

Abstract
HLA-G is a nonclassical class I human leukocyte antigen (HLA) involved in mechanisms of immune tolerance. The objective of this study was to determine whether N-(3-oxododecanoyl)-l-homoserine lactone (3O-C12-HSL), a quorum sensing molecule produced by Pseudomonas aeruginosa, could modify HLA-G expression to control the host immune response. We evaluated the ability of 3O-C12-HSL to induce HLA-G expression in primary immune cells, monocytes (U937 and THP1), and T-cell lines (Jurkat) in vitro and analyzed the cellular pathway responsible for HLA-G expression. We studied the HLA-G promoter with a luciferase assay and interleukin-10 (IL-10) and p38/CREB signaling with enzyme-linked immunosorbent assay and immunofluorescence, respectively. We observed that 3O-C12-HSL is able to induce HLA-G expression in human monocytes and T cells. We showed that the induction of HLA-G by 3O-C12-HSL is p38/CREB and IL-10 dependent. 3O-C12-HSL treatment is able to arrest only the U937 cell cycle, possibly due to the peculiar expression of the ILT2 receptor in the U937 cell line. Our observations suggest HLA-G as a mechanism to create a protected niche for the bacterial reservoir, similar to the role of HLA-G molecules during viral infections.
INTRODUCTION
Bacterial diseases can result in serious or life-threatening complications, such as bacteremia, kidney failure, and toxic shock syndrome. For this, the fight against bacterial infection represents one of the high points of modern medicine. Lack of progress in controlling mortality and morbidity associated with severe bacterial infections in part reflects our limited understanding of the complex biological pathways that bacteria use to regulate host immune response. Many bacteria are capable of forming a well-organized bacterial population during host infection, and Pseudomonas aeruginosa is one of the most commonly studied. As the cell population increases, P. aeruginosa increases the expression of quorum sensing (QS) molecules that bind to the transcriptional activators, enabling the expression of target genes involved in P. aeruginosa virulence (1). P. aeruginosa has two well-studied QS systems, las and rhl (2–4). The las system consists of the LasR transcriptional regulator and the LasI synthase protein, which is essential for the production of the signal molecule N-(3-oxododecanoyl)-l-homoserine lactone (3O-C12-HSL) that also is required for LasR activation (4). Recent studies have shown that 3O-C12-HSL has the potential to modify the functions of host immune cells (5–9). In particular, 3O-C12-HSL inhibits professional immune cells, such as dendritic and T cells (10), promotes immune cell apoptosis (11–13), and blocks the response of macrophages and monocytes to toll-like receptor (TLR) signals (14). However, the mechanism of this immune-regulatory activity has yet to be fully characterized.
HLA-G is a nonclassical class I human leukocyte antigen (HLA) characterized by the presence of membrane-bound (G1-G4) and soluble (G5-G7) isoforms (15). HLA-G is involved in mechanisms of immune tolerance under several conditions, including pregnancy, organ transplantation, and autoimmune and inflammatory diseases by inhibiting cytolytic functions of natural killer cells, cytotoxic T lymphocytes, and T and dendritic cells, as well as by inhibiting alloproliferative responses (16). Both soluble and membrane-bound HLA-G isoforms have similar functions and interact with specific inhibitory receptors (ILT-2 and ILT-4) expressed by immune cells. During viral infections, HLA-G molecules are upregulated by the virus as a mechanism of immune escape (16), inhibiting the host immune response. Few data are available on the effect of bacterial infections on HLA-G expression.
The objective of the present study was to determine whether P. aeruginosa 3O-C12-HSL could modify HLA-G expression by immune cells, supporting the hypothesis of a direct involvement of HLA-G molecules in P. aeruginosa's ability to control host immune response.
MATERIALS AND METHODS
Cell lines.
U937 (ATCC CRL1593.2) and THP-1 (ATCC TIB202) monocyte cell lines, a Jurkat (ATCC TIB152) T-cell line, and the 721.221 (ATCC CRL1885) B-cell line were grown in RPMI 1640 (Gibco) with 10% fetal bovine serum (FBS), 10% HEPES buffer, 5% penicillin-streptomycin at 37°C with 5% CO2.
PBMC purification.
Peripheral blood mononuclear cells (PBMCs) were isolated from whole blood of 10 control subjects by Ficoll gradient (Cederlane, Hornby, Ontario, Canada) and resuspended in RPMI medium (EuroClone, Milan, Italy) with 10% fetal calf serum (FCS), 100 U/ml penicillin, and 100 U/ml streptomycin (Sigma-Aldrich, St. Louis, MO). Ethical approval was obtained from the University of Ferrara Review Board.
Cell treatments.
The cells were treated with 3O-C12-HSL (Sigma-Aldrich) at concentrations of 10, 25, and 50 μM, as suggested by the literature (17), at different time points and then analyzed.
For the inhibitory experiments, Fc receptors first were blocked using human serum, and cells were incubated for 30 min at 37°C with a final concentration of 10 μg/ml isotype control monoclonal antibody (MAb) or blocking anti-interleukin-10 (IL-10; BioLegend) monoclonal antibody prior to use.
Cytometric analysis.
For flow cytometric analysis, 106 cells were washed and incubated for 30 min on ice in 100 μl of phosphate-buffered saline (PBS) containing 1% FBS, 10 mM sodium azide plus appropriately diluted fluorescent MAb. After two washes with cold washing buffer, cells were washed, fixed in 2% formaldehyde, and analyzed by flow cytometry with a FACSVantage flow cytometer (Becton Dickinson, San Jose, CA) using standard settings and CellQuest software (Becton Dickinson, San Jose, CA) for data analysis. The membrane-bound HLA-G antigens were detected by anti-HLA-G fluorescein isothiocyanate (FITC) MAb (87G; Exbio, Prague, Czech Republic), and ILT2 and ILT4 receptors were analyzed by specific MAbs (Becton Dickinson). Isotype controls (Exbio, Prague, Czech Republic) were performed. PBMCs were analyzed using anti-CD3-peridinin chlorophyll protein, anti-CD14-phycoerythrin (PE), anti-CD45-PE, and anti-CD56-PE (BD) MAbs.
The cell cycle was analyzed by propidium iodide (PI) staining. Cells were resuspended cells in 500 μl PI–Triton X-100 staining solution (0. 1% [vol/vol] Triton X-100 [Sigma] in PBS, 2 mg DNase-free RNase A [Sigma], 0.40 ml of 500 μg/ml PI [Roche]), incubated for 30 min at RT, and analyzed by flow cytometry.
Immunofluorescence microscopy.
For immunofluorescence microscopy, 106 cells were washed and incubated for 30 min on ice in 100 μl of PBS containing 1% FBS, 10 mM sodium azide, and the appropriately diluted fluorescent MAb. After two washes with cold washing buffer, cells were washed again, fixed in 2% formaldehyde, and analyzed by fluorescence microscopy. Cyclic AMP (cAMP) response element-binding protein (CREB) and phosphorylated CREB (pCREB) (Immunological Sciences), as well as p38 and phosphorylated p38 (pp38) (Santa Cruz), were detected by indirect immunofluorescence with the secondary antibody goat anti-mouse FITC (Dako).
Western blot analysis.
Cell pellets were treated with lysis buffer with fresh protease inhibitors, biotinylated with 0.2 mg/ml EZ-Link sulfo-NHS-LC-biotin (Pierce, Rockford, IL) in 1× PBS (pH 8.0) for 30 min at 4°C. Samples then were immunoprecipitated for 2 h at RT with CREB/pCREB (Immunological Sciences) and p38/pp38 (SantaCruz) MAbs or actin MAb (Santa Cruz) as a positive loading control, washed twice in 1× PBS, and incubated overnight with protein G-Sepharose beads (Santa Cruz, CA) at 4°C. The samples were washed twice and resuspended in 20 μl of Laemmli buffer (Bio-Rad, Segrate, Italy). Immunoprecipitates were denatured at 100°C for 5 min. Proteins were loaded with native or reducing (in the presence of SDS) running buffers in 10% TGX-precast gel (Bio-Rad, Segrate, Italy), with subsequent electroblotting transfer onto a polyvinylidene difluoride (PVDF) membrane (Millipore, MA) (1). The membrane was incubated with a horseradish peroxidase (HRP)-conjugated anti-mouse antibody (1:5,000; Amersham Biosciences, NJ) and developed with the ECL kit (Amersham Biosciences, NJ). The images were acquired by GelDoc (Bio-Rad).
ELISA.
Soluble HLA-G5 levels in cell culture supernatants were measured by enzyme-linked immunosorbent assay (ELISA) using as capture antibody the MAb 5A6G7 (Exbio, Prague, Czech Republic), which recognizes the HLA-G5 molecule. The intra-assay coefficient of variation (CV) was 1.3%, and the interassay CV was 3.7%. The limit of sensitivity was 1.0 ng/ml (18).
IL-10 levels were analyzed using a human IL-10 ELISA detection kit (EBioscience).
MTT cell viability assay.
One hundred microliters of cells at a density of 1 × 106/ml were seeded into 96-well plates and treated with 3O-C12-HSL (10 μM and 25 μM) for 6, 12, and 24 h. After incubation, 10 μl of MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide; Sigma-Aldrich] was added for 4 h at 37°C. Cells then were lysed by adding 100 μl of MTT solvent. Plates then were read at 570 nm for viability evaluation using an ELISA reader (Victor; Perkin-Elmer).
mRNA preparation.
Total cellular RNA was prepared from each cell culture with TRIzol reagent (Life Technologies, NY). The RNA samples were digested with DNase. The quality and quantity of RNA samples were assessed by a 1% agarose gel electrophoresis, followed by ethidium bromide staining. These mRNA samples were immediately used for cDNA synthesis or stored frozen at −80°C until use.
Real-time PCR.
To analyze the presence of HLA-G mRNA, 2 μg mRNA was reverse transcribed for each sample using a SuperScript first-strand synthesis system (Invitrogen, San Giuliano Milanese, Italy) according to the manufacturer's instructions. The primers and the detection probe for HLA-G gene expression analysis were the following: forward primer HLAG-F, 5′-CCCACCATCCCCATCATG-3′; reverse primer HLA-G-R, 5′-CCAGTGACTACAGCTGCAAGGA-3′; and the MGB probe, 5′-TATCGTTGCTGGCCTGG-6-carboxyflourescein-3′ (Applied Biosystems) (19). Fold changes in expression were determined by the 2−ΔΔCT method. Amplification was performed with 100 ng of RNA converted into cDNA with TaqMan 2× universal PCR master mix in a final volume of 50 μl (Applied Biosystems) by using the following protocol: 2 min at 50°C for AmpErase UNG activation, 20 s at 95°C for initial denaturation, and then 40 cycles of 20 s at 95°C and 60 s at 60°C for amplification. All reactions were performed in triplicate.
Reporter constructs and expression vectors.
Luciferase reporter plasmids were generated by cloning genomic promoter fragments into pGL3-Basic (Promega, Madison, WI). These constructs contain a 1,438-bp promoter fragment of HLA-G (pGL3-G1500) and a 269-bp AspI-AhaII-HLA-B7 promoter fragment (pGL3-HLA-B) (kind gift of Sam J. P. Gobin) (20). All inserts were verified by sequence analysis.
The Renilla luciferase control plasmid pRL-actin was used as a transfection efficiency control.
Transient transfection.
721.221 cells were transfected by Amaxa nucleofector technology (Lonza) with a DNA precipitate of 1 μg of pGL3 reporter plasmid, 1 or 0.5 μg of expression vector, and 0.1 μg of Renilla luciferase control plasmid (pRL-actin) per well. Luciferase activity was determined using a luminometer (Victor; PerkinElmer) and corrected for transfection efficiency with the Renilla luciferase activity values.
Statistical analysis.
Since the values presented a normal distribution (Kolmogorov-Smirnov test), the differences were evaluated by Student t test using Stat View software (SAS Institute Inc., Cary, NC). The P value was considered to be statistically significant when it was <0.05.
RESULTS
P. aeruginosa 3O-C12-HSL induces HLA-G expression in human monocytes and T cells.
We first analyzed the ability of P. aeruginosa 3O-C12-HSL to induce HLA-G transcription and transduction in human primary immune cells. We exposed peripheral blood mononuclear cells (PBMCs) from 10 control subjects to P. aeruginosa 3O-C12-HSL (17). PBMCs were negative for HLA-G staining before the treatment. Both CD3+ and CD14+ cells induced membrane-bound HLA-G expression, with the highest levels occurring after 12 h of incubation with 25 μM 3O-C12-HSL (2.7% ± 0.3% CD3+ HLA-G+ and 6.4% ± 0.6% CD14+ HLA-G+ cells) (Fig. 1a) that decreased after 24 h of incubation (0.6% ± 0.1% CD3+ HLA-G+ and 1.6% ± 0.4% CD14+ HLA-G+). As a confirmation, we performed a real-time PCR quantification of HLA-G mRNA in PBMCs after 3O-C12-HSL treatment. We observed a 6-fold increase in HLA-G mRNA transcription 12 h after the incubation with 3O-C12-HSL (Fig. 1b) (P < 0.0001) that was lost after 24 h. The analysis of B and NK cells showed no HLA-G induction (data not shown).
FIG 1.

(a) Membrane HLA-G expression in PBMCs from 10 healthy subjects. Cells were treated with 10 and 25 μM 3O-C12-HSL for 12 h (left) and 24 h (right). CD3+ and CD14+ cell results are reported. (b) HLA-G mRNA expression analysis in PBMCs after 12 h of treatment with 25 μM 3O-C12-HSL. RQ, relative quantitation. Means ± standard deviations (SD) are reported. **, P < 0.05 by Student t test.
To confirm our data in a standardized in vitro model, we used Jurkat T-cell and monocyte cell lines that differ in maturation stages (less mature THP-1 and U937 cells are at a more advanced stage of differentiation). We treated these cell lines with 10 and 25 μM 3O-C12-HSL (17) at the 12-h time point, selected on the basis of the results obtained with PBMC treatment (Fig. 1a). We confirmed the increased expression of HLA-G molecules on T and monocyte cells (Fig. 2a, b, and c), as previously observed in PBMCs (Fig. 1a), with a significantly lower upregulation in THP1 cells (Fig. 2c). These results also were confirmed by mRNA analysis. We observed a 6-fold increase in HLA-G mRNA transcription 12 h after the incubation of Jurkat and U937 cells with 3O-C12-HSL (Fig. 2b) (P < 0.0001), while THP1 induced lower levels of HLA-G mRNA after 3O-C12-HSL treatment (Fig. 2b). These results also confirmed the effect of 3O-C12-HSL on human immune cells in a standardized in vitro model.
FIG 2.

HLA-G membrane (87G-FITC MAb) (a) and mRNA HLA-G (b) expression in Jurkat, U937, and THP1 cell lines. Cells were treated with 10 and 25 μM 3O-C12-HSL for 12 h. (c) Luciferase assay of HLA-B and HLA-G promoter activation in 721.221-transfected cell lines after treatment with 10 and 25 μM 3O-C12-HSL for 12 h. Means ± SD are reported. **, P < 0.05 by Student t test.
P. aeruginosa 3O-C12-HSL induces HLA-G via p38/CREB and IL-10 pathways.
We wanted to investigate the mechanisms used by 3O-C12-HSL to induce HLA-G expression. To account for the direct effect of 3O-C12-HSL on the HLA-G gene promoter, we transfected the 721.221 cell line (classical and nonclassical HLA-I-negative cells) with HLA-G or HLA-B promoters (20). These cells were treated with 3O-C12-HSL, and a luciferase assay was performed. We observed an increased activation of HLA-G promoter after 3O-C12-HSL treatment. In particular, the promoter activation was dose dependent, with an increased activation of 21- and 39-fold with 10 μM and 25 μM 3O-C12-HSL, respectively (Fig. 2d, black histograms). On the contrary, the HLA-B promoter was not affected by 3O-C12-HSL treatment (Fig. 2d, gray histograms). These data suggest a specific effect of 3O-C12-HSL on the HLA-G promoter, with the implication of specific transcription factors. It is known that 3O-C12-HSL modifies transduction pathways (21), in particular inducing p38, a mitogen-activated protein kinase (MAPK) that phosphorylates and activates CREB (22), a transcription factor that regulates HLA-G gene expression (21). We hypothesized that the p38/CREB pathway could be implicated in the increased HLA-G expression via 3O-C12-HSL. We evaluated the expression of p38 and CREB and their phosphorylated isoforms after 3O-C12-HSL treatment in U937 and THP1 cells. We observed a clear increase in the amount of p38 and phosphorylated p38 after 15 min (Fig. 3a) and a consequent increase in CREB and phosphorylated CREB after 30 min of 10 μM 3O-C12-HSL treatment of U937 cells (Fig. 3b). Western blot analysis confirmed an increased expression of p38 (Fig. 3b) and CREB (Fig. 3d) protein with a consequent increase in the phosphorylated isoforms. Jurkat cells showed a similar pattern of activation (data not shown). THP1 cells reached a lower rate of CREB phosphorylation (Fig. 3e and f), accounting for the lower HLA-G expression observed in this cell line (Fig. 2c). These data suggest that the induction of the p38/CREB pathway is one of the mechanisms used by 3O-C12-HSL to induce HLA-G expression.
FIG 3.

Intracellular pathway activation analysis in U937 and THP1 cell lines. U937 and THP1 cells were treated with 25 μM 3O-C12-HSL. p38 and CREB analysis was performed by immunofluorescence for total (p38, CREB) and phosphorylated (pp38, pCREB) status in U937 (a and c) and THP1 (e and f) cells. Western blot analysis for total (p38; 38 kDa), CREB (37 kDa), phosphorylated (pp38, pCREB), and actin (loading control; 42 kDa) status was performed in U937 cells (b and d). Shown are the most representative results at 0 and 15 s for p38 and at 0 and 30 s for CREB after 25 μM 3O-C12-HSL treatment.
Since IL-10 is one of the major inducers of soluble HLA-G5 expression (23), and since it is known that 3O-C12-HSL is able to increase IL-10 expression in macrophages (17), we evaluated if this cytokine could be implicated in the induction of HLA-G5 secretion via 3O-C12-HSL. We analyzed the levels of IL-10 and HLA-G5 expression in Jurkat, U937, and THP1 cell culture supernatants after 3O-C12-HSL treatment. We observed an increased secretion of IL-10 already 6 h after 3O-C12-HSL addition in both Jurkat and U937 cells (Fig. 4a, white histograms), while HLA-G5 levels increased after 12 h of treatment (Fig. 4, gray histograms). On the contrary, THP1 presented no IL-10 induction after 3O-C12-HSL treatment (Fig. 4a), coinciding with the absence of HLA-G5 upregulation in THP1 cells (Fig. 4b). To evaluate the role of IL-10, we pretreated U937 cells with anti-IL-10 MAb and observed the reduction of HLA-G5 secretion in 3O-C12-HSL-treated U937 cell culture supernatants (Fig. 5, white histograms). This blocking treatment was not able to completely inhibit HLA-G5 expression, even at increased anti-IL-10 MAb concentrations (data non shown), demonstrating that IL-10 is only one of the mechanisms used by 3O-C12-HSL to induce HLA-G5 secretion.
FIG 4.

IL-10 (a) and HLA-G5 (b) levels in Jurkat, U937, and THP1 cell culture supernatants. Cells were treated with 10 and 25 μM 3O-C12-HSL for 6, 12, and 24 h. Means ± SD are reported. **, P < 0.05 obtained by Student t test.
FIG 5.
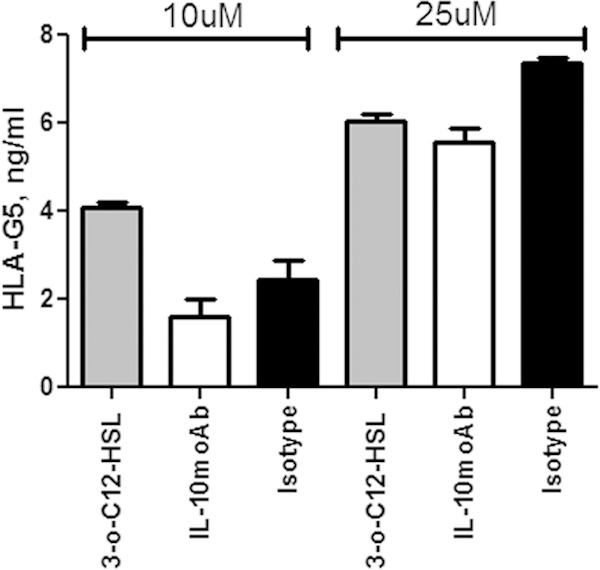
HLA-G5 levels in cell culture supernatants of U937 cells after 10 or 25 μM 3O-C12-HSL treatment for 12 h (gray histogram) with anti-IL-10 (white histogram) or anti-isotype (black histogram) MAb addition. Means ± SD are reported. **, P < 0.05 obtained by Student t test.
P. aeruginosa 3O-C12-HSL effect on monocyte and T-cell viability.
Since 3O-C12-HSL treatment was reported to induce cell apoptosis (13), we verified if 3O-C12-HSL treatment could affect monocyte and T cell viability. We treated Jurkat, U937, and THP1 cells with 10, 25, or 50 μM 3O-C12-HSL for 6, 12, and 24 h. After the treatments, cell viability was evaluated by MTT assay. We observed no significant decrease in cell viability in both Jurkat and THP1 cells (Fig. 6a and c). On the contrary, we found a 40% decrease in U937 cell viability after 12 h of incubation with all concentrations of 3O-C12-HSL (Fig. 6b). Interestingly, U937 cells reconstituted their viability after 24 h of incubation. The analysis of the U937 cell cycle showed that the treatment with 3O-C12-HSL for 12 h blocked 62% of cells in the G0/G1 stage (Fig. 6d).
FIG 6.

MTT cell viability assay. Cells were treated with 10, 25, and 50 μM 3O-C12-HSL for 6, 12, or 24 h. Jurkat (a), U937 (b), and THP1 (c) cell lines are shown. Means ± SD are reported. **, P < 0.05 obtained by Student t test. (d) Propidium iodide staining for cell cycle analysis in U937 cells treated with 25 M 3O-C12-HSL for 12 h. The percentage of cells in each cell cycle stage is reported. The most representative results are shown.
Since HLA-G interaction with its ligands (ILT2 and ILT4) is able to modify immune cell proliferation (24, 25), we tested U937, THP1, and Jurkat cells for ILT2 and ILT4 expression. We observed no ILT4 expression on any of the three cell lines, while ILT2 was expressed only on the surface of U937 cells (Fig. 7).
FIG 7.

ILT2 membrane expression, evaluated by flow cytometry (anti-ILT2 MAb) in Jurkat (a), U937 (b), and THP1 (c) cell lines. The most representative results are shown.
DISCUSSION
The inability of the host to contain an invading pathogen is associated with a systemic inflammatory response that, when deregulated, can lead to organ injury. Recently, it has been established that QS molecules not only are important in the regulation of bacterial virulence genes but also interact with eukaryotic cells and modulate immune responses (5–14). The investigations reported here demonstrate that 3O-C12-HSL, a P. aeruginosa QS molecule, is able to induce HLA-G expression in human monocytes and T cells. Moreover, we showed that the induction of HLA-G by 3O-C12-HSL is p38/CREB and IL-10 dependent. The implication of IL-10 and p38/CREB pathways has been previously reported in bacterial infections. In particular, IL-10 expression previously has been related to the role of 3O-C12-HSL in P. aeruginosa infection by simultaneously upregulating the anti-inflammatory cytokine IL-10 and downregulating the proinflammatory cytokine tumor necrosis factor alpha (17). IL-10 expression is prompted during bacterial infections (26) as a mechanism of immune suppression, and CREB phosphorylation is induced to modify cellular patterns of protein expression (27). As a proof of concept of the implication of both IL-10 and p38/CREB pathways, the THP1 cell line showed a lower increase of HLA-G expression than U937 after 3O-C12-HSL treatment with corresponding lower p38/CREB expression and IL-10 induction. It is worth noting that U937 and THP1 cell lines already have been found to behave in a different way after exposure to compounds (28, 29) and microbial infections (30), sustaining the data obtained in our study. In particular, another bacterium, Chlamydia pneumoniae, already has been shown to downregulate genes related to cell division only in U937 cells (31), supporting the peculiar effect of 3O-C12-HSL treatment in arresting the U937 cell cycle. Moreover, only U937 cells express the ILT2 receptor, and interacting with HLA-G molecules in the cell culture supernatant might modify cell activation status (24). However, we are aware that the effects of 3O-C12-HSL on cell viability are still to be understood.
The results obtained suggest HLA-G as a mechanism of immune privilege that creates a protected niche for a bacterial reservoir, similar to the role of HLA-G molecules during viral infections (16). This hypothesis is in agreement with the previous observations on the implications of HLA-G expression in bacterial infections. First, HLA-G expression at the extravillous cytotrophoblast surface seems to increase the risk of Listeria monocytogenes infection (32). We suggest that this bacteria exploits cytotrophoblast constitutive HLA-G expression to evade host immune cells. Moreover, soluble HLA-G molecules are expressed during septic shock and are predictive of better survival (33). The patients with sepsis are characterized by a deregulated immune response (10), impaired chemotactic responses, and alterations in neutrophil functions (34) that could benefit from HLA-G–ILT2 interaction, allowing the restoration of a controlled immune activation (35).
In conclusion, we present for the first time the effect of a bacterial QS compound, 3O-C12-HSL, on HLA-G induction in human immune cells. These results are important to understanding bacterial virulence mechanisms and in evaluating the role of HLA-G expression during bacterial infections. In fact, HLA-G could be considered on the one hand as a natural way to create an immune tolerance condition that could allow bacterial persistence in the host and, on the other hand, as an immune factor that could control impaired immune activation during bacterial infection that is frequently harmful to patient health. As a future perspective, it will be of importance to recognize the cellular characteristics that make a cell responsive to 3O-C12-HSL treatment for HLA-G expression and the specific pathological contests that could benefit or be damaged by the modulation of HLA-G molecules.
ACKNOWLEDGMENTS
Daria Bortolotti, Roberta Rizzo, Claudio Trapella and Dario Di Luca are inventors of the patent FE2014A000005 (submitted November 2014), which is relevant to this work.
We thank Iva Pivanti for technical support, Marina Daouya for laboratory support, and Linda M. Sartor for language revision of the manuscript.
REFERENCES
- 1.Williams P. 2007. Quorum sensing, communication and cross-kingdom signalling in the bacterial world. Microbiology 153:3923–3938. doi: 10.1099/mic.0.2007/012856-0. [DOI] [PubMed] [Google Scholar]
- 2.Davenport P, Griffin JL, Welch M. 2015. Quorum sensing is accompanied by global metabolic changes in the opportunistic human pathogen, Pseudomonas aeruginosa. J Bacteriol 197:2072–2082. doi: 10.1128/JB.02557-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee J, Zhang L. 2015. The hierarchy quorum sensing network in Pseudomonas aeruginosa. Protein Cell 6:26–41. doi: 10.1007/s13238-014-0100-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Williams P, Camara M. 2009. Quorum sensing and environmental adaptation in Pseudomonas aeruginosa: a tale of regulatory networks and multifunctional signal molecules. Curr Opin Microbiol 12:182–191. doi: 10.1016/j.mib.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 5.Smith RS, Kelly R, Iglewski BH, Phipps RP. 2002. The Pseudomonas autoinducer N-(3-oxododecanoyl) homoserine lactone induces cyclooxygenase-2 and prostaglandin E2 production in human lung fibroblasts: implications for inflammation. J Immunol 169:2636–2642. doi: 10.4049/jimmunol.169.5.2636. [DOI] [PubMed] [Google Scholar]
- 6.Ritchie AJ, Yam AOW, Tanabe KM, Rice SA, Cooley MA. 2003. Modification of in vivo and in vitro T and B cell mediated immune responses by the Pseudomonas aeruginosa quorum sensing molecule N-(3-oxododecanoyl)-l-homoserine lactone (OdDHL). Infect Immun 71:4421–4431. doi: 10.1128/IAI.71.8.4421-4431.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Telford G, Wheeler D, Williams P, Tomkins PT, Appleby P, Sewell H, Stewart GS, Bycroft BW, Pritchard DI. 1998. The Pseudomonas aeruginosa quorum-sensing signal molecule N-(3-oxododecanoyl)-L-homoserine lactone has immunomodulatory activity. Infect Immun 66:36–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hooi DS, Bycroft BW, Chhabra SR, Williams P, Pritchard DI. 2004. Differential Immune modulatory activity of Pseudomonas aeruginosa quorum-sensing signal molecules. Infect Immun 72:6463–6470. doi: 10.1128/IAI.72.11.6463-6470.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith RS, Harris SG, Phipps R, Iglewski B. 2002. The Pseudomonas aeruginosa quorum sensing molecule N-(3-oxododecanoyl) homoserine lactone contributes to virulence and induces inflammation in vivo. J Bacteriol 184:1132–1139. doi: 10.1128/jb.184.4.1132-1139.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boontham P, Robins A, Chandran P, Pritchard D, Cámara M, Williams P, Chuthapisith S, McKechnie A, Rowlands BJ, Eremin O. 2008. Significant immunomodulatory effects of Pseudomonas aeruginosa quorum-sensing signal molecules: possible link in human sepsis. Clin Sci 115:343–351. doi: 10.1042/CS20080018. [DOI] [PubMed] [Google Scholar]
- 11.Li H, Wang L, Zhang C, Gong F, Li H, Xie X, Xia C, Chen J, Song Y, Shen A, Song J. 2009. Influence of Pseudomonas aeruginosa quorum sensing signal molecule N-(3-oxododecanoyl) homoserine lactone on mast cells. Med Microbiol Immunol 198:113–121. doi: 10.1007/s00430-009-0111-z. [DOI] [PubMed] [Google Scholar]
- 12.Jacobi C, Schiffner F, Henkel M, Waibel M, Stork B, Daubrawa M, Eberl L, Gregor M, Wesselborg S. 2009. Effects of bacterial N-acyl homoserine lactones on human Jurkat T lymphocytes-OdDHL induces apoptosis via the mitochondrial pathway. Int J Med Microbiol 299:509–519. doi: 10.1016/j.ijmm.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 13.Tateda K, Ishii Y, Horikawa M, Matsumoto T, Miyairi S, Pechere JC, Standiford TJ, Ishiguro M, Yamaguchi K. 2003. The Pseudomonas aeruginosa autoinducer N-3-oxododecanoyl homoserine lactone accelerates apoptosis in macrophages and neutrophils. Infect Immun 71:5785–5793. doi: 10.1128/IAI.71.10.5785-5793.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kravchenko VV, Kaufmann GF, Mathison JC, Scott DA, Katz AZ, Grauer DC, Lehmann M, Meijler MM, Janda KD, Ulevitch RJ. 2008. Modulation of gene expression via disruption of NF-kappaB signaling by a bacterial small molecule. Science 321:259–263. doi: 10.1126/science.1156499. [DOI] [PubMed] [Google Scholar]
- 15.Rizzo R, Bortolotti D, Baricordi OR, Fainardi E. 2012. New insights into HLA-G and inflammatory diseases. Inflamm Allergy Drug Targets 11:448–463. doi: 10.2174/187152812803590037. [DOI] [PubMed] [Google Scholar]
- 16.Rizzo R, Bortolotti D, Bolzani S, Fainardi E. 2014. HLA-G molecules in autoimmune diseases and infections. Front Immunol 5:592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glucksam-Galnoy Y, Sananes R, Silberstein N, Krief P, Kravchenko VV, Meijler MM, Zor T. 2013. The bacterial quorum-sensing signal molecule N-3-oxo-dodecanoyl-l-homoserine lactone reciprocally modulates pro- and anti-inflammatory cytokines in activated macrophages. J Immunol 191:337–344. doi: 10.4049/jimmunol.1300368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rizzo R, Malagutti N, Bortolotti D, Gentili V, Rotola A, Fainardi E, Pezzolo T, Aimoni C, Pelucchi S, Di Luca D, Pastore A. 2014. Infection and HLA-G molecules in nasal polyposis. J Immunol Res 2014:407–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yao YQ, Barlow DH, Sargent IL. 2005. Differential expression of alternatively spliced transcripts of HLA-G in human preimplantation embryos and inner cell masses. J Immunol 175:8379–8385. doi: 10.4049/jimmunol.175.12.8379. [DOI] [PubMed] [Google Scholar]
- 20.Gobin SJP, Biesta P, de Steenwinkel JEM, Datema G, Van den Elsen PJ. 2002. HLA-G transactivation by cAMP-response element-binding protein (CREB). An alternative transactivation pathway to the conserved major histocompatibility complex (MHC) class I regulatory routes. J Biol Chem 277:39525–39531. [DOI] [PubMed] [Google Scholar]
- 21.Kravchenko VV, Kaufmann GF, Mathison JC, Scott DA, Katz AZ, Wood MR, Brogan AP, Lehmann M, Mee JM, Iwata K, Pan Q, Fearns C, Knaus UG, Meijler MM, Janda KD, Ulevitch RJ. 2006. N-(3-oxo-acyl)homoserine lactones signal cell activation through a mechanism distinct from the canonical pathogen-associated molecular pattern recognition receptor pathways. J Biol Chem 281:28822–28830. doi: 10.1074/jbc.M606613200. [DOI] [PubMed] [Google Scholar]
- 22.Deak M, Clifton AD, Lucocq LM, Alessi DR. 1998. Mitogen and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. EMBO J 17:4426–4441. doi: 10.1093/emboj/17.15.4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rizzo R, Hviid TV, Stignani M, Balboni A, Grappa MT, Melchiorri L, Baricordi OR. 2005. The HLA-G genotype is associated with IL-10 levels in activated PBMCs. Immunogenetics 57:172–181. doi: 10.1007/s00251-005-0788-0. [DOI] [PubMed] [Google Scholar]
- 24.Shiroishi M, Tsumoto K, Amano K, Shirakihara Y, Colonna M, Braud VM, Allan DS, Makadzange A, Rowland-Jones S, Willcox B, Jones EY, van der Merwe PA, Kumagai I, Maenaka K. 2003. Human inhibitory receptors Ig-like transcript 2 (ILT2) and ILT4 compete with CD8 for MHC class I binding and bind preferentially to HLA-G. Proc Natl Acad Sci U S A 100:8856–8861. doi: 10.1073/pnas.1431057100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carosella ED, Gregori S, LeMaoult J. 2011. The tolerogenic interplay (s) among HLA-G, myeloid APCs, and regulatory cells. Blood 118:6499–6505. doi: 10.1182/blood-2011-07-370742. [DOI] [PubMed] [Google Scholar]
- 26.McNab FW, Ewbank J, Howes A, Moreira-Teixeira L, Martirosyan A, Ghilardi N, Saraiva M, O'Garra A. 2014. Type I IFN induces IL-10 production in an IL-27-independent manner and blocks responsiveness to IFN-γ for production of IL-12 and bacterial killing in Mycobacterium tuberculosis-infected macrophages. J Immunol 193:3600–3612. doi: 10.4049/jimmunol.1401088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim YO, Jung MJ, Choi JK, Ahn do W, Song KS. 2011. Peptidoglycan from Staphylococcus aureus increases MUC5AC gene expression via RSK1-CREB pathway in human airway epithelial cells. Mol Cells 32:359–365. doi: 10.1007/s10059-011-0118-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Panzarini E, Ramires PA, Miccoli MA, Tenuzzo B, Scordari A, Dini L. 2006. Differentiation of THP-1 and U937 cells in presence of synthetic hydrogels. Caryologia 59:395–402. [Google Scholar]
- 29.Legdeur MC, Bontje PM, Ossenkoppele GJ, Beelen RH, van de Loosdrecht AA, Broekhoven MG, Langenhuijsen MM, Thijsen SF, Hofstee H, Schuurhuis GJ. 1996. The role of BCL-2 and bax protein in monocyte-mediated apoptosis in human leukemic cell lines. Exp Hematol 24:1530–1539. [PubMed] [Google Scholar]
- 30.Nonaka T, Kuwabara T, Mimuro H, Kuwae A, Imajoh-Ohmi S. 2003. Shigella-induced necrosis and apoptosis of U937 cells and J774 macrophages. Microbiology 149:2513–2527. doi: 10.1099/mic.0.26341-0. [DOI] [PubMed] [Google Scholar]
- 31.Virok D, Loboda A, Kari L, Nebozhyn M, Chang C, Nichols C, Endresz V, Gonczol E, Berencsi K, Showe MK, Showe LC. 2003. Infection of U937 monocytic cells with Chlamydia pneumoniae induces extensive changes in host cell gene expression. J Infect Dis 188:1310–1321. doi: 10.1086/379047. [DOI] [PubMed] [Google Scholar]
- 32.Cao B, Mysorekar IU. 2014. Intracellular bacteria in placental basal plate localize to extravillous trophoblasts. Placenta 35:139–142. doi: 10.1016/j.placenta.2013.12.007. [DOI] [PubMed] [Google Scholar]
- 33.Monneret G, Voirin N, Krawice-Radanne I, Bohé J, Lepape A, Rouas-Freiss N, Carosella ED. 2007. Soluble human leukocyte antigen-G5 in septic shock: marked and persisting elevation as a predictor of survival. Crit Care Med 35:1942–1947. doi: 10.1097/01.CCM.0000277039.84372.1C. [DOI] [PubMed] [Google Scholar]
- 34.Kovach MA, Standiford TJ. 2012. The function of neutrophils in sepsis. Curr Opin Infect Dis 25:321–327. doi: 10.1097/QCO.0b013e3283528c9b. [DOI] [PubMed] [Google Scholar]
- 35.Bankey PE, Banerjee S, Zucchiatti A, De M, Sleem RW, Lin CF, Miller-Graziano CL, De AK. 2010. Cytokine induced expression of programmed death ligands in human neutrophils. Immunol Lett 129:100–107. doi: 10.1016/j.imlet.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]