Blue line, CXCL12 + AMD3100.
Both CXCR4 and CXCR7 are known receptors for CXCL12. To elucidate which receptor was responsible for effects of CXCL12 on alveolar epithelial cell migration, we used AMD3100, an antagonist for CXCR4 but an allosteric agonist for CXCR7. AMD3100 reversed the effects of CXCL12 in both cell lines suggesting that inhibition of CXCR4 abrogates the effects of CXCL12 on alveolar epithelial cell migration (Fig 2).
CXCL12 promotes alveolar epithelial cell migration
We next determined whether enhanced wound closure by CXCL12 occurs as a result of increased alveolar epithelial cell migration or proliferation. Alveolar epithelial cell migration was studied using transwell assays in Boyden chambers. The upper chamber contained FBS-depleted media and the lower chamber had 10% FBS. By using these conditions, cells migrate from the upper chamber toward the FBS-rich lower chamber. Migration of both A549 and MLE12 cell types was determined at 4 hours with or without CXCL12. In parallel, AMD3100 (6 μg/mL) was used to block CXCR4 for 15 minutes prior to the addition of recombinant CXCL12. As shown in Fig 3, B, CXCL12 increased the migration of A549 cells by 57 ± 6%, (P = .0008) and MLE12 cells by 87 ± 5% (P < .0001). The effect of CXCL12 on cell migration was inhibited by AMD3100 again suggesting that this effect occurs through the CXCR4 receptor and not CXCR7.
Fig 3.

C-X-C motif chemokine 12 (CXCL12) promotes transwell migration of alveolar epithelial cells. Recombinant CXCL12 (100 ng/mL) enhanced transwell migration from serum-free medium (upper chamber) to fetal bovine serum–rich medium (lower chamber) over 4 hours. A, Representative images of transwell migration in both A549 and MLE12 cells lines. B, Addition of CXCL12 promoted A549 migration by 56.9 ± 6.2% (P = .0008) and MLE12 migration by 86.7 ± 5.2% (P < .0001).
CXCL12 does not affect alveolar epithelial cell proliferation
Wound closure is dependent on both cell proliferation and migration.12,14 CXCL12 promoted wound closure (Fig 2) and cell migration (Fig 3). However, this does not eliminate the possibility that CXCL12 promoted cell proliferation. Therefore, we tested whether the effect of CXCL12 on wound closure was also owing to an increase in cellular proliferation. A549 as well as MLE12 cells were exposed to CXCL12, and cell proliferation determined at various time points using BrdU incorporation as well as cell counting. At 24 hours, CXCL12 did not increase BrdU incorporation in A549 cells (90.9 ± 1.0% vs 92.5 ± 0.8%; P = .3; Fig 4). Similarly, there was no difference in BrdU incorporation in MLE12 cells with CXCL12 (86.1 ± 1.9% vs 86.5 ± 1.0%; P = .9). Furthermore, cell counts at 24, 48, and 72 hours were not different with or without CXCL12 in both A549 and MLE12 cells (Fig 4, B).
Fig 4.

C-X-C motif chemokine 12 (CXCL12) does not affect alveolar epithelial cell proliferation. Recombinant CXCL12 (100 ng/mL) did not affect BrdU incorporation at 24 hours or cell counts over 72 hours. A, Representative images of bromodeoxyuridine (BrdU) staining (red) and Hoechst 33342 staining (blue). Nuclei completely lacking red color were considered negative. B, BrdU incorporation for A549 and MLE12 cells with and without CXCL12. C, Relative cell count results over 72 hours for A549 and MLE12 cells with (red) and without (blue) CXCL12.
CXCL12 activates Rac1 and cofilin in alveolar epithelial cells
Binding of CXCL12 to its receptor CXCR4 can lead to a variety of intracellular effects. We specifically determined whether CXCL12 promoted the activation of Rac1, which is important for phosphorylation of cofilin, a downstream protein necessary for cytoskeleton rearrangement and cell migration.17 A549 as well as MLE cells were treated with CXCL12 for 5 minutes, and total and activated Rac1 determined using a pull-down Western Blot assays. In A549 cells, adding CXCL12 led to a 124 ± 44% increase in activated Rac1 density compared with controls (corrected with total Rac1 levels; P = .049). In MLE12 cells, CXCL12 led to a 90.5 ± 13.1% increase in activated Rac1 density compared with controls (corrected with total Rac1 levels; P = .02). Total Rac1 levels were not altered in the presence of CXCL12.
We then investigated the effect of CXCL12 on the phosphorylation of cofilin, which is essential for actin depolymerization and lamellipodia formation in cell migration. As shown in Fig 5, B, in the presence of CXCL12, A549 cells showed a 36.4 ± 6.5% increase in phospho-cofilin (corrected with beta-tubulin) compared with controls (P = .0051). MLE12 cells also revealed a 35.8 ± 11.1% increase in phospho-cofilin compared with controls (P = .0329). As with Rac1, total cofilin levels did not change with the addition of CXCL12.
Fig 5.
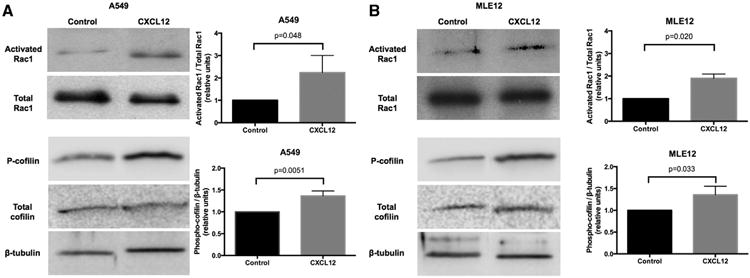
C-X-C motif chemokine 12 (CXCL12) leads to Rac1 activation and cofilin phosphorylation. A, Representative blots displaying the effect of recombinant CXCL12 (100 ng/mL) on activated Rac1 and total Rac1, as well as on phospho-cofilin and total cofilin, in A549 and MLE12 cells after a 5-minute treatment. Total Rac1 and cofilin levels were not affected. B, Average change in densities upon addition of CXCL12. The graphs demonstrate relative activated Rac1/total Rac1 and phospho-cofilin/beta-tubulin ratios for both A549 and MLE12 cells.
Discussion
The pathogenesis of PAL is not understood fully, and there is a paucity of mechanistic studies investigating lung healing after surgical injury. The optimal pleural biochemical milieu for lung healing remains undefined. We found that CXCL12 could play a role in the lung injury response. CXCL12, also known as SDF-1, is a member of the CXC chemokine family that binds to its receptors CXCR4 and CXCR7. It has been shown to have pleiotropic effects in hematopoiesis, tumorigenesis, and chemotaxis. CXCL12 has been shown to induce proliferation in a number of cancer cell lines.16,18 Interestingly, CXCL12 knockout mice die in utero, implicating a critical role in development.18-20 In fact, CXCL12 has been shown to stimulate cell migration across various cell lines stemming from several different organs.
Patients with PAL were found to have decreased pleural CXCL12 levels after surgery. CXCL12 promoted wound closure in alveolar epithelial cells after scratch wounding in vitro, and this effect was owing to increased cell migration but not cell proliferation. Although the BrdU incorporation levels were high for both control and CXCL12 treatment, cell count levels over a 72-hour period were not different between those 2 groups. Prior data shows that CXCL12 can promote cell proliferation,19 and this might be related to the differential effects of CXCL12 on different tissue or cell types. CXCL12 binds to both CXCR4 and CXCR7. Although CXCR4 leads to activation of Rac1 and promotes cell migration, CXCR7 triggers cell proliferation and differentiation.21 Therefore, the differential expression of these receptors on tissue types might dictate the predominant effects of CXCL12 on that tissue. The bicyclam AMD3100 is an antagonist for CXCR4, but acts as an allosteric agonist for CXCR7,22 promoting CXCL12-CXCR7 binding. Blocking CXCR4 using AMD3100 reversed the effects of CXCL12 in the wound closure and transwell migration assays, indicating that CXCR4, and not CXCR7, is responsible for CXCL12 mediated cell migration in alveolar epithelial cells.
It has been previously shown that injury to alveolar epithelial cells leads to autologous production of CXCL12,14 and we have corroborated this finding in our own laboratory by analyzing media of control and scratched cells using ELISA as well as CXCL12 mRNA using real-time polymerase chain reaction (data not shown). In addition, it has been shown that CXCL12 is constitutively expressed by stromal and endothelial cells, and that it can be produced by dendritic cells and macrophages.18,19,23 A recent study has also implicated platelets in the production of CXCL12 after lung injury.24 All of these cells are involved at the site of lung injury. More recently, macrophages have emerged as potent scavengers at the site of injury, clearing apoptotic cells by a process called efferocytosis that leads to chemokine secretion.25 We have previously demonstrated that hypercapnia suppresses cytokine and chemokine production by macrophages.26 Therefore, suppression of macrophages by intrapleural hypercapnia might lead to suppression of efferocytosis and expression of CXCL12 and other growth factors necessary for lung injury repair. However, this hypothesis needs to be validated in subsequent studies.
It is important to note that both cell proliferation and migration play important roles in lung repair. Additionally, clearance of apoptotic cells at the site of injury by local scavenger cells is important. Therefore, there are multiple components of the lung injury response that can contribute to the pathogenesis of PAL. Unfortunately, the present literature lacks mechanistic studies on lung injury repair. Further studies need to be performed to determine the factors that alter the pleural chemical milieu and to elucidate the role of different cellular mechanisms that lead to poor lung repair and PAL.
References
- 1.Brunelli A, Monteverde M, Borri A, Salati M, Marasco RD, Fianchini A. Predictors of prolonged air leak after pulmonary lobectomy. Ann Thorac Surg. 2004;77:1205–10. doi: 10.1016/j.athoracsur.2003.10.082. [DOI] [PubMed] [Google Scholar]
- 2.Abolhoda A, Liu D, Brooks A, Burt M. Prolonged air leak following radical upper lobectomy: an analysis of incidence and possible risk factors. Chest. 1998;113:1507–10. doi: 10.1378/chest.113.6.1507. [DOI] [PubMed] [Google Scholar]
- 3.Cerfolio RJ, Tummala RP, Holman WL, Zorn GL, Kirklin JK, McGiffin DC, et al. A prospective algorithm for the management of air leaks after pulmonary resection. Ann Thorac Surg. 1998;66:1726–31. doi: 10.1016/s0003-4975(98)00958-8. [DOI] [PubMed] [Google Scholar]
- 4.Lackey A, Mitchell JD. The cost of air leak: physicians' and patients' perspectives. Thorac Surg Clin. 2010;20:407–11. doi: 10.1016/j.thorsurg.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 5.Gomez-Caro A, Calvo MJ, Lanzas JT, Chau R, Cascales P, Parrilla P. The approach of fused fissures with fissureless technique decreases the incidence of persistent air leak after lobectomy. Eur J Cardiothorac Surg. 2007;31:203–8. doi: 10.1016/j.ejcts.2006.11.030. [DOI] [PubMed] [Google Scholar]
- 6.Keller CA. Lasers, staples, bovine pericardium, talc, glue and…suction cylinders? Tools of the trade to avoid air leaks in lung volume reduction surgery. Chest. 2004;125:361–3. doi: 10.1378/chest.125.2.361. [DOI] [PubMed] [Google Scholar]
- 7.DeCamp MM, Blackstone EH, Naunheim KS, Krasna MJ, Wood DE, Meli YM, et al. Patient and surgical factors influencing air leak after lung volume reduction surgery: lessons learned from the National Emphysema Treatment Trial. Ann Thorac Surg. 2006;82:197–206. doi: 10.1016/j.athoracsur.2006.02.050. [DOI] [PubMed] [Google Scholar]
- 8.Antanavicius G, Lamb J, Papasavas P, Caushaj P. Initial chest tube management after pulmonary resection. Am Surg. 2005;71:416–9. [PubMed] [Google Scholar]
- 9.Coughlin SM, Emmerton-Coughlin HM, Malthaner R. Management of chest tubes after pulmonary resection: a systematic review and meta-analysis. Can J Surg. 2012;55:264–70. doi: 10.1503/cjs.001411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mueller MR, Marzluf BA. The anticipation and management of air leaks and residual spaces post lung resection. J Thorac Dis. 2014;6:271–84. doi: 10.3978/j.issn.2072-1439.2013.11.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Venuta F, Rendina EA, De Giacomo T, Coloni GF. Postoperative strategies to treat permanent air leaks. Thorac Surg Clin. 2010;20:391–7. doi: 10.1016/j.thorsurg.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 12.Castranova V, Rabovsky J, Tucker JH, Miles PR. The alveolar type II epithelial cell: a multifunctional pneumocyte. Toxi-col Appl Pharmacol. 1988;93:472–83. doi: 10.1016/0041-008x(88)90051-8. [DOI] [PubMed] [Google Scholar]
- 13.Mason RJ. Biology of alveolar type II cells. Respirology. 2006;11(Suppl):S12–5. doi: 10.1111/j.1440-1843.2006.00800.x. [DOI] [PubMed] [Google Scholar]
- 14.Ghosh MC, Makena PS, Gorantla V, Sinclair SE, Waters CM. CXCR4 regulates migration of lung alveolar epithelial cells through activation of Rac1 and matrix metalloproteinase-2. Am J Physiol Lung Cell Mol Physiol. 2012;302:L846–56. doi: 10.1152/ajplung.00321.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sisson TH, Mendez M, Choi K, Subbotina N, Courey A, Cunningham A, et al. Targeted injury of type II alveolar epithelial cells induces pulmonary fibrosis Am J Respir Crit Care Med. 2010;181:254–63. doi: 10.1164/rccm.200810-1615OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu J, Mora A, Shim H, Stecenko A, Brigham KL, Rojas M. Role of the SDF-1/CXCR4 axis in the pathogenesis of lung injury and fibrosis. Am J Respir Cell Mol Biol. 2007;37:291–9. doi: 10.1165/rcmb.2006-0187OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vitriol EA, Wise AL, Berginski ME, Bamburg JR, Zheng JQ. Instantaneous inactivation of cofilin reveals its function of F-actin disassembly in lamellipodia Mol Biol Cell. 2013;24:2238–47. doi: 10.1091/mbc.E13-03-0156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kryczek I, Wei S, Keller E, Liu R, Zou W. Stroma-derived factor (SDF-1/CXCL12) and human tumor pathogenesis. Am J Physiol Cell Physiol. 2007;292:C987–95. doi: 10.1152/ajpcell.00406.2006. [DOI] [PubMed] [Google Scholar]
- 19.Ratajczak MZ, Zuba-Surma E, Kucia M, Reca R, Wojakowski W, Ratajczak J. The pleiotropic effects of the SDF-1-CXCR4 axis in organogenesis, regeneration and tumorigenesis. Leukemia. 2006;20:1915–24. doi: 10.1038/sj.leu.2404357. [DOI] [PubMed] [Google Scholar]
- 20.Zou YR, Kottmann AH, Kuroda M, Taniuchi I, Littman DR. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature. 1998;393:595–9. doi: 10.1038/31269. [DOI] [PubMed] [Google Scholar]
- 21.Sanchez-Martin L, Sanchez-Mateos P, Cabanas C. CXCR7 impact on CXCL12 biology and disease. Trends Mol Med. 2013;19:12–22. doi: 10.1016/j.molmed.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 22.Kalatskaya I, Berchiche YA, Gravel S, Limberg BJ, Rosenbaum JS, Heveker N. AMD3100 is a CXCR7 ligand with allosteric agonist properties. Mol Pharmacol. 2009;75:1240–7. doi: 10.1124/mol.108.053389. [DOI] [PubMed] [Google Scholar]
- 23.Sanchez-Martin L, Estecha A, Samaniego R, Sanchez-Ramon S, Vega MA, Sanchez-Mateos P. The chemokine CXCL12 regulates monocyte-macrophage differentiation and RUNX3 expression. Blood. 2011;117:88–97. doi: 10.1182/blood-2009-12-258186. [DOI] [PubMed] [Google Scholar]
- 24.Rafii S, Cao Z, Lis R, Siempos II, Chavez D, Shido K, et al. Platelet-derived SDF-1 primes the pulmonary capillary vascular niche to drive lung alveolar regeneration. Nat Cell Biol. 2015;17:123–36. doi: 10.1038/ncb3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martin CJ, Peters KN, Behar SM. Macrophages clean up: efferocytosis and microbial control. Curr Opin Microbiol. 2014;17:17–23. doi: 10.1016/j.mib.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang N, Gates KL, Trejo H, Favoreto S, Jr, Schleimer RP, Sznajder JI, et al. Elevated CO2 selectively inhibits interleukin-6 and tumor necrosis factor expression and decreases phagocytosis in the macrophage. FASEB J. 2010;24:2178–90. doi: 10.1096/fj.09-136895. [DOI] [PMC free article] [PubMed] [Google Scholar]