

Abstract
Iron regulatory proteins 1 and 2 (IRP1 and IRP2) are two cytosolic proteins that maintain cellular iron homeostasis by binding to RNA stem loops known as iron responsive elements (IREs) that are found in the untranslated regions of target mRNAs that encode proteins involved in iron metabolism. IRPs modify expression of iron metabolism genes, and global and tissue-specific knockout mice have been made to evaluate the physiological significance of these iron regulatory proteins (Irps). Here, we will discuss the results of the studies that have been performed with mice engineered to lack expression of one or both Irps, and made in different strains using different methodologies. Both Irp1 and Irp2 knockout mice are viable, but the double knockout (Irp1−/−Irp2−/−) mice die before birth, indicating that these Irps play a crucial role in maintaining iron homeostasis. Irp1−/− mice develop polycythemia and pulmonary hypertension, and when these mice are challenged with a low iron diet, they die early of abdominal hemorrhages, suggesting that Irp1 plays an essential role in erythropoiesis and in the pulmonary and cardiovascular systems. Irp2−/− mice develop microcytic anemia, erythropoietic protoporphyria and a progressive neurological disorder, indicating that Irp2 has important functions in the nervous system and erythropoietic homeostasis. Several excellent review articles have recently been published on Irp knockout mice that mainly focus on Irp1−/− mice (referenced in the introduction). In this review, we will briefly describe the phenotypes and physiological implications of Irp1−/− mice, and will discuss the phenotypes observed for Irp2−/− mice in detail with a particular emphasis on the neurological problems of these mice.
Keywords: Iron, Iron regulatory protein, polycythemia, pulmonary hypertension, anemia, neurodegeneration, axonal degeneration, amino cupric silver stain, erythropoietic protoporphyria, motor neuron
Introduction
In mammals and most organisms, iron is indispensable because of its essential role in DNA synthesis, mitochondrial respiration, hemoglobin synthesis and formation of iron sulfur clusters. Iron is a transition metal with two stable oxidation states, +2 (Ferrous, Fe2+) and +3 (Ferric, Fe3+). Since the redox potential of aqueous Fe3+/Fe2+ system (0.77 V) is neither too high, nor too low, conversion from one oxidation state of iron to the other does not require much energy. Thus, iron has a unique ability to act both as an electron acceptor and an electron donor, making it indispensible for many biological reactions involving electron transfer and oxygen transport.
Maintenance of iron homeostasis is extremely vital. When cellular iron concentrations are low, the functions of numerous iron proteins are impaired, causing diseases such as anemia and cognitive deficits that affect millions of people worldwide. On the contrary, excess cellular iron catalyzes the formation of reactive oxygen species that are associated with diseases like hemochromatosis. In addition, excess iron in the brain is associated with diseases like Alzheimer’s, Parkinson’s and Friedreich’s ataxia (Zecca et al., 2004). Thus, organisms and cells must tightly regulate iron metabolism to ensure that sufficient, but not excess, iron is supplied to heme, the iron-sulfur prosthetic groups of mitochondrial respiratory chain complexes, and other cellular iron proteins. This elegant regulation of iron is largely orchestrated at the cellular level by two iron regulatory proteins, IRP1 and IRP2, and at the systemic level by iron regulatory hormone, hepcidin (reviewed in Rouault, 2006, 2013; Hentze et al., 2010; Anderson et al., 2012; Ganz et al., 2012; Wilkinson et al., 2014; Zhang et al, 2014).
Cellular iron metabolism
Iron absorption in the enterocyte
Dietary ferric iron is first reduced in duodenum by the ferrireductase, duodenal cytochrome B (DcytB) (McKie et al., 2001; Choi et al., 2012). The divalent ferrous iron is then transported by a membrane iron transporter, divalent metal transporter 1 (DMT1) into the enterocyte (Gunshin et al., 1997). Internalized ferrous iron either remains accessible in a labile iron pool (LIP) by binding to hitherto unknown molecules, is stored in the iron storage protein ferritin, or exits the enterocyte through the basolateral iron exporter, ferroportin 1 (FPN1) (Abboud et al. 2000; Donovan et al., 2000; McKie at al., 2000). Exported ferrous iron is then oxidized by hephaestin to ferric iron (Vulpe et al., 1999) which binds to transferrin (Tf), and the resulting diferric transferrin complex (diferric-Tf) circulates through the plasma to deliver iron to the tissues (Fig. 1).
Fig. 1.

Iron uptake and systemic distribution. Dietary iron, predominantly in the form of Fe3+ is absorbed in the enterocyte through a concerted action between DcytB, which reduces Fe3+ to Fe2+, and DMT1, which imports this Fe2+ into the enterocyte. After internalization, Fe2+ passes through a poorly characterized labile iron pool, from which it is stored in ferritin or exported across the basolateral membrane by FPN1. Exported Fe2+ is then oxidized by the membrane-bound ferroxidase, hephaestin, to Fe3+ which binds to Tf, and the diferric transferrin complex then circulates through the plasma. This holo-Tf binds to TfR1 on the plasma membrane of most cells, and the resulting complex is endocytosed. Fe3+ is released from the complex in the acidic environment of endosome, and is reduced by Steap3 to Fe2+ which is transported by DMT1 into cytosol. Fe2+ is utilized by several cellular organelles, or stored in ferritin, and unused Fe2+ is exported out by FPN1. This Fe2+ is then oxidized by hephaestin or ceruloplasmin to Fe3+ which then binds to Tf and is recycled back to plasma.
Iron transport
In most tissues, the circulating pool of diferric-Tf serves as the major source of iron. This diferric-Tf binds to transferrin receptor 1 (TfR1) on the plasma membrane, and the resulting diferric-Tf -TfR1 complex internalizes into the endosome where V-ATPase-mediated acidification causes conformational changes in Tf and TfR1, facilitating release of ferric iron from diferric-Tf. Ferric iron is reduced to ferrous iron by the endosomal ferrireductase known as six-transmembrane epithelial antigen of the prostate 3 (Steap3) (Ohgami et al., 2005). Ferrous iron is then transported by DMT1 into the cytosol (Fleming et al., 1998). The apo-Tf -TfR1 complex is unstable because apo-Tf has a very low affinity for TfR1. Thus, after the release of iron, both Tf and TfR1 recycle back to the cell surface. DMT1 can also directly transport non-transferrin-bound iron (NTBI) into cells, especially in conditions of hemochromatosis and hemolytic anemia when serum iron concentrations exceed the binding ability of Tf (Chua et al., 2004; Sarkar 1970). ZIP14, a member of the SLC39A zinc transporter family, also transports NTBI in iron overload conditions (Liuzzi et al., 2006; Gao et al., 2008; Bishop et al., 2010; Pinilla-Tenas et al., 2011). In the cytosol, iron is incorporated into iron-proteins or transported to cellular organelles, and excess iron is sequestered and stored in the iron storage protein, ferritin. The iron that is not used or stored, is exported by the iron exporter, FPN1 into plasma where it is oxidized to ferric iron by ceruloplasmin or hephaestin (Wolf et al., 2006; Vulpe et al., 1999). Tf binds ferric iron and circulates iron to other cells and tissues (Fig. 1).
Regulation of cellular iron metabolism by IRPs
Cells maintain optimum cytosolic iron levels by regulating expression of the iron import protein TfR1, the iron transport protein, DMT1, the iron storage protein, ferritin, and the iron export protein, FPN1. The expression levels of TfR1, DMT1, ferritin and FPN1 are post-transcriptionally regulated by binding of IRP1 or IRP2 to IREs in the transcripts that encode these iron metabolism proteins.
IRP1 and IRP2 are homologous proteins with 56% sequence identity (Pantopoulos, 2004). IRP2 has an additional cysteine-rich 73 amino acid domain with unknown function (Bourdon et al., 2003; Wang et al, 2004). Both IRP1 and IRP2 are expressed ubiquitously. The expression of Irp1 is dominant in kidney, liver and brown fat, whereas the expression of Irp2 is dominant in the central nervous system (Meyron-Holtz et al., 2004). The activities of these iron regulatory proteins are also regulated by iron, but through different mechanisms (Rouault, 2006).
IRP1 is a bifunctional protein that exists in an equilibrium between the [4Fe-4S] containing holo-form which has cytosolic aconitase activity, and its apo-form which binds to IREs. At high cellular iron concentrations, this equilibrium shifts towards cytosolic aconitase, and at low cellular iron levels, the equilibrium shifts towards the IRE-binding form. Thus, iron changes the IRE-binding activity of IRP1, but usually not the protein concentration. In contrast, at high iron concentrations, IRP2 undergoes proteasomal degradation by an E3 ubiquitin ligase complex that contains an F-box protein, FBXL5, which is activated when iron and oxygen bind to a hemerythrin domain in FBXL5 (Salahudeen et al., 2009; Vashisht et al., 2009; reviewed in Rouault, 2009). Therefore, both the activity and protein level of IRP2 decrease when the cells are iron-replete (reviewed in Rouault, 2006).
Thus, at low intracellular iron concentrations, both IRP1 and IRP2 bind to IREs with high affinity. If an IRE is located in the 5′UTR of target mRNAs, binding of iron regulatory proteins inhibits the translation of target mRNAs. Ferritin (both L and H chain transcripts) (Hentze et al., 1987; Theil, 1990), FPN1 (Abboud et al., 2000; Donovan et al., 2000; McKie et al., 2000), mitochondrial aconitase (ACO2) (Kim et al., 1996; Schalinske et al., 1998), erythroid 5-aminolevulinic acid synthase (eALAS) (Dandekar et al., 1991) and hypoxia inducible factor 2α (HIF2α) (Sanchez et al., 2007; Zimmer et al., 2008) have IREs in the 5′UTR of their mRNAs. In contrast, if the IRE is located in the 3′UTR of the target mRNAs, binding of IRPs can increase protein expression by stabilizing the mRNA. TfR1 (Casey et al, 1988; Mullner et al., 1988) and DMT1 (Garrick et al., 2003) have the IREs in the 3′UTRs of their transcripts. Thus, when the cells are iron depleted, expression levels of the mRNAs of the iron import proteins TfR1 and DMT1 increase, whereas translation of the iron storage protein ferritin, the iron export protein FPN1, the first enzyme for heme synthesis eALAS, the protein involved in energy production ACO2, and the hypoxia and erythropoiesis sensor and effector protein HIF2α decreases. These changes lead to an increase in iron absorption and decrease in iron sequestration, thus restoring intracellular iron hemostasis.
Regulation of iron metabolism by hepcidin
While cellular iron hemostasis is regulated by IRP1 and IRP2, systemic iron hemostasis is regulated by hepcidin, a peptide hormone that is mainly secreted by the liver (Ganz, 2013). Circulating hepcidin regulates the expression of FPN1 on the plasma membrane by binding to it and inducing its internalization and degradation (Nemeth et al., 2004). Thus, overproduction of hepcidin causes reduction of iron influx into blood, leading to hypoferremia and anemia of inflamation, and deficiency of hepcidin causes iron overload, leading to hemochromatosis (Ganz and Nemeth, 2011, 2012).
Ablation of Irp1 and Irp2 in mice
To evaluate the physiological significance of Irp1 and Irp2, mice with ablations of Irp1 (Irp1−/−) or Irp2 (Irp2−/−) were created. Although life expectancies of these Irp1−/− and Irp2−/− mice at standard conditions were not remarkably different from those of wild type (WT) animals (Ghosh et al., 2006, 2013), the double knockouts, Irp1−/−Irp2−/− mice, did not survive through the blastocyst stage (Smith et al., 2006), highlighting the physiological significance of these iron regulatory proteins. Early death (within 1 month) of conditional knockout mice that lacked both Irp1 and Irp2 in intestine or in hepatocytes (Galy et al, 2008; 2010) further established the essential physiological role of these iron regulatory proteins. The fact that the life-span and fertility of either global Irp1−/− or Irp2−/− mice do not differ remarkably from the WT animals (Ghosh et al, 2006, 2013; Meyron-Holtz et al, 2004a) indicated that each Irp can compensate for the loss of the other, at least partially, and these Irps are functionally redundant.
Irp1 knockout mice
The physiological significance of Irp1 remained elusive in the initial years of research since Irp1−/− mice did not show overt phenotypes. Misregulation of TfR1 and ferritin was initially observed only in kidney and brown fat, the two tissues in which the expression level of Irp1 exceeded that of Irp2 (Meyron-Holtz et al., 2004a, b).
However, three papers were published in 2013 by the Rouault, Eisenstein and Pantopoulos labs, and all these three groups reported that Irp1−/− mice develop polycythemia (Ghosh et al., 2013; Anderson et al., 2013; Wilkinson et al., 2013). HIF2α has an IRE that is located in the 5′UTR, and binding of iron regulatory proteins with this IRE inhibits the translation of HIF2α. Since Irp1 is generally more abundant in kidney than Irp2, deletion of Irp1 enhanced HIF2α protein expression in the kidney lysates of Irp1−/− mice, and this led to increased erythropoietin (EPO) expression causing polycythemia and concomitant tissue iron deficiency. Derepression of HIF2α was particularly apparent in renal interstitial fibroblasts, the cells that sense oxygen tension and accordingly synthesize EPO. Interestingly, when Irp1−/− mice were fed with a low iron diet, their hematocrit increased further to 60%, compared to the normal level of 45% in WT animals, serum EPO levels increased seven-fold, and the mice died prematurely at an average age of 10 months due to abdominal hemorrhages (Ghosh et al., 2013). The observed polycythemia in Irp1−/− mice and its exacerbation by a low iron diet establishes an important and crucial role of Irp1 in regulation of systemic iron homeostasis and erythropoiesis.
In addition to polycythemia, Irp1−/− mice developed pulmonary hypertension and cardiac hypertrophy (Ghosh et al., 2013), two serious human diseases for which the pathogenesis is not yet clear in humans. Both mRNA and protein levels of endothelin-1, another transcription target of HIF2α, were increased about 2-fold in lungs of Irp1−/− mice, and HIF2α protein levels were significantly increased in primary pulmonary endothelial cells isolated from Irp1−/− mice compared to those isolated from WT controls. Interestingly, although the iron-deficient diet increased EPO expression and exacerbated the polycythemia of Irp1−/− mice, probably due to stabilization of HIF2α, it did not change endothelin-1 levels, and did not exacerbate pulmonary hypertension in Irp1−/− mice (Ghosh et al., 2013). Similarly, when Irp1−/− mice were placed in hypoxia chambers (10% O2) for 23 days, the hematocrits increased dramatically, but there was no further increase in right ventricular pressure, which increases in response to pulmonary hypertension (Ghosh et al., unpublished data) emphasizing the difference, at least in part, between the molecular pathophysiology of polycythemia vs. pulmonary hypertension.
The first Irp2 knockout mouse model
Iron misregulation and neurodegeneration
The first study on Irp2 knockout mice was reported by the Rouault lab in 2001 (LaVaute et al., 2001). Global Irp2−/− mice were generated by inserting a PGK-neomycin gene into exon 3/4 of the Irp2 gene. These Irp2−/− mice developed progressive neurodegeneration after 6 months of age characterized initially by abnormal gait and subtle kyphosis followed by gradual onset of pronounced gait difficulties, tremor, and postural abnormalities. When these mice were subjected to hanging wire tests, their balance and grip strength were impaired compared to WT controls. These phenotypes became more pronounced in the animals that were bred to lack one copy of Irp1 in addition to lacking both copies of Irp2 (Irp1+/−Irp2−/−) (Smith et al., 2004), suggesting that there was a dose-dependent effect.
Increased iron accumulation in different parts of the brain was observed when the neuropathology of these Irp2−/− mice was evaluated (Fig. 2). Since Irp2 is the predominant iron regulatory protein in many cells of the brain, loss of Irp2, but not Irp1, caused increased levels of iron deposition in axons and oligodendrocytes (Fig. 2). Amino cupric silver staining of the brain sections of Irp2−/− mice, but not Irp1−/− animals, showed axonal degeneration in comparison to WT controls, and the signs of axonal degeneration were more prominent in Irp1+/−Irp2−/− animals (Fig. 3). High ferritin expression and reduced levels of TfR1 were observed in tissue lysates from the cerebellum and other tissues of these mice, and ferritin iron overload was detected in the magnetic resonance imaging (MRI) of Irp2−/− mouse brain (Grabill et al., 2003). Total iron levels in the liver and duodenal mucosa were higher in 8–14 months old Irp2−/− mice compared with WT controls (Table 1), and Prussian blue staining showed more ferric iron in epithelial cells of the duodenal mucosa. Serum ferritin levels were elevated (Table 2), and ferritin expression levels in epithelial cells of the distal villi of 12 month-old Irp2−/− mice were higher compared to WT animals (LaVaute et al., 2001). Misregulation of ferritin and TfR1 was even more pronounced in Irp1+/−Irp2−/− animals (Smith et al., 2004). More than three-fold increases of ferritin were directly observed in the 3D distribution of ferritin by electron tomography in different regions of brains of Irp1+/−Irp2−/− mice, and structural damage was observed within the axons (Zhang et al., 2005). The Kirsch group found that both loosely bound iron and non-heme iron increased during development between 6 and 12 weeks of age in Irp2−/− mice, whereas the reverse was observed for WT mice (Magaki et al., 2007).
Fig. 2.

Increased iron staining in the cerebellums of Irp2 deficient mice. Perls’DAB stain of brain sections of WT (Irp1+/+Irp2+/+), Irp1+/+Irp2−/−, Irp1+/−Irp2−/−, Irp1−/−Irp2+/+, Irp1−/− Irp2+/− mice showed iron accumulation in different parts of Irp1+/+Irp2−/− and Irp1+/−Irp2−/− mouse brains. One-year old mice were perfused with 4% paraformaldehyde/PBS. Gelatin-embedded brains were sectioned, and sequential sections were stained with DAB as described (LaVaute et al., 2001; Smith et al., 2004). Abbreviations: CDN - Cerebellar deep nuclei, W – White matter, G – Granular cell layer, P – Purkinje cell layer, M – Molecular layer, the blue dotted lines represent the Purkinje cell layers of cerebellar folia.
Fig. 3.

Evidence for axonal degeneration in cerebellum of Irp2 deficient mice. Amino cupric silver stains of brain sections of WT (Irp1+/+Irp2+/+), Irp1+/+Irp2−/−, Irp1+/−Irp2−/−, Irp1−/−Irp2+/+, Irp1−/−Irp2+/− mice showed axonal degeneration in white matter tracts of Irp1+/+Irp2−/− and Irp1+/−Irp2−/− mouse brains, illustrated here in the cerebellum. One-year old mice were perfused with 4% paraformaldehyde/PBS. Gelatin-embedded brains were sectioned, and sequential sections were stained with amino cupric silver stain for detection of degenerating axons as previously described (LaVaute et al., 2001; Smith et al., 2004). Note the black strands, which represent axons that have lost integrity and allowed silver to stain their neurofilaments, particularly in the Irp1+/− Irp2−/− animals. Abbrebiations: CDN - Cerebellar deep nuclei, W – White matter, G – Granular cell layer, P – Purkinje cell layer, M – Molecular layer.
Table 1.
Tissue iron reported for WT and Irp2−/− mice
| Cooperman et al (40–64 weeks old) | Galy et al (8–10 Weeks old) | Zumbrennen-Bullough et al (49–75 wks old) | |||||
|---|---|---|---|---|---|---|---|
| Total iron in ppm (ICP-MS) | Non-heme iron in mg/ga | Total iron in μg/mg (ICP-OES)b | |||||
| WT | Irp2−/− | Irp1+/− Irp2−/− | WT | Irp2−/− | WT males | Irp2−/− males | |
| Brain | 102 ± 13 | 108 ± 5 | 93 ± 13 | 157 ± 11 | 148 ± 13 | 1.98 ± 0.08 | 1.84 ± 0.07 |
|
| |||||||
| Spleen | 1852 ± 697 | 1554 ± 388 | 1841 ± 635 | 2191 ± 191 | 1331 ± 122*** | 15.4 ± 2.4 | 12.5 ± 1.1 |
|
| |||||||
| Liver | 360 ± 72 | 748 ± 204 | 1279 ± 342* | 195 ± 9 | 300 ± 15*** | 2.77 ± 0.21 | 5.81 ± 0.39*** |
|
| |||||||
| Duodenum | NR | NR | NR | 571 ± 41 | 1058 ± 113*** | 6.01 ± 1.68 | 15.4 ± 1.8* |
|
| |||||||
| Heart | NR | NR | NR | NR | NR | 5.49 ± 0.05 | 4.92 ± 0.13* |
|
| |||||||
| Kidney | NR | NR | NR | NR | NR | 2.25 ± 0.10 | 3.37 ± 0.15*** |
|
| |||||||
| Lung | NR | NR | NR | NR | NR | 3.01 ± 0.40 | 2.80 ± 0.35 |
p<0.05,
p <0.01,
p<0.001,
NR = Not Reported
The unit for non-heme iron in the paper published by Galy et al. (2005) will probably be μg/g instead of mg/g.
Values for total iron in the paper published by Zumbrennen-Bullough et al. are roughly 10-fold higher than the values obtained by Cooperman et al.
Table 2.
Serum iron parameters reported for WT and Irp2−/− mice
| Cooperman et al (40–64 weeks old) | Galy et al (8–10 Weeks old) | Zumbrennen-Bullough (49–75 wks old) | |||||
|---|---|---|---|---|---|---|---|
| WT | Irp2−/− | Irp1+/− Irp2−/− | WT | Irp2−/− | WT males | Irp2−/− males | |
| Serum iron (μg/dL) | 118 ± 16 | 121 ± 26 | 184 ± 25** | 212 ± 18 | 212 ± 14 | 140 ± 7 | 139 ± 8 |
|
| |||||||
| Serum Ferritin (ng/ml) | 182 ± 61 | 1283 ±514* | 1547 ± 562** | NR | NR | 58 ± 13 | 219 ± 7*** |
|
| |||||||
| Transferrin (mg/dL) | NR | NR | NR | NR | NR | 168 ± 6 | 159 ± 3 |
|
| |||||||
| Transferrin saturation (%) | 39 ± 6 | 44 ± 11 | 48 ± 10** | 50.6 ± 3.0 | 51.3 ± 1.9 | 32.7 ± 1.0 | 35.0 ± 1.4 |
|
| |||||||
| TIBC (μg/dL) | 305 ± 30 | 278 ± 40 | 389 ± 39*** | 416 ± 20 | 412 ± 20 | NR | NR |
|
| |||||||
| UIBC (μg/dL) | NR | NR | NR | NR | NR | 287 ± 14 | 258 ± 6 |
p<0.05,
p <0.01,
p<0.001,
NR = Not Reported
Myelin dense bodies (MDB), which develop when axons degenerate and the myelin sheaths collapse to fill the region formerly occupied by an intact axon, accumulated in the ventral and lateral white matter in spinal cords of these Irp2−/− mice (Jeong et al., 2011). Loss of Irp2 caused lower motor neuronal degeneration in these mice with more swollen axons and fewer myelinated fibers in the ventral nerve roots. In addition, motor neurons of these mice showed a number of hallmarks of retrograde cell body degeneration including distorted shapes, with rounding of cell bodies and loss of multipolarity. Since the lower motor neurons are involved in control of movements, lower motor degeneration was consistent with the hind limb weakness and the abnormal gait observed in these mice. Irp2−/− mice likely also had abnormalities in the upper motor neurons. Histochemical stains revealed some chromatolysis of upper motor neurons, a sign of nuclear dysfunction, but this observation needs more firm support. Increased anti-ubiquitin immunoreactivity in the motor neuronal cell bodies of these mice also indicated stress in the neurons. Loss of Irp2 caused decreased TfR1 and increased ferritin expression in motor neurons. The resulting functional iron deficiency led to decreased activities of iron-sulfur containing Complex I and Complex II (but not Complex IV which does not contain an Fe-S cluster). As a result, Irp2−/− mice had decreased mitochondrial function and swollen mitochondria (Jeong et al., 2011). Intact mitochondria are particularly important in axons, because they generate the energy that drives pumps important in axonal function. Our hypothetical model to explain axonal degeneration of Irp2 deficient mice is shown in Fig. 4.
Fig. 4.

Hypothetical model to explain axonal degeneration of Irp2 deficient mice. IRP2 is the predominant IRP protein in many neurons, where it stabilizes TfR1 mRNA and represses ferritin translation, thus maintaining intracellular iron homeostasis. IRP2 deficiency decreases iron absorption by reducing TfR1 expression and increases iron sequestration by enhancing ferritin translation, resulting in a deficiency of available cytosolic iron, even though much iron can be sequestered in ferritin, leading to “functional iron deficiency”. Deficiency of bioavailable iron impairs the synthesis of iron-sulfur clusters, heme and other iron proteins, which compromises mitochondrial function. Consequently, mitochondria cannot provide enough ATP to support activity of ion channels and pumps in axons, leading to the potential loss of electrical potential and inability to maintain axonal integrity and function.
To characterize the dopamine (DA) system in these Irp2−/− mice, Salvatore et al harvested CNS tissue from 16–19 month-old WT and Irp2−/− animals, and observed a modest decrease of tyrosine hydroxylase (TH) and an increase in phosphorylation of serine 40 of TH in both the dorsal and ventral striata of Irp2−/− mice in comparison to those of WT controls (Salvatore et al., 2005). These researchers also found a moderate decrease in dopamine transporter (DAT) and vesicular monoamine transporter (VMAT2) levels and a mild decrease in DA levels in the ventral striatum, but not in the dorsal striatum of Irp2−/− mice, supporting that misregulation of iron in Irp2−/− animals affects DA regulation in the striatum. Chen et al (2010) observed 8.4–11 fold increases in striatal ferritin in Irp2−/− mice, but not in Irp1−/− mice in comparison to the WT animals 3 days after steriotactic injection of artificial cerebrospinal fluid (CSF) or autologous blood, and they suggested that the IRE-binding activity of Irp2 reduces ferritin expression in the striatum after intracerebral hemorrhage (ICH). This is consistent with the observation made by Regan et al (2008) in cortical cell cultures that Irp2−/− neurons with high ferritin expression are protected from iron-dependent injuries produced by hemoglobin and hydrogen peroxide.
Partial protection of Irp2−/− mice from neuronal abnormality through pharmacological activation of Irp1 by Tempol
Oral treatment with Tempol (4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl), a membrane-permeable stable nitroxide, prevented the symptoms of neurodegeneration of these Irp2−/− mice, and the Tempol-treated Irp2−/− axons were found to be partially spared from degeneration (Ghosh et al., 2008; Jeong et al., 2011). Tempol converted Irp1 from the cytosolic aconitase to the IRE-binding form of the protein, resulting in stabilization of the TfR1 transcript and repression of ferritin systhesis. Thus, Tempol treatment restored iron homeostasis in the cerebellums, brain stem and forebrains of these mice (Ghosh et al., 2008). Moreover, Tempol treatment restored the Complex I activity of these Irp2−/− mice back almost to the WT level, suggesting that the motor neuronal degeneration was probably caused by functional iron starvation, and that Tempol exerted its therapeutic effect partly by increasing TfR1 expression (Jeong et al., 2011).
Interestingly, Tempol treatment did not prevent disease progression in Irp1+/−Irp2−/− mice, suggesting that Tempol protects Irp2−/− animals by recruiting the latent IRE-binding activity of Irp1, and activation of one allele of Irp1 is not enough to compensate for the loss of Irp2 activity. These findings strongly suggested that the observed neurodegeneration of these Irp2−/− mice results from the loss of most cellular IRE-binding activity in affected cells (Ghosh et al., 2008). The neurodegenerative symptoms of Irp1+/−Irp2−/− mice were also improved by deletion of one allele of ferritin-H chain (Jeong et al., 2011), which would be expected to reduce ferritin sequestration of iron and increase bioavailability of intracellular iron.
Anemia and erythropoietic protoporphyria
These Irp2−/− mice developed microcytic anemia which was more severe in Irp1+/−Irp2−/− animals (Cooperman et al., 2005). Levels of hemoglobin, hematocrit and mean cell volume (MCV) were 25–30% lower in these Irp2−/− mice in comparison to WT animals, but RBC counts remained unchanged (Table 3). Tf saturations were normal in these mice, distinguishing the phenotype from iron deficiency anemia (Table 2). Serum ferritin levels were more than 8-fold increased, and TfR1 levels were significantly decreased in erythroid precursors in Irp2−/− mice, and these effects were more pronounced in Irp1+/−Irp2−/− animals (Cooperman et al., 2005). Although Tempol treatment prevented symptoms of neurodegeneration, Tempol did not correct the anemia in Irp2−/− mice (Ghosh et al., 2008). To determine why Tempol failed to improve the anemia in Irp2−/− mice, the authors isolated erythroblasts from WT mice, and observed that erythroblasts have much lower relative Irp1 activity compared to brain. In addition, β-mercaptoethanol, which converts Irp1 from the cytosolic aconitase to IRE binding form, did not increase the IRE-binding activity of Irp1 in erythroblasts, although it increased IRE binding about 5-fold in brain lysates, suggesting that erythroblasts lack a significant amount of Irp1 in the cytosolic aconitase form that can be converted to the IRE binding form by treatment with Tempol (Ghosh et al., 2008).
Table 3.
Hematological parameters reported for WT and Irp2−/− mice
| Cooperman et al | Galy et al | Zumbrennen-Bullough et al | |||||||
|---|---|---|---|---|---|---|---|---|---|
| WT | Irp2−/− | Irp1+/− Irp2−/− | WT males | Irp2−/− males | WT females | Irp2−/− females | WT males | Irp2−/− males | |
| RBC (106/μL) | 9.0 ± 0.3 | 9.0 ± 0.3 | 8.7 ± 0.6 | 8.4 ± 0.3 | 8.0 ± 0.2 | 7.5 ± 0.2 | 7.3 ± 0.2 | 9.6 ± 0.2 | 9.5± 0.2 |
|
| |||||||||
| Hemoglobin (g/dL) | 13.2 ± 0.9 | 11.4 ± 0.5** | 10.1 ± 0.8*** | 13.8 ± 0.3 | 11.5 ± 0.3*** | 12.8 ± 0.6 | 10.8 ± 0.3** | 14.3 ± 0.2 | 12.3 ± 0.2** |
|
| |||||||||
| Hematocrit (%) | 45.2 ± 1.2 | 36.0 ± 1.3*** | 32.9 ± 2.2*** | 45 ± 1 | 37 ± 1*** | 39 ± 1 | 34 ± 1** | 44.2 ± 0.6 | 38.0 ± 0.6** |
|
| |||||||||
| MCV (fL) | 50.3 ± 0.9 | 40.2 ± 1.2*** | 37.9 ± 1.3*** | 53.0 ± 0.6 | 46.8 ± 0.75*** | 51.1 ± 0.6 | 47.2 ± 0.5*** | 46.3 ± 0.4 | 40.9 ± 0.2** |
|
| |||||||||
| MCHC (g/dL) | 29.3 ± 2.3 | 31.5 ± 1.0 | 30.1 ± 1.0 | NR | NR | NR | NR | 32.4 ± 0.2 | 31.5 ± 0.1** |
|
| |||||||||
| MCH (pg) | NR | NR | NR | NR | NR | NR | NR | 14.9 ± 0.2 | 12.9 ± 0.1** |
|
| |||||||||
| WBC (103/μL) | NR | NR | NR | 6.32 ± 0.64 | 6.99 ± 0.88 | 5.04 ± 0.42 | 5.32 ± 0.40 | 7.8 ± 0.7 | 7.3 ± 0.4 |
|
| |||||||||
| PLT (103/μL) | NR | NR | NR | NR | NR | NR | NR | 1311 ± 36 | 1072 ± 32** |
|
| |||||||||
| RDW (% of MCV) | NR | NR | NR | NR | NR | NR | NR | 15.4 ± 0.2 | 16.0 ± 0.1* |
p<0.05,
p <0.01,
p<0.001,
NR = Not Reported
Interestingly, these Irp2−/− mice also developed erythropoietic protoporphyria. Alas2 (5-aminolevulinic acid synthase 2), a transcript which has an IRE in the 5′ end, was highly overexpressed in Irp2−/− mice. Since Alas2 encodes the rate-limiting enzyme in the heme biosynthetic pathway, levels of the downstream intermediate, protoporphyrin IX, increased tremendously in red cells of these mice (Cooperman et al., 2005). Iron deficiency would then interfere with insertion of iron into protoporphyrin IX to generate heme.
A second Irp2 knockout mouse model
In 2004, three years after the Rouault group published their first study on Irp2−/− mice, the Hentze lab (Galy et al., 2004) reported a second Irp2−/− mouse model that was generated differently by using 129P2/OlaHsd ES cell line and backcrossing to C57Bl/6J mice. As was observed by the Rouault group, these researchers also found decreased TfR1 expression and increased ferritin expression in brain lysates (Galy et al., 2006). These Irp2−/− mice displayed significantly lower self-grooming activity and a tendency towards reduced rearing compared to WT animals, suggesting a trend toward decreased vertical locomotor activity. In addition, these Irp2−/− mice showed significant compromise in motor ability when tested on a rotarod, indicating that these mice had impaired motor coordination and strength. However, Hentze and his colleages did not find any iron deposits in the brain of their Irp2−/− mice compared to WT controls. This group also did not detect any tremors, gait abnormalities or postural problems in Irp2−/− animals, and they concluded that their Irp2−/− mice displayed only a mild clinical phenotype without pathological signs of neurodegeneration (Galy et al., 2006).
However, in agreement with the observation made by Rouault group in their mouse model, Hentze and co-workers found that their Irp2−/− mice had a mild microcytic anemia with decreased serum hematocrit and hemoglobin levels (Table 3). These Irp2−/− animals had unchanged serum iron and transferrin saturation, indicating that the microcytosis was not due to systemic iron deficiency (Table 2). The observed reduction of TfR1 expression was also suggested to cause microcytosis in Irp2−/− mice (Galy et al., 2005).
A third Irp2 knockout mouse model
The third total body Irp2−/− mouse model reported by the Leibold group in 2014 was generated by inserting a self-excision cassette containing neomycin linked to Cre-recombinase into exon 3 of the mouse Irp2 gene (Zumbrennen-Bullough et al., 2014). These Irp2−/− mice showed reduced locomotion, speed of movement and vertical exploratory activities. Rotarod performances of old Irp2−/− mice (45–63 weeks), but not young ones (20 weeks), were slightly impaired indicating that neuromuscular compromise could be a progressive disorder. In addition, they observed nociceptive heat tolerance in these Irp2−/− animals, indicating that sensory impairments were present. However, these mice did not show tremors, kyphosis or abnormal gait, even at 45–63 weeks of age (Zumbrennen-Bullough et al., 2014).
As reported by Rouault group and Hentze group, these authors also did not see any significant change in total brain iron content in their Irp2−/− mice in comparison to WT controls (Table 1). Moreover, in agreement with the Rouault group, the Leibold group found marked iron deposition in various regions of the brain including in the cortex, thalamus and cerebellum of their Irp2−/− mice. They also observed iron accumulation in axonal tracts, cerebellar white matter and oligodendrocytes, and detected significant iron deposition in the cerebral cortex and corpus callosum of Irp2−/− mice. These researchers concluded that abnormal iron metabolism in their Irp2−/− mice was associated with mild neurological and behavioral impairments (Zumbrennen-Bullough et al., 2014).
Similar to the Rouault lab mice and Hentze lab mice, the Irp2−/− mice of the Leibold lab also had significantly lower hemoglobin and hematocrit levels, unchanged RBC counts, transferrin saturation and serum iron levels, and increased serum ferritin levels (Tables 2, 3). As observed by the Rouault group, these researchers also found dramatic increases in protoporphyrin IX level in serum, as well as in the liver and bile duct of these Irp2−/− animals, where protoporphyin deposits were visible at autopsy (Zumbrennen-Bullough et al., 2014).
Notably, both the Rouault and Leibold groups concluded that certain cells of the CNS showed histological abnormalities. The Rouault group found that lower motor neuron counts were low, and axon tracts were degenerating throughout the brain (Jeong et al., 2011; Smith et al., 2004). The Leibold group reported that iron was reduced in Purkinje neurons and in CA1 pyramidal neurons in Irp2−/− mice, but they did not perform silver stains, and could not detect any degeneration, though their mice showed neuromuscular impairments (Zumbrennen-Bullough et al., 2014). Both the Rouault and Leibold groups concluded that Irp2−/− animals are adversely affected because they develop functional iron deficiency. As a result of synthesizing too much ferritin, they sequester iron rather than making it available for mitochondria and cytosolic and nuclear proteins. Moreover, they express less TfR1, and reduced iron uptake further exacerbates the problem of low iron availability. Axons are particularly dependent on good mitochondrial function, because mitochondria provide the ATP that energizes pump activities at the nodes of Ranvier along the axon (Fig. 4). Without functional healthy mitochondria, it is not surprising that axons cannot maintain their integrity. Loss of axonal integrity appears to be the first sign of neuronal problems in Irp2−/− mice (LaVaute et al., 2001), and it manifests when silver penetrates the degenerating axonal membrane and binds to the negatively charged neurofilaments that give shape to axons (Fig. 4).
Tissue specific ablation of Irp2 in mouse
The Hentze group made tissue-specific ablations of Irp2 in enterocytes, hepatocytes or macrophages using Cre/Lox technology to evaluate the effect of local Irp2 deficiency (Ferring-Appel et al., 2009). Red blood cell and plasma iron parameters in all three of these enterocyte-, hepatocyte-, or macrophage-specific Irp2 deficient mice were normal. In contrast to enterocyte- and hepatocyte-specific Irp2 deficiency, selective Irp2 ablation in macrophages did not recapitulate the splenic iron deficiency phenotype observed in global Irp2−/− mice. They concluded that splenic iron misregulation probably resulted from deletion of Irp2 in other cell types in global Irp2−/− mice (Ferring-Appel et al., 2009).
Tissue specific ablation of both Irp1 and Irp2 in mouse
Since Irp1 and Irp2 double knockout mice are embryonically lethal, Hentze and coworkers (Galy et al., 2008; 2010; 2013) made mice lacking expression of both Irp1 and Irp2 in specific tissues using Cre/Lox technology to study the function of the Irp/IRE regulatory network in vivo. For simplicity, we will call these mice tissue specific double knockouts.
Intestine specific double knockout mice died at 4 weeks of age. Two weeks-old mice showed weakness and signs of dehydration with wrinkled skin and sparse fur. Interestingly, serum iron levels and transferrin saturations were normal. However, as expected, protein and mRNA levels of TfR1 were decreased, and those of ferritin H and ferritin L were increased. Ferroportin protein levels dramatically increased in these mice, although Fpn1 mRNA levels remained normal, and hepcidin expression levels increased (Galy et al., 2008).
Liver specific double knockout mice died within 8 to 12 days after birth due to liver failure. Eight day-old mice had mitochondrial iron deficiency and suffered from mitochondrial dysfunction. Surprisingly, hematological and blood parameters were normal in these mice, although hepcidin expression in the livers was 30% down-regulated in comparison to the WT controls. As expected, the activities of the iron-sulfur cluster containing electron transport chain complexes I, II and III were decreased, and that of complex IV which does not contain a [Fe-S] cluster, remained unchanged. Activities of citric acid cycle enzymes were also reduced. Non-heme iron levels decreased, post-transcriptional expression of ferritin (both H and L) increased, and protein and mRNA levels of TfR1 were reduced in the liver of these mice. In addition, the activity of ferrochelatase, the enzyme that catalyzes iron insertion into protoporphyrin to make heme was also reduced, resulting in decreased hepatic heme levels. Interestingly, duodenal iron contents and ferritin levels remained unaffected in the liver-specific double knockout mice, indicating that dysregulated iron homeostasis in the duodenum of global Irp2−/− mice is probably cell autonomous (Galy et al., 2010).
Duodenal enterocyte specific adult double knockout mice generated by ligand induced deletion of both Irp1 and Irp2 are viable (Galy et al., 2013). Although expression levels of ferritin and ferroportin of these mice were high and TfR1 expression was low as expected, DMT1 was upregulated in the intestine. The stimulation of DMT1 was possibly mediated by HIF2α, the translational derepression of which was observed in Irp1−/− mice (Ghosh et al., 2013; Anderson et al., 2013; Wilkinson and Pantapoulos, 2013). Even though there was upregulation of both the apical and basolateral iron transporters DMT1 and FPN1, these enterocyte-specific adult double knockout mice displayed reduced iron absorption. This has been explained by ferritin-mediated “mucosal block” which prevents iron delivery to the plasma (Galy et al., 2013). In an intestine specific ferritin H knockout mouse model, ferritin H has been shown to control iron absorption (Vanoaica et al., 2010).
Mouse model for ablation of Fbxl5 and Irp2
The importance of Irp2 in cellular iron metabolism was further established by F-box and leucine-rich repeat protein 5 (Fbxl5) deficient mice. Fbxl5 is an E3 ubiquitin ligase that contains an iron- and oxygen-binding hemerythrin domain, senses iron and oxygen availability, and facilitates proteasomal degradation of Irp2 in iron-replete cells. Deletion of Fbxl5 was embryonically lethal, and was associated with excessive iron accumulation and oxidative stress in embryos. This embryonic lethality of Fbxl5−/− mouse was prevented by additional ablation of Irp2, but not Irp1, suggesting that impaired Irp2 degradation was primarily responsible for the death of Fbxl5−/− animals. Fbxl5−/−Irp2−/− mice developed normally, and were fertile and phenotypically indistinguishable from WT littermates. Iron metabolism in Fbxl5−/−Irp2−/− mice was similar to that in Irp2−/− mice with indistinguishable hematologic parameters (Moroishi et al., 2011; Ruiz et al., 2014).
Iron regulatory proteins and Alzheimer disease
Studies from several research groups have implicated the involvement of Irps in Alzheimer’s disease. Smith and colleagues showed abnormal localization of IRP2 in the brain tissue of Alzheimer patients more than a decade ago (Smith et al., 1998). Kirsch and his collaborators reported decreased levels of copper and increased expression of amyloid β precursor protein (AβPP) in the hippocampus of Irp2−/− mice suggesting that IRP2 may have a significant role in Alzheimer’s disease (Mueller et al., 2009). Recently, Rogers and his colleagues found that the amyloid precursor protein (APP) also has an IRE-like RNA stem loop in its 5′ untranslated region, and that IRP1 selectively binds to this APP IRE in human neural cells (Bandyopadhyay et al., 2013, 2014; Cho et al., 2010). High expression of Irp1 in the cortex suggests that Irp1 might play an important role in Alzheimer’s disease.
Conclusions
Irp1 knockout mice
All three research groups that worked with Irp1−/− mice, found that Irp1−/− mice developed polycythemia. In addition, the Rouault lab observed that these mice also developed pulmonary hypertension, and died early of abdominal hemorrhages when they were fed with a low iron diet. All three groups attributed the phenotype to changes in expression of HIF2α, which has a 5′IRE and is regulated by iron regulatory proteins.
Irp2 knockout mice
Three research labs independently made complete Irp2 knockout mice, and discovered that these mice develop microcytic anemia. The Rouault and Leibold groups found erythropoietic protoporphyria in these mice. All three groups observed increased ferritin and decreased TfR1 expression in brain lysates of these mice. However, different observations and conclusions were reached by these groups on whether there was iron deposition and neurodegeneration in the brains of Irp2−/− mice. The Rouault and Leibold groups found marked iron deposition in different parts of the brain, but the Hentze group did not find iron deposits in Irp2−/− mouse brain. The Rouault group reported a progressive neurodegenerative phenotype in their mice, whereas Hentze lab concluded that their Irp2−/− mice show ‘only a mild clinical phenotype without pathological signs of neurodegeneration’, and Leibold group observed mild neurological and behavioral phenotype in their Irp2−/− mice. Genetic background has been suggested as one of the few possible reasons for the observed differences in these three lines of Irp2−/− mice (Galy et al., 2006; Zumbrennen-Bullough et al., 2014). However, the observation of anemia was made in all three of these models, and protoporphyria was reported in the mouse models of Rouault lab and Leibold lab. In addition, Galy et al (2005) did not observe significantly increased ferroportin and DMT1 expression in duodenum of their mixed C57BL6/Sv129 genetic background mice whereas LaVaute et al (2001) found increased FPN1 and DMT1 in duodenum of their Irp2−/− mice of similar background, suggesting that genetic background may not be responsible for the differences observed in these mouse models. The iron content of the foods provided to the mice in Rouault lab, Hentze lab and Leibold lab are 206 mg/kg, 200 mg/kg and 270 mg/kg respectively. This suggests that the differences in the neurological phenotype observed in the mice of these three labs probably were not caused by differences of the iron in the diet.
Different methods of creation of mice using targeting strategies that differed from those of the Rouault group were suggested to be another possible reason for the differences in neurological problems observed in their Irp2−/− mice (Galy et al., 2006). If the neurodegenerative phenotype observed in the Rouault lab mice were not due to ablation of Irp2, loss of one allele of Irp1 would not have worsened neurodegeneration in Irp1+/−Irp2−/− animals. In addition, the observation that activation of Irp1 by Tempol reduced the symptoms of neurodegeneration, restored iron homeostasis in cerebellum, brain stem and forebrain, and restored the Complex I activity of these Irp2−/− mice back almost to the WT level, suggests that more activity of Irp1, the homologous protein of Irp2, can partially compensate for the loss of Irp2. The Hentze group and Leibold group have not made Irp1+/−Irp2−/− mice yet. Generating Irp1+/−Irp2−/− animals using the Irp2−/− mice of the Hentze lab and Leibold labs could help with analysis of phenotypes by using genetic dosage effects to substantiate conclusions.
Why do Irp1−/− mice suffer from polycythemia and pulmonary hypertension, whereas Irp2−/− mice develop neurological problems and anemia?
To understand why Irp1−/− mice suffered from polycythemia and pulmonary hypertension whereas Irp2−/− mice developed neurological impairments, it was important to know whether both Irp1 and Irp2 can regulate HIF2α expression through their binding to the HIF2α IRE. Although Zimmer et al (2008) reported binding of HIF2α IRE to only Irp1, but not Irp2, Sanchez et al (2007) and Ghosh et al (Fig. 5) have found that both Irp1 and Irp2 bind efficiently to HIF2α IRE. Since Irp1 is the major contributor towards IRE-binding in specific cells of kidney and lung tissues, whereas Irp2 is the major contributor towards IRE-binding in neurons, Ghosh et al (2013) concluded that tissue and cell-specific differences in relative expression levels of Irp1 and Irp2 are responsible for different phenotypes of Irp1−/− and Irp2−/− mice.
Fig. 5.
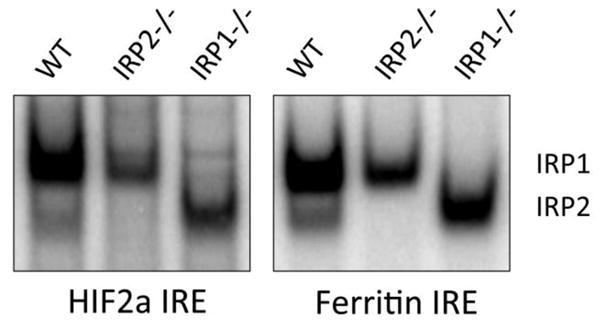
Both Irp1 and Irp2 bind to the HIF2α IRE. Band-shift experiments were done with MEF cells to measure the HIF2α IRE and ferritin IRE binding activities of Irp1 and Irp2 following a published method (Ghosh et al., 2008). HIF2α is highly expressed in brain endothelial cells (Tian et al., 1997).
Possible human diseases from mutation in iron regulatory proteins and future directions
Although clear phenotypes have been observed for both Irp1−/− and Irp2−/− mice, no human patient has yet been identified with mutation in either of the iron regulatory proteins. Since human genomes contain at least 20 loss-of-function mutations per genome (MacArthur et al., 2012), and there are seven billion people in the world, there is a high probability that there are human patients with loss of function mutations in either of the two IRPs, but they are yet to be found. Patients suffering from both polycythemia and pulmonary hypertension should be tested for mutations in IRP1, and when any such patient is detected, iron deficiency should be avoided to prevent exacerbation of the polycythemia. Patients with combined microcytic anemia, erythropoietic protoporphyria and neurological disorders should be tested for mutations in IRP2, and when these patients are found, they could be treated with a drug like Tempol that can activate the IRE binding of IRP1, and can thereby partially compensate for the loss of IRP2.
Acknowledgments
This work was supported by the intramural program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development. We thank Dan Crooks for making the HIF2α IRE.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abboud S, Haile DJ. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. The Journal of biological chemistry. 2000;275:19906–12. doi: 10.1074/jbc.M000713200. [DOI] [PubMed] [Google Scholar]
- Anderson CP, Shen M, Eisenstein RS, Leibold EA. Mammalian iron metabolism and its control by iron regulatory proteins. Biochim Biophys Acta. 2012;1823:1468–1483. doi: 10.1016/j.bbamcr.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson SA, Nizzi CP, et al. The IRP1-HIF-2alpha Axis Coordinates Iron and Oxygen Sensing with Erythropoiesis and Iron Absorption. Cell Metab. 2013;17(2):282–290. doi: 10.1016/j.cmet.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhyay S, Cahill C, et al. Novel 5′ untranslated region directed blockers of iron-regulatory protein-1 dependent amyloid precursor protein translation: implications for down syndrome and Alzheimer’s disease. PLoS One. 2013;8(7):e65978. doi: 10.1371/journal.pone.0065978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhyay S, Rogers JT. Alzheimer’s disease therapeutics targeted to the control of amyloid precursor protein translation: maintenance of brain iron homeostasis. Biochem Pharmacol. 2014;88(4):486–494. doi: 10.1016/j.bcp.2014.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop GM, Scheiber IF, et al. Synergistic accumulation of iron and zinc by cultured astrocytes. J Neural Transm. 2010;117:809–817. doi: 10.1007/s00702-010-0420-9. [DOI] [PubMed] [Google Scholar]
- Bourdon E, et al. The role of endogenous heme synthesis and degradation domain cysteines in cellular iron-dependent degradation of IRP2. Blood Cells Mol Dis. 2003;31:247–255. doi: 10.1016/s1079-9796(03)00161-x. [DOI] [PubMed] [Google Scholar]
- Casey JL, Hentze MW, Koeller DM, Caughman SW, Rouault TA, Klausner RD, Harford JB. Iron-responsive elements: regulatory RNA sequences that control mRNA levels and translation. Science. 1988;240:924–928. doi: 10.1126/science.2452485. [DOI] [PubMed] [Google Scholar]
- Chen M, Awe OO, et al. Iron regulatory protein-2 knockout increases perihematomal ferritin expression and cell viability after intracerebral hemorrhage. Brain Res. 2010;1337:95–103. doi: 10.1016/j.brainres.2010.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho HH, Cahill CM, et al. Selective translational control of the Alzheimer amyloid precursor protein transcript by iron regulatory protein-1. J Biol Chem. 2010;285(41):31217–31232. doi: 10.1074/jbc.M110.149161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Masaratana P, et al. Duodenal reductase activity and spleen iron stores are reduced and erythropoiesis is abnormal in Dcytb knockout mice exposed to hypoxic conditions. J Nutr. 2012;142:1929–1934. doi: 10.3945/jn.112.160358. [DOI] [PubMed] [Google Scholar]
- Chua AC, Olynyk JK, et al. Nontransferrin-bound iron uptake by hepatocytes is increased in the Hfe knockout mouse model of hereditary hemochromatosis. Blood. 2004;104(5):1519–1525. doi: 10.1182/blood-2003-11-3872. [DOI] [PubMed] [Google Scholar]
- Cooperman S, Meyron-Holtz EG, et al. Microcytic anemia, erythropoietic protoporphyria, and neurodegeneration in mice with targeted deletion of iron-regulatory protein 2. Blood. 2005;106(3):1084–1091. doi: 10.1182/blood-2004-12-4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dandekar T, Stripecke R, et al. Identification of a novel iron-responsive element in murine and human erythroid delta-aminolevulinic acid synthase mRNA. EMBO J. 1991;10(7):1903–1909. doi: 10.1002/j.1460-2075.1991.tb07716.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan A, Brownlie A, et al. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature. 2000;403:776–81. doi: 10.1038/35001596. [DOI] [PubMed] [Google Scholar]
- Ferring-Appel D, Hentze MW, et al. Cell-autonomous and systemic context-dependent functions of iron regulatory protein 2 in mammalian iron metabolism. Blood. 2009;113(3):679–687. doi: 10.1182/blood-2008-05-155093. [DOI] [PubMed] [Google Scholar]
- Fleming MD, Romano MA, et al. Nramp2 is mutated in the anemic Belgrade (b) rat: evidence of a role for Nramp2 in endosomal iron transport. Proc Natl Acad Sci U S A. 1998;95(3):1148–1153. doi: 10.1073/pnas.95.3.1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galy B, Ferring D, et al. Targeted mutagenesis of the murine IRP1 and IRP2 genes reveals context-dependent RNA processing differences in vivo. RNA. 2004;10(7):1019–1025. doi: 10.1261/rna.7220704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galy B, Ferring D, et al. Altered body iron distribution and microcytosis in mice deficient in iron regulatory protein 2 (IRP2) Blood. 2005;106(7):2580–2589. doi: 10.1182/blood-2005-04-1365. [DOI] [PubMed] [Google Scholar]
- Galy B, Holter SM, et al. Iron homeostasis in the brain: complete iron regulatory protein 2 deficiency without symptomatic neurodegeneration in the mouse. Nat Genet. 2006;38(9):967–969. doi: 10.1038/ng0906-967. discussion 969–970. [DOI] [PubMed] [Google Scholar]
- Galy B, Ferring-Appel D, et al. Iron regulatory proteins are essential for intestinal function and control key iron absorption molecules in the duodenum. Cell metabolism. 2008;7(1):79–85. doi: 10.1016/j.cmet.2007.10.006. [DOI] [PubMed] [Google Scholar]
- Galy B, Ferring-Appel D, et al. Iron Regulatory Proteins Secure Mitochondrial Iron Sufficiency and Function. Cell Metab. 2010;12(2):194–201. doi: 10.1016/j.cmet.2010.06.007. [DOI] [PubMed] [Google Scholar]
- Galy B, Ferring-Appel D, et al. Iron regulatory proteins control a mucosal block to intestinal iron absorption. Cell Rep. 2013;3(3):844–857. doi: 10.1016/j.celrep.2013.02.026. [DOI] [PubMed] [Google Scholar]
- Ganz T, Nemeth E. Hepcidin and Disorders of Iron Metabolism. Annu Rev Med. 2011;62:347–360. doi: 10.1146/annurev-med-050109-142444. [DOI] [PubMed] [Google Scholar]
- Ganz T, Nemeth E. Hepcidin and iron homeostasis. Biochim Biophys Acta. 2012;1823(9):1434–1443. doi: 10.1016/j.bbamcr.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganz T. Systemic iron homeostasis. Physiol Rev. 2013;93(4):1721–1741. doi: 10.1152/physrev.00008.2013. [DOI] [PubMed] [Google Scholar]
- Garrick MD, Dolan KG, et al. DMT1: a mammalian transporter for multiple metals. Biometals. 2003;16(1):41–54. doi: 10.1023/a:1020702213099. [DOI] [PubMed] [Google Scholar]
- Ghosh MC, Ollivierre-Wilson H, Cooperman S, Rouault TA. Reply to “Iron homeostasis in the brain: complete iron regulatory protein 2 deficiency without symptomatic neurodegeneration in the mouse”, Nat. Genet. 2006;38:969–970. doi: 10.1038/ng0906-967. [DOI] [PubMed] [Google Scholar]
- Ghosh MC, Tong WH, et al. Tempol-mediated activation of latent iron regulatory protein activity prevents symptoms of neurodegenerative disease in IRP2 knockout mice. Proc Natl Acad Sci U S A. 2008;105(33):12028–12033. doi: 10.1073/pnas.0805361105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh MC, Zhang DL, et al. Deletion of Iron Regulatory Protein 1 Causes Polycythemia and Pulmonary Hypertension in Mice through Translational Derepression of HIF2alpha. Cell Metab. 2013;17(2):271–281. doi: 10.1016/j.cmet.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabill C, Silva AC, et al. MRI detection of ferritin iron overload and associated neuronal pathology in iron regulatory protein-2 knockout mice. Brain Res. 2003;971(1):95–106. doi: 10.1016/s0006-8993(03)02366-7. [DOI] [PubMed] [Google Scholar]
- Gunshin HB, Mackenzie U, et al. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature. 1997;388:482–8. doi: 10.1038/41343. [DOI] [PubMed] [Google Scholar]
- Guo J, Zhao N, et al. The hereditary hemochromatosis protein, HFE, inhibits iron uptake via down-regulation of Zip14 in HepG2 cells. J Biol Chem. 2008;283:21462–21468. doi: 10.1074/jbc.M803150200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentze MW, Caughman SW, et al. Identification of the iron-responsive element for the translational regulation of human ferritin mRNA. Science. 1987;238(4833):1570–1573. doi: 10.1126/science.3685996. [DOI] [PubMed] [Google Scholar]
- Hentze MW, Muckenthaler MU, Galy B, Camaschella C. Two to tango: regulation of Mammalian iron metabolism. Cell. 2010;142:24–38. doi: 10.1016/j.cell.2010.06.028. [DOI] [PubMed] [Google Scholar]
- Jeong SY, Crooks DR, et al. Iron insufficiency compromises motor neurons and their mitochondrial function in irp2-null mice. PLoS One. 2011;6(10):e25404. doi: 10.1371/journal.pone.0025404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HY, LaVaute T, et al. Identification of a conserved and functional iron-responsive element in the 5′-untranslated region of mammalian mitochondrial aconitase. J Biol Chem. 1996;271(39):24226–24230. doi: 10.1074/jbc.271.39.24226. [DOI] [PubMed] [Google Scholar]
- LaVaute T, Smith S, et al. Targeted deletion of the gene encoding iron regulatory protein-2 causes misregulation of iron metabolism and neurodegenerative disease in mice. Nat Genet. 2001;27(2):209–214. doi: 10.1038/84859. [DOI] [PubMed] [Google Scholar]
- Liuzzi JP, Aydemir F, et al. Zip14 (Slc39a14) mediates non-transferrin-bound iron uptake into cells. Proc Natl Acad Sci U S A. 2006;103(37):13612–13617. doi: 10.1073/pnas.0606424103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur DG, Balasubramanian S, et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science. 2012;335(6070):823–828. doi: 10.1126/science.1215040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magaki S, Mueller C, et al. Regional dissection and determination of loosely bound and non-heme iron in the developing mouse brain. Brain Res. 2007;1158:144–150. doi: 10.1016/j.brainres.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKie AT, Marciani P, et al. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Molecular cell. 2000;5:299–309. doi: 10.1016/s1097-2765(00)80425-6. [DOI] [PubMed] [Google Scholar]
- McKie AT, Barrow D, et al. An iron-regulated ferric reductase associated with the absorption of dietary iron. Science. 2001;291:1755–9. doi: 10.1126/science.1057206. [DOI] [PubMed] [Google Scholar]
- Meyron-Holtz EG, Ghosh MC, et al. Genetic ablations of iron regulatory proteins 1 and 2 reveal why iron regulatory protein 2 dominates iron homeostasis. EMBO J. 2004a;23(2):386–395. doi: 10.1038/sj.emboj.7600041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyron-Holtz EG, Ghosh MC, et al. Mammalian tissue oxygen levels modulate iron-regulatory protein activities in vivo. Science. 2004b;306(5704):2087–2090. doi: 10.1126/science.1103786. [DOI] [PubMed] [Google Scholar]
- Mueller C, Magaki S, et al. Iron regulatory protein 2 is involved in brain copper homeostasis. J Alz Dis. 2009;18:201–210. doi: 10.3233/JAD-2009-1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullner EW, Kuhn LC. A stem-loop in the 3′ untranslated region mediates iron-dependent regulation of transferrin receptor mRNA stability in the cytoplasm. Cell. 1988;53(5):815–825. doi: 10.1016/0092-8674(88)90098-0. [DOI] [PubMed] [Google Scholar]
- Nemeth E, Tuttle MS, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306(5704):2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- Ohgami RS, Campagna DR, et al. Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat Genet. 2005;37(11):1264–1269. doi: 10.1038/ng1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantopoulos K. Iron metabolism and the IRE/IRP regulatory system: an update. Ann N Y Acad Sci. 2004;1012:1–13. doi: 10.1196/annals.1306.001. [DOI] [PubMed] [Google Scholar]
- Pinilla-Tenas JJ, Sparkman BK, et al. Zip14 is a complex broad-scope metal ion transporter whose functional properties support roles in the cellular uptake of zinc and nontransferrin-bound iron. Am J Physiol Cell Physiol. 2011;301:C862–C871. doi: 10.1152/ajpcell.00479.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regan RF, Chen M, et al. Neurons lacking iron regulatory protein-2 are highly resistant to the toxicity of hemoglobin. Neurobiol Dis. 2008;31(2):242–249. doi: 10.1016/j.nbd.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouault TA. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nature chemical biology. 2006;2(8):406–414. doi: 10.1038/nchembio807. [DOI] [PubMed] [Google Scholar]
- Rouault TA. Cell biology. An ancient gauge for iron. Science. 2009;326:676–677. doi: 10.1126/science.1181938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouault TA. Iron metabolism in the CNS: implications for neurodegenerative diseases. Nat Rev Neurosci. 2013;14(8):551–564. doi: 10.1038/nrn3453. [DOI] [PubMed] [Google Scholar]
- Salahudeen AA, Thompson JW, et al. An E3 ligase possessing an iron-responsive hemerythrin domain is a regulator of iron homeostasis. Science. 2009;326(5953):722–726. doi: 10.1126/science.1176326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvatore MF, Fisher B, et al. Neurochemical investigations of dopamine neuronal systems in iron-regulatory protein 2 (IRP-2) knockout mice. Brain Res Mol Brain Res. 2005;139(2):341–347. doi: 10.1016/j.molbrainres.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Sanchez M, Galy B, et al. Iron-regulatory proteins limit hypoxia-inducible factor-2alpha expression in iron deficiency. Nature structural & molecular biology. 2007;14(5):420–426. doi: 10.1038/nsmb1222. [DOI] [PubMed] [Google Scholar]
- Sarkar B. State of iron(III) in normal human serum: low molecular weight and protein ligands besides transferrin. Can J Biochem. 1970;48(12):1339–1350. doi: 10.1139/o70-208. [DOI] [PubMed] [Google Scholar]
- Schalinske KL, Chen OS, et al. Iron differentially stimulates translation of mitochondrial aconitase and ferritin mRNAs in mammalian cells. Implications for iron regulatory proteins as regulators of mitochondrial citrate utilization. J Biol Chem. 1998;273(6):3740–3746. doi: 10.1074/jbc.273.6.3740. [DOI] [PubMed] [Google Scholar]
- Smith MA, Wehr K, et al. Abnormal localization of iron regulatory protein in Alzheimer’s disease. Brain Res. 1998;788(1–2):232–236. doi: 10.1016/s0006-8993(98)00002-x. [DOI] [PubMed] [Google Scholar]
- Smith SR, Cooperman S, et al. Severity of neurodegeneration correlates with compromise of iron metabolism in mice with iron regulatory protein deficiencies. Ann N Y Acad Sci. 2004;1012:65–83. doi: 10.1196/annals.1306.006. [DOI] [PubMed] [Google Scholar]
- Smith SR, Ghosh MC, et al. Complete loss of iron regulatory proteins 1 and 2 prevents viability of murine zygotes beyond the blastocyst stage of embryonic development. Blood cells, mol & dis. 2006;36(2):283–287. doi: 10.1016/j.bcmd.2005.12.006. [DOI] [PubMed] [Google Scholar]
- Theil EC. Regulation of ferritin and transferrin receptor mRNAs. J Biol Chem. 1990;265(9):4771–4774. [PubMed] [Google Scholar]
- Tian H, McKnight SL, et al. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 1997;11:72–82. doi: 10.1101/gad.11.1.72. [DOI] [PubMed] [Google Scholar]
- Vanoaica L, Darshan D, et al. Intestinal ferritin H is required for an accurate control of iron absorption. Cell Metab. 2010;12:273–282. doi: 10.1016/j.cmet.2010.08.003. [DOI] [PubMed] [Google Scholar]
- Vashisht AA, Zumbrennen KB, et al. Control of iron homeostasis by an iron-regulated ubiquitin ligase. Science. 2009;326(5953):718–721. doi: 10.1126/science.1176333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vulpe CD, Kuo YM, et al. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet. 1999;21(2):195–199. doi: 10.1038/5979. [DOI] [PubMed] [Google Scholar]
- Wang J, Chen G, et al. Iron-mediated degradation of IRP2, an unexpected pathway involving a 2-oxoglutarate-dependent oxygenase activity. Mol Cell Biol. 2004;24(3):954–965. doi: 10.1128/MCB.24.3.954-965.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson N, Pantopoulos K. The IRP/IRE system in vivo: insights from mouse models. Front Pharmacol. 2014;5(176):1–15. doi: 10.3389/fphar.2014.00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson N, Pantopoulos K. IRP1 regulates erythropoiesis and systemic iron homeostasis by controlling HIF2alpha mRNA translation. Blood. 2013;122(9):1658–1668. doi: 10.1182/blood-2013-03-492454. [DOI] [PubMed] [Google Scholar]
- Wolf TL, Kotun J, Meador-Woodruff JH. Plasma copper, iron, ceruloplasmin and ferroxidase activity in schizophrenia. Schizophr Res. 2006;86(1–3):167–71. doi: 10.1016/j.schres.2006.05.027. [DOI] [PubMed] [Google Scholar]
- Zecca L, Youdim MB, et al. Iron, brain ageing and neurodegenerative disorders. Nat Rev Neurosci. 2004;5(11):863–873. doi: 10.1038/nrn1537. [DOI] [PubMed] [Google Scholar]
- Zhang D-L, Ghosh MC, Rouault TA. The physiological functions of iron regulatory proteins in iron homeostasis – an update. Front Pharmacol. 2014;5(124):1–12. doi: 10.3389/fphar.2014.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Land W, et al. Electron tomography of degenerating neurons in mice with abnormal regulation of iron metabolism. J Struct Biol. 2005;150(2):144–153. doi: 10.1016/j.jsb.2005.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer M, Ebert BL, et al. Small-molecule inhibitors of HIF-2a translation link its 5′UTR iron-responsive element to oxygen sensing. Mol Cell. 2008;32(6):838–848. doi: 10.1016/j.molcel.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zumbrennen-Bullough KB, Becker L, et al. Abnormal brain iron metabolism in Irp2 deficient mice is associated with mild neurological and behavioral impairments. PLoS One. 2014;9(6):e98072. doi: 10.1371/journal.pone.0098072. [DOI] [PMC free article] [PubMed] [Google Scholar]