

Abstract
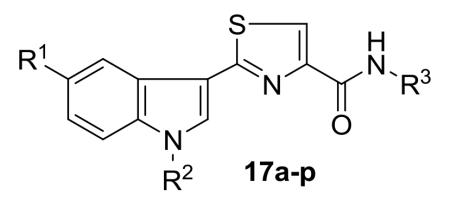
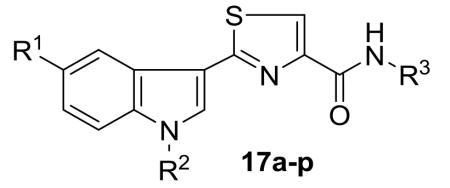
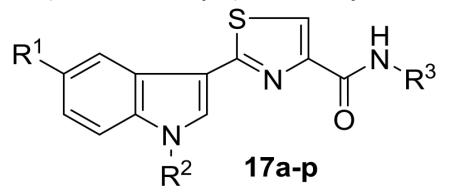
A new series of 2-(3′-indolyl)-N-arylthiazole-4-carboxamides 17a-p has been designed and synthesized. Initial reaction of readily available thioamides 15 with bromopyruvic acid under refluxing conditions produced different thiazole carboxylic acids 16 which upon coupling with arylamines by using EDCI.HCl and HOBt afforded diverse arylthiazole-4-carboxamides 17a-p in 78-87% yields. Antibacterial activity evaluation against Gram-positive and Gram-negative bacterial strains led to compounds 17i-k and 17o as potent and selectively (Gram-negative) antibacterial agents. The cytotoxicity of thiazole carboxamides 17a-p was also evaluated on a panel of human cancer cell lines. Among the tested derivatives, compounds 17i (IC50 = 8.64 μM; HEK293T) and 17l (IC50 = 3.41 μM; HeLa) were identified as the most potent analogues of the series. Preliminary mechanism of action studies of thiazole carboxamide 17i suggested that its cytotoxicity against HeLa cells involves the induction of cell death by apoptosis.
Keywords: Indoles, Thiazole carboxamides, Antibacterial, Cancer, Cytotoxicity, Apoptosis
Graphical Abstract
Indole derivatives are recognized as a class of important heterocycles for their useful pharmacological properties.1 Existing studies have proved their wide range of biological activities such as antiviral, antimicrobial and antitumor agents. Indole ring system is found in a number of clinically useful therapeutic agents such as, indomethacin, indoramin, and indorenate.2 Moreover, several indole containing molecules have been documented for their interesting antibacterial and anticancer activities. For example, 3-thiazol-2′-ylindole (Camalexin) 1 is the characteristic phytoalexin of Arabidopsis thaliana, which is induced by a variety of plant pathogens as well as human tumor cell lines.3 Isolated from marine sponges, the bis(indole)alkaloids Topsentins4 (2) and Nortopsentins5 (structure not shown) have received increasing attention due to their interesting anticancer properties. In 2012, Li et al. explored 2-indolyl-4-benzoylimidazole (3) as a tubulin targeting anticancer agent with an average IC50 value of 3.8 nM against the tested cancer cell lines (Fig. 1).6 Ki-Bong Oh group explored the antibacterial activity of bis(indole) class of alkaloids including topsentins and hamacanthins (4), which exhibited significant antibacterial activity against Gram-positive and Gram-negative bacteria in addition to anticancer activity.7 Hoemann et al has prepared a combinatorial library of 2-(1H-indol-3-yl)quinolines (5) which were effective against methicillin-resistant Staphylococcus aureus with an MIC < 1.0 μg/mL.8 More recently, Singh et al. synthesized bis(indole)glyoxamide (6) and evaluated as potent antibacterial candidates.9
Figure 1.

Representative indole-based antibacterial and anticancer agents
Similarly, over the past few years, various substituted thiazole analogues have been reported to demonstrate diverse biological activities including antimicrobial, antimalarial, antitubercular, antiviral and anticancer activities.10,11,12 Thiazole nucleus is also an essential part of all the available penicillins.13 Among the thiazoles, substituted thiazolecarboxamides exhibited encouraging antibacterial and anticancer activities.14 For examples; thiazolecarboxamide scaffold bearing analogues of Distamycin (7) were reported by Suckling et al. as an antibacterial agent. Very recently, Rostom and co-worker synthesized 2-amino-4-methylthiazole-5-carboxylates (8) and evaluated for their preliminary in vitro antimicrobial and anticancer activities. Most of synthesized analogues were found to be active against various bacterial strains and malignant cells.15 Hofle and co-workers isolated peptides namely Tubulysins A and D (9) from the myxobacteria Archangium gephyra and Angiococcus disciformis, respectively,16,17 were found to display a broad range of anticancer activity.18 In 2012, Huang and co-worker identified 2-aryl-N-(2-(piperazin-1-yl)phenyl)-thiazole-4-carboxamide (ALIS hit 10), as a potent CHK1 inhibitor.19 Various 2-phenylthiazole-4-carboxamides 11 (IC50 = 2-10 μM) and 12 (IC50 = 10-25 μM) synthesized by Foroumadi et al. were found to be potent anticancer agents against a panel of human cancerous cells.20, 21
In the light of above mentioned observations regarding potential of indole-based antibacterial and anticancer agents, thiazole carboxamides and in pursuit of previous results on indole derivatives, 22 we designed a new library of 2-(3′-indolyl)-N-arylthiazole-4-carboxamides 17a-p by incorporating crucial structural features of both the bioactive scaffolds (Fig. 3). The goal was to develop potential small molecules that display either antibacterial or anticancer properties, or react with both species. Such molecules provide a unique opportunity to uncover drug mechanism across divergent species. More importantly, the resulting SAR studies could be further used to design specific anti-bacterial agents that do not cross-react with mammalian targets and vice versa. Such kind of specificity is extremely desirable for developing anti-infective agents which do not target human tissues, and thus likely to exhibit minimal collateral toxicity.
Figure 3.

Rational approach to 2-(3′-indolyl)-N-arylthiazole-4-carboxamides 17a-p
The 2-(3′-indolyl)-N-arylthiazole-4-carboxamides 17a-p were prepared according to Scheme 1. Precursor thioamides 15 were prepared from the corresponding indole by known methods.23,24 Reaction of thioamide 15 with bromopyruvic acid under refluxing conditions led to thiazole carboxylic acids 16 in good yields. Diverse carboxylic acids 16 were coupled with various arylamines in the presence of coupling reagent, EDCI.HCl and HOBt to prepare arylthiazole-4-carboxamides 17a-p in 78-87% yields.25 Structures of thiazole-carboxamides 17a-p were elucidated through their IR, NMR (1H &13C) and Mass spectral analysis. In IR spectra, a characteristic sharp peak at ~1660 cm−1 was observed due to C=O stretching of an amide functional group. Carbon of an amide moiety (CONH) was resonated at ~163 ppm in the 13C NMR spectra of compounds 17a-p.
Scheme 1.

Reagents and conditions: (a) Bromopyruvic acid, 1,4-dioxane, 100 °C, 2 h; (b) EDCI.HCl, HOBt, NEt3, THF, rt, 10 h.
Initially, we screened all the synthesized compounds for their in vitro antibacterial activity against two Gram-positive bacteria Staphylococcus aureus (MTCC 96), Bacillus subtilis (MTCC 121) and two Gram-negative bacteria Escherichia coli (MTCC 1652), Pseudomonas putida (MTCC 102) bacterial strains. Ciprofloxacin was used as a standard drug and the resulting activity (zone of inhibition (ZOI) and minimum inhibitory concentration (MIC)) are given in Table 1.
Table 1.
In vitro antibacterial activity of 2-(3′-indolyl)-N-arylthiazole-4-carboxamides 17a–p
|
ZOI (mm) and MIC (μg/mL) values | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| Compound | Gram-positive bacteria | Gram-negative bacteria | |||||||||
|
| |||||||||||
| S. auresus | B. cereus | E. coli | P. putida | ||||||||
|
| |||||||||||
| R1 | R2 | R3 | ZOI | MIC | ZOI | MIC | ZOI | MIC | ZOI | MIC | |
| 17a | H | H | C6H5 | 14 | 50 | - | - | 13 | 100 | 13 | 100 |
| 17b | H | H | 4-CH3C6H4 | - | - | - | - | 12 | 100 | 12 | 100 |
| 17c | H | H | 4-CH3OC6H4 | 11 | 100 | 12 | 100 | 14 | 50 | 12 | 100 |
| 17d | H | H | 3,4-(CH3O)2C6H3 | 14 | 50 | 14 | 50 | 16 | 25 | 16 | 25 |
| 17e | H | H | 3,4,5-(CH3O)3C6H2 | 12 | 100 | 14 | 50 | 14 | 50 | 13 | 100 |
| 17f | H | H | 4-FC6H4 | 12 | 100 | 10 | 100 | 13 | 100 | 12 | 100 |
| 17g | H | H | 4-(CH3)2NC6H4 | 13 | 100 | 14 | 50 | 12 | 100 | 14 | 50 |
| 17h | H | H | CH2C6H5 | - | - | - | - | 13 | 100 | 13 | 100 |
| 17i | OCH3 | H | 3,4,5-(CH3O)3C6H2 | 13 | 100 | 14 | 50 | 18 | 12.5 | 18 | 12.5 |
| 17j | Br | H | 4-CH3OC6H4 | 14 | 50 | 15 | 50 | 15 | 12.5 | 16 | 12.5 |
| 17k | Br | H | 3,4,5-(CH3O)3C6H2 | 15 | 50 | 16 | 12.5 | 14 | 50 | 16 | 12.5 |
| 17l | F | H | 4-CH3OC6H4 | 12 | 100 | 13 | 100 | 14 | 50 | 13 | 100 |
| 17m | F | H | 3,4,5-(CH3O)3C6H2 | 12 | 100 | 10 | 100 | 14 | 50 | 14 | 50 |
| 17n | H | 4-ClC6H4CH2 | C6H5 | 12 | 100 | 13 | 100 | 15 | 50 | 15 | 50 |
| 17o | H | 4-ClC6H4CH2 | 4-CH3OC6H4 | 14 | 100 | 15 | 50 | 16 | 12.5 | 16 | 12.5 |
| 17p | H | 4-ClC6H4CH2 | 3,4,5-(CH3O)3C6H2 | 13 | 100 | 12 | 100 | 15 | 50 | 14 | 100 |
| Ciprofloxacin | 23 | 6.25 | 24 | 6.25 | 23 | 6.25 | 21 | 12.5 | |||
The zone of inhibition and MIC values for compounds with significant activity are shown in bold
Antibacterial activity results (Table 1) demonstrated that the analogue 17i possessing 5-methoxyindole and trimethoxyphenyl substituents displayed excellent activity against Gram-negative bacteria (ZOI = 18 mm and MIC = 12.5 μg/mL); comparable to standard drug Ciprofloxacin but almost inactive against Gram-positive bacteria. Interestingly, replacement of a C-5 methoxy group on indole ring with bromine in compound 17i led to 17k endowed with good antibacterial activity against tested bacterial strains (ZOI = 14-16 mm; MIC = 12.5-50 μg/mL). Methoxyphenyl analogue 17j also exhibited high antibacterial activity with MIC value of 12.5 μg/mL in P. putida strain. Further, we also observed that the protection of indole N-H as 4-chlorobenzyl group was beneficial for the activity (17a vs 17n and 17c vs 17o), especially 17o had MIC values (12.5 μg/mL) similar to Ciprofloxacin. Remaining compounds (17a-h, 17l-n and 17p) of the series displayed moderate activity against the tested bacterial strains. Among the synthesized compounds, 17i-j and 17o were found to be most potent and selective against Gram-negative bacteria. Compound 17k was also found to be equally effective against Gram-negative bacteria (MIC = 12.5 μg/mL; P. putida) in addition to moderate activity against B. cereus strain. Results from antibacterial activity study shows that substitution on indole (5-OMe/Br), methoxyphenyl substitution on arylamino part and protection of indole N-H with 4-chlorobenzyl moiety, were beneficial for their antibacterial activity.
Once we identified the most potent antibacterial agents, compounds 17i and 17j were selected to evaluate their time-dependent killing effect against tested bacterial strains E. coli (MTCC 1652) and P. putida (MTCC 102). Compounds 17i and 17j with higher than MIC were incubated with log phase culture of E. coli and P. putida at 37 °C and change in optical density (OD600) was monitored at different time intervals (Fig. 4). The results showed a significant reduction in number of bacterial cells after addition of 17i and 17j at 4×MIC. Against E. coli compound 17i was found to be more effective as compared to 17j even up to 6 h. Similarly against P.putida, compound 17j was found to be less effective as compared to 17i, however, both the compounds inhibited bacterial growth from 2-3 h and arrested till 6 h. At 4×MIC compound 17i exhibited the potent inhibition of growth as compared to 17j. Thus the present study results illustrated that 17i and 17j were capable of inhibiting the bacterial growth within few hours of initial interactions.
Figure 4.

Time dependent killing of (A) E.coli and (B) P.putida upon treated with compounds 17i and 17j at 4×MIC
After determining the antibacterial activities of 2-(3′-indolyl)-N-arylthiazole-4-carboxamides 17a-p, we next evaluated their potential anticancer activities in human embryonic kidney 293 cells (HEK 293T), human prostate (PC3, LNCaP and castration-resistant prostate cancer cell line C4-2), cervical (HeLa), and breast (MDA-MB-231) cancer cell lines using MTT assay. Doxorubicin was used as a reference drug. We initially analyzed potential cytotoxicity of these compounds in aforementioned cancer cell lines in the presence of FBS (Table 1).
The cells growing in 10% FBS were treated with varying concentrations of (3′-indolyl)-N-arylthiazole-4-carboxamides 17a-p for 48 h and cytotoxicity evaluated using MTT assay. The cytotoxicity results of compounds 17a-p are expressed as IC50 values in micromolar (Table 2).
Table 2.
In vitro cytotoxicity (with FBS) of 2-(3′-indolyl)-N-arylthiazole-4-carboxamides 17a-p
|
IC50(μM)a,b | ||||||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| Compd | R1 | R2 | R3 | HEK 293T | C4-2 | HeLa | PC3 | MDA- MB-231 |
LNCaP |
| 17a | H | H | C6H5 | >100 | >100 | >100 | >100 | >100 | >100 |
| 17b | H | H | 4-CH3C6H4 | >100 | >100 | >100 | >100 | >100 | >100 |
| 17h | H | H | CH2C6H5 | 74.50±1.19 | >100 | 41.38±2.03 | >100 | >100 | >100 |
| 17i | OCH3 | H | 3,4,5-(CH3O)3C6H2 | 8.74±1.26 | >100 | 9.98±0.01 | >100 | >100 | 84.80±1.21 |
| 17k | Br | H | 3,4,5-(CH3O)3C6H2 | 32.82±0.75 | >100 | 93.03±3.96 | >100 | >100 | >100 |
| 17l | F | H | 4-CH3OC6H4 | >100 | >100 | 33.48±0.98 | >100 | >100 | 75.42±1.88 |
| 17m | F | H | 3,4,5-(CH3O)3C6H2 | 66.02±3.28 | >100 | 48.85±1.35 | >100 | >100 | 88.51±3.25 |
| 17n | H | 4-ClC6H4CH2 | C6H5 | >100 | >100 | 32.07±0.87 | >100 | >100 | 68.94±2.49 |
| 17o | H | 4-ClC6H4CH2 | 4-CH3OC6H4 | >100 | >100 | 90.85±3.73 | >100 | >100 | >100 |
| 17p | H | 4-ClC6H4CH2 | 3,4,5-(CH3O)3C6H2 | >100 | >100 | >100 | >100 | >100 | >100 |
| Doxorubicin | 0.75±0.03 | 0.57±0.08 | 0.43±0.10 | 9.8±0.40 | 6.29±0.24 | 7.40±1.10 | |||
IC50 values are the mean of three different experiments performed in duplicate;
17c-g and 17j: ND
Surprisingly, most of the compounds showed minimal cytotoxic effect following 48 h exposure of the micromolar concentrations of the compounds. A few exceptions, including compound 17i (Table 2) was highly potent and selective for HEK293T and HeLa cells. Compound 17k, which contains a bromo group instead of methoxy at R1 position compared to 17i, selectively inhibited HEK293T cells, but had negligible effect on HeLa cells.
These results suggested that either most of these (3′-indolyl)-N-arylthiazole-4-carboxamides are ineffective as anti-cancer agents or they are not readily available to cells. The latter concern was also prompted by the fact that compounds 17c-g and 17j in this series displayed poor water solubility and were precipitated out of solution when added to cells in an overall DMSO concentration of 0.05-0.1% (Table 2). Similar to fatty acids, several water-insoluble compounds are known to avidly bind serum proteins, which drastically reduces the effective concentration of free molecules to enter the cells.26 Further, serum in growth media can also interfere with MTT reagent leading to overestimation of cell growth and an underestimation of potential cytotoxicity of compounds.27,28 As a result, these compounds are rendered ineffective in the presence of serum, although when serum is removed, they promote significant cell death.
Therefore, we examined the potential cytotoxicity of these compounds in cells in the absence of serum. The cells were freshly plated and grown in serum containing media. After 12h, the media was replaced with serum free media and varying concentrations of the compounds were added. After 48h, cell viability was analyzed using MTT assay. As expected, we observed ~10-20% cell growth with no cell death in DMSO-treated cells after 48 h of treatment, thereby ruling out any artifact that can potentially interfere with cytotoxicity assays. Importantly, in the absence of serum, a number of compounds showed significant and selective cytotoxicity, suggesting that this set of compounds indeed bind serum proteins, which interfere with their ability to penetrate the cells (Table 3).
Table 3.
In vitro cytotoxicity (without FBS) of 2-(3′-indolyl)-N-arylthiazole-4-carboxamides 17a-p
|
IC50 (μM)a | ||||||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| Compd | R1 | R2 | R3 | HEK 293T | C4-2 | HeLa | PC3 | MDA-MB-231 | LNCaP |
| 17a | H | H | C6H5 | 41.07±2.64 | >100 | >100 | >100 | 79.37±3.98 | >100 |
| 17b | H | H | 4-CH3C6H4 | >100 | >100 | >100 | >100 | >100 | >100 |
| 17h | H | H | CH2C6H5 | >100 | >100 | 9.51±0.22 | >100 | >100 | >100 |
| 17i | OCH3 | H | 3,4,5-(CH3O)3C6H2 | 8.60±0.91 | >100 | 29.64±1.44 | 94.34±2.04 | >100 | >100 |
| 17k | Br | H | 3,4,5-(CH3O)3C6H2 | >100 | >100 | 55.79±4.73 | >100 | >100 | >100 |
| 17l | F | H | 4-CH3OC6H4 | 12.10±0.61 | >100 | 3.41±0.07 | >100 | 49.41±5.91 | 66.41±4.94 |
| 17m | F | H | 3,4,5-(CH3O)3C6H2 | >100 | >100 | >100 | 89.00±0.81 | >100 | >100 |
| 17n | H | 4-ClC6H4CH2 | C6H5 | >100 | >100 | 24.27±2.93 | 76.40±3.22 | 50.51±4.92 | 64.07±4.16 |
| 17o | H | 4-ClC6H4CH2 | 4-CH3OC6H4 | >100 | >100 | >100 | >100 | 84.90±2.38 | >100 |
| 17p | H | 4-ClC6H4CH2 | 3,4,5-(CH3O)3C6H2 | 58.69±1.90 | >100 | 91.39±3.31 | >100 | >100 | 97.70±1.76 |
| Doxorubicin | 0.84±0.05 | 1.02±0.06 | 0.45±0.06 | 7.65±0.11 | 6.03±0.22 | 6.46±0.09 | |||
IC50 values are the mean of three different experiments performed in duplicate
From the cytotoxicity results (Table 3) it was found that compounds 17a and 17b with unsubstituted indole and R3 as phenyl and p-tolyl groups were inactive. Replacement of a phenyl moiety in 17a with a benzyl substituent led to compound 17h with selective cytotoxicity against HeLa cells (IC50 = 9.51 μM). Analogue 17i having 5-methoxyindole and trimethoxyphenyl substituent was found to exhibit selective cytotoxic against the HEK293T cells (IC50 = 8.60 μM). Replacement of a 5-methoxyindole with 5-bromoindole (17i vs 17k) was unfavorable for the activity. Interestingly, analogue 17l with 5-fluoroindole and methoxyphenyl moieties found to be the most potent compound of the series and selectively cytotoxic towards HEK293T and HeLa cells with IC50 values of 12.10 and 3.41 μM, respectively. Introduction of additional methoxy groups in compound 17l, resulted in an inactive analogue 17m. Protection of an indole ring nitrogen with p-chlorobenzyl moiety led to compounds 17n-p endowed with moderate activity against the tested cancer cell lines. As noted before, when the MTT assay was performed in the presence of FBS, only compound 17i was found to exhibit selective cytotoxicity against HEK293T (IC50 = 8.74 μM) and HeLa (IC50 = 9.98 μM) cells. Compounds 17h, 17k, 17l and 17n showed moderate activity (IC50 = 32-41 μM) against the tested cell lines.
We next examined whether these compounds induce cytotoxicity by inducing apoptosis in cancer cell lines. As compound 17i exhibited high potency in HeLa cells (IC50 = 9.98 μM), these cells were treated with 17i for 24 h, fixed and stained with propidium iodide, and their nuclear morphology was analyzed using fluorescence microscopy. DMSO-treated cells were used as negative control and doxorubicin, which exerts cytotoxicity by promoting apoptosis, was used as a positive control. As shown in figure 5, nuclei in DMSO-treated HeLa cells retained their normal size and shape while HeLa cells treated with 17i or doxorubicin showed large percentage of apoptotic nuclei, thereby confirming that 17i indeed induces apoptosis in HeLa cells.
Figure 5.

Propidium iodide staining of HeLa cell treated with compound 17i for 48 h. DMSO was used as a control.
Comparison of antibacterial and anticancer activities of 2-(3′-indolyl)-N-arylthiazole-4-carboxamides 17a-p yielded either very specific antibacterial agents or anticancer agents or analogues possessing both activities. Analogue 17l, which displayed high potency and specificity for HEK293T and HeLa cells, was largely ineffective as antibacterial agent both in Gram-positive and Gram-negative bacteria, suggesting that 17l likely targets specific mammalian protein(s), which may be key to survival in these cells. Similarly, analogue 17h was extremely specific and potent for HeLa cells, but did not affect other cancer cell lines even at 100 μM concentration. This compound displayed minimal activity in Gram-positive and Gram-negative bacteria as well, further underscoring its specificity towards mammalian targets. On the other hand, analogue 17i, which too was highly potent against HEK293T and HeLa cells, but not against other cell lines, was among the most effective antibacterial agent against Gram-negative bacteria. Importantly, 17i was inactive against Gram-positive bacteria, suggesting it binds to specific targets in Gram-negative bacteria. It would be interesting to identify these proteins in E. coli and P. putida and analyze whether their orthologues exist in mammalian cells particularly in HEK293T and HeLa cells. We also identified 17o to be potent and active against Gram-negative bacteria, but was completely inactive in mammalian cells. Thus, our library of compounds offer either highly specific antibacterial agents or equally specific anticancer agents and analogues which target both the species. This set of compounds provide an opportunity to dissect the drug mechanism which is either conserved across diverse species, or has diverged to specifically target one species in the presence of other.
In summary, we synthesized a diverse series of 2-(3′-indolyl)-N-arylthiazole-4-carboxamides 17a-p from the initial reaction of thioamides and bromopyruvic acid to afford thiazole carboxylic acids 16, which were coupled with appropriate arylamines. Antibacterial study of 2-(3′-indolyl)-N-arylthiazole-4-carboxamides led to identified 17i-k and 17o as potent compounds against Gram-negative bacteria, P. putida bacterial strain (MIC = 12.5 μg/mL). Moreover, in vitro cytotoxicity study of 2-(3′-indolyl)-N-arylthiazole-4-carboxamides were resulted in 17i and 17l as the most potent compounds of the series. Our preliminary mechanism of action studies further indicated that thiazole carboxamide 17i induces apoptosis in HeLa cells. Overall, these results suggest that appropriate substituents in indole and arylamide moieties of (3′-indolyl)-N-arylthiazole-4-carboxamides 17a-p are crucial for their targeted selectivity and potency for their antibacterial as well anticancer activities. Further, it may be necessary to perform MTT or XTT assays for testing potential cytotoxicity of test compounds both in the absence and presence of FBS to rule out any interference from serum binding proteins which may drastically impact cellular availability. Importantly, analogues 17h and 17l not only displayed high specificity among the cancer cell lines, they were also largely ineffective as antibacterial agents. In contrast, 17i was highly potent in HeLa cells and Gram-negative bacteria, but inactive in other cancer cell lines and Gram-positive bacteria, suggesting that 2-(3′-indolyl)-N-arylthiazole-4-carboxamides display a broad range of bioactivities. As exemplified by our SAR studies, this series of compounds are very versatile and can be exploited to develop either highly specific and potent antibacterial agents or anticancer agents, or if required both of these properties could be incorporated in the same molecule. Finally, it would be important to identify their targets in bacteria and human cell lines to uncover their molecular mechanism, which in turn can be exploited to develop potential therapeutic interventions against various human diseases.
Supplementary Material
Figure 2.

Antibacterial and anticancer agents with thiazole-carboxamide scaffold
Acknowledgments
We gratefully acknowledge the financial support from DBT, New Delhi (DK) and NIH R03 CA166912-2 (KS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary data (experimental procedures and characterization data for compounds (17a–p) associated with this article can be found, in the online version, at http://dx.doi.org/
References and notes
- 1.El-Sayed NS, Shirazi AN, El-Meligy MG, El-Ziaty AK, Rowley D, Sun J, Nagib ZA, Parang K. Tetrahedron Lett. 2014;55:1154. doi: 10.1016/j.tetlet.2013.12.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li W-T, Hwang D-R, Chen C-P, Shen C-W, Huang C-L, Chen T-W, Lin C-H, Chang Y-L, Chang Y-Y, Lo Y-K. J. Med. Chem. 2003;46:1706. doi: 10.1021/jm020471r. [DOI] [PubMed] [Google Scholar]
- 3.Glawischnig E. Phytochemistry. 2007;68:401. doi: 10.1016/j.phytochem.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 4.Bartik K, Braekman J-C, Daloze D, Stoller C, Huysecom J, Vandevyver G, Ottinger R. Can. J. Chem. 1987;65:2118. [Google Scholar]
- 5.Kawasaki I, Yamashita M, Ohta S. Chem. Pharm. Bull. 1996;44:1831. [Google Scholar]
- 6.Chen J, Ahn S, Wang J, Lu Y, Dalton JT, Miller DD, Li W. J. Med. Chem. 2012;55:7285. doi: 10.1021/jm300564b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oh K, Mar W, Kim S, Kim J, Lee T, Kim J, Shin D, Sim CJ, Shin J. Biol Pharm Bull. 2006;29:570. doi: 10.1248/bpb.29.570. [DOI] [PubMed] [Google Scholar]
- 8.Hoemann MZ, Kumaravel G, Xie RL, Rossi RF, Meyer S, Sidhu A, Cuny GD, Hauske JR. Bioorg. Med. Chem. Lett. 2000;10:2675. doi: 10.1016/s0960-894x(00)00542-4. [DOI] [PubMed] [Google Scholar]
- 9.Singh P, Verma P, Yadav B, Komath SS. Bioorg. Med. Chem. Lett. 2011;21:3367. doi: 10.1016/j.bmcl.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 10.Rostom SAF, Faidallah HM, Radwan MF, Badr MH. Eur. J. Med. Chem. 2014;76:170. doi: 10.1016/j.ejmech.2014.02.027. [DOI] [PubMed] [Google Scholar]
- 11.Kamal A, Balakrishna M, Nayak VL, Shaik TB, Faazil S, Nimbarte VD. ChemMedChem. 2014;9:2766. doi: 10.1002/cmdc.201402310. [DOI] [PubMed] [Google Scholar]
- 12.Chauhan K, Sharma M, Trivedi P, Chaturvedi V, Chauhan PMS. Bioorg. Med. Chem. Lett. 2014;24:4166. doi: 10.1016/j.bmcl.2014.07.061. [DOI] [PubMed] [Google Scholar]
- 13.(a) Oncü S, Punar M, Eraksoy H. Chemotherapy. 2004;50:98. doi: 10.1159/000077810. [DOI] [PubMed] [Google Scholar]; (b) Bondock S, Naser T, Ammar YA. Eur. J. Med. Chem. 2013;62:270. doi: 10.1016/j.ejmech.2012.12.050. [DOI] [PubMed] [Google Scholar]
- 14.(a) Rodriguez-Lucena D, Gaboriau F, Rivault F, Schalk IJ, Lescoat G, Mislin GL. Bioorg. Med. Chem. 2010;18:689. doi: 10.1016/j.bmc.2009.11.057. [DOI] [PubMed] [Google Scholar]; (b) Ayati A, Emami S, Asadipour A, Shafiee A, Foroumadi A. Eur. J. Med. Chem. 2015 doi: 10.1016/j.ejmech.2015.04.015. [DOI] [PubMed] [Google Scholar]
- 15.Rostom SA, Faidallah HM, Radwan MF, Badr MH. Eur. J. Med. Chem. 2014;76:170. doi: 10.1016/j.ejmech.2014.02.027. [DOI] [PubMed] [Google Scholar]
- 16.Steinmetz H, Glaser N, Herdtweck E, Sasse F, Reichenbach H, Höfle G. Angew. Chem. Int. Ed. 2004;43:4888. doi: 10.1002/anie.200460147. [DOI] [PubMed] [Google Scholar]
- 17.Sasse F, Steinmetz H, Heil J, Hoefle G, Reichenbach H. J. Antibiot. 2000;53:879. doi: 10.7164/antibiotics.53.879. [DOI] [PubMed] [Google Scholar]
- 18.Balasubramanian R, Raghavan B, Begaye A, Sackett DL, Fecik RA. J. Med. Chem. 2008;52:238. doi: 10.1021/jm8013579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang X, Cheng CC, Fischmann TO, Duca JS, Yang X, Richards M, Shipps GW., Jr ACS Med. Chem. Lett. 2012;3:123. doi: 10.1021/ml200249h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aliabadi A, Shamsa F, Ostad SN, Emami S, Shafiee A, Davoodi J, Foroumadi A. Eur. J. Med. Chem. 2010;45:5384. doi: 10.1016/j.ejmech.2010.08.063. [DOI] [PubMed] [Google Scholar]
- 21.Mohammadi-Farani A, Foroumadi A, Kashani MR, Aliabadi A. Iran J Basic Med Sci. 2014;17:502. [PMC free article] [PubMed] [Google Scholar]
- 22.(a) Kumar D, Maruthi Kumar N, Chang K-H, Shah K. Eur. J. Med. Chem. 2010;45:4664. doi: 10.1016/j.ejmech.2010.07.023. [DOI] [PubMed] [Google Scholar]; (b) Kumar D, Narayanam MK, Chang KH, Shah K. Chem. Biol. Drug Des. 2011;77:182. doi: 10.1111/j.1747-0285.2010.01051.x. [DOI] [PubMed] [Google Scholar]; (c) Tantak MP, Kumar A, Noel B, Shah K, Kumar D. ChemMedChem. 2013;8:1468. doi: 10.1002/cmdc.201300221. [DOI] [PubMed] [Google Scholar]; (d) Kumar D, Kumar NM, Noel B, Shah K. Eur. J. Med. Chem. 2012;55:432. doi: 10.1016/j.ejmech.2012.06.047. [DOI] [PubMed] [Google Scholar]; (e) Kumar D, Kumar NM, Tantak MP, Ogura M, Kusaka E, Ito T. Bioorg. Med. Chem. Lett. 2014;24:5170. doi: 10.1016/j.bmcl.2014.09.085. [DOI] [PubMed] [Google Scholar]
- 23.Manaka A, Sato M. Synth. Commun. 2005;35:761. [Google Scholar]
- 24.Kumar D, Kumar NM, Chang K-H, Gupta R, Shah K. Bioorg. Med. Chem. Lett. 2011;21:5897. doi: 10.1016/j.bmcl.2011.07.089. [DOI] [PubMed] [Google Scholar]
- 25.General procedure for the synthesis of thiazole carboxamide (17a-p). To a mixture of thiazole carboxylic acid 16 (0.8 mmol) in dry THF (3 mL) was added EDCI.HCl (0.9 mmol), HOBt (0.9 mmol) and triethylamine (1.6 mmol) and stirred at 25 °C for 30 min. Arylamine (0.8 mmol) was added and resulting mixture was stirred for 10 h at 25 °C. Upon completion of reaction as indicated by TLC, solvent was evaporated in vaccuo, water was added (15 mL) and extracted with ethyl acetate (2 × 20 mL). Combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure to yield crude product which was purified through column chromatography to afford pure thiazole carboxamides 17a-p in 78-87% yields.Spectral data for selected compounds: 2-(1H-Indol-3-yl)-N-phenylthiazole-4-carboxamide (17a). Yield 80%; off white solid; M.p 206-208 °C; IR (KBr, v cm−1): 3348, 3263, 1666, 1597, 1545, 1435, 1126, 741, 687; 1H NMR (400 MHz, DMSO-d6) δ 11.91 (s, 1H), 10.11 (s, 1H), 8.35–8.26 (m, 3H), 7.87 (d, J = 7.8 Hz, 2H), 7.54 (dd, J = 5.9, 3.1 Hz, 1H), 7.40 (t, J = 7.8 Hz, 2H), 7.27 (dd, J = 6.0, 3.1 Hz, 2H), 7.15 (t, J = 7.3 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 163.3, 159.3, 149.5, 138.3, 136.6, 128.6, 127.6, 124.0, 124.0, 122.5, 121.4, 121.0, 120.6, 120.4, 112.2, 109.9; ESI(FAB) m/z calcd for C18H13N3NaOS: 342.07 (M + Na)+, found 342.05. 2-(1H-Indol-3-yl)-N-p-tolylthiazole-4-carboxamide (17b). Yield 82%; pale yellow solid; M.p 221-223 °C; IR (KBr, v cm−1): 3340, 3256, 1666, 1548, 1242, 1126, 810, 741, 671; 1H NMR (400 MHz, DMSO-d6) δ 11.90 (s, 1H), 10.03 (s, 1H), 8.31 (dd, J = 6.3, 2.7 Hz, 1H), 8.28 (d, J = 2.4 Hz, 1H), 8.24 (s, 1H), 7.72 (d, J = 8.4 Hz, 2H), 7.54–7.50 (m, 1H), 7.28–7.24 (m, 2H), 7.20 (d, J = 8.3 Hz, 2H), 2.30 (s, 3H).; 13C NMR (101 MHz, DMSO-d6) δ 163.7, 159.6, 150.1, 137.1, 136.3, 133.4, 129.5, 128.1, 124.5, 123.0, 121.7, 121.5, 121.0, 120.9, 112.7, 110.4, 21.0.; ESI(FAB) m/z calcd for C19H16N3OS: 334.10 (M + H)+, found 334.05. 2-(1H-indol-3-yl)-N-(4-methoxyphenyl)thia-zole-4-carboxamide (17c). Yield 80%; off white solid; M.p 200-202 °C; IR (KBr, v cm−1): 3364, 3232, 1666, 1512, 1242, 1111, 748, 617; 1H NMR (300 MHz, DMSO-d6) δ 11.94 (s, 1H), 10.02 (s, 1H), 8.36–8.29 (m, 1H), 8.28 (d, J = 2.6 Hz, 1H), 8.23 (s, 1H), 7.77 (s, 2H), 7.55–7.51 (m, 1H), 7.29–7.22 (m, 2H), 6.97 (d, J = 9.0 Hz, 2H), 3.76 (s, 3H).; 13C NMR (75 MHz, DMSO d6) δ 163.3, 159.0, 155.8, 149.7, 136.6, 131.3, 127.6, 124.0, 122.5, 122.3, 121.0, 121.0, 120.4, 113.8, 112.2, 109.9, 55.2.; ESI(FAB) m/z calcd for C19H14N3O2S: 348.09 (M - H)+, found 348.15. N-(3,4-Dimethoxyphenyl)-2-(1H-indol-3-yl)thiazole-4-carboxamide (17d). Yield 79%; off white solid; M.p 176-178 °C; IR (KBr, v cm−1): 3340, 3132, 1643, 1520, 1458, 1219, 1018, 741, 633; 1H NMR (300 MHz, DMSO-d6) δ 11.90 (s, 1H), 9.99 (s, 1H), 8.29 (s, 2H), 8.23 (s, 1H), 7.54 (dd, J = 6.8, 3.2 Hz, 2H), 7.43 (d, J = 7.1 Hz, 1H), 7.26 (dd, J = 5.8, 2.9 Hz, 2H), 6.97 (d, J = 8.7 Hz, 1H), 3.80 (s, 3H), 3.76 (s, 3H).; 13C NMR (75 MHz, DMSO-d6) δ 163.3, 159.0, 149.7, 148.5, 145.4, 136.6, 131.8, 127.5, 124.0, 122.5, 121.1, 121.0, 120.3, 112.7, 112.3, 111.9, 111.0, 105.9, 69.7, 55.7, 55.5.; ESI(FAB) m/z calcd for C20H16N3O3S: 378.10 (M - H)+, found 378.20.
- 26.Bojesen IN, Hansen HS. J. Lipid Res. 2003;44:1790. doi: 10.1194/jlr.M300170-JLR200. [DOI] [PubMed] [Google Scholar]
- 27.Funk DS, Frei HH. Biotechniques. 2007;43:178. doi: 10.2144/000112528. E. [DOI] [PubMed] [Google Scholar]
- 28.Bilmin K, Kopczyńska B, Grieb P. Folia Neuropathol. 2013;51:44. doi: 10.5114/fn.2013.34195. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.