

Abstract
Toll-like receptor 4 (TLR4) has been implicated as a locus for initiation of paclitaxel related chemotherapy induced peripheral neuropathy (CIPN). This project explores the involvement of the immediate down-stream signal molecules in inducing paclitaxel CIPN. Mitogen-activated protein kinases (MAPK) and nuclear factor-κB (NFκB) were measured in dorsal root ganglia (DRG) and the spinal cord over time using Western blot and immunohistochemistry in a rat model of paclitaxel CIPN. The effects of MAPK inhibitors in preventing and reversing behavioral signs of CIPN were also measured (group sizes 4–9). Extracellular signal related kinase (ERK1/2) and p38 but not c-Jun N terminal kinase (JNK) or PI3K-Akt signaling expression was increased in DRG. Phospho-ERK1/2 staining was co-localized to small CGRP-positive DRG neurons in cell profiles surrounding large DRG neurons consistent with satellite glial cells. The expression of phospho-P38 was co-localized to small IB4-positive and CGRP-positive DRG neurons. The TLR4 antagonist LPS derived from R. sphaeroides (LPS-RS) inhibited paclitaxel-induced phosphorylation of ERK1/2 and P38. The MAPK inhibitors PD98059 (MEK1/2), U0126 (MEK1/2) and SB203580 (P38) prevented but did not reverse paclitaxel-induced behavioral hypersensitivity. Paclitaxel treatment resulted in phosphorylation of Inhibitor α of NFκB (IκBα) in DRG resulting in an apparent release of NFκB from the IκBα-NFκB complex as increased expression of nuclear NFκB was also observed. LPS-RS inhibited paclitaxel-induced translocation of NFκB in DRG. No change was observed in spinal NFκB. These results implicate TLR4 signaling via MAP kinases and NFκB in the induction and maintenance of paclitaxel-related CIPN.
Keywords: Dorsal root ganglion, spinal dorsal horn, chemotherapy, ERK1/2, p38, NFκB
1.0 Introduction
Paclitaxel is the frontline chemotherapeutic agent for many of the most common solid tumors, including those of the breast, ovary, and lung (Hagiwara and Sunada 2004). Peripheral neuropathy is the major dose-limiting side effect of paclitaxel and can prompt dose reductions or even the discontinuation of therapy, thus impacting survival in cancer patients (Chaudhry et al., 1994). Additionally, chemotherapy-induced peripheral neuropathy (CIPN) often persists long after cancer treatment is complete and is commonly refractory to current treatment strategies, thus impacting rehabilitation, the return to productivity and quality of life in cancer survivors (Boyette-Davis et al., 2012; Kosturakis et al., 2014).
Pro-inflammatory immune responses are thought to play an important role in the underlying basic pathophysiology of neuropathic pain (Austin and Moalem-Taylor 2010; Vallejo et al., 2010; Zhang et al., 2013), and evidence implicates similar mechanisms in CIPN (Boyette-Davis and Dougherty 2011; Boyette-Davis et al., 2011a). Of note, paclitaxel engages the same signaling pathway in monocytes in vitro via toll-like receptor 4 (TLR4) as the very well-known pro-inflammatory agent lipopolysaccharide (LPS) (Byrd-Leifer et al., 2001; Guha and Mackman 2001). Paclitaxel binds to and activates TLR4 in monocytes resulting in the activation of the nuclear factor-κB (NF-κB) and MAPK signaling cascades downstream to TLR4 including ERK1/2, P38, and JNK. These pathways directly or indirectly phosphorylate and activate various transcription factors (Guha and Mackman 2001); that lead to the induction and release of proinflammatory cytokine expression identical to that produced by LPS (Byrd-Leifer et al., 2001; Han et al., 1994; Karin and Ben-Neriah 2000; Li et al., 2013). Specifically, the cytokines IFNα/γ, TNFα, IL-1, and IL-6 are increased in vitro by paclitaxel (O’Brien, Jr. et al., 1995; Zaks-Zilberman et al., 2001) and cisplatin (Basu and Sodhi 1992; Gan et al., 1992; Pai and Sodhi 1991). The binding site for LPS on human TLR4 includes an interaction with the accessory protein MD-2 that paclitaxel also binds in an overlapping fashion with LPS (Resman et al., 2008).
Recent work has shown that paclitaxel also interacts with TLR4 in dorsal root ganglion and in the spinal dorsal horn (Li et al., 2014b). Paclitaxel treatment resulted in increased expression of TLR4 and its canonical immediate downstream signaling molecules Myeloid-differentiation response gene 88 (MyD88) and TIR-domain-containing adapter-inducing interferon-β (TRIF) in DRG neurons that paralleled the development of chemotherapy-related mechanical hyper-responsiveness. Moreover, co-treatment of rats with the TLR-4 antagonist LPS derived from R. sphaeroides (LPS-RS) during chemotherapy prevented both the up-regulation of TLR4, MyD88 and TRIF as well as the development of the behavioral CIPN phenotype (Li et al., 2014b). Yet, in addition to MyD88 and TRIF, mitogen-activated protein kinases (MAPKs) are also activated downstream to TLR4, and activation of MAPKs in sensory neurons contribute to behavioral hypersensitivity in a rodent model of neuropathic pain (Ji et al., 2009). In addition, MAPK activation has also been shown to modulate the activities of ion channels such as sodium channel Nav1.7 (Black et al., 2008; Dib-Hajj et al., 2009; Hudmon et al., 2008) and TRPV1 (Han et al., 2012; Ji et al., 2002) that have also been implicated as contributing to paclitaxel-related CIPN (Hara et al., 2013; Zhang and Dougherty 2014). Hence MAPKs may be engaged in the generation or maintenance of paclitaxel-related CIPN.
We hypothesized that MAPKs and NFκB signal pathways are induced by TLR4 activation by paclitaxel and play important downstream roles to induce and maintain CIPN. So we sought to determine the effects of paclitaxel on the expression of MAPKs and NFκB in DRG and spinal cord and the effects of antagonists to MAPKs in reducing paclitaxel-induced neuropathic pain.
2.0 Materials and Methods
2.1 Animals
Male Sprague-Dawley rats weighing 250–300 g (Harlan, Houston, TX) were housed in temperature- and light-controlled (12-hour light/dark cycles) conditions with food and water available ad libitum. All 248 rats used in the study were included in the behavioral analysis portions and then used in follow-up pharmacologic, immunohistochemistry, or Western blot analyses. Animals were randomly assigned to different groups for these experiments. The numbers of rats in each of these studies are detailed in the relevant sections of the Results. All experimental protocols were approved by the Institutional Animal Care and Use Committee at The University of Texas MD Anderson Cancer Center and were performed in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. Every procedure was designed to minimize discomfort to the animals and to use the fewest animals needed for statistical analysis.
2.2 Paclitaxel-Induced Neuropathy Model
Rats were treated with paclitaxel (TEVA Pharmaceuticals, Inc., North Wales, PA) as previously described (Zhang et al., 2012) based on the protocol of Polomano et al. (Polomano et al., 2001). Briefly, pharmaceutical grade paclitaxel (Taxol®) was diluted with sterile saline from the original stock concentration of 6 mg/mL (in 1:1 Cremophor EL: ethanol) to 1 mg/mL and given at a dosage of 2 mg/kg intraperitoneally (i.p.) every other day for a total of four injections (days 0, 2, 4, and 6), resulting in a final cumulative dose of 8 mg/kg. Control animals received an equivalent volume of the vehicle only, which consisted of equal amounts of Cremophor EL® and ethanol diluted with saline to reach a concentration of vehicle similar to the paclitaxel concentration. No abnormal spontaneous behavioral changes were noted during or after paclitaxel or vehicle treatment.
2.3 TLR4 Antagonist LPS-RS and MAPK Signal Pathway Antagonist Administration
The TLR4 inhibitor LPS derived from R. sphaeroides (LPS-RS) and MAPK inhibitors were injected intrathecally via L5 puncture. Twenty μg of LPS-RS was injected in 20 μl PBS (InvivoGen, San Diego, CA) beginning 2 days before paclitaxel treatment and then daily until 2 days after the completion of paclitaxel treatment. To assess the role of the MAPK signaling pathways on maintenance of paclitaxel CIPN, rats were injected via L5 puncture with single dose of 10 μg (or 30 μg) or 5 consecutive daily doses of 30 μg of the MEK1/2 inhibitor PD98059 (Tocris, Bristol, UK), MEK1/2 inhibitor U0126 (Tocris, Bristol, UK) or the P38 inhibitor SB203580 (Tocris, Bristol, UK) in 30 μl PBS at day 14 after paclitaxel treatment in rats with behaviorally confirmed paclitaxel-induced mechanical hyperalgesia. The rats were briefly anesthetized with 3% isoflurane and flexed over a tube and a 27-gauge needle inserted between the L5-S1 vertebrae with a deflection of the tail indicating entry to the subarachnoid space. The doses of inhibitors were chosen based on previously published studies (Cao et al., 2013; Lim et al., 2007; Svensson et al., 2005). The same volume of DMSO (U0126, PD98059 solution) or ethanol (SB203580 solution) in PBS (30 μl) was used as the vehicle control. To determine whether PD98059, U0126 and SB203580 can prevent paclitaxel-induced CIPN, we treated rats with 10 μg PD98059, U0126 or SB203580 in 30 μl PBS by intrathecal injection beginning 2 days before paclitaxel treatment and then daily until 2 days after the completion of paclitaxel treatment.
2.4 Mechanical withdrawal threshold measurements
Mechanical withdrawal thresholds were assessed before, during, and after paclitaxel treatment by an experimenter blinded to treatment groups. The 50% paw withdrawal threshold in response to a series of eight von Frey filaments (041–15.10 g) was assessed using the up-down method as described previously, beginning with a filament with a bending force of 2.0 g (Chaplan et al., 1994). Animals were placed in clear acrylic cages atop a wire mesh floor and allowed to acclimate for 30 min. The filaments were applied to the paw just below the pads with no acceleration at a force just sufficient to produce a bend and held for 6–8 s. A quick flick or full withdrawal was considered a response.
2.5 Immunohistochemical Analysis
The rats were deeply anesthetized with sodium pentobarbital (100 mg/kg i.p., Lundbeck, Inc., Deerfield, IL) and perfused through the ascending aorta with warm saline followed by cold 4% paraformaldehyde in 0.1 M PB. The L4 and L5 DRG were removed, fixed in 4% paraformaldehyde for 6 hours, and then cryo-protected in 30% sucrose solution. The L4 and L5 spinal cord segments were also removed, fixed in 4% paraformaldehyde for 12 hours, and then cryo-protected in 30% sucrose solution. Transverse spinal cord sections (15 μm) and longitudinal DRG sections (8 μm) were cut in a cryostat, mounted on gelatin-coated glass slides (Southern Biotech, Birmingham, AL), and processed for immunofluorescence staining (Li et al., 2014a; Zhang et al., 2013). After blocking in 5% normal donkey serum and 0.2% Triton X-100 in PBS for 1 hour at room temperature, the sections were incubated overnight at 4°C in 1% normal donkey serum and 0.2% Triton X-100 in PBS containing primary antibodies against the following targets: phospho-ERK1/2 (rabbit, 1:500; Cell Signaling Technology, Beverly, MA), phospho-P38 (rabbit, 1:500; Cell Signaling Technology, Beverly, MA), IB4 (1:1000 BS-Isolectin B4 FITC conjugate, Sigma, St. Louis, MO), and CGRP (mouse, 1:1000; Abcam). After being washed with PBS, the sections were then incubated with Cy3-, Cy5-, or FITC-conjugated secondary antibodies overnight at 4°C. Sections were viewed under a fluorescent microscope (Eclipse E600; Nikon, Japan). For a given experiment, all images were taken using identical acquisition parameters. To measure cell size, we graphically highlighted each neuron, including the nuclear region. For each positive staining of phospho-ERK1/2 and phospho-P38 with IB4 or CGRP co-localization, the numbers of total and positively co-localized neurons from 3 sections of each DRG of 3 rats were counted. The percentages of positive neurons to total neurons were calculated and statistically analyzed. The intensity of positively stained neurons was measured after the background fluorescence was subtracted; the intensity of positively stained neurons was at least 4 times higher than the background by setting a threshold according to each individual set of slices. The staining intensity of the vehicle groups for each of the different time points (sets of slices) were designated as 100%; fold changes of paclitaxel groups compare to vehicle groups were calculated and described as percent changes. All images were analyzed using NIC Elements imaging software by an experimenter blinded to treatment groups (Nikon, Japan).
2.6 Western Blot Analysis
L4 and L5 DRG and L4–L5 spinal cord segments were collected from rats that were deeply anesthetized with sodium pentobarbital (100 mg/kg i.p.) and snap frozen in liquid nitrogen. The tissues were later disrupted in RIPA lysis buffer (20 mM Tris-HCl, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, and 1 μg/mL leupeptin) mixed with 1 mM dithiothreitol, protease inhibitor cocktail (P8340; Sigma), and phosphatase inhibitor cocktails (P0044 and P5726; Sigma) on ice. The supernatant was then collected and denatured with sample buffer (X5) consisting of 0.25 M Tris-HCl, 52% glycerol, 6% SDS, 5% β-Mercaptoethanol, and 0.1% bromophenol blue for 10 minutes at 70°C. NE-PER Nuclear Protein Extraction Kit was used for the nucleus protein collection (78835; Thermo scientific). Lysates (total protein: 20 μg) were separated on SDS-PAGE gels and transferred to PVDF membranes (Bio-Rad). After blocking with 5% fat-free milk in TBST (137 mM sodium chloride, 20 mM Tris, and 0.1% Tween-20) for 1 hour at room temperature and then incubated with mouse anti-phospho-ERK1/2 and rabbit anti-ERK1/2 antibody (1:2000; Cell Signaling Technology, Beverly, MA), rabbit anti-GAPDH antibody (1:5000; Cell Signaling Technology, Beverly, MA), rabbit anti-NFκB antibody (1:2000; Abcam, Cambridge, MA) and anti-phospho-NFκB antibody (1:500; Cell Signaling Technology, Beverly, MA), rabbit anti-phospho-P38 and anti-P38 antibody (1:1000; Cell Signaling Technology, Beverly, MA), rabbit anti-phospho-JNK (1:2000; Abcam, Cambridge, MA) and anti-JNK antibody (1:5000; Cell Signaling Technology, Beverly, MA), rabbit anti-phospho-Akt and anti-Akt antibody (1:5000; Cell Signaling Technology, Beverly, MA), rabbit anti-histone H4 (1:2000; Cell Signaling Technology, Beverly, MA) and β-actin (1:10,000; Sigma) in 5% fat-free milk in TBST overnight at 4°C. After being washed with TBST, the membranes were incubated with goat anti-rabbit or anti-mouse antibody (labeled with horseradish peroxidase; Calbiochem, CA) diluted with 5% fat-free milk in TBST for 1 hour at room temperature and were detected with ECL reagents (GE Healthcare, Little Chalfont, UK). The blots were scanned with Spot Advanced and Adobe Photoshop 8.0 (Adobe, Inc., San Jose, CA), and the band densities were detected and compared using ImageJ (NIH, Bethesda, MD).
2.7 Data Analysis
Data were expressed as mean ± SEM and analyzed with GraphPad Prism 6. Behavioral data were analyzed with 2-way analysis of variance followed by a Bonferroni post hoc test (two-tailed). The cell counts percentages of neurons with phospho-ERK1/2 and phospho-P38 positive staining, the intensity of phospho-ERK1/2 and phospho-P38 positive staining, and the percentages of neurons that co-localized with IB4 or CGRP, and Western blot results were analyzed by using the Mann Whitney test (two-tailed). P< 0.05 was considered statistically significant.
3.0 Results
3.1 Expression and distribution of MAPK in the DRG after paclitaxel treatment
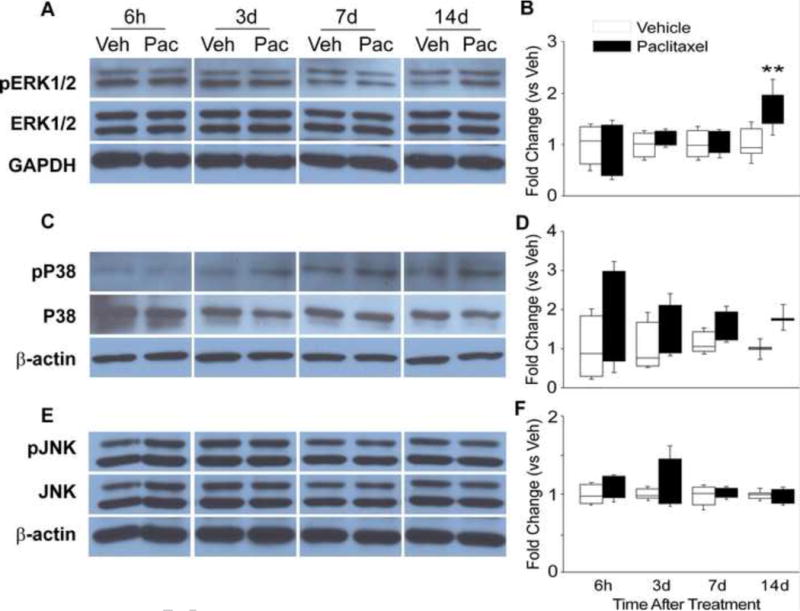
Initial screening of MAPK involvement in paclitaxel-related CIPN was done using Western blot and this was then followed using immunohistochemistry (IHC). Paclitaxel induced the activation of ERK1/2 and p38 but not JNK MAPK signaling (Figures 1–5). Western blot showed that the expression of both phospho-ERK1/2 (pERK1/2) and phospho-p38 was increased both at day 7 and 14 in the DRG (Figure 1A–D) (n=4, p<0.05 at day 7, p<0.01 and p<0.05 at day 14 for pERK1/2 and PP38 respectively). Consistent with the Western blot results, IHC analysis showed an increase in the expression of pERK1/2 at days 7 and 14 after paclitaxel treatment in the DRG (Figure 2D,E, p<0.001), but as well a significant increase in the expression of in pERK1/2 was also detected at day 3 after paclitaxel treatment (Fig 2C, p<0.001). Phospho-ERK1/2 was present at only very low levels in rats that received vehicle alone (Figure 2A) (n=6). Further IHC analysis of the localization of pERK1/2 at day 14 after paclitaxel treatment showed that pERK1/2-positive staining was localized in small (<30μm in diameter) CGRP-positive DRG neurons (p=0.01), with lesser expression in small IB4-positive (p=0.01) and CGRP/IB4 negative DRG neurons (Figure 3, p=0.001) and in narrow bands surrounding large neurons (>45μm) in profiles consistent with satellite glial cells (Figure 2) with little or no localization to large or medium (30–45μm) sized neurons.
Figure 1.
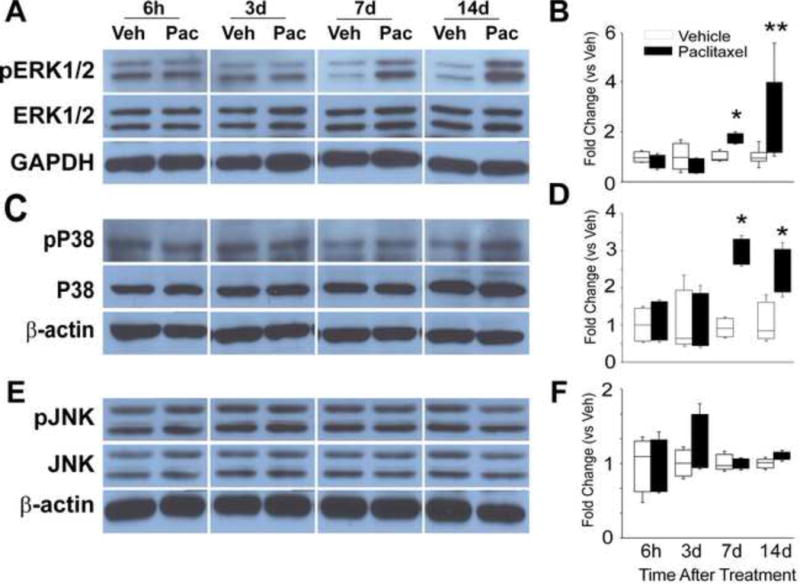
The representative Western blot images shown in (A) illustrate that the expression of phospho-ERK1/2 (pERK1/2) was low in vehicle treated rats, but increased expressions at day 7 and 14 of chemotherapy treatment in DRG. The bar graphs in (B) indicate that the change in expression of pERK1/2 was significantly different in the/paclitaxel-treated group (black bars) versus the vehicle-treated group (open bars). The representative Western blot images shown in (C) illustrate that the expression of phospho-P38 (pP38) was increased expressed at day 7 and 14 of chemotherapy treatment in DRG. The bar graphs in (D) indicate that the change in expression of pP38 was significantly different in the paclitaxel-treated group (black bars) versus the vehicletreated group (open bars). The representative Western blot images shown in (E) illustrate that the expression of neither JNK nor phosho-JNK (pJNK) showed any change in expression through day 14 of chemotherapy treatment in either the DRG. The bar graphs in (F) confirm a lack of statistical differences between the groups of paclitaxel-treated rats (black bars) compared to the vehicle-treated rats (open bars).* = p<0.05, ** = p<0.01.
Figure 5.
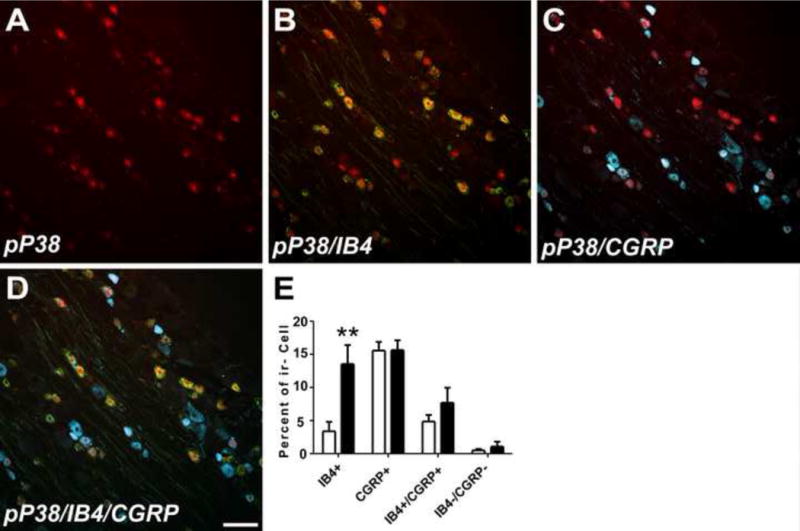
Double IHC revealed that pP38 (red color, A) was most especially expressed in small IB4 positive neurons (green for IB4, yellow for double label, B) with only sparse localization to small CGRP positive neurons (blue for CGRP, white for double label, C). The bar graph in (E) shows that a significant increased expression in pP38 was observed in small IB4 positive neurons while expression did not change in the small percentage of IB4+/CGRP+ or IB4/CGRP negative neurons that also expressed pP38. No localization was observed in either large (>45μm) or medium sized (30–45μm) neurons (not shown). Scale bar= 100 μm. ** = P < 0.01.
Figure 2.
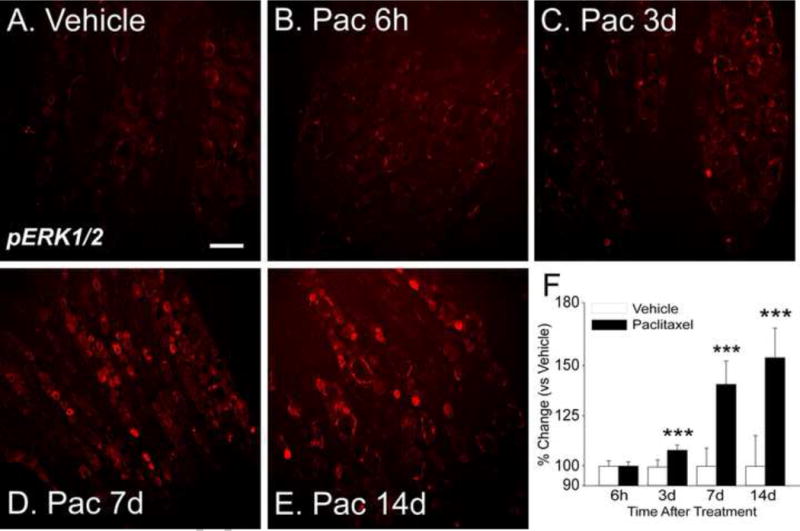
Representative IHC images show that expression of pERK1/2 in rat DRG is quite low in vehicle treated rats (A). Paclitaxel treatment results in no change in pERK1/2 expression at six hours (B) but by day 3 (24 hours after the second paclitaxel dose) there is increased expression (C). The increase in expression was more pronounced at day 7 (D, 24 hours after the fourth paclitaxel dose) and even more so at day 14 (E). The bar graphs (F) show the summarized data for all groups and indicate that the increases in expression were significant at days 3, 7 and 14 for the paclitaxeltreated group (black bars) versus the vehicle treated controls (open bars). Scale bar= 100 μm. *** = p <0.001.
Figure 3.
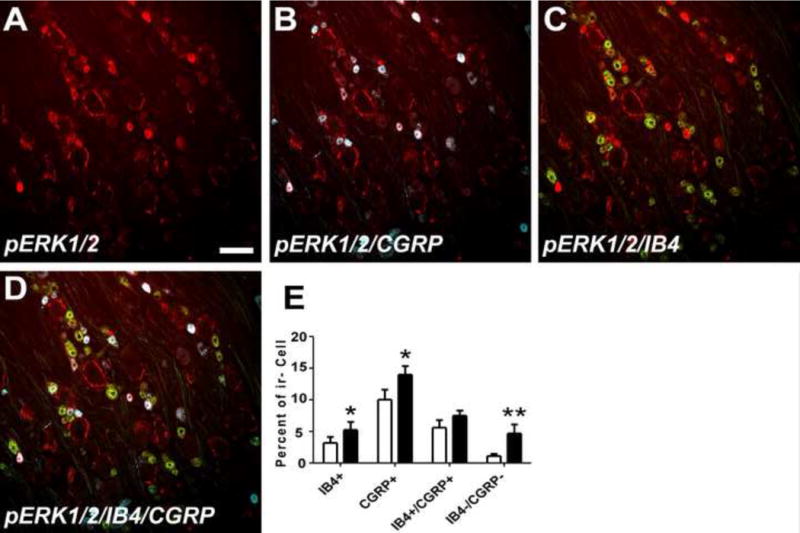
Double IHC revealed that pERK1/2 (red color, A) was most especially expressed in small (<30μm) CGRP positive neurons (blue color, double label is white, B) with only sparse localization to small IB4-positive neurons (green color, double label is yellow, C) and also found in ring shaped cellular profiles around large DRG neurons consistent with localization to satellite glial cells (A–D). The bar graph in (E) shows that a significant increased expression in pERK1/2 was also observed in small cells negative for both CGRP and IB4 while expression did not change in the small percentage or IB4+/CGRP+ that also expressed pERK1/2. No localization was observed in either large (>45μm) or medium sized (30–45μm) neurons. Scale bar= 100 μm. * = p <0.05, ** = P < 0.01.
IHC analysis confirmed a significant increase in the expression of pP38 at days 7 and 14 after paclitaxel treatment in the DRG (Figure 4D–F, p<0.001) that was present at only very low levels in rats treated with vehicle (Figure 4A) (n=6). Further IHC analysis of the localization of pP38 at day 14 after paclitaxel treatment, showed that pP38 was co-localized primarily in small IB4-positive neurons with sparse co-localization to CGRP+ neurons (Figure 5). Histography showed that the proportion of pP38-positive/IB4-positive neurons was significantly increased with little change in the proportions of other subgroups of neurons (Figure 5E, p=0.005). There was no localization of pP38 to either large or medium DRG neurons.
Figure 4.
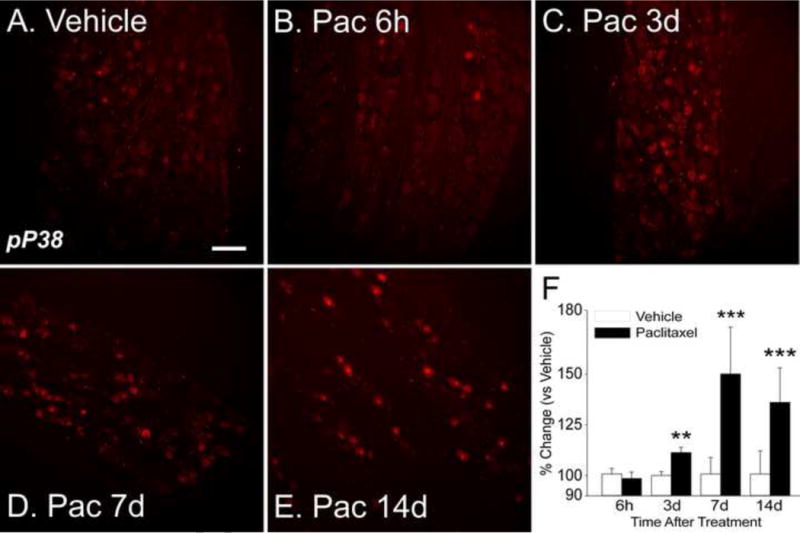
Representative IHC images show that expression of pp38 is expressed at low levels in DRG in vehicle treated rats (A). Paclitaxel treatment resulted in no change in expression at six hours (B) but by day 3 (24 hours after the second paclitaxel dose) there is increased expression (C). The increase in expression was more pronounced at day 7 (D, 24 hours after the fourth paclitaxel dose) and remained elevated at day 14 (E). The bar graphs (F) show the summarized data for all groups and indicate that the increases in expression were significant at days 3, 7 and 14 for the paclitaxel-treated group (black bars) versus the vehicle treated controls (open bars). Scale bar= 100 μm. ** = p <0.01, *** = p <0.001.
3.2 Expression and distribution of MAPK in the dorsal horn after paclitaxel treatment
Western blot indicated that the expression of pERK1/2 alone was increased in the spinal dorsal horn after paclitaxel treatment only at day 14 (Figure 6A–B, p<0.01) (n=4). There was no change in expression of either pP38 or pJNK in the dorsal horn at any time point observed (n=4). IHC revealed that the pattern of pERK1/2 in spinal cord was confined to the most superficial layers consistent with localization to primary afferent terminals with no apparent localization to spinal neurons (data not shown).
Figure 6.
The representative Western blot images shown in (A) illustrate that the expression of phospho-ERK1/2 (pERK1/2) was increased expressed at day 14 of chemotherapy in the spinal cord. The bar graphs in (B) show that the level of expression of pERK1/2 was significantly higher at day 14 in the group of paclitaxel-treated rats (black bars) compared to the vehicle-treated rats (open bars). The representative Western blot in (C) and bar graph summaries in (D) show that pP38 showed no change in expression in the spinal cord over the interval measured. Finally, the Western blots in (E) and bar graphs in (F) show that JNK signaling also was not altered in spinal cord over the interval observed. ** = p <0.01.
3.3 Expression of TLR4-IκBα-NFκB and PI3K-Akt in the DRG and spinal cord at day 14 after paclitaxel treatment
Based on the data in Figures 1–5 additional study of other potential down-stream TLR4 signals, here IκBα-NFκB and PI3K-Akt signaling, was focused only on day 14 after treatment. Paclitaxel induced no change in total NFκB in DRG (Figure 7A), but there was a significant increase in the expression of nuclear NFκB (Figure 7B, p<0.05) and phospho-NFκB (Figure 7C, p<0.05) (n=4). HSP90 staining revealed little crosscontamination between the cytoplasm and nuclear proteins (data not shown). The levels of neither total nor nuclear NFκB expression in the spinal cord showed any change (data not shown). Paclitaxel also did not increase phospho-Akt expression in either DRG or spinal cord (Figure 8A, B) (n=4).
Figure 7.
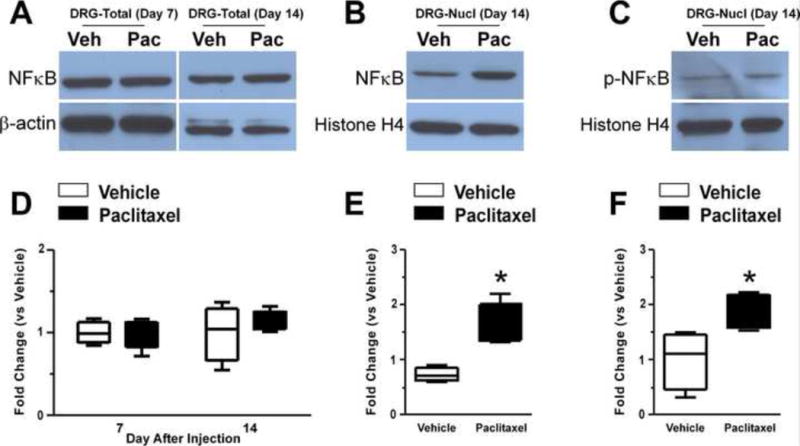
The representative Western blot images shown in (A) illustrate that the expression of total NFκB showed no change at day 14 of chemotherapy in either DRG (A) or spinal cord (data not shown). However, nuclear NFκB and nuclear phospho-NFκB (pNFκB) showed increased expressions at day 14 (B and C). There was no change of nuclear NFκB or pNFκB in spinal cord (data not shown). The bar graphs indicate that the level of expression of total NFκB was not changed (D) whereas nuclear NFκB (E) and nuclear pNFκB (F) were statistically different between the group of paclitaxeltreated rats (black bars) versus vehicle-treated rats (open bars), with NFκB and pNFκB significantly increased expressed in DRG nuclei. There were no statistically significant changes in any of these measures in spinal cord. * = p <0.05.
Figure 8.
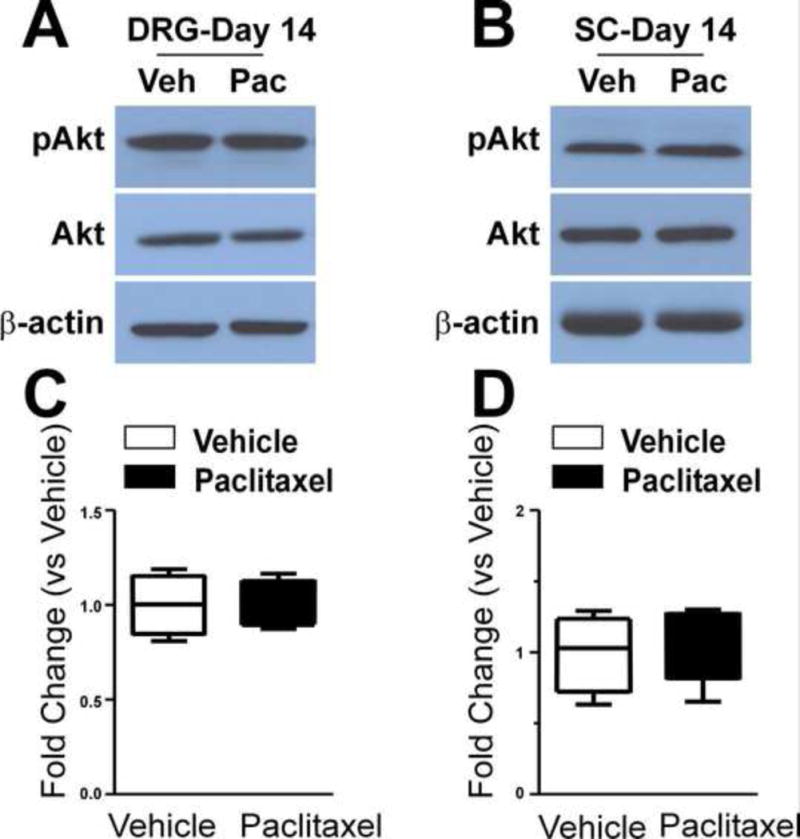
The representative western blot images shown in (A) and (B) show that the expression of Akt and phospho-Akt (pAkt) were not changed at day 14 of chemotherapy in either DRG (A) or spinal cord (B) and the bar graphs show the summaries of this data for the DRG (C) and spinal cord (D) group of paclitaxel-treated rats (black bars) compared to the vehicle-treated rats (open bars).
3.4 LPS-RS inhibits paclitaxel-induced phosphorylation of ERK1/2 and P38
LPS-RS was used to test whether activation of the MAPK signaling pathways by paclitaxel treatment was downstream to TLR4 activation. Rats were treated with 20 μg LPS-RS in 20 μl PBS intrathecally via L5 puncture beginning 2 days before paclitaxel treatment and then daily until 2 days after the completion of paclitaxel treatment (n=4). LPS-RS reduced ERK1/2 phosphorylation in both the DRG (Figure 9A, p<0.05) and spinal cord (Figure 9B, p<0.05) at day 14 compared to vehicle-paclitaxel-treated controls. LRS-RS also blocked the increased expression of P38 phosphorylation in the DRG (Figure 9C, p<0.05) at day 14. Treatment with LPS-RS alone for 10 days in naïve rats had no effect on the expression of P-ERK or pP38 compared to naïve rats receiving saline vehicle (data not shown).
Figure 9.
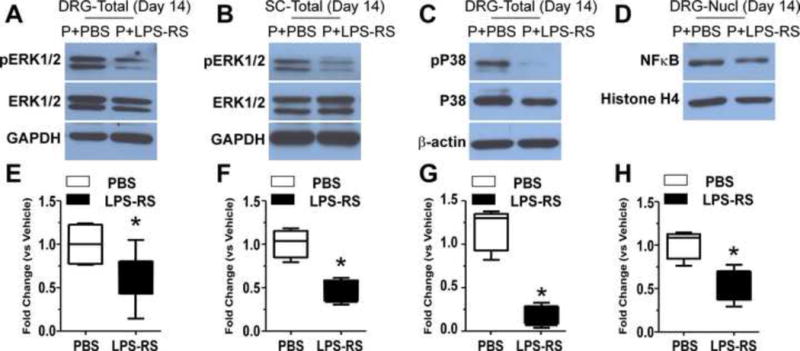
The representative Western blots show that LPS-RS co-treatment with paclitaxel prevented the increased expressions in ERK1/2 phosphorylation both in DRG (A) and spinal cord (B), prevented the increased expression of P38 phosphorylation in DRG (C) and also prevented the increased expression in nuclear NFκB (D) at day 14 after chemotherapy. The level of pERK1/2, pP38 and nuclear NFκB in paclitaxel-PBS (P+PBS) treated rats is in the left hand column of each representative blot while the paclitaxel-LPS-RS (P+LPS-RS) data is shown in the right hand column. The bar graphs show the level of expression of phospho-ERK1/2 in the DRG (E) and spinal cord (F), phospho-p38 (G) and nuclear NFκB in the DRG (H) respectively was significantly lower in the paclitaxel-treated rats co-treated with LRS-RS (black bars) compared to the rats co-treated with PBS vehicle (open bars). * = p <0.05.
3.5 LPS-RS inhibits paclitaxel-induced internalization of NFκB
LPS-RS co-treatment during paclitaxel administration blocked the nuclear translocation of NFκB in DRG neurons (Figure 9D) at day 14 after paclitaxel treatment (n=4). The levels of nuclear NFκB was significantly less in the LPS-RS-paclitaxel group than in the PBS-paclitaxel group (Figure 9D, p<0.05). Treatment with LPS-RS alone for 10 days in naïve rats had no effect on the translocation of NFκB to the nucleus compared to naïve rats receiving saline vehicle (data not shown).
3.6 Prevention but not reversal of paclitaxel-induced behavioral hypersensitivity by inhibitors of MAPK signaling pathway
The contribution of MAPK signaling to paclitaxel-induced behavioral hypersensitivity to mechanical stimulation of the hind paws was assessed by testing the effects of intrathecally administered inhibitors of the MAPK signaling pathway, PD98059 (MEK1/2), U0126 (MEK1/2) and SB203580 (P38), on both pre-established paclitaxel CIPN and on the induction of paclitaxel CIPN (Figure 10A–I). In the first set of three experiments, wherein PD98059, U0126 or SB203580 was tested to reverse paclitaxel-induced behavioral hypersensitivity, two groups in each experiment were first treated with paclitaxel, and hypersensitivity to mechanical stimuli was confirmed in each group such that there was a statistical difference between the baseline measurement and that at day 14 for both while neither paclitaxel treatment group was different from the other (Figure 10A–C). ANOVA for the PD98059 experiment showed significant effects for both treatment (F (4,192)=654.8, p<0.0001) and time (F (7,192)=92.90, p<0.0001) and for the treatment-time interaction (F (28,192)=14.94, p<0.001). The same analysis for the U0126 experiment also showed significant effects for treatment (F (4,208)=420.1, p<0.0001), time (F (7, 208)=55.98, p<0.0001), and the treatment-time interaction (F (28, 208)=8.624, p<0.0001). Finally, the analysis in the SB20380 experiment showed significant effects for treatment (F (4,192)=473.7, p<0.0001), time (F (7,192)=68.22, p<0.0001), and the treatment-time interaction (F (28, 192)=11.17, p<0.0001). Rats in both groups were then given a single intrathecal dose of either 10 μg or 30 μg PD98059, U0126 or SB203580 (n=7 or 8 each) in 30 μl PBS or 30 μl PBS with vehicle diluent solution alone (n=4, each). Neither, PD98059 or SB203580 at a dose of 10 μg reversed paclitaxel induced hypersensitivity. Similarly, animals treated with higher doses (30 μg) of PD98059 and SB203580 showed no differences compared to the paclitaxel-vehicle treated controls. Given that PD98059 is a relatively weak MEK/ERK inhibitor, U0126 was also tested. Again, no differences in the mechanical withdrawal threshold in either the inhibitor-treated or vehicle-treated animals were observed. Given that repeated dosing is often required to reverse existing clinical neuropathic pain, this was also tested here (Figure 10D–F). Again, three experiments, one for each inhibitor, were run, though here only two paclitaxel groups were include and the design did not include the vehicle-drug or vehicle-vehicle groups as used in the first set of experiments. Hyper-responsiveness was confirmed in the paclitaxel groups at day 14 and then animals were intrathecally injected with MAPK inhibitors (30 μg) for 5 consecutive days. ANOVA for the PD90859 experiment only showed a significant effect for time (F (6, 85)= 75.52, p <0.0001) wherein the paclitaxel treatment induced hyperalgesia in both groups compared to the baseline withdrawal value, but no effect for treatment (F (1, 84)= 0.3530, p=0.5540) or the treatment-time interaction (F (6, 84)= 0.2047, p= 0.9745). Similar results were observed in the U0126 repeat dose experiment for time (F (6, 84)=267.0, p<0.0001), treatment (F (1,84)= 1.255, p=0.2659) and the treatment-time interaction (F (6,84)=1.782, p= 0.1126); and in the SB203580 experiment for time (F (6,84)=129.4, p< 0.0001), treatment (F (1,84)=0.3820, p= 0.5382) and the treatment-time interaction (F (6,84)= 1.985, p= 0.0768). Hence, like in the first set of experiments the repeated dosing of MAPK inhibitors also failed to reverse paclitaxel-induced behavioral hypersensitivity.
Figure 10.
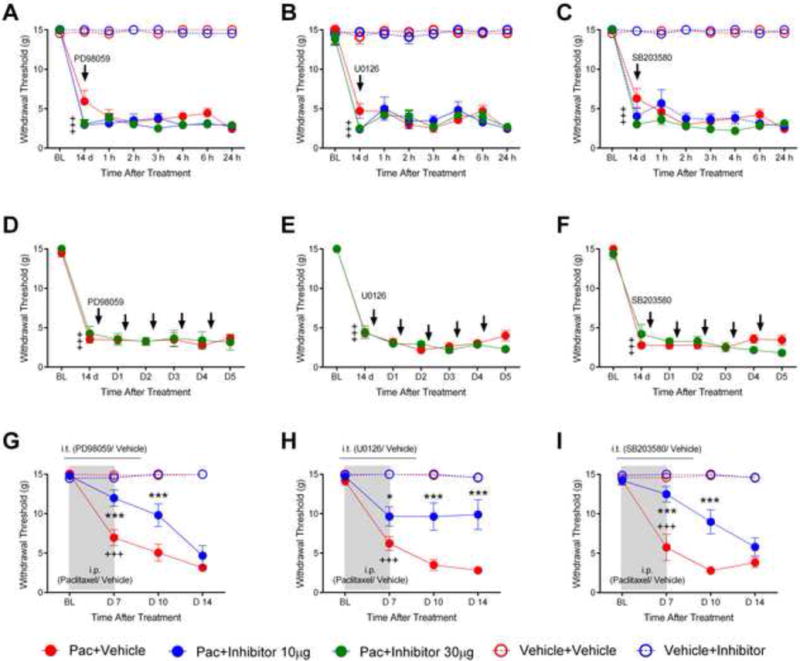
The scatter and line plots in (A to F) show the effects of two MEK1/2 (PD98059 and U0126) and a p38 inhibitor (SB203580) on established paclitaxelinduced mechanical withdrawal threshold hypersensitivity at 14 days after chemotherapy. The data for paclitaxel-vehicle treated rats is shown in red filled circles, vehicle-vehicle data is in red open circles, vehicle-inhibitor data is in blue open circles and paclitaxel-inhibitor data is shown in blue or green filled circles (two doses). In (A to C) rats were treated with a single intrathecal dose of the inhibitors (or vehicle) indicated by the arrow and then re-tested hourly for six hours and then the following day. In (D to F) rats were treated daily for five days as indicated by the arrows and tested after each dose as well as the day following the final dose. Crosses indicate significant decrease in mechanical withdrawal threshold compared to baseline (BL; +++ p<0.001). In (G–I) rats were intrathecally treated daily with 10μg PD98059 (U0126 or SB203580) beginning 2 days before and continuing to 2 days after paclitaxel (blue filled circles) and showed a significant partial prevention of mechanical hypersensitivity compared with the paclitaxel-vehicle group (red filled circles) at day 7 and day 10. Vehicle-treated rats receiving i.t vehicle solution (red opened circles) or inhibitors (blue opened circles) showed no changes from the baseline. The paclitaxel-treated rats that received inhibitor vehicle solution only had an expected decrease in mechanical withdrawal threshold that was significant from the vehicle treated rats by day 7 (G–I) (BL; baseline, +++ p<0.001). * = p<0.05; *** = p<0.001. Asterisks indicate significant differences between the paclitaxel-PD98059 (U0126 or SB203580) groups versus the paclitaxel-vehicle groups.
PD98059, U0126 and SB203580 were also evaluated to determine whether either might be useful in preventing paclitaxel-induced behavioral hypersensitivity. Rats were given PD98059, U0126 or SB203580 (10 μg in 30 μl) or vehicle control solution (30 μl) via i.t. injection every day beginning 2 days before paclitaxel treatment and then daily until 2 days after the completion of paclitaxel treatment. ANOVA in the PD98059 experiment showed significant effects for treatment (F (3,84)= 51.26, p<0.0001), time (F (3,84)= 19.19, p< 0.0001), and the treatment-time interaction (F (9, 84)= 8.610, p< 0.0001). Similarly in the U0126 experiment there was a significant effect for treatment (F (3,72)= 57.19, p<0.0001), time (F (3,72)= 13.16, p<0.0001) and the treatment-time interaction (F (9,72)= 5.804, p<0.0001). Likewise in the SB203580 experiment a significant effect was found for treatment (F (3,84)= 52.84, p< 0.0001), time (F (3,84)= 14.63, p< 0.0001), and the treatment-time interaction (F (9,84)= 7.109, p<0.0001). Post-hoc analyses revealed that PD98059, U0126 or SB203580 had no effect on the baseline mechanical withdrawal threshold and showed no interaction with the paclitaxel vehicle over time (Figure 10G–I). The paclitaxel-treated rats that received vehicle solution only (n=8) had an expected decrease in mechanical withdrawal threshold that was significant from the vehicle treated rats by day 7 (Figure 10 G–I, p<0.001). In contrast the paclitaxel-U0126 rats showed only partial development of mechanical hypersensitivity that was significantly less when compared with the paclitaxel-vehicle treated rats at days 7, 10 and 14 (Figure 10H, n=7, p< 0.05 at day 7, and p <0.001 at days 10 and 14). Similarly, the paclitaxel-PD98059 and paclitaxel-SB203580 rats showed only partial development of mechanical hypersensitivity, but this was evident only at day 7 and 10 (Figure 10G and I, p <0.001 at each time point) (n=9).
4.0 Discussion
Previous publications indicated that TLR4 in the central nervous system plays a role in the development of behavioral hypersensitivity in a rodent model of neuropathic pain (Hutchison et al., 2008; Tanga et al., 2005). Rats that lacked functional TLR4 and those that received intrathecally administrated TLR4 antisense oligonucleotides showed attenuated nerve injury–induced behavioral hypersensitivity in both the initiation and maintenance phases (Bettoni et al., 2008).
Canonical signal transduction following TLR4 activation in monocytes/macrophages occurs via two distinct pathways. One pathway results in the induction of a MyD88-dependent cascade leading to NF-κB activation and subsequent increased synthesis and release of multiple cytokines and chemokines (O’Neill and Bowie 2007; Palsson-McDermott and O’Neill 2004). The second cascade is TRIF dependent and leads to delayed NF-κB activation and interferon-β production (O’Neill and Bowie 2007; Palsson-McDermott and O’Neill 2004). In addition to the canonical signal pathways, non-canonical MAPK signaling is also associated with activation of TLR4 (Guha and Mackman 2001). Numerous studies have shown that LPS activates ERK1/2 in monocytes/macrophages (Geppert et al., 1994; Marie et al., 1999; Swantek et al., 1997; van der Bruggen et al., 1999), and the MEK1/2 inhibitor, PD98059, blocked LPS induced TNFα activation in human monocytes (van der Bruggen et al., 1999). Likewise, the p38 signaling pathway is recruited by TLR4 activation and the p38 inhibitor, SB203580, reduces LPS-stimulated IL-1 and TNFα expression (Lee et al., 1994).
The combined results of a previous study (Li et al., 2014b) with the present implicates both the canonical and non-canonical MAPK signaling pathways of TLR4 in DRG and spinal cord in the generation of paclitaxel CIPN. Here we detected increased expression of pERK1/2 and pP38 in DRG following paclitaxel treatment that was prevented by co-treatment with the TLR4 inhibitor LPS-RS that itself was previously shown to prevent development of paclitaxel related CIPN when given in concert with chemotherapy. The contribution of ERK1/2 and p38 signaling in CIPN were in turn demonstrated by the partial reduction of the behavioral phenotype by co-treatment with the inhibitors PD98059, U0126 and SB203580 during chemotherapy, whereas previously a MyD88 inhibitor peptide was also shown to inhibit paclitaxel CIPN (Li et al., 2014b). At a more granular level, TLR4 is found predominantly in small IB4+ and CGRP+ DRG neurons with only low levels of expression in medium and large size DRG neurons. Paclitaxel treatment induces an increase of expression of TLR4 in large, medium and small sized DRG neurons, but most especially in small IB4+ and CGRP+ DRG neurons. The downstream signaling of TLR4 is distinct between the two subtypes of small DRG neurons with the CGRP+ neurons showing increased expression in pERK1/2, MyD88 and TRIF whereas the IB4+ neurons show increased expression in pERK1/2, pP38 and TRIF. Interestingly, the small numbers of large and medium sized neurons expressing TLR4 show increased expression in TRIF signaling alone following paclitaxel treatment. It remains unknown whether individual DRG cells show increased expression in both canonical and non-canonical MAPK signaling or whether these occur in isolated subsets of cells. Given that intraplantar protein kinase (PK) A and ε antagonists suppress CIPN-related hyperalgesia (Dina et al., 2001), subsets of DRG neurons may also show increased expression of these signal molecules. Finally, the relationship between the increased expression in these signaling pathways and the increased excitability and changes in the expression of ion channels in DRG neurons remains to be determined (Li et al., 2014b).
TLR4 signaling at the spinal level may also contribute to paclitaxel-related CIPN. TLR4 was found co-localized only in astrocytes (GFAP+ cell profiles), but not in either microglia or neurons (OX-42+ or NeuN+ cell profiles, respectively) (Li et al., 2014b). Although neuron glial interactions have become well recognized as involved in neuropathic pain this recognition is largely confined to microglia. CIPN appears unusual in this context in that microglia appear to play little if any role while astrocytes appear to contribute a major role (Li et al., 2014b; Zhang et al., 2012). The data shown here that pERK1/2 is increased in spinal cord, but pP38 is not reinforces the findings of a minimal role for activated microglia in CIPN in that p38 signaling is essential in the activation of spinal microglia. Yet, it should be noted that others have reported increased CD11b at very late time points following taxol treatment beyond that surveyed here (Ledeboer et al., 2007). Hence microglia may play important roles in the resolution phase of CIPN.
CIPN shows both similarities as well as differences to other types of neuropathic pain. While many types of neuropathic pain are characterized by pronounced touch evoked pain this is uncommon in CIPN (Boyette-Davis et al., 2013; Boyette-Davis et al., 2011b). Touch evoked pain in CRPS types I and II, by example, are produced by activation of Aβ myelinated fibers (Campbell et al., 1988);(Gracely et al., 1992), whereas thresholds to similar stimuli in CIPN patients are elevated on average by more than two orders of magnitude (Boyette-Davis et al., 2013; Boyette-Davis et al., 2011b). Importantly however, touch evoked pain in CPRS type II is driven by on-going activity in nociceptors (Gracely et al., 1992) and possibly also in mechanoreceptors (Devor 2009) and spontaneous activity also develops in DRG neurons in CIPN (Zhang and Dougherty 2014). Although the animal models in CIPN show decreased mechanical withdrawal thresholds, this is not mirrored by a lowering of threshold in spinal neurons (Cata et al., 2006a; Cata et al., 2006b; Cata et al., 2008; Robinson et al., 2014a; Weng et al., 2003). What really stand out in the animal studies are the pronounced increases in afterdischarges observed following cutaneous stimuli. These findings are exciting in that these also parallel the behavioral observations of rats with CIPN (Boyette-Davis et al., 2011b; Cata et al., 2007; Dougherty et al., 2007; Dougherty et al., 2004) where exaggerated paw withdrawal to mechanical stimuli, often licking the paw, maintaining an elevated posture and often rolling over to avoid additional stimuli are a prominent part of the phenotype. Similarly, CIPN patients often report that one of the most disturbing symptoms they suffer is that once pain is provoked in their hands or feet affected by CIPN the pain will linger for many minutes, hours, or even days. Hence, the specific modality or modalities of fibers driving the animals’ behavioral responses are unclear. Ablation studies in mice show that mechanical pain is signaled by CGRP+ or Mrgprd+/IB4+ nociceptors, whereas TRPV1+ nociceptors signal heat pain and others have reported a key role for IB4+ DRG neurons in CIPN (Joseph et al., 2008). The observations here that signaling increased in IB4+ and CGRP+ nociceptors further suggests an important role for these neurons.
The role for MAP kinase signaling as shown here also illustrates similarities and differences of CIPN with other types of neuropathic pain. The MAP kinases p38 and ERK1/2 are associated with the activation of microglia and in mediating changes in conduction through multiple subtypes of neuronal ion channels, the two effects resulting in increased primary afferent and spinal neuron excitability in models of neuropathic pain following peripheral nerve damage (Black et al., 2008; Dib-Hajj et al., 2009; Hudmon et al., 2008; Janes et al., 2015). Increased signaling of NFκB also contributes to the activation of microglia in models of direct peripheral nerve damage and suppression of glial activation and MAP kinase signaling prevents and reverses signs of behavioral hyper-responsiveness (Ledeboer et al., 2005; Ledeboer et al., 2007). CIPN is shown here to involve a key role for p38 and ERK1/2 in dorsal root ganglion neurons and elsewhere has been shown to play a key role in the responses of spinal neurons in CIPN(Janes et al., 2015). Yet, CIPN does not involve activation of microglia, but rather astrocytes play a key role at spinal levels in generating behavioral hyperresponsiveness (Robinson et al., 2014b; Yoon et al., 2013; Zhang et al., 2012; Zheng et al., 2011). Thus, CIPN is not only a clinically important problem, but as well a unique model of neuropathic pain that may reveal novel mechanisms of glial-neuronal interactions in chronic pain.
In summary, previous studies have implicated TLR4 and its canonical signal pathways in CIPN while this study provides important new data concerning the involvement of the non-canonical MAPK signal pathways. These signaling pathways may provide novel targets for the prevention of CIPN.
Highlights.
Paclitaxel increases the expression of ERK1/2, pP38 and NFκB
Co-application of a TLR4 antagonist blocks paclitaxel-induced MAP kinase increases
MAP kinase inhibitors partially prevent paclitaxel-induced hyperalgesia
Acknowledgments
The work was supported by NIH grant NS046606 and the H.E.B. Cancer Research Professorship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Austin PJ, Moalem-Taylor G. The neuro-immune balance in neuropathic pain: involvement of inflammatory immune cells, immune-like glial cells and cytokines. J Neuroimmunol. 2010;229:26–50. doi: 10.1016/j.jneuroim.2010.08.013. [DOI] [PubMed] [Google Scholar]
- Basu S, Sodhi A. Increased release of interleukin-1 and tumour necrosis factor by interleukin-2-induced lymphokine-activated killer cells in the presence of cisplatin and FK-565. Immunol Cell Biol. 1992;70(Pt 1):15–24. doi: 10.1038/icb.1992.3. [DOI] [PubMed] [Google Scholar]
- Bettoni I, Comelli F, Rossini C, Granucci F, Giagnoni G, Costa B. Glial TLR4 receptor as new target to treat neuropathic pain: Efficacy of a new receptor antagonist in a model of peripheral nerve injury in mice. GLIA. 2008;56:1312–1319. doi: 10.1002/glia.20699. [DOI] [PubMed] [Google Scholar]
- Black JA, Nikolajsen L, Kroner K, Jensen TS, Waxman SG. Multiple sodium channel isoforms and mitogen-activated protein kinases are present in painful human neuromas. Annals of Neurology. 2008;64:644–653. doi: 10.1002/ana.21527. [DOI] [PubMed] [Google Scholar]
- Boyette-Davis J, Dougherty PM. Protection against oxaliplatin-induced mechanical hyperalgesia and intraepidermal nerve fiber loss by minocycline. Exp Neurol. 2011;229:353–357. doi: 10.1016/j.expneurol.2011.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyette-Davis J, Xin W, Zhang H, Dougherty PM. Intraepidermal nerve fiber loss corresponds to the development of Taxol-induced hyperalgesia and can be prevented by treatment with minocycline. Pain. 2011a;152:308–313. doi: 10.1016/j.pain.2010.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyette-Davis JA, Cata JP, Driver LC, Novy DM, Bruel BM, Mooring DL, Wendelschafer-Crabb G, Kennedy WR, Dougherty PM. Persistent chemoneuropathy in patients receiving the plant alkaloids paclitaxel and vincristine. Cancer Chemother Pharmacol. 2013;71:619–626. doi: 10.1007/s00280-012-2047-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyette-Davis JA, Cata JP, Zhang H, Driver LC, Wendelschafer-Crabb G, Kennedy WR, Dougherty PM. Follow-up psychophysical studies in bortezomib-related chemoneuropathy patients. J Pain. 2011b;12:1017–1024. doi: 10.1016/j.jpain.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyette-Davis JA, Eng C, Wang XS, Cleeland CS, Wendelschafer-Crabb G, Kennedy WR, Simone DA, Dougherty PM. Subclinical peripheral neuropathy is a common finding in colorectal cancer patients prior to chemotherapy. Clin Cancer Res. 2012 doi: 10.1158/1078-0432.CCR-12-0205. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd-Leifer CA, Block EF, Takeda K, Akira S, Ding A. The role of MyD88 and TLR4 in the LPS-mimetic activity of Taxol. Eur J Immunol. 2001;31:2448–2457. doi: 10.1002/1521-4141(200108)31:8<2448::aid-immu2448>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Campbell JN, Raja SN, Meyer RA, Mackinnon SE. Myelinated afferents signal the hyperalgesia associated with nerve injury. Pain. 1988;32:89–94. doi: 10.1016/0304-3959(88)90027-9. [DOI] [PubMed] [Google Scholar]
- Cao Y, Li K, Fu KY, Xie QF, Chiang CY, Sessle BJ. Central sensitization and MAPKs are involved in occlusal interference-induced facial pain in rats. J Pain. 2013;14:793–807. doi: 10.1016/j.jpain.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cata JP, Weng HR, Chen JH, Dougherty PM. Altered discharges of spinal wide dynamic range neurons and down-regulation of glutamate transporter expression in rats with paclitaxel-induced hyperalgesia. Neuroscience. 2006a;138:329–338. doi: 10.1016/j.neuroscience.2005.11.009. [DOI] [PubMed] [Google Scholar]
- Cata JP, Weng HR, Burton AW, Villareal H, Giralt S, Dougherty PM. Quantitative sensory findings in patients with bortezomib-induced pain. J Pain. 2007;8:296–306. doi: 10.1016/j.jpain.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Cata JP, Weng HR, Dougherty PM. Clinical and experimental findings in humans and animals with chemotherapy-induced peripheral neuropathy. Minerva Anes. 2006b;72:151–169. [PubMed] [Google Scholar]
- Cata JP, Weng HR, Dougherty PM. Behavioral and electrophysiological studies in rats with cisplatin-induced chemoneuropathy. Brain Res. 2008;1230:91–98. doi: 10.1016/j.brainres.2008.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Chaudhry V, Rowinsky EK, Sartorius SE, Donehower RC, Cornblath DR. Peripheral neuropathy from taxol and cisplatin combination chemotherapy: clinical and electrophysiological studies. Ann Neurol. 1994;35:304–311. doi: 10.1002/ana.410350310. [DOI] [PubMed] [Google Scholar]
- Devor M. Ectopic discharge in Ab afferents as a source of neuropathic pain. Exp Brain Res. 2009;196:115–128. doi: 10.1007/s00221-009-1724-6. [DOI] [PubMed] [Google Scholar]
- Dib-Hajj SD, Binshtok AM, Cummins TR, Jarvis MF, Samad T, Zimmermann K. Voltage-gated sodium channels in pain states: role in pathophysiology and targets for treatment. Brain Research Reviews. 2009;60:65–83. doi: 10.1016/j.brainresrev.2008.12.005. [DOI] [PubMed] [Google Scholar]
- Dina OA, Chen X, Reichling D, Levine JD. Role of protein kinase C[epsi] and protein kinase A in a model of paclitaxel-induced painful peripheral neuropathy in the rat. Neuroscience. 2001;108:507–515. doi: 10.1016/s0306-4522(01)00425-0. [DOI] [PubMed] [Google Scholar]
- Dougherty PM, Cata JP, Burton AW, Vu K, Weng HR. Dysfunction in multiple primary afferent fiber subtypes revealed by quantitative sensory testing in patients with chronic vincristine-induced pain. J Pain Symptom Manage. 2007;33:166–179. doi: 10.1016/j.jpainsymman.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Dougherty PM, Cata JP, Cordella JV, Burton A, Weng HR. Taxolinduced sensory disturbance is characterized by preferential impairment of myelinated fiber function in cancer patients. Pain. 2004;109:132–142. doi: 10.1016/j.pain.2004.01.021. [DOI] [PubMed] [Google Scholar]
- Gan XH, Jewett A, Bonavida B. Activation of human peripheral-blood-derived monocytes by cis-diamminedichloroplatinum: enhanced tumoricidal activity and secretion of tumor necrosis factor-alpha. Nat Immun. 1992;11:144–155. [PubMed] [Google Scholar]
- Geppert TD, Whitehurst CE, Thompson P, Beutler B. Lipopolysaccharide signals activation of tumor necrosis factor biosynthesis through the ras/raf-1/MEK/MAPK pathway. Mol Med. 1994;1:93–103. [PMC free article] [PubMed] [Google Scholar]
- Gracely RH, Lynch SA, Bennett GJ. Painful neuropathy: altered central processing maintained dynamically by peripheral input. Pain. 1992;51:175–194. doi: 10.1016/0304-3959(92)90259-E. [DOI] [PubMed] [Google Scholar]
- Guha M, Mackman N. LPS induction of gene expression in human monocytes. Cell Signal. 2001;13:85–94. doi: 10.1016/s0898-6568(00)00149-2. [DOI] [PubMed] [Google Scholar]
- Hagiwara H, Sunada Y. Mechanism of taxane neurotoxicity. Breast Cancer. 2004;11:82–85. doi: 10.1007/BF02968008. [DOI] [PubMed] [Google Scholar]
- Han J, Lee JD, Bibbs L, Ulevitch RJ. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science. 1994;265:808–811. doi: 10.1126/science.7914033. [DOI] [PubMed] [Google Scholar]
- Han Y, Li Y, Xiao X, Liu J, Meng XL, Liu FY, Xing GG, Wan Y. Formaldehyde up-regulates TRPV1 through MAPK and PI3K signaling pathways in a rat model of bone cancer pain. Neurosci Bull. 2012;28:165–172. doi: 10.1007/s12264-012-1211-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Chiba T, Abe K, Makabe A, Ikeno S, Kawakami K, Utsunomiya I, Hama T, Taguchi K. Effect of paclitaxel on transient receptor potential vallinoid 1 in rat dorsal root ganglion. Pain. 2013;154:882–889. doi: 10.1016/j.pain.2013.02.023. [DOI] [PubMed] [Google Scholar]
- Hudmon A, Choi JS, Tyrrell L, Black JA, Rush AM, Waxman SG, Dib-Hajj SD. Phosphorylation of sodium channel Na(v)1.8 by p38 mitogen-activated protein kinase increases current density in dorsal root ganglion neurons. J Neurosci. 2008;28:3190–3201. doi: 10.1523/JNEUROSCI.4403-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison MR, Zhang Y, Brown K, Coats BD, Shridhar M, Sholar PW, Patel SJ, Crysdale NY, Harrison JA, Maier SF, Rice KC, Watkins LR. Nonstereoselective reversal of neuropathic pain by naloxone and naltrexone: Involvement of toll-like receptor 4 (TLR4) Eur J Neurosci. 2008;28:20–29. doi: 10.1111/j.1460-9568.2008.06321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janes K, Esposito E, Doyle T, Cuzzocrea S, Tosh DK, Jacobson KA, Salvemini D. A3 adenosine receptor agonist prevents the development of paclitaxel-induced neuropathic pain by modulating spinal glial-restricted redox-dependent signaling pathways. Pain. 2015;155:2560–2567. doi: 10.1016/j.pain.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Gereau RW, Malcangio M, Strichartz GR. MAP kinase and pain. Brain Res Rev. 2009;60:135–148. doi: 10.1016/j.brainresrev.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Samad TA, Jin SX, Schmoll R, Woolf CJ. p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron. 2002;36:57–68. doi: 10.1016/s0896-6273(02)00908-x. [DOI] [PubMed] [Google Scholar]
- Joseph EK, Chen X, Bogen O, Levine JD. Oxaliplatin acts on IB4-positive nociceptors to induce an oxidative stress-dependent acute painful peripheral neuropathy. J Pain. 2008;9:463–472. doi: 10.1016/j.jpain.2008.01.335. [DOI] [PubMed] [Google Scholar]
- Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol. 2000;18:621–663. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- Kosturakis AK, He Z, Li Y, Boyette-Davis JA, Shah N, Thomas SK, Zhang H, Vichaya EG, Wang XS, Wendelschafer-Crabb G, Kennedy WR, Simone DA, Cleeland CS, Dougherty PM. Subclinical peripheral neuropathy in patients with multiple myeloma before chemotherapy is correlated with decreased fingertip innervation density. J Clin Oncol. 2014;32:3156–3162. doi: 10.1200/JCO.2013.54.5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledeboer A, Gamanos M, Lai W, Martin D, Maier SF, Watkins LR, Quan N. Involvement of spinal cord nuclear factor kappaB activation in rat models of proinflammatory cytokine-mediated pain facilitation. Eur J Neurosci. 2005;22:1977–1986. doi: 10.1111/j.1460-9568.2005.04379.x. [DOI] [PubMed] [Google Scholar]
- Ledeboer A, Jekich BM, Sloane EM, Mahoney JH, Langer SJ, Milligan ED, Martin D, Maier SF, Johnson KW, Leinwand LA, Chavez RA, Watkins LR. Intrathecal interleukin-10 gene therapy attenuates paclitaxel-induced mechanical allodynia and proinflammatory cytokine expression in dorsal root ganglia in rats. Brain Behav Immun. 2007;21:686–698. doi: 10.1016/j.bbi.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JC, Laydon JT, McDonnell PC, Gallagher TF, Kumar S, Green D, McNulty D, Blumenthal MJ, Heys JR, Landvatter SW. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372:739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- Li D, Fu Y, Zhang W, Su G, Liu B, Guo M, Li F, Liang D, Liu Z, Zhang X, Cao Y, Zhang N, Yang Z. Salidroside attenuates inflammatory responses by suppressing nuclear factor-kappaB and mitogen activated protein kinases activation in lipopolysaccharide-induced mastitis in mice. Inflamm Res. 2013;62:9–15. doi: 10.1007/s00011-012-0545-4. [DOI] [PubMed] [Google Scholar]
- Li Y, Cai J, Han Y, Xiao X, Meng XL, Su L, Liu FY, Xing GG, Wan Y. Enhanced function of TRPV1 via up-regulation by insulin-like growth factor-1 in a rat model of bone cancer pain. Eur J Pain. 2014a;18:774–784. doi: 10.1002/j.1532-2149.2013.00420.x. [DOI] [PubMed] [Google Scholar]
- Li Y, Zhang H, Zhang H, Kosturakis AK, Jawad AB, Dougherty PM. Toll-like receptor 4 signaling contributes to paclitaxel-induced peripheral neuropathy. J Pain. 2014b;15:712–725. doi: 10.1016/j.jpain.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim EJ, Jeon HJ, Yang GY, Lee MK, Ju JS, Han SR, Ahn DK. Intracisternal administration of mitogen-activated protein kinase inhibitors reduced mechanical allodynia following chronic constriction injury of infraorbital nerve in rats. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31:1322–1329. doi: 10.1016/j.pnpbp.2007.05.016. [DOI] [PubMed] [Google Scholar]
- Marie C, Roman-Roman S, Rawadi G. Involvement of mitogen-activated protein kinase pathways in interleukin-8 production by human monocytes and polymorphonuclear cells stimulated with lipopolysaccharide or Mycoplasma fermentans membrane lipoproteins. Infect Immun. 1999;67:688–693. doi: 10.1128/iai.67.2.688-693.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien JM, Jr, Wewers MD, Moore SA, Allen JN. Taxol and colchicine increase LPS-induced pro-IL-1 beta production, but do not increase IL-1 beta secretion. A role for microtubules in the regulation of IL-1 beta production. J Immunol. 1995;154:4113–4122. [PubMed] [Google Scholar]
- O’Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007;7:353–364. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- Pai K, Sodhi A. Effect of cisplatin, rIFN-Y, LPS and MDP on release of H2O2, O2- and lysozyme from human monocytes in vitro. Indian J Exp Biol. 1991;29:910–915. [PubMed] [Google Scholar]
- Palsson-McDermott EM, O’Neill LA. Signal transduction by the lipopolysaccharide receptor, Toll-like receptor-4. Immunology. 2004;113:153–162. doi: 10.1111/j.1365-2567.2004.01976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polomano RC, Mannes AJ, Clark US, Bennett GJ. A painful peripheral neuropathy in the rat produced by the chemotherapeutic drug, paclitaxel. Pain. 2001;94:293–304. doi: 10.1016/S0304-3959(01)00363-3. [DOI] [PubMed] [Google Scholar]
- Resman N, Gradisar H, Vasl J, Keber MM, Pristovsek P, Jerala R. Taxanes inhibit human TLR4 signaling by binding to MD-2. FEBS Lett. 2008;582:3929–3934. doi: 10.1016/j.febslet.2008.10.037. [DOI] [PubMed] [Google Scholar]
- Robinson CR, Zhang H, Dougherty PM. Altered discharges of spinal wide dynamic range neurons parallel the behavioral phenotype shown by rats with bortezomib related chemotherapy induced peripheral neuropathy. Brain Res. 2014a;1574:6–13. doi: 10.1016/j.brainres.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson CR, Zhang H, Dougherty PM. Astrocytes, but not microglia, are activated in oxaliplatin and bortezomib-induced peripheral neuropathy. Neuroscience. 2014b;274:308–317. doi: 10.1016/j.neuroscience.2014.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson CI, Fitzsimmons B, Azizi S, Powell HC, Hua XY, Yaksh TL. Spinal p38 beta isoform mediates tissue injury-induced hyperalgesia and spinal sensitization. J Neurochem. 2005;92:1508–1520. doi: 10.1111/j.1471-4159.2004.02996.x. [DOI] [PubMed] [Google Scholar]
- Swantek JL, Cobb MH, Geppert TD. N-terminal kinase/stress-activated protein kinase (JNK/SAPK) is required for lipopolysaccharide stimulation of tumor necrosis factor alpha (TNF-alpha) translation: glucocorticoids inhibit TNF-alpha translation by blocking JNK/SAPK. Mol Cell Biol. 1997 Jun;17:6274–6282. doi: 10.1128/mcb.17.11.6274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanga FY, Nutile-McMenemy N, DeLeo JA. The CNS role of Toll-like receptor 4 in innate neuroimmunity and painful neuropathy. Proc Natl Acad Sci U S A. 2005;102:5856–5861. doi: 10.1073/pnas.0501634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallejo R, Tilley DM, Vogel L, Benyamin R. The role of glia and the immune system in the development and maintenance of neuropathic pain. Pain Pract. 2010;10:167–184. doi: 10.1111/j.1533-2500.2010.00367.x. [DOI] [PubMed] [Google Scholar]
- van der Bruggen T, Nijenhuis S, van RE, Verhoef J, van Asbeck BS. Lipopolysaccharide-induced tumor necrosis factor alpha production by human monocytes involves the raf-1/MEK1-MEK2/ERK1-ERK2 pathway. Infect Immun. 1999;67:3824–3829. doi: 10.1128/iai.67.8.3824-3829.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng H-R, Cordella JV, Dougherty PM. Changes in sensory processing in the spinal dorsal horn accompany vincristine-induced hyperalgesia and allodynia. Pain. 2003;103:131–138. doi: 10.1016/s0304-3959(02)00445-1. [DOI] [PubMed] [Google Scholar]
- Yoon SY, Robinson CR, Zhang H, Dougherty PM. Gap junction protein connexin 43 is involved in the induction of oxaliplatin-related neuropathic pain. J Pain. 2013;14:205–214. doi: 10.1016/j.jpain.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaks-Zilberman M, Zaks TZ, Vogel SN. Induction of proinflammatory and chemokine genes by lipopolysaccharide and paclitaxel (Taxol) in murine and human breast cancer cell lines. Cytokine. 2001;15:156–165. doi: 10.1006/cyto.2001.0935. [DOI] [PubMed] [Google Scholar]
- Zhang H, Boyette-Davis JA, Kosturakis AK, Li Y, Yoon SY, Walters ET, Dougherty PM. Induction of monocyte chemoattractant protein-1 (MCP-1) and its receptor CCR2 in primary sensory neurons contributes to paclitaxel-induced peripheral neuropathy. J Pain. 2013;14:1031–1044. doi: 10.1016/j.jpain.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Dougherty PM. Enhanced excitability of primary sensory neurons and altered gene expression of neuronal ion channels in dorsal root ganglion in paclitaxelinduced peripheral neuropathy. Anesthesiology. 2014;120:1463–1475. doi: 10.1097/ALN.0000000000000176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Yoon SY, Zhang H, Dougherty PM. Evidence that spinal astrocytes but not microglia contribute to the pathogenesis of paclitaxel-induced painful neuropathy. J Pain. 2012;13:293–303. doi: 10.1016/j.jpain.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng FY, Xiao WH, Bennett GJ. The response of spinal microglia to chemotherapy-evoked painful peripheral neuropathies is distinct from that evoked by traumatic nerve injuries. Neuroscience. 2011;176:447–454. doi: 10.1016/j.neuroscience.2010.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]