

Abstract
Induction of cell cycle arrest in lymphocytes following exposure to the Aggregatibacter actinomycetemcomitans cytolethal distending toxin (Cdt) is dependent upon the integrity of lipid membrane microdomains. Moreover, we have previously demonstrated that the association of Cdt with target cells involves the CdtC subunit which binds to cholesterol via a cholesterol recognition amino acid consensus sequence (CRAC site). In this study, we demonstrate that the active Cdt subunit, CdtB, also is capable of binding to large unilamellar vesicles (LUVs) containing cholesterol. Furthermore, CdtB binding to cholesterol involves a similar CRAC site as that demonstrated for CdtC. Mutation of the CRAC site reduces binding to model membranes as well as toxin binding and CdtB internalization in both Jurkat cells and human macrophages. A concomitant reduction in Cdt-induced toxicity was also noted, indicated by reduced cell cycle arrest and apoptosis in Jurkat cells and a reduction in the proinflammatory response in macrophages (interleukin 1β [IL-1β] and tumor necrosis factor alpha [TNF-α] release). Collectively, these observations indicate that membrane cholesterol serves as an essential ligand for both CdtC and CdtB and, further, that this binding is necessary for both internalization of CdtB and subsequent molecular events leading to intoxication of cells.
INTRODUCTION
Aggregatibacter actinomycetemcomitans is a Gram-negative organism that is associated with aggressive forms of periodontitis and other systemic infections (1–5). Periodontitis is a chronic infectious inflammatory disorder that ultimately leads to the destruction of tooth-supporting tissue. While the exact nature of the pathogenesis of periodontal disease and the contribution of bacteria to this process are not known, it is becoming increasingly clear that A. actinomycetemcomitans produces several potential virulence factors; these include adhesins and fimbria which have been shown to contribute to colonization of the human oral cavity as well as two exotoxins, cytolethal distending toxin (Cdt) and leukotoxin, both of which are capable of killing and/or altering the function of host immune cells (4, 6–8).
The Cdts are a family of heat-labile protein cytotoxins produced by several additional bacterial species, including Campylobacter jejuni, Shigella species, Haemophilus ducreyi, and diarrheal disease-causing enteropathogens such as some Escherichia coli isolates (9–15). There is clear evidence that Cdts are encoded by three genes, designated cdtA, cdtB, and cdtC, which are arranged as an apparent operon (7, 15–20). The Cdt holotoxin consists of three subunits, CdtA, CdtB, and CdtC, that form a heterotrimeric complex. Furthermore, there is considerable agreement among investigators that, regardless of the microbial source of Cdt, the heterotrimeric holotoxin functions as an AB2 toxin where CdtB is the active (A) unit and the complex of CdtA and CdtC comprises the binding (B) unit (18, 21, 22). Indeed, several investigators have demonstrated that the internalization of CdtB requires the presence of both CdtA and CdtC (21, 23, 24).
While several cell types and cell lines have been shown to be susceptible to the toxic actions of Cdt, tropism for specific cells and/or tissue remains to be identified. In this regard, we have demonstrated that lymphocytes in vitro are most susceptible, requiring very low concentrations of Cdt (picograms/milliliter) to induce cell cycle arrest and apoptosis versus other cell types that typically require as much as microgram quantities (25). Typically, susceptibility to bacterial toxins is dependent upon the expression of specific receptors or moieties that enable the toxin to preferentially associate with target host cells. Structural analysis of CdtA and CdtC identified ricin-like lectin domains, suggesting that these units interact with cell surface carbohydrate moieties (18). Several investigators have further demonstrated that, depending on the Cdt source, toxin binding to target cells was dependent upon cell surface N-linked glycoproteins, fucose, glycans, or glycosphingolipid (26, 27).
In previous studies we have demonstrated that A. actinomycetemcomitans Cdt subunits CdtA and CdtC are not only required for the toxin to associate with lymphocytes but also responsible for localizing the toxin to lipid membrane microdomains (28, 29). Furthermore, Cdt-mediated toxicity was found to be dependent upon the integrity of these lipid domains. Previously, we demonstrated that toxin association with lymphocytes, delivery of CdtB to intracellular targets, and the induction of both cell cycle arrest and apoptosis were dependent upon cholesterol (29). Specifically, we have shown that the CdtC subunit contains a cholesterol recognition amino acid consensus (CRAC) site that binds to cholesterol in the context of lipid membrane microdomains. More recently, other investigators have also demonstrated CRAC sites on Cdt produced by Haemophilus parasuis and C. jejuni (30, 31). We now report that in addition to CdtC, the active subunit CdtB also contains a CRAC site that is required for its internalization and the induction of toxicity in both lymphocytes and macrophages.
MATERIALS AND METHODS
Cell culture and analysis for cell cycle and apoptosis.
The human leukemic T cell line, Jurkat (E6-1), was maintained in RPMI 1640 medium supplemented with 10% fetal calf serum (FCS), 2 mM glutamine, 10 mM HEPES, 100 U/ml penicillin, and 100 μg/ml streptomycin. Cells were harvested in mid-log growth phase and plated at 5 × 105 cells/ml in 24-well tissue culture plates. Cells were exposed to medium, CdtA, and CdtC along with wild-type CdtB (CdtBWT) or CdtB mutants for 18 h (cell cycle) or 48 h (apoptosis). To measure Cdt-induced cell cycle arrest, cells were incubated for the time indicated in the figures and then washed and fixed for 60 min with cold 80% ethanol (28). The cells were stained with 10 μg/ml propidium iodide containing 1 mg/ml RNase (Sigma Chemical) for 30 min. Samples were analyzed on a Becton Dickinson LSRII flow cytometer (BD Biosciences) as previously described (28). A minimum of 15,000 events were collected for each sample; cell cycle analysis was performed using Modfit (Verity Software House).
DNA fragmentation in Cdt-treated Jurkat cells was employed to determine the percentage of apoptotic cells using a terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) assay (In Situ Cell Death Detection Kit; Boehringer Mannheim, Indianapolis, IN). Jurkat cell cultures were prepared as described above; at the end of the incubation period cells were centrifuged, resuspended in 1 ml of freshly prepared 4% formaldehyde, and vortexed gently. After 30 min at room temperature (RT), the cells were washed with phosphate-buffered saline (PBS) and permeabilized in 0.1% Triton X-100 for 2 min at 4°C. The cells were then washed with PBS and incubated in a solution containing fluorescein isothiocyanate (FITC)-labeled nucleotide and terminal deoxynucleotidyl transferase (TdT) according to the manufacturer's specifications. Following the final wash, the cells were resuspended in PBS and analyzed by flow cytometry.
The human acute monocytic leukemia cell line THP-1 was obtained from ATCC; cells were maintained in RPMI 1640 medium containing 10% fetal bovine serum (FBS), 1 mM sodium pyruvate, 20 μM 2-mercaptoethanol, and 2% penicillin-streptomycin at 37°C with 5% CO2 in a humidified incubator. THP-1 cells were differentiated into macrophages by incubating cells in the presence of 50 ng/ml phorbol myristate acetate (PMA) for 48 h, at which time the cells were washed and incubated for an additional 24 h in medium prior to use.
Construction and expression of plasmid containing CdtB mutant genes.
Amino acid substitutions were introduced into the cdtB gene by in vitro site-directed mutagenesis using oligonucleotide primer pairs containing appropriate base changes (Table 1). Site-directed mutagenesis was performed using a QuikChange II site-directed mutagenesis kit (Stratagene) according to the manufacturer's directions. Amplification of the mutant plasmid was carried out using Pfu Ultra HF DNA polymerase (Stratagene). All mutant constructs utilized pGEMCdtB as a template; construction and characterization of this plasmid were previously described (28).
TABLE 1.
CdtB CRAC site mutant constructs
| Plasmid | Primer | Sequence |
|---|---|---|
| pGEMCdtBV104P | P1 | CCGTCCAAATATGCCCTATATTTATTATTCCCG |
| P2 | CGGGAATAATAAATATAGGGCATATTTGGACGG | |
| pGEMCdtBY105P | P3 | GGTACTCGCTCCCGTCCAAATATGGTCCCTATTTATTATTCCCG |
| P4 | CGGGAATAATAAATAGGGACCATATTTGGACGGGAGCGAGTACC | |
| pGEMCdtBY107P | P5 | GGTCTATATTCCCTATTCCCGTTTAGATGTTGG |
| P6 | CCAACATCTAAACGGGAATAGGGAATATAGACC | |
| pGEMCdtBR110P | P7 | GGTCTATATTTATTATTCCCCTTTAGATGTTGG |
| P8 | CCAACATCTAAAGGGGAATAATAAATATAGACC |
In vitro expression of Cdt peptides and CdtB mutants was performed using a rapid translation system (RTS 500 ProteoMaster; Roche Applied Science) as previously described (28). Reactions were run according to the manufacturer's specifications (Roche Applied Science) using 10 to 15 μg of template DNA. After 20 h at 30°C, the reaction mix was removed, and the expressed Cdt peptides were purified by nickel affinity chromatography as described previously (28). Each CdtB peptide contains a His tag on its C terminus; previous studies have demonstrated that the presence of the His does not interfere with biological activity (24).
Immunofluorescence and flow cytometry.
Jurkat cells or THP-1-derived macrophages (2 × 106) were incubated for 30 min (surface staining) or 1 h (intracellular staining) in the presence of medium or with 2 μg/ml of CdtBWT or mutant in the presence of CdtA and CdtC at 37°C (internalization) or 5°C (surface binding). Surface CdtC was detected by washing cells and exposing them to normal mouse IgG (Zymed Labs; San Francisco, CA); cells were then stained (30 min) for CdtC peptides with anti-CdtC subunit monoclonal antibody (MAb) conjugated to Alexa Fluor 488 (Molecular Probes, Eugene, OR) according to the manufacturer's directions. After cells were washed, they were fixed in 2% paraformaldehyde and analyzed by flow cytometry as previously described (29). Intracellular CdtB was detected after exposure of cells to toxin (above) and fixation with 2% formaldehyde for 30 min, followed by permeabilization with 0.1% Triton X-100 in 0.1% sodium citrate and staining with anti-CdtB MAb conjugated to Alexa Fluor 488 (Molecular Probes).
Measurement of cytokine production.
Cytokine production was measured in THP-1-derived macrophages (2 × 105 cells) incubated for 5 h. Culture supernatants were collected and analyzed by enzyme-linked immunosorbent assay (ELISA) for interleukin-1β (IL-1β) (Quantikine ELISA kit; R&D Systems) and tumor necrosis factor alpha (TNF-α; Peprotech) using commercially available kits according to the manufacturers' instructions. In each instance, the amount of cytokine present in the supernatant was determined using a standard curve.
Preparation of LUVs and biolayer interferometry (BLI).
Phosphatidylcholine (PC), phosphatidylethanolamine (PE)-biotin, phosphatidylserine (PS), sphingomyelin (SM), and cholesterol were obtained from Avanti Polar Lipids; lipids were stored in chloroform at −20°C with desiccation. Briefly, large unilamellar vesicles (LUVs) were prepared with a PC/SM/PE lipid ratio of 50:13:37 (mol%), or as indicated in the figure legends, in the presence of various amounts of cholesterol as previously described (32, 33). For some experiments LUV cholesterol was replaced with stigmasterol as described in the figure legends. Lipids were cosolubilized in chloroform and dried under N2, and trace amounts of residual solvent were removed under high vacuum. The lipid mixtures were rehydrated at a concentration of 8 mM (total phospholipid) in 20 mM Tris-HCl, pH 7.4, containing 50 mM NaCl for 1 h; the suspensions were then vortexed and freeze-thawed in liquid nitrogen, and LUVs were prepared by extrusion by 11 passes through a 100-nm-pore-size membrane (Mini Extruder; Avanti Polar Lipids).
BLI analyses were performed using an Octet QKe system (FortéBio) with LUVs comprised of biotinylated PE immobilized on streptavidin sensors in PBS containing 1 mM n-octyl-β-d-glucopyranoside (OG). Assays were performed in black 96-well plates with a total working volume of 200 μl per well at 30°C with an rpm setting of 1,000. Following immobilization of the substrate (LUVs), free streptavidin moieties on the BLI sensors were blocked with biocytin. Measurements of interactions between LUVs and CdtB (and mutants) were performed by incubating the immobilized substrate with various concentrations of CdtBWT (or mutants) in PBS containing 1 mM OG. CdtB was allowed to associate with LUVs for 10 min, followed by a 10-min dissociation in binding buffer alone. After subtraction of the signal from a buffer-alone blank sensor, the data were analyzed using Octet data analysis software using a 1:1 global fit model to calculate the affinity constant (KD).
Phosphatase assay.
Phosphatase activity was assessed by monitoring the dephosphorylation of phosphatidylinositol-3,4,5-triphosphate (PI-3,4,5-P3) as previously described (25, 34). Briefly, the reaction mixture (20 μl) consisted of 100 mM Tris-HCl (pH 8.0), 10 mM dithiothreitol, 0.5 mM diC16-phosphatidylserine (Avanti), 25 μM PI-3,4,5-P3 (diC16; Echelon), and the amount of CdtB or mutant as indicated in the figures. Appropriate amounts of lipid solutions were deposited in 1.5-ml tubes, organic solvent was removed, the buffer was added, and a lipid suspension was formed by sonication. Phosphatase assays were carried out at 37°C for 30 min; the reactions were terminated by the addition of 15 μl of 100 mM N-ethylmaleimide. Inorganic phosphate levels were then measured using a malachite green assay. Malachite green solution (Biomol Green; Biomol) was added to 100 μl of the enzyme reaction mixture, and color was developed for 20 min at RT. Absorbance at 650 nm was measured, and phosphate release was quantified by comparison to inorganic phosphate standards.
Statistical analysis.
Means ± standard errors of the means (SEM) were calculated for replicate experiments. Significance was determined using Student's t test; differences between multiple treatments were compared by analysis of variance (ANOVA) paired with Tukey's honestly significant difference (HSD) posttest. A P value of less than 0.05 was considered to be statistically significant.
RESULTS
It is generally accepted that the association of Cdt holotoxin with target cell membranes involves a binding component comprised of the CdtA and CdtC subunits; moreover, this interaction is required for subsequent delivery of the active subunit, CdtB, to intracellular compartments. We have previously demonstrated that the CdtC subunit contains a CRAC region that is involved in CdtC binding to cholesterol within the plasma membrane as well as to model membranes (33). In addition to CdtC, we have now determined that CdtB also contains a CRAC site, 104VYIYYSR110. The CRAC site conforms to the pattern (L/V)-X1–5-Y-X1–5-(R/K) in which X1–5 represents between one and five residues of any amino acid. Figure 1 shows the position (yellow) of the CdtB CRAC site based upon CdtB's crystal structure; this putative cholesterol-binding region is clearly spatially separated from either of the Cdt binding subunits, CdtA or CdtC. Two edges of the site are exposed to the surface while the middle is buried within the structure, suggesting that it would not be very accessible. While the CRAC site would appear to have minimal access for binding, there is a loop, 81IQHGGTPI88, that is potentially flexible enough to move out of the way and provide access to the CRAC site. Moreover, the hydrophobic stretch of the CRAC site, 104VYIYY108, is compatible with hydrophobic binding that might occur in the context of membrane lipid microdomains. Based upon these molecular models and mutational analysis of critical residues performed on CRAC regions of other proteins, we decided to generate four single-point mutants, CdtBV104P, CdtBY105P, CdtBY107P, and CdtBR110P, to enable us to study the contribution of the CRAC region to toxin interaction with model membranes and host cells.
FIG 1.
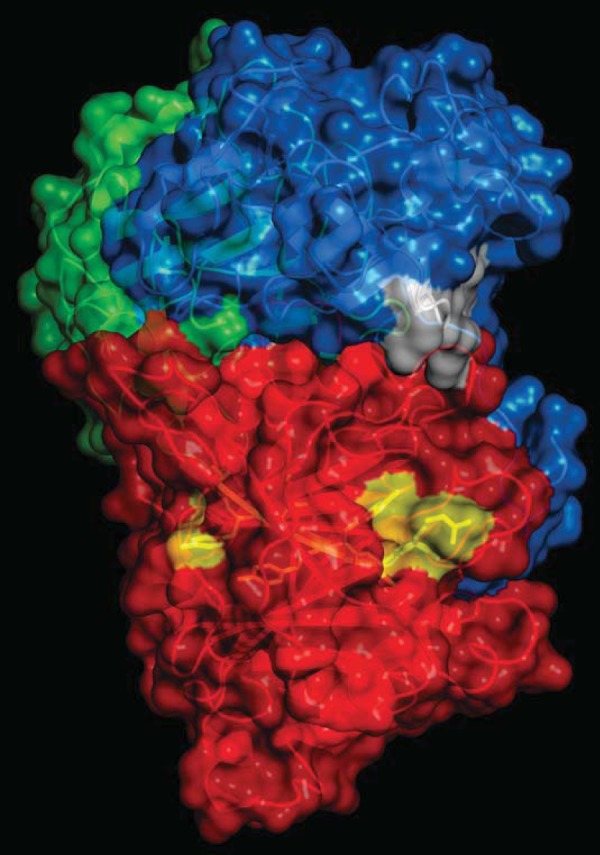
Localization of the CRAC sites on Cdt. A surface representation of Cdt holotoxin is shown indicating the accessibility of the CdtB CRAC site (yellow) and the CdtC CRAC site (white). CdtA is shown in green, CdtC is in blue, and CdtB is in red.
We previously employed surface plasmon resonance (SPR) to assess the role of the CRAC region within the CdtC subunit and its interaction with cholesterol-containing LUVs. In our current study, biolayer interferometry (BLI) was employed to assess the CdtB-associated CRAC region; BLI is a label-free technology that measures molecular interactions in real time for the purpose of detecting and quantifying them and performing kinetic analysis (35). We initially employed the Cdt holotoxin and compared the affinity constant (KD) obtained with BLI to that previously determined with SPR. Cholesterol-containing LUVs prepared with PE-biotin were immobilized on streptavidin biosensor plates; Cdt holotoxin (0 to 50 μM) was added to the wells, and the association and dissociation between toxin and LUV were determined (see Fig. S1 in the supplemental material). The KD was determined to be 2.03 × 10−6 M, in contrast to the value 3.09 × 10−6 M previously determined by SPR (33). We next assessed the binding of the CdtB subunit by adding various concentrations of CdtBWT (0 to 50 μM) to wells. The association and dissociation between toxin and LUV were then measured over a 10-min period. Figure 2A shows a representative series of overlay responses (both real data and modeled data are plotted) for various concentrations of CdtBWT; based on these responses, the KD was calculated to be 1.37 × 10−6 ± 0.2 × 10−6 M (Fig. 2E). Previously, we determined that the CdtC subunit binding to LUV was not only cholesterol dependent but also sterol specific. Therefore, to determine if CdtB exhibits similar properties to CdtC, LUVs were prepared with 20% cholesterol or 20% stigmasterol or without sterol (33). As shown in Fig. 2D, CdtBWT binding to LUV was sterol dependent and specific; maximum binding to LUV containing cholesterol was 0.24 ± 0.02 nm, a measurement of increased sensor thickness, whereas binding to LUV containing stigmasterol or to LUV prepared without sterol resulted in reduced sensor thickness (binding) (0.12 ± 0.02 nm or 0.04 ± 0.01 nm, respectively).
FIG 2.

CdtB binding to LUVs containing cholesterol is dependent upon the CRAC site. LUVs containing PE-biotin were immobilized on streptavidin sensors, and CdtBWT or CdtB CRAC site-containing mutants served as the analytes. (A to C) Representative BLI sensorgrams for the interaction of various concentrations of CdtBWT, CdtBV104P, and CdtBR110P with LUVs containing 20% cholesterol; the experimental line (black) is shown along with the model fit (red line). (D) Relative binding of 25 μM CdtBWT to LUVs containing 20% cholesterol, 20% stigmasterol, or 0% cholesterol; results from three experiments are plotted as the means ± SEM. (E) KD (means ± SEM) values determined from three independent experiments.
We next compared the binding affinity of CdtB containing CRAC site mutants to that of the wild-type protein. Three of the mutants, CdtBV104P, CdtBY105P, and CdtBY107P, exhibited significantly reduced ability to bind to cholesterol-containing LUVS; the KD for CdtBV104P was determined to be 2.08 × 10−5 ± 0.8 × 10−5 M; CdtBY105P and CdtBY107P binding was too weak, and the KD value could not be calculated even when higher protein concentrations were employed (Fig. 2B and E). In contrast to the these three mutants, a fourth CRAC site mutant, CdtBR110P, retained its ability to bind to cholesterol-containing LUVs; this mutant exhibited an increase in binding, with a KD of 3.35 × 10−8 ± 1.9 × 10−8 M (Fig. 2C and E). It should be noted that changes in the ability of the CdtB mutant proteins to bind to LUVs were not likely due to alterations in structure; circular dichroism (CD) analysis of these proteins failed to demonstrate significant differences among these mutants relative to CdtBWT (see Fig. S2 in the supplemental material).
In previous studies, we demonstrated that Cdt holotoxin binds to target cells with outcomes that are cell type specific. For example, exposure of lymphocytes to Cdt results in cell cycle arrest and apoptosis; in contrast, toxin-treated macrophages derived from either the human THP-1 cell line or monocytes do not become apoptotic but instead are induced to synthesize and secrete proinflammatory cytokines (17, 28, 36). It should be pointed out that toxin binding to both lymphocytes and macrophages was dependent upon cholesterol and the CRAC region on CdtC. Therefore, we next determined if the CRAC site on CdtB was also important for internalization of this subunit in both lymphocytes and macrophages. Jurkat cells were treated with Cdt subunits as described in Materials and Methods and then permeabilized, stained with anti-CdtB MAb conjugated to Alexa Fluor 488, and analyzed by flow cytometry. Cells treated with subunits CdtA and CdtC as well as untreated cells served as controls and exhibited minimal fluorescence; mean channel fluorescence (MCF) was 5.7 and 7.2, respectively (Fig. 3A). Intracellular CdtB was detected in cells exposed to CdtBWT in the presence of CdtA and CdtC subunits exhibiting an MCF of 24.9 (Fig. 3B). The three CdtB CRAC site mutants, CdtBV104P, CdtBY105P, and CdtBY107P, which failed to interact with cholesterol-containing LUVs also were unable to enter Jurkat cells; MCF values for these proteins were 4.3, 6.1, and 3.6, respectively (Fig. 3C to E). In contrast, cells exposed to the mutant that retained cholesterol-binding capability with LUVs, CdtBR110P, contained detectable protein (Fig. 3F) (MCF of 29). Pooled results from multiple experiments are shown in Fig. 4A. MCF values were reduced to 17.2% ± 0.6% (CdtBV104P), 34.1% ± 5.6%(CdtBY105P), and 16.9% ± 0.8% (CdtBY107P) of the value observed for CdtBWT while CdtBR110P exhibited an MCF of 104.4% ± 11.7%. Similar results were observed with THP-1-derived macrophages (Fig. 5); control cells and cells exposed to CdtA and CdtC exhibited background fluorescence of 8.6, whereas cells exposed to CdtBWT contained detectable intracellular CdtB (MCF of 33.9). CdtB was not detectable in macrophages treated with CdtBV104P (MCF of 8.5), CdtBY105P (MCF of 6.7), and CdtBY107P (MCF of 11.3) while cells treated with CdtBR110P exhibited increased immunofluorescence (MCF of 37). As shown in Fig. 4B, results from multiple experiments demonstrate significant reductions in intracellularly associated immunofluorescence of 25.4% ± 11.6% (CdtBV104P), 28.1% ± 3.8% (CdtBY105P), and 29.1% ± 13.5% of that observed with CdtBWT, while CdtBR110P exhibited an increase (144.3% ± 25.1%) in fluorescence that was not statistically significant.
FIG 3.

Immunofluorescence analysis of internalization of CdtB CRAC site mutants in Jurkat cells. Jurkat cells were exposed to medium alone (gray curves), CdtA and CdtC alone (A), and CdtA and CdtC in the presence of CdtBWT (B), CdtBV104P (C), CdtBY105P (D), CdtBY107P (E), or CdtBR110P (F) for 1 h and then analyzed by immunofluorescence and flow cytometry for the presence of CdtB following fixation, permeabilization, and staining with anti-CdtB MAb conjugated to Alexa Fluor 488. Fluorescence is plotted versus relative cell number. Numbers represent the mean channel fluorescence (MCF). Note that the MCF for cells not exposed to any Cdt peptide was 5.7. At least 10,000 cells were analyzed per sample; results are representative of three experiments.
FIG 4.
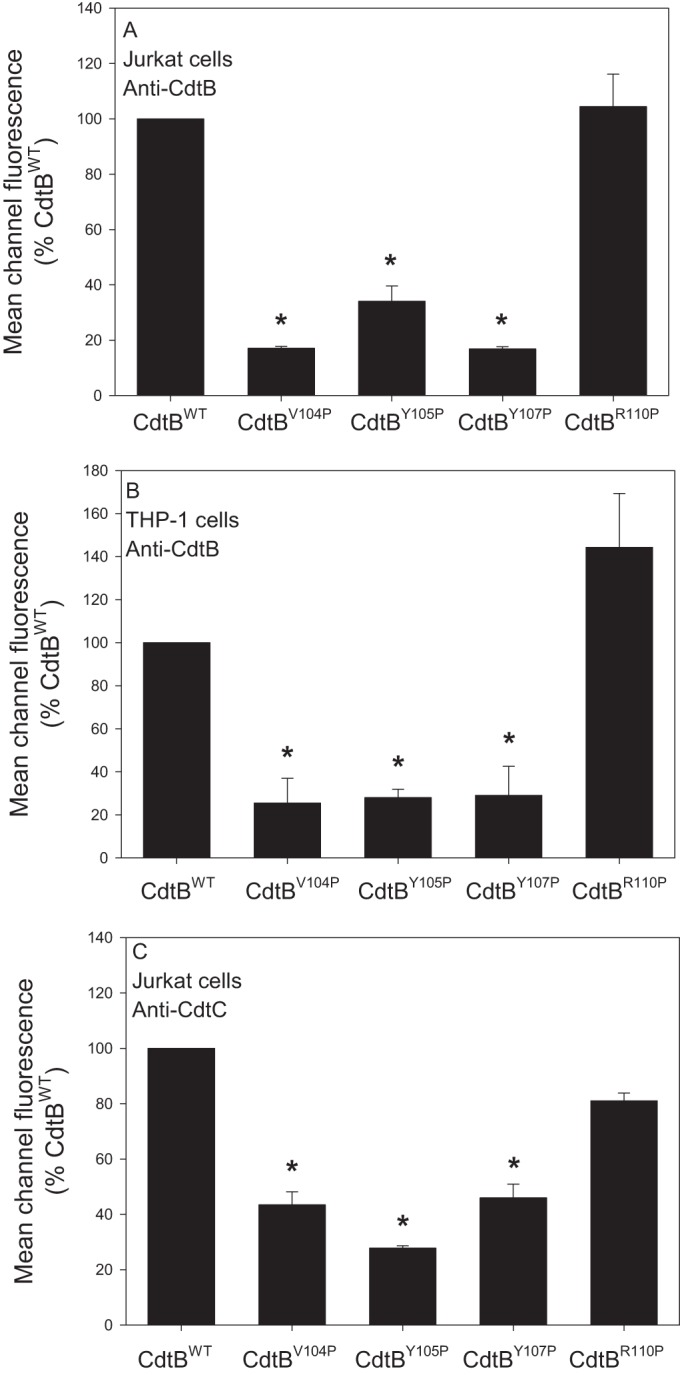
Cumulative results of immunofluorescence assessment of CdtB internalization and Cdt binding. (A) Results obtained from three experiments for internalization of CdtB in Jurkat cells. (B) Results obtained from three experiments for internalization of CdtB in THP-1 cells. (C) Results obtained from three experiments for holotoxin binding to the cell surface determined by immunofluorescence staining for the presence of CdtC in the absence of permeabilization. Results in each panel are expressed as a percentage of the MCF observed in CdtBWT and represent the means ± SEM *, P < 0.05, compared to results with the wild-type protein.
FIG 5.

Immunofluorescence analysis of internalization of CdtB CRAC site mutants in THP-1-derived macrophages. Macrophages were exposed to medium alone (gray curves), CdtA and CdtC alone (A), and CdtA and CdtC in the presence of CdtBWT (B), CdtBV104P (C), CdtBY105P (D), CdtBY107P (E), or CdtBR110P (F) for 1 h and then analyzed by immunofluorescence and flow cytometry for the presence of CdtB following fixation, permeabilization, and staining with anti-CdtB MAb conjugated to Alexa Fluor 488. Fluorescence is plotted versus relative cell number. Numbers represent the mean channel fluorescence (MCF). Note that the MCF for cells not exposed to any Cdt peptide was 8.6. At least 10,000 cells were analyzed per sample; results are representative of three experiments.
We have previously demonstrated that non-cholesterol-binding CdtC CRAC site mutants prevent toxin binding to target cells (33). Therefore, we wanted to determine if, in addition to preventing internalization, the CdtB CRAC site mutants also affected the ability of holotoxin to associate with cells. For these studies, Jurkat cells were treated as described above except that the incubation was performed at 5°C, which we have previously shown blocks CdtB internalization but does not interfere with toxin binding to the cell surface. Cells were then analyzed for the presence of the CdtC subunit on the cell surface as cells were stained without permeabilization. Results are shown in Fig. 6 and demonstrate that CdtC was detectable on cells exposed to CdtBWT; the MCF was 16.2 (Fig. 6B) versus 5.5 and 5.9 in control cells exposed to medium only or Cdt subunits CdtA and CdtC only (Fig. 6A). Cells exposed to Cdt containing either CdtBV104P, CdtBY105P, or CdtBY107P did not contain detectable CdtC on the surface, with MCF values of 5.6, 4.6, and 5.9 (Fig. 6C, D, and E), respectively. In contrast, cells treated with toxin containing CdtBR110P did contain detectable CdtC on the surface (MCF of 12.2). Figure 4C shows pooled results from multiple experiments which demonstrate that the reductions in toxin binding were significant for CdtBV104P (43.5% ± 4.78%), CdtBY105P (27.8% ± 0.8%), and CdtBY107P (45.9% ± 5.0%).
FIG 6.

Immunofluorescence analysis of surface-associated CdtC in Jurkat cells. Jurkat cells were exposed to medium alone (gray curves), CdtA and CdtC alone (A), and CdtA and CdtC in the presence of CdtBWT (B), CdtBV104P (C), CdtBY105P (D), CdtBY107P (E), or CdtBR110P (F) for 30 min and then analyzed by immunofluorescence and flow cytometry for the presence of CdtC following fixation and staining with anti-CdtC MAb conjugated to Alexa Fluor 488. Fluorescence is plotted versus relative cell number. Numbers represent the mean channel fluorescence (MCF). Note that the MCF for cells not exposed to any Cdt peptide was 5.5. At least 10,000 cells were analyzed per sample; results are representative of three experiments.
It is well established that toxin binding and subsequent internalization of CdtB are required for intoxication of cells (37, 38) although the specific intracellular target is somewhat controversial (i.e., nuclear versus cytoplasmic versus membrane). Nonetheless, to corroborate that the three CdtB CRAC site mutants were unable to enter target cells, we next assessed their ability to induce toxicity in both lymphocytes and macrophages. The effect of CdtB mutants on Jurkat cell cycle arrest is shown in Fig. 7. Cells exposed to medium alone exhibited 9% G2 cells. In contrast, cells treated with CdtBWT exhibited an increasing percentage of G2 cells as the dose of toxin increased: 21% with 0.8 ng/ml CdtBWT, 29% with 4.0 ng/ml CdtBWT, and 42% with 20 ng/ml CdtBWT. Likewise, cells treated with the same doses of toxin comprised of CdtBR110P also exhibited increases in the percentage of G2 cells (11%, 17%, and 26%, respectively). However, Jurkat cells treated with CdtBV104P, CdtBY105P, or CdtBY107P failed to exhibit cell cycle arrest as the percentage of G2 cells was similar to that observed in control cells regardless of protein concentration.
FIG 7.

Assessment of CdtB CRAC site mutants for their ability to induce G2 arrest in Jurkat cells. Jurkat cells were exposed to medium alone or 10 ng/ml each of CdtA and CdtC in the presence of 0.8 to 20.0 ng/ml CdtBWT (A to C), CdtBV104P (D to F), CdtBY105P (G to I), CdtBV107P (J to L), or CdtBR110P (M to O). Cells were analyzed for cell cycle distribution 18 h after exposure to toxin subunits using flow cytometric analysis of propidium iodide fluorescence (28). The numbers in each panel represent the percentages of cells in the G2/M phase of the cell cycle. Cells exposed to medium alone exhibit 9.2% G2 cells, and cells treated with only 10 ng/ml each of CdtA and CdtC exhibited 9.0% G2 cells (data not shown). Results are representative of three experiments.
In addition to cell cycle analysis, toxin-treated Jurkat cells were assessed for apoptosis using a TUNEL assay 48 h following exposure to the same protein concentrations utilized for cell cycle analysis. Control cells (medium only) as well as cell cultures exposed to the CdtA and CdtC subunits exhibited only 19% apoptosis. As shown in Fig. 8, Jurkat cells treated with CdtBWT exhibited dose-dependent increases in the percentages of apoptotic cells: 42% ±13.9% with 4 ng/ml CdtB and 53.1% ± 10.2% with 20 ng/ml CdtB. The CRAC site mutant that retained the ability to bind to cholesterol-containing LUVs, CdtBR110P, also retained the ability to induce Jurkat cell apoptosis; exposure to this mutant resulted in 25.6% ± 9.2% (4 ng/ml CdtBR110P) and 41.6% ± 11.4% (20 ng/ml CdtBR110P) apoptotic cells. In contrast, treatment of lymphocytes with either of the non-cholesterol-binding CdtB CRAC site mutants (0.4 to 20 ng/ml), CdtBV104P, CdtBY105P, or CdtBY107P, failed to result in apoptosis.
FIG 8.
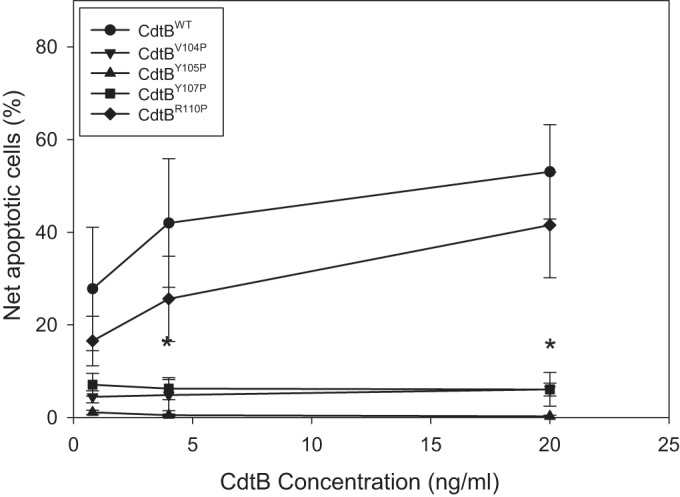
Effect of CdtB CRAC site mutants on DNA fragmentation in Jurkat cells. Jurkat cells were treated with 10 ng/nl each of CdtA and CdtC in the presence of 0 to 20 ng/ml CdtBWT (●), CdtBV104P (▼), CdtBY105P (▲), CdtBY107P (■), or CdtBR110P (◆) for 48 h. The cells were then analyzed by flow cytometry for the presence of DNA fragmentation as described in Materials and Methods. Results are plotted as net percent apoptotic cells versus CdtB concentration and represent the means ± SEM of three experiments. *, P < 0.05, compared to results for the wild-type protein.
In previous studies, we have identified macrophages as another potential target of Cdt (36). However, macrophages derived from either human blood monocytes or the monocytic leukemic cell line THP-1 were not susceptible to Cdt-induced apoptosis. Instead, Cdt induced a proinflammatory cytokine response in these cells within 2 h. Moreover, toxin interaction with these cells was shown to be cholesterol dependent as toxin comprised of CdtC containing a CRAC site mutant failed to bind to macrophages and was unable to induce cytokine synthesis and release. Therefore, we also assessed the ability of CdtB-containing CRAC site mutants to induce a proinflammatory cytokine response in THP-1-derived macrophages. As shown in Fig. 9, treatment of macrophages with CdtBWT resulted in increased release of IL-1β; control cells produced 24.6 ± 1.3 pg/ml while exposure to 20 ng/ml CdtB resulted in 336.4 ± 29.3 pg/ml IL-1β. Similar results were obtained for TNF-α; control cells released 168.2 ± 17.9 pg/ml, and cells exposed to 20 ng/ml CdtB induced the release of 1,676.8 ± 261.0 pg/ml TNF-α. Treatment of macrophages with CdtBV104P, CdtBY105P, or CdtBY107P resulted in a significantly lower cytokine response than that observed with CdtBWT: 83.9 ± 12.0 pg/ml IL-1β and 481 ± 125.7 pg/ml TNF-α in the presence of CdtBV104P, 86.6 ± 14.7 pg/ml IL-1β and 477.1 ± 74.6 pg/ml TNF-α in the presence of CdtBY105P, and 96.2 ± 7.5 pg/ml IL-1β and 486.2 ± 88.3 pg/ml TNF-α in the presence of CdtBY107P. Although these values are significantly lower than those observed with CdtBWT, they do represent increases over background levels, suggesting that the mutants retained residual low-level binding. Macrophages exposed to CdtBR110P exhibited a robust dose-dependent cytokine response relative to the other two CdtB mutants but somewhat less than that observed with CdtBWT. CdtBR110P induced the release of 216.0 ± 11.9 pg/ml IL-1β and 1,091.4 ± 117.6 pg/ml TNF-α; these differences were not statistically different from those values obtained with CdtBWT.
FIG 9.
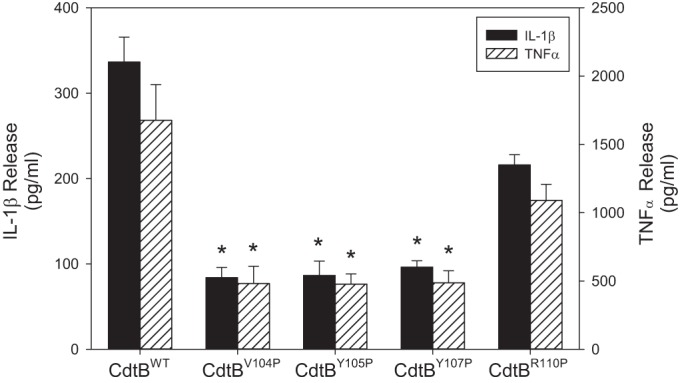
Effect of CdtB CRAC site mutants on macrophage release of IL-1β and TNF-α release. THP-1-derived macrophages were treated with 10 ng/ml each of CdtA and CdtC in the presence of 20 ng/ml CdtBWT, CdtBV104P, CdtBY105P, CdtBY107P, or CdtBR110P for 5 h, and the supernatants were analyzed by ELISA for IL-1β and TNF-α. Results are the means ± SEM for three experiments, each performed in triplicate. *, P < 0.05, compared to CdtBWT values. Cells exposed to medium alone released 24.6 ± 1.3 pg/ml IL-1β and 168.2 ± 17.9 pg/ml TNF-α.
In previous studies we have demonstrated that Cdt-induced toxicity in lymphocytes (cell cycle arrest and apoptosis) and macrophages (proinflammatory cytokine response) were dependent upon CdtB's ability to function as a PIP3 phosphatase (25, 36). Therefore, we next confirmed that our observations regarding the loss of toxicity associated with the CdtB CRAC site mutants was not due to altered phosphatase activity. CdtBWT was initially assessed for its ability to dephosphorylate PI-3,4,5-P3; as shown in Fig. 10, the wild-type subunit exhibits dose-dependent phosphate release, with amounts of 0.24 ± 0.04, 0.73 ± 0.20, and 1.39 ± 0.34 nmol/30 min in the presence of 0.25, 0.5, and 1.0 μM CdtB, respectively. All four CdtB mutants exhibited similar activities that were slightly, but not significantly, lower than those observed with CdtBWT. In the presence of 0.5 μM protein, phosphatase release was 0.42 ± 0.17 (CdtBV104P), 0.43 ± 0.3 (CdtBY105P), 0.50 ± 0.28 (CdtBY107P), and 0.52 ± 0.22 (CdtBR110P) nmol; phosphatase activity increased in the presence of 1.0 μM protein to 0.94 ± 0.21 (CdtBV104P), 0.94 ± 0.25 (CdtBY105P), 1.12 ± 0.20 (CdtBY107P), and 1.01 ± 0.21 (CdtBR110P) nmol.
FIG 10.
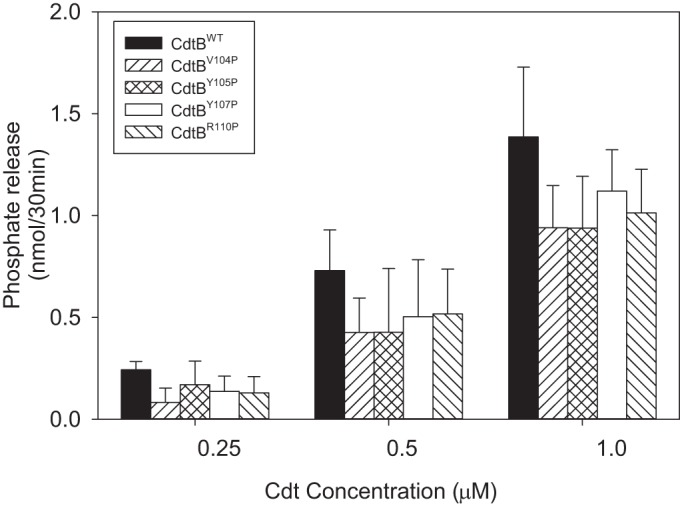
Assessment of CdtB CRAC site mutants for PIP3 phosphatase activity. Various amounts (0.25 to 1.0 μM) of CdtBWT, CdtBV104P, CdtBY105P, CdtBY107P, or CdtBR110P were assessed for their abilities to hydrolyze PI-3, 4, 5-P3 as described in Materials and Methods. The amount of phosphate release was measured using a malachite green binding assay. Data are plotted as phosphate release (nmol/30 min) versus protein concentration; results represent the means ± SEM for three experiments, each performed in triplicate. Phosphatase activity exhibited by CdtB CRAC site mutants was not statistically different from that of CdtBWT.
DISCUSSION
It is generally accepted that all three Cdt subunits, CdtA, CdtB, and CdtC, are required to achieve maximal toxic activity, regardless of the target cell. Thus, the holotoxin is believed to function as an AB2 toxin where the cell binding unit, B, is responsible for toxin binding to the cell surface and thereby delivers the active subunit, A, to intracellular compartments. In the context of Cdt, binding activity is considered to be the result of cooperative activities of both the CdtA and CdtC subunits (reviewed in reference 39). Moreover, it has been proposed that these subunits share structural homology with lectin-like proteins and, further, that fucose moieties might be involved in toxin association with the cell surface (18, 26). In other studies, glycosphingolipids have been implicated as possible binding sites since inhibitors of glycosphingolipid synthesis reduce toxin intoxication (27). It should be pointed out that in a more recent study in which Cdts from different microbial species were studied simultaneously, fucosylated structures as well as N- and O-glycans were found not to be required for Cdt-host cell interaction (40). In contrast, these authors observed that Cdt derived from three sources, A. actinomycetemcomitans, C. jejuni, and H. ducreyi, were each dependent upon membrane cholesterol, thereby confirming the previous observations of Boesze-Battaglia et al. (33) and Guerra et al. (37) and more recently those of Zhou et al. (30) and Lai et al. (31). These studies are opposed by other investigators who have reported that cholesterol depletion failed to alter toxin subunit internalization (41). It should be noted that this study, by Damek-Poprawa et al., is difficult to assess with respect to the role of cholesterol as the experimental protocol utilized a long exposure time to methyl-β-cyclodextrin, and, further, cholesterol repletion was not employed to demonstrate specificity.
We have previously demonstrated that the Cdt holotoxin colocalizes on the cell surface with the ganglioside GM1 in the context of cholesterol-rich membrane microdomains, or lipid rafts (29). Furthermore, disruption of lipid rafts by cholesterol depletion reduced toxin binding, CdtB internalization, and cell susceptibility to Cdt intoxication (33). We have also demonstrated cholesterol-specific binding of Cdt to both model membranes and lymphocyte membranes; furthermore, this binding is dependent upon a cholesterol recognition sequence within the CdtC subunit. In this regard, numerous proteins have been shown to bind cholesterol; these include the benzodiazepine receptor, the HIV transmembrane protein gp41, caveolin, G protein-coupled receptors, components of Ca++- and voltage-gated K+ channels, apolipoproteins, the translocator protein TSPO, and, more recently, the leukotoxin produced by A. actinomycetemcomitans (42–50). Each of these cholesterol-binding proteins contains a CRAC sequence, L/V-X1–5-Y-X1–5-R/K, where X1–5 represents one to five residues of any amino acid. Indeed, we have shown that the CdtC subunit of the A. actinomycetemcomitans Cdt also contains such a CRAC site, with the sequence 68LIDYKGK74. Mutation of residues within this region reduced toxin binding to LUVs and cells, CdtB internalization, and toxicity. In the current study, we have analyzed the active subunit of the A. actinomycetemcomitans Cdt, CdtB, for its ability to bind to LUVs containing cholesterol. Indeed, CdtBWT demonstrated high affinity for cholesterol-containing LUVs, and binding was significantly reduced when LUVs either did not contain any sterol or when stigmasterol was utilized as a substitute for cholesterol. These observations led us to consider the possibility that, like CdtC, CdtB also might contain a CRAC site; indeed motif analysis of CdtB confirmed the presence of the CRAC site 104VYIYYSR110.
In order to study the requirement for the CdtB CRAC site in binding to LUVs and cells, we mutated four residues within this site (104VYIYYSR110; mutations are shown in bold); we predicted that three of these would be critical to its binding function. First, CdtBV104P and CdtBY107P were selected because they represent critical residues within the CRAC motif. Also, they are very well conserved in all CdtB proteins with the exception that CdtBV104 is not conserved in toxin from bacteria that are facultative intracellular pathogens, such as Salmonella. Thus, Salmonella-derived CdtB most likely utilizes a different pathway to reach its intracellular target than that used by CdtB produced by bacteria that are associated with extracellular infection, such as A. actinomycetemcomitans. This raises the likelihood that CdtBV104P is a critical residue for cholesterol interaction since it is unlikely that the Salmonella-derived toxin is dependent upon a pathway that has a requirement for binding to plasma membrane cholesterol. Second, CdtBY105P and CdtBY107P were selected as they are among the most conserved residues in the entire CRAC site, and, therefore, there was a high likelihood that they would be critical to the function of the CRAC site. Finally, CdtBR110P was selected as this is the least conserved CRAC site residue among the CdtB proteins. In fact, several CdtB proteins from Helicobacter species have a proline conserved at this position instead of arginine. Therefore, we expected not only that this residue was not critical to CRAC site function but also that the proline substitution would also not contribute to any change in cholesterol binding. Thus, CdtBR110P served as a control for the other mutations.
Analysis of the CdtBV104P, CdtBY105P, and CdtBY107P mutants demonstrated reduced ability to bind to LUVs as well as decreased internalization in both lymphocytes and macrophages. Moreover, analysis of the effect of CdtB CRAC site mutants on Cdt holotoxin binding to the cell surface indicated that these CdtB mutations did not permit holotoxin association with target cells as well. These observations are consistent with our previous studies involving mutation of the CRAC region within CdtC (33) and with those of other investigators who made similar CRAC site mutations of residues within the CdtC subunit of Cdts produced by other bacterial species as well as to other cholesterol-binding proteins (30, 31, 42, 50). In each instance CRAC site mutations resulted in a loss of cholesterol binding and associated downstream biological effects; in contrast, CdtBR110P had no effect on Cdt binding and CdtB internalization, confirming our expectation for the relative role of each of these residues within the CRAC site and overall role in the CdtB-cholesterol interaction. It should be noted that we do not have a clear explanation for the observed increase in affinity for this mutant.
The CRAC domain is a short linear motif that proceeds from the N terminus to the C terminus and represents the most popular cholesterol-binding domain. More recently, Baier et al. (51) identified another cholesterol-binding motif, CARC, with a sequence similar to that of the CRAC domain but oriented along the polypeptide chain in the opposite direction. CARC constitutes an inverted CRAC domain, (K/R)-X1–5-(Y/F)-X1–5-(L/V), from the N terminus to the C terminus. Interestingly, we found that a CARC site exists in CdtB and is embedded within the CRAC site (shown in bold), with the sequence 100RPNMVYIYYSRL111. While we cannot entirely exclude the possibility that the CARC motif may also contribute to CdtB-cholesterol binding, we believe that the analysis of the CRAC site mutants is more consistent with a functional role for the CRAC motif. Furthermore, CdtBR100 is only moderately conserved among CdtB proteins.
The location of the CRAC site based upon molecular models of the crystal structure of CdtB indicates that the region has two exposed edges and a buried middle region. Our mutation analysis suggests that the lower region (Fig. 1) and buried middle region are critical to binding. These results would suggest that the structure of CdtB and the availability of the CRAC site are likely altered in the context of lipid rafts as opposed to the rigid crystal structure.
It should also be noted that in previous studies we demonstrated that depletion of cholesterol from lymphocyte membranes reduced Cdt holotoxin binding, CdtB internalization, and toxicity (33). Likewise, repletion studies in which membrane cholesterol was restored also resulted in reestablishment of toxin binding, CdtB internalization, and susceptibility to Cdt intoxication. We initially interpreted these results as evidence for the role of cholesterol and the CdtC CRAC site in toxin-cell interactions; however, it should now be noted that these observations are also consistent with our current findings for a critical role for cholesterol and the CdtB CRAC site as well.
Finally, we assessed the CdtB CRAC site mutants for their ability to intoxicate cells. For these studies we employed two host target cells: lymphocyte and macrophages. Jurkat cells treated with the three CdtB cholesterol-binding-defective mutants, CdtBV104P, CdtBY105P, and CdtBY107P, also exhibited reduced toxicity, which was reflected in fewer cells in G2 arrest at 18 h and fewer apoptotic cells at 48 h than amounts with CdtBWT. Likewise, the proinflammatory cytokine response that is induced in macrophages treated with wild-type toxin was reduced in cells treated with the defective cholesterol-binding mutants; this was reflected in reduced secretion of both IL-1β and TNF-α. The fourth CRAC site mutant, CdtBR110P, was unaltered in its ability to bind to cholesterol-containing LUVs and to deliver CdtB to intracellular compartments in both Jurkat cells and macrophages. Likewise, the CdtBR110P mutant exhibited toxic activity comparable to that of CdtBWT as it induced both G2 arrest and apoptosis in lymphocytes and cytokine release in macrophages. While CdtBR110P was able to bind, internalize, and intoxicate cells, it did so at levels less than those observed with CdtBWT although these differences were not statistically significant.
In previous studies we demonstrated that toxic effects of Cdt on both lymphocytes and macrophages were dependent upon the ability of CdtB to function as a phosphatidylinositol-3,4,5-triphosphate phosphatase (25, 36). Thus, we also assessed the CdtB mutants for lipid phosphatase activity; indeed, all four CdtB CRAC site mutants retained enzymatic activity at levels comparable to those of the wild-type protein. This finding provides further evidence that reduced toxicity associated with these mutants is the result of changes in cell binding and internalization rather than of altered structure and/or the molecular mode of action associated with the active subunit; lack of altered structure was confirmed by CD analysis.
Collectively, our current findings, along with previous observations, provide strong support for the notion that Cdt association with target cells is dependent upon the ability of both the CdtC and CdtB subunits to bind to cholesterol. Furthermore, our observations demonstrate that Cdt-host cell association involves membrane microdomains enriched in cholesterol and is dependent upon the integrity of these so-called lipid rafts. Lipid rafts represent liquid-ordered microdomains which are distributed in the plasma membrane and whose lipid composition and high cholesterol content differ from the those of the rest of the membrane (52). Generally, lipid rafts are regarded as scaffolds for a number of molecular entities, which include ion channels, receptors, and signaling platforms; thus, these membrane regions provide an ideal structure to facilitate communication of extracellular stimuli to the intracellular milieu, leading to signaling events that regulate cell growth, proliferation, and survival (53, 54).
It is becoming increasingly evident that membrane rafts facilitate target cell interaction with several microbial toxins (55, 56); in addition to binding and clustering, these interactions may contribute to toxin internalization and/or a molecular mode of action. For instance, lipid membrane rafts may provide a mechanism by which receptors are concentrated and thereby promote ligand or pathogen binding. One such example is cholera toxin, which is pentameric and binds to targets cells via the ganglioside GM1. It is likely that cholera toxin simultaneously binds with high affinity to multiple receptors as a result of receptor concentration within the raft (56, 57). Likewise, the pore-forming leukotoxin from A. actinomycetemcomitans recognizes cholesterol via a CRAC site, which facilitates clustering to its receptor in the context of lipid rafts (50). Another pore-forming toxin, the aerolysin from Aeromonas hydrophila, which binds to glycosylphosphatidylinositol (GPI)-anchored proteins, also utilizes the concentrating properties of rafts to facilitate oligomerization, a requisite for channel formation (56, 58). These findings are consistent with the high cholesterol content of lipid rafts, which we propose facilitate Cdt binding as well.
Association with lipid rafts may also provide access to endocytic processes and intracellular trafficking routes. For example, Shiga toxin and cholera toxin bind to glycosphingolipids, which results in lipid clustering and changes in membrane properties that facilitate internalization and endocytic uptake (59). Likewise, several pathogens enter host cells in a cholesterol-dependent manner (55, 56); for example, the uptake of E. coli strains which express FimH has been shown to involve cholesterol-rich rafts. Similarly, Shigella invades cells via interaction between the invasin, IpaB, and the raft-associated receptor CD44 (60). Several enveloped and nonenveloped viruses (for example, simian virus 40 [SV40], HIV, and herpes simplex virus [HSV]) also require lipid rafts for binding or entry by endocytosis (60). It is interesting that Cdt associates with cholesterol in the context of lipid rafts; several investigators have also demonstrated that CdtB is internalized by endocytic mechanisms, and in the case of H. ducreyi Cdt, this appears to be dynamin dependent (38). This last observation is critical as disruption of the cholesterol-binding CRAC region for baculovirus has also been shown to compromise dynamin-dependent viral endocytosis (61).
In conclusion, we propose that binding of cholesterol by the CRAC regions contained within the CdtC and CdtB subunits results in the association of the Cdt holotoxin with membrane lipid rafts. It is likely that lipid raft association is critical for not only holotoxin binding but also the internalization and, possibly, the function of the active subunit, CdtB. These studies predict that cholesterol disposition within the membrane influences binding of CdtC and CdtB to the cell surface; therefore, we propose that Cdt favors raft-associated cholesterol, resulting in localized toxin-rich regions. This association may also be critical to the mode of action of the toxin, thereby allowing it to hijack lipid raft-associated signaling platform(s) and, perhaps, provide access to intracellular pools of PI-3,4,5-P3. Furthermore, perturbation of signaling cascades likely contributes to cell cycle arrest and eventual cell death in lymphocytes; similar events likely contribute to the toxin-induced proinflammatory cytokine response in macrophages as well. While these studies do not exclude the possibility of the existence of additional receptors that might be recognized by CdtA, they do clearly demonstrate that cholesterol recognition via CRAC sites and mutation of these regions are sufficient to block Cdt-mediated toxicity in target cells.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge the expertise of the staff of the School of Dental Medicine Flow Cytometry Facility in helping to carry out this study.
This work was supported by the National Institutes of Health grants DE06014 and DE023071.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00788-15.
REFERENCES
- 1.Zambon JJ. 1985. Actinobacillus actinomycetemcomitans in human periodontal disease. J Clin Periodontol 12:1–20. doi: 10.1111/j.1600-051X.1985.tb01348.x. [DOI] [PubMed] [Google Scholar]
- 2.van Winkelhoff A, Slots J. 1999. Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis in non oral infections. Periodontol 2000 20:122–135. doi: 10.1111/j.1600-0757.1999.tb00160.x. [DOI] [PubMed] [Google Scholar]
- 3.Fine D, Kaplan J, Kachlany S, Schreiner H. 2006. How we got attached to Actinobacillus actinomycetemcomitans: a model for infectious diseases. Periodontol 2000 42:114–157. doi: 10.1111/j.1600-0757.2006.00189.x. [DOI] [PubMed] [Google Scholar]
- 4.Henderson B, Ward J, Ready D. 2010. Aggregatibacter (Actinobacillus) actinomycetemcomitans: a triple A* periodontopathogen? Periodontol 2000 54:78–105. doi: 10.1111/j.1600-0757.2009.00331.x. [DOI] [PubMed] [Google Scholar]
- 5.Rahamat-Langendoen J, van Vonderen M, Engstrom L, Manson W, van Winkelhoff A, Mooi-Kokenberg E. 2011. Brain abscess associated with Aggregatibacter actinomycetemcomitans: case report and review of literature. J Clin Periodontol 38:702–706. doi: 10.1111/j.1600-051X.2011.01737.x. [DOI] [PubMed] [Google Scholar]
- 6.Rabie G, Lally ET, Shenker BJ. 1988. Immunosuppressive properties of Actinobacillus actinomycetemcomitans leukotoxin. Infect Immun 56:122–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shenker BJ, McKay TL, Datar S, Miller M, Chowhan R, Demuth DR. 1999. Actinobacillus actinomycetemcomitans immunosuppressive protein is a member of the family of cytolethal distending toxins capable of causing a G2 arrest in human T cells. J Immunol 162:4773–4780. [PubMed] [Google Scholar]
- 8.Korostoff J, Yamaguchi N, Miller M, Kieba I, Lally E. 2000. Perturbation of mitochondrial structure and function plays a central role in Actinobacillus actinomycetemcomitans leukotoxin-induced apoptosis. Microb Pathog 29:267–278. doi: 10.1006/mpat.2000.0390. [DOI] [PubMed] [Google Scholar]
- 9.Comayras C, Tasca C, Peres SY, Ducommun B, Oswald E, De Rycke J. 1997. Escherichia coli cytolethal distending toxin blocks the HeLa cell cycle at the G2/M transition by preventing cdc2 protein kinase dephosphorylation and activation. Infect Immun 65:5088–5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Okuda J, Fukumoto M, Takeda Y, Nishibuchi M. 1997. Examination of diarrheagenicity of cytolethal distending toxin: suckling mouse response to the products of the cdtABC genes of Shigella dysenteriae. Infect Immun 65:428–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Okuda J, Kurazono H, Takeda Y. 1995. Distribution of the cytolethal distending toxin A gene (cdtA) among species of Shigella and Vibrio, and cloning and sequencing of the cdt gene from Shigella dysenteriae. Microb Pathog 18:167–172. doi: 10.1016/S0882-4010(95)90022-5. [DOI] [PubMed] [Google Scholar]
- 12.Scott DA, Kaper JB. 1994. Cloning and sequencing of the genes encoding Escherichia coli cytolethal distending toxin. Infect Immun 62:244–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pickett CL, Cottle DL, Pesci EC, Bikah G. 1994. Cloning, sequencing, and expression of the Escherichia coli cytolethal distending toxin genes. Infect Immun 62:1046–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mayer M, Bueno L, Hansen E, DiRienzo JM. 1999. Identification of a cytolethal distending toxin gene locus and features of a virulence-associated region in Actinobacillus actinomycetemcomitans. Infect Immun 67:1227–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pickett CL, Whitehouse CA. 1999. The cytolethal distending toxin family. Trends Microbiol 7:292–297. doi: 10.1016/S0966-842X(99)01537-1. [DOI] [PubMed] [Google Scholar]
- 16.Shenker BJ, Hoffmaster RH, McKay TL, Demuth DR. 2000. Expression of the cytolethal distending toxin (Cdt) operon in Actinobacillus actinomycetemcomitans: evidence that the CdtB protein is responsible for G2 arrest of the cell cycle in human T-cells. J Immunol 165:2612–2618. doi: 10.4049/jimmunol.165.5.2612. [DOI] [PubMed] [Google Scholar]
- 17.Shenker BJ, Hoffmaster RH, Zekavat A, Yamguchi N, Lally ET, Demuth DR. 2001. Induction of apoptosis in human T cells by Actinobacillus actinomycetemcomitans cytolethal distending toxin is a consequence of G2 arrest of the cell cycle. J Immunol 167:435–441. doi: 10.4049/jimmunol.167.1.435. [DOI] [PubMed] [Google Scholar]
- 18.Nesic D, Hsu Y, Stebbins CE. 2004. Assembly and function of a bacterial genotoxin. Nature 429:429–433. doi: 10.1038/nature02532. [DOI] [PubMed] [Google Scholar]
- 19.De Rycke J, Oswald E. 2001. Cytolethal distending toxin (CDT): a bacterial weapon to control host cell proliferaton? FEMS Microbiol Lett 203:141–148. doi: 10.1111/j.1574-6968.2001.tb10832.x. [DOI] [PubMed] [Google Scholar]
- 20.Thelestam M, Frisan T. 2004. Cytolethal distending toxins. Rev Physiol Biochem Pharmacol 152:111–133. [DOI] [PubMed] [Google Scholar]
- 21.Lara-Tejero M, Galan JE. 2001. CdtA, CdtB, and CdtC form a tripartite complex that is required for cytolethal distending toxin activity. Infect Immun 69:4358–4365. doi: 10.1128/IAI.69.7.4358-4365.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Elwell CA, Chao K, Patel K, Dreyfus LA. 2001. Escherichia coli CdtB mediates cytolethal distending toxin cell cycle arrest. Infect Immun 69:3418–3422. doi: 10.1128/IAI.69.5.3418-3422.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mao X, DiRienzo JM. 2002. Functional studies of the recombinant subunits of a cytolethal distending toxin. Cell Microbiol 4:245–255. doi: 10.1046/j.1462-5822.2002.00186.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shenker BJ, Besack D, McKay TL, Pankoski L, Zekavat A, Demuth DR. 2004. Actinobacillus actinomycetemcomitans cytolethal distending toxin (Cdt): evidence that the holotoxin is composed of three subunits: CdtA, CdtB, and CdtC. J Immunol 172:410–417. doi: 10.4049/jimmunol.172.1.410. [DOI] [PubMed] [Google Scholar]
- 25.Shenker BJ, Dlakic M, Walker L, Besack D, Jaffe E, Labelle E, Boesze-Battaglia K. 2007. A novel mode of action for a microbial-derived immunotoxin: the cytolethal distending toxin subunit B exhibits phosphatidylinositol 3,4,5-triphosphate phosphatase activity. J Immunol 178:5099–5108. doi: 10.4049/jimmunol.178.8.5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McSweeney L, Dreyfus LA. 2005. Carbohydrate-binding specificity of the Escherichia coli cytolethal distending toxin CdtA-II and CdtC-II subunits. Infect Immun 73:2051–2060. doi: 10.1128/IAI.73.4.2051-2060.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mise K, Akifusa S, Watarai S, Ansai T, Nishihara T, Takehara T. 2005. Involvement of ganglioside GM3 in G2/M cell cycle arrest of human monocytic cells induced by Actinobacillus actinomycetemcomitans cytolethal distending toxin. Infect Immun 73:4846–4852. doi: 10.1128/IAI.73.8.4846-4852.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shenker BJ, Besack D, McKay TL, Pankoski L, Zekavat A, Demuth DR. 2005. Induction of cell cycle arrest in lymphocytes by Actinobacillus actinomycetemcomitans cytolethal distending toxin requires three subunits for maximum activity. J Immunol 174:2228–2234. doi: 10.4049/jimmunol.174.4.2228. [DOI] [PubMed] [Google Scholar]
- 29.Boesze-Battaglia K, Besack D, McKay TL, Zekavat A, Otis L, Jordan-Sciutto K, Shenker BJ. 2006. Cholesterol-rich membrane microdomains mediate cell cycle arrest induced by Actinobacillus actinomycetemcomitans cytolethal distending toxin. Cell Microbiol 8:823–836. doi: 10.1111/j.1462-5822.2005.00669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou M, Zhang Q, Zhao J, Jin M. 2012. Haemophilus parasuis encodes two functional cytolethal distending toxins: CdtC contains an aytpical cholesterol recognition/interaction region. PLoS One 7:e32580. doi: 10.1371/journal.pone.0032580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lai C, Lai C, Lin Y, Hung C, Chu C, Feng C, Chang C, Su H. 2013. Characterization of putative cholesterol recognition/interaction amino acid consensus-like motif of Campylobacter jejuni cytolethal distending toxin C. PLoS One 8:e66202. doi: 10.1371/journal.pone.0066202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hekman M, Hamm H, Villar A, Bader B, Kuhlmann J, Nickel J, Rapp UR. 2002. Associations of B- and C-Raf with cholesterol, phosphatidylserine, and lipid second messengers. J Biol Chem 277:24090–24102. doi: 10.1074/jbc.M200576200. [DOI] [PubMed] [Google Scholar]
- 33.Boesze-Battaglia K, Brown A, Walker L, Besack D, Zekavat A, Wrenn S, Krummenacher C, Shenker BJ. 2009. Cytolethal distending toxin-induced cell cycle arrest of lymphocytes is dependent upon recognition and binding to cholesterol. J Biol Chem 284:10650–10658. doi: 10.1074/jbc.M809094200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maehama T, Taylor G, Slama J, Dixon J. 2000. A sensitive assay for phosphoinositide phosphatases. Anal Biochem 279:248–250. doi: 10.1006/abio.2000.4497. [DOI] [PubMed] [Google Scholar]
- 35.Wijeyesakere SJ, Rizvi SM, Raghavan M. 2013. Glycan-dependent and -independent interactions contribute to cellular substrate recruitment by calreticulin. J Biol Chem 288:35104–35116. doi: 10.1074/jbc.M113.507921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shenker B, Walker L, Zekavat A, Dlakic M, Boesze-Battaglia K. 2014. Blockade of the PI-3K signaling pathway by the Aggregatibacter actinomycetemcomitans cytolethal distending toxin induces macrophages to synthesize and secrete pro-inflammatory cytokines. Cell Microbiol 16:1391–1404. doi: 10.1111/cmi.12299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guerra L, Teter K, Lilley B, Stenerlow B, Holmes R, Ploegh H, Sandvik JA, Thelastam M, Frisan T. 2005. Cellular internalization of cytolethal distending toxin: a new end to a known pathway. Cell Microbiol 7:921–934. doi: 10.1111/j.1462-5822.2005.00520.x. [DOI] [PubMed] [Google Scholar]
- 38.Cortes-Bratti X, Chaves-Olarte E, Lagergard T, Thelastam M. 2000. Cellular internalization of cytolethal distending toxin from Haemophilus ducreyi. Infect Immun 68:6903–6911. doi: 10.1128/IAI.68.12.6903-6911.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gargi A, Reno M, Blanke S. 2012. Bacterial toxin modulation of the eukaryotic cell cycle: are all cytolethal distending toxins created equally? Front Cell Infect Microbiol 2:124. doi: 10.3389/fcimb.2012.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eshraghi A, Maldonado-Arocho F, Gargi A, Cardwell M, Prouty M, Blanke S, Bradley K. 2010. Cytolethal distending toxin family members are differentially affected by alterations in host glycans and membrane cholesterol. J Biol Chem 285:18199–18207. doi: 10.1074/jbc.M110.112912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Damek-Poprawa M, Jang J, Volgina A, Korostoff J, DiRienzo J. 2012. Localization of Aggregatibacter actinomycetemcomitans cytolethal distending toxin subunits during intoxication of live cells. Infect Immun 80:2761–2770. doi: 10.1128/IAI.00385-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jamin N, Neumann J, Ostuni M, Vu T, Yao Z, Murail S, Robert J, Fiatzakis C, Papadopoulos V, Lacapere J. 2005. Characterization of the cholesterol recognition amino acid consensus sequence of the peripheral-type benzodiazepine receptor. Mol Endocrinol 19:588–594. doi: 10.1210/me.2004-0308. [DOI] [PubMed] [Google Scholar]
- 43.Li H, Papadopoulos V. 1998. Peripheral-type benzodiazepine receptor function in cholesterol transport. Identification of a putative cholesterol recognition/interaction amino acid sequence and consensus pattern. Endocrinology 139:4991–4997. [DOI] [PubMed] [Google Scholar]
- 44.Epand R, Sayer B, Epand R. 2005. Caveolin scaffolding region and cholesterol-rich domains in membranes. J Mol Biol 345:339–350. doi: 10.1016/j.jmb.2004.10.064. [DOI] [PubMed] [Google Scholar]
- 45.Vincent N, Genin C, Malvoisin E. 2002. Identification of a conserved domain of the HIV-1 transmembrane protein gp41 which interacts with cholesteryl groups. Biochim Biophys Acta 1567:157–164. doi: 10.1016/S0005-2736(02)00611-9. [DOI] [PubMed] [Google Scholar]
- 46.Oddi S, Dainese E, Fezza F, Lanuti M, Barcaroli D, DeLaurenzi V, Centonze D, Maccarrone M. 2011. Functional characterization of putative cholesterol binding sequence (CRAC) in human type-1 cannabinoid receptor. J Neurochem 116:858–865. doi: 10.1111/j.1471-4159.2010.07041.x. [DOI] [PubMed] [Google Scholar]
- 47.Singh A, McMillan J, Bukiya A, Burton B, Parrill A, Dopico A. 2012. Multiple cholesterol recognition/interaction amino acid consensus (CRAC) motifs in cytosolic C tail of Slo1 subunit determine cholesterol sensitivity of Ca2+- and voltage-gated K+ (BK) channels. J Biol Chem 287:20509–20521. doi: 10.1074/jbc.M112.356261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lecanu L, Yao Z, McCourty A, Sidahmed E, Orellana M, Burnier M, Papadopoulos V. 2013. Control of hypercholesterolemia and atherosclerosis using the cholesterol recognition/interaction amino acid sequence of the translocator protein TSPO. Steroids 78:137–146. doi: 10.1016/j.steroids.2012.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jafurulla M, Tiwari S, Chattopadhyay A. 2011. Identification of cholesterol recognition amino acid consensus (CRAC) motif in G-protein coupled receptors. Biochem Biophys Res Commun 404:569–573. doi: 10.1016/j.bbrc.2010.12.031. [DOI] [PubMed] [Google Scholar]
- 50.Brown A, Balashova N, Epand R, Epand R, Bragin A, Kachlany S, Walters M, Du Y, Boesze-Battaglia K, Lally E. 2013. Aggregatibacter actinomycetemcomitans leukotoxin utilizes a cholesterol recognition amino acid consensus site for membrane association. J Biol Chem 288:23607–23621. doi: 10.1074/jbc.M113.486654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baier C, Fantini J, Barrantes F. 2011. Disclosure of cholesterol recognition motifs in transmembrane domains of the human nicotinic acetylcholine receptor. Sci Rep 1:69. doi: 10.1038/srep00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Head B, Patel H, Insel P. 2014. Interaction of membrane/lipid rafts with the cytoskeleton: impact on signaling and function. Biochim Biophys Acta 1838:532–545. doi: 10.1016/j.bbamem.2013.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dykstra M, Cherukuri A, Sohn H, Tzeng S, Pierce S. 2003. Location is everything: lipid rafts and immune cell signaling. Annu Rev Immunol 21:457–481. doi: 10.1146/annurev.immunol.21.120601.141021. [DOI] [PubMed] [Google Scholar]
- 54.Cherukuri A, Dykstra M, Pierce S. 2001. Floating the raft hypothesis: lipid rafts play a role in immune cell function. Immunity 14:657–660. doi: 10.1016/S1074-7613(01)00156-X. [DOI] [PubMed] [Google Scholar]
- 55.Lencer W. 2001. Microbes and microbial toxins: paradigms for microbial-mucosal toxins. V. cholera: invasion of the intestinal epithelial barrier by a stably folded protein toxin. Am J Physiol Gastrointest Liver Physiol 280:G781–G786. [DOI] [PubMed] [Google Scholar]
- 56.van der Goot FG, Harder T. 2001. Raft membrane domains: from a liquid-ordered membrane phase to site of pathogen attack. Semin Immunol 13:89–97. doi: 10.1006/smim.2000.0300. [DOI] [PubMed] [Google Scholar]
- 57.Montecucco C, Papini E, Schiavo G. 1994. Bacterial protein toxins penetrate cells via a four-step mechanism. FEBS Lett 346:92–98. doi: 10.1016/0014-5793(94)00449-8. [DOI] [PubMed] [Google Scholar]
- 58.Abrami L, van der Goot FG. 1999. Plasma membrane microdomains act as concentration platforms to facilitate intoxication by aerolysin. J Cell Biol 147:175–184. doi: 10.1083/jcb.147.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ewers H, Helenius A. 2011. Lipid-mediated endocytosis. Cold Spring Harb Perspect Biol 3:a004721. doi: 10.1101/cshperspect.a004721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lafont F, Tran Van Nhieu G, Hanada K, Sansonetti P, van der Goot FG. 2002. Initial steps of Shigella infection depend on the cholesterol/sphingolipid raft-mediated CD44-IpaB interaction. EMBO J 21:4449–4457. doi: 10.1093/emboj/cdf457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Luz-Madrigal A, Asanov A, Camacho-Zarco A, Sampieri A, Vaca L. 2013. A cholesterol recognition amino acid consensus domain in GP64 fusion protein facilitates anchoring of baculovirus to mammalian cells. J Virol 87:11894–11907. doi: 10.1128/JVI.01356-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.