

Abstract
Th17 immunity in the gastrointestinal tract is regulated by the intestinal microbiota composition, particularly the presence of segmented filamentous bacteria (sfb), but the role of the intestinal microbiota in pulmonary host defense is not well explored. We tested whether altering the gut microbiota by acquiring sfb influences the susceptibility to staphylococcal pneumonia via induction of type 17 immunity. Groups of C57BL/6 mice which differed in their intestinal colonization with sfb were challenged with methicillin-resistant Staphylococcus aureus in an acute lung infection model. Bacterial burdens, bronchoalveolar lavage fluid (BALF) cell counts, cell types, and cytokine levels were compared between mice from different vendors, mice from both vendors after cohousing, mice given sfb orally prior to infection, and mice with and without exogenous interleukin-22 (IL-22) or anti-IL-22 antibodies. Mice lacking sfb developed more severe S. aureus pneumonia than mice colonized with sfb, as indicated by higher bacterial burdens in the lungs, lung inflammation, and mortality. This difference was reduced when sfb-negative mice acquired sfb in their gut microbiota through cohousing with sfb-positive mice or when given sfb orally. Levels of type 17 immune effectors in the lung were higher after infection in sfb-positive mice and increased in sfb-negative mice after acquisition of sfb, as demonstrated by higher levels of IL-22 and larger numbers of IL-22+ TCRβ+ cells and neutrophils in BALF. Exogenous IL-22 protected mice from S. aureus pneumonia. The murine gut microbiota, particularly the presence of sfb, promotes pulmonary type 17 immunity and resistance to S. aureus pneumonia, and IL-22 protects against severe pulmonary staphylococcal infection.
INTRODUCTION
Staphylococcus aureus continues to be one of the most common pathogens causing invasive life-threatening infections (1). Methicillin-resistant S. aureus (MRSA) currently accounts for 20 to 40% of hospital-acquired and ventilator-associated pneumonias (2) and 9% of community-acquired pneumonias (3), and MRSA pneumonia is associated with very high mortality rates (3, 4).
The Th17 pathway plays an important role in mucosal host defense against a wide range of bacterial pathogens (reviewed in reference 5). Defects in human Th17 signaling (e.g., in hyper-IgE or Job's syndrome) are associated with immunodeficiency syndromes characterized by increased susceptibility to staphylococcal infections of the lung and skin, suggesting a specific role for Th17 immunity in the host defense against S. aureus (6, 7). Additionally, mice with defects in Th17 signaling have impaired bacterial clearance from the lung after infection with Klebsiella pneumoniae (8). More recently, the Th17 pathway was implicated in the defense against S. aureus pneumonia as well (9–11). Mice lacking the interleukin-17 (IL-17) receptor or IL-22 or mice that were coinfected with influenza A virus and thereby deficient in type 17 immunity displayed impaired bacterial clearance of S. aureus compared to wild-type or influenza virus-free mice (10). Type 17 immunity has also been reported to contribute to mucosal vaccine responses against Pseudomonas aeruginosa and Mycobacterium tuberculosis (12–14).
The gastrointestinal (GI) tract of mammals is inhabited by thousands of species of commensal microorganisms that exist in a mutualistic relationship with the host. How the commensal microbiota influences the host immune system is poorly understood, but it appears clear that the microbiota is a major regulator of the immune system and that bacterial signals have profound influences on antibacterial defenses in the GI tract, and also in other organs (15, 16). Ivanov et al. showed that colonization of the GI tract of mice with a commensal microbe, the segmented filamentous bacterium (sfb), was sufficient to induce the appearance of Th17 cells in the small intestine, leading to increased expression of genes associated with inflammation and antimicrobial defenses, and resulted in enhanced resistance to the murine intestinal pathogen Citrobacter rodentium (17–19). The influence of the GI microbiota on lung immunity, the so-called gut-lung axis, has recently become the focus of more interest, but the underlying mechanisms are still incompletely understood (20). Commensal organisms of the GI tract contribute to the host defense against Escherichia coli pneumonia via Toll-like receptor (TLR) signaling (21), and germfree mice have a strikingly higher mortality rate than that of conventional mice following P. aeruginosa pneumonia (22). Little is known regarding the role of specific organisms in modulating pulmonary immunity and whether the gastrointestinal microbiota has any influence on Gram-positive lung pathogens, or S. aureus in particular. We hypothesized that the intestinal microbiota can affect S. aureus pneumonia and that the presence of sfb in the mouse intestine specifically influences type 17 immunity in the lung and increases resistance to S. aureus pneumonia. To test this hypothesis, we compared mice with different intestinal microbiota in a murine staphylococcal pneumonia model.
C57BL/6 mice from The Jackson Laboratory and Taconic Biosciences differ in their gastrointestinal microbiota, most notably in colonization with the commensal sfb (with Jackson mice being sfb negative and Taconic mice generally being sfb positive) (18). We demonstrate here that mice from The Jackson Laboratory are more susceptible to intranasal (i.n.) challenge with the MRSA strain LAC than mice from Taconic Biosciences. This difference in resistance diminishes when mice from both vendors are cohoused, indicating an environmental rather than a genetic or inherent factor influencing the variation in pulmonary phenotype. Following cohousing, sfb commensals are passed from sfb-positive mice to their sfb-negative cagemates, leading to increased resistance to S. aureus pneumonia. Furthermore, when sfb-negative mice are given sfb via gavage 2 weeks prior to infection, they also become more resistant to S. aureus pneumonia. Bronchoalveolar lavage fluid (BALF) from the more resistant sfb-positive mice as well as from initially sfb-negative mice that acquired sfb via GI colonization contained more IL-22, more IL-22+ TCRβ+ cells, and more neutrophils, all of which are indicators of increased type 17 immunity activation. We also show that neutralization of the Th17 cytokine IL-22 with anti-IL-22 antibody prior to challenge with S. aureus leads to increased susceptibility and, conversely, that i.n. administration of IL-22 at the time of infection with S. aureus renders mice more resistant to infection, suggesting that IL-22 is protective against S. aureus pneumonia.
MATERIALS AND METHODS
Bacterial strains.
Staphylococcus aureus strain LAC (a USA300 MRSA strain), initially isolated from a patient suffering from necrotizing pneumonia, was obtained from the Network on Antimicrobial Resistance in S. aureus (NARSA). To prepare bacterial inocula for in vivo challenge studies, frozen bacterial stocks of S. aureus were plated on Columbia agar (CBA) supplemented with 2% sodium chloride (Columbia salt agar [CSA]) and grown at 37°C overnight. Subsequently, colonies were grown in Columbia salt broth (CSB; Columbia broth supplemented with 2% sodium chloride) at 37°C, with shaking at 200 rpm, to an optical density at 650 nm of 0.5. Bacteria were washed and resuspended in phosphate-buffered saline (PBS) to yield the intended calculated inoculum (2 × 108 to 4 × 108 per mouse for experiments measuring bacterial burdens and 7.5 × 108 to 8 × 108 per mouse for survival experiments). The inoculum was verified after serial dilution in tryptic soy broth (TSB) supplemented with 0.05% Tween and enumeration of growth on 5% sheep blood agar plates (BAP) after overnight incubation at 37°C. Twenty-microliter aliquots of bacterial suspensions were used for i.n. challenge in the mouse experiments.
Mice.
All animal experiments were approved by the Harvard Medical Area Institutional Animal Care and Use Committee. Four- to 6-week-old C57BL/6 mice were purchased from The Jackson Laboratory or Taconic Biosciences. All groups of mice were age, gender, and weight matched for comparison experiments. Mice were housed under virus-free conditions in microisolator-topped cages.
Cohousing and sfb detection.
For cohousing experiments, two sfb-negative mice and two sfb-positive mice were transferred to one common cage after ear tagging of sfb-negative mice to enable identification. Prior to cohousing and 12 to 18 days into cohousing, fecal pellets (freshly produced after temporary transfer of mice to a new cage) were collected from each mouse group. DNA from feces was purified using a QIAamp DNA stool kit following the manufacturer's instructions (Qiagen, Valencia, CA). The presence of sfb was examined by PCR using the primers 736F (GACGCTGAGGCATGAGAGCAT) and 844R (GACGGCACGGATTGTTATTCA) and the following PCR conditions: 94°C for 5 min; 30 cycles of 94°C for 30 s, 52°C for 40 s, and 72°C for 90 s; and 72°C for 7 min. The presence of sfb resulted in an ∼150-bp amplicon.
sfb acquisition.
sfb-containing stools were obtained from initially germfree mice that were monocolonized with sfb (kindly provided by Neil Surana), and the samples were suspended in PBS and passed through a cell strainer to remove particulate matter. Fifty microliters of this suspension was given orogastrically via gavage to 4-week-old female C57BL/6 mice sedated with isoflurane. Control mice were given PBS. Prior to challenge with S. aureus 2 weeks later, stools from these mice were collected, bacterial DNAs extracted, and the presence or absence of sfb confirmed by PCR.
Murine pneumonia model.
Mice were anesthetized via intraperitoneal (i.p.) injection with ketamine and xylazine. After inoculation with 10 μl of the S. aureus suspension in each nostril, mice were observed until they recovered from anesthesia. For survival studies, mice were observed closely for signs of illness and impending death (i.e., a moribund state) at least three times daily. If they appeared moribund (based on hunched posture, an inability to move, eat, or drink, ruffled fur, labored breathing, and crusted eyes), mice were euthanized by carbon dioxide inhalation followed by cervical dislocation and were considered nonsurvivors. For quantification of bacteria and analysis of cells and cytokines in BALF, mice were euthanized by i.p. injection with pentobarbital at the indicated time points. For experiments with IL-22 administration, recombinant IL-22 (rIL-22; R&D Systems) suspended in PBS plus 0.1% bovine serum albumin (BSA) was mixed with the bacterial inoculum and administered to anesthetized mice via i.n. inhalation. Control mice were given the bacterial inoculum mixed with PBS plus 0.1% BSA.
Mice given the anti-IL-22 antibody i.n. were sedated with ketamine and xylazine and then given 25 μl containing 25 μg polyclonal anti-IL-22 goat IgG or polyclonal goat IgG (all from R&D Systems) (as a control) 4 h prior to bacterial challenge.
BALF.
The tracheas of euthanized mice were cannulated with a 20-gauge angiocatheter, and BALF was obtained following two instillations of 350 μl of ice-cold PBS containing 0.5 mM EDTA. BALF was centrifuged and the supernatant stored at −80°C before measurement of cytokines by use of a Luminex magnetic assay with a mouse cytokine 20-plex panel (Life Technologies, Grand Island, NY) or, for IL-17 and IL-22 concentrations, by enzyme-linked immunosorbent assay (ELISA; R&D Systems, Minneapolis, MN). Erythrocytes in the cell pellets were lysed using a mouse erythrocyte lysis kit (R&D Systems) according to the manufacturer's instructions. Total cell counts were determined by trypan blue staining in a Countess automated cell counter (Life Technologies, Grand Island, NY).
Determination of viable bacterial counts.
The right lung and the spleen were removed, weighed, and homogenized in TSB with 0.05% Tween by use of an Omni TissueMaster homogenizer-125 (Omni International, Marietta, GA). Serial 10-fold dilutions of the lung and spleen homogenates were then plated onto 5% sheep blood agar plates and incubated overnight at 37°C prior to counting CFU and calculating the number of CFU per gram of tissue.
Flow cytometry.
BALF cells were washed with flow cytometry buffer (2% fetal calf serum [FCS] in PBS) and adjusted to a concentration of 5 × 105/ml. Cells were treated with FcγR blocker (anti-CD16/32; clone 2.4G2) in flow cytometry buffer for 20 min on ice to block nonspecific staining and then stained with appropriate isotype-matched antibodies to the following cell surface markers: CD45 (30-F11), Ly6G (1A8) (both from Biolegend, San Diego, CA), and T cell receptor beta (TCRβ) (H57-597; eBioscience, San Diego, CA). For intracellular cytokine staining of IL-17A (using the monoclonal antibody [MAb] TC11-18H10.1 [Biolegend, San Diego, CA]) and IL-22 (using the MAb 1H8PWSR [eBioscience, San Diego, CA]), cells were stimulated ex vivo for 6 h with phorbol 12-myristate 13-acetate (PMA) and ionomycin in the presence of brefeldin A, followed by fixation, permeabilization with saponin, and staining (all using a Cytofix/Cytoperm fixation/permeabilization kit [BD Biosciences, San Jose, CA]) and then analysis on a FACSCalibur flow cytometer.
Confocal microscopy.
Paraffin-embedded lung sections were deparaffinized using EZ-Dewax per the manufacturer's protocol. Deparaffinized samples were blocked with 1% BSA-PBS overnight at 4°C, followed by labeling with a 1:200 dilution of rabbit anti-IL-22 antibody (Abcam), fluorescein isothiocyanate (FITC)-conjugated CD3e antibody (eBioscience), allophycocyanin (APC)-conjugated RORγt antibody (eBioscience), or the appropriate isotype control antibodies (Biolegend or eBioscience) in 0.5% BSA-PBS overnight in a humidified chamber at 4°C. The samples were washed, and a 1:400 dilution of the secondary antibody, Alexa Fluor 568-conjugated donkey anti-rabbit IgG (Invitrogen), was added to the relevant samples for 2 h at room temperature. Samples were washed, mounted, and analyzed by confocal microscopy with a Zeiss LSM5 Pa instrument equipped with a krypton-argon laser.
Histology.
The trachea was cannulated with an angiocatheter, and Bouin's fixative solution was injected directly into the lung. The left lung was then removed and placed in Bouin's fixative solution prior to paraffin embedding, thin sectioning, and hematoxylin-eosin (H&E) staining by the Harvard Rodent Histopathology Core. The lung tissue sections were scored for the percentage of lung tissue affected by inflammation by a veterinary pathologist who was blinded to the experimental conditions.
Data analysis and interpretation.
The significance of differences in lethal outcomes and bacterial burdens in lungs and spleens between groups of mice was analyzed by parametric or nonparametric analysis of variance (ANOVA) or t tests, as appropriate, depending on whether the data were normally distributed, followed by post hoc analyses for significant differences between paired groups. We considered P values of <0.05 to be statistically significant (*, P < 0.05; **, P < 0.01). We performed survival analysis using the Kaplan-Meier method and the log rank test. All analyses were performed using GraphPad Prism software.
RESULTS
sfb-negative C57BL/6 mice are more susceptible to S. aureus pneumonia than sfb-positive C57BL/6 mice.
Following intranasal challenge with the MRSA strain LAC, sfb-negative mice were found to have larger infectious bacterial burdens than those of age- and gender-matched sfb-positive mice. sfb-negative mice had 21-fold larger bacterial burdens in the lungs 18 h after infection than those of sfb-positive mice (mean of 1.4 × 109 CFU/g lung tissue versus 6.6 × 107 CFU/g lung tissue) (Fig. 1A). This was accompanied by higher rates of translocated bacteria, as the spleens of sfb-negative mice had about 70 times more bacteria than the spleens of sfb-positive mice (Fig. 1B).
FIG 1.

sfb-negative mice exhibit increased susceptibility to MRSA pneumonia compared to sfb-positive mice. C57BL/6 mice from The Jackson Laboratory (sfb negative) and Taconic Biosciences (sfb positive) were challenged with 5 × 108 CFU of MRSA intranasally and sacrificed after 18 h for determination of the number of bacterial CFU per gram of lung (A) or spleen (B) tissue. Symbols represent individual animals, and horizontal bars represent the medians. P values were determined by the Mann-Whitney U test. The histological appearance of lung tissue is shown for representative sfb-negative (C and D) and sfb-positive (E and F) mice 18 h after infection with MRSA Lac. The sfb-negative mice showed higher percentages of lung tissue involved in inflammation (60% affected [C and D] versus 10% affected [E and F]). Magnification, ×40 (C and E) or ×100 (D and F). Bars, 1 mm.
Analysis of the lung histology of MRSA-infected mice showed dense inflammatory cell infiltration as visualized by H&E staining, which was more pronounced in sfb-negative mice (Fig. 1C and D) than in sfb-positive mice (Fig. 1E and F). These results indicate a higher susceptibility of sfb-negative mice than sfb-positive mice to S. aureus pneumonia.
Resistance to S. aureus pneumonia is influenced by an environmental factor.
To examine whether sfb-negative mice are more susceptible to S. aureus lung infection due to an environmental or genetic factor, we cohoused female, age-matched, sfb-negative (purchased from The Jackson Laboratory) and sfb-positive (purchased from Taconic Biosciences) mice in the same cages for 2 weeks, from 4 to 6 weeks of age, prior to respiratory challenge with S. aureus. While the median levels of S. aureus in the lungs of the initially sfb-negative mice cohoused with sfb-positive mice were somewhat higher than bacterial levels in the continuously sfb-positive mice, this difference was no longer statistically significant (Fig. 2). In addition, mice that became sfb positive during cohousing were significantly more resistant to S. aureus lung infection than sfb-negative mice that were not cohoused. Cohousing of sfb-positive mice with sfb-negative mice did not alter their response to S. aureus lung infection, as they showed the same bacterial levels as those of mice that were sfb positive and not cohoused. To confirm the presence of sfb in the mice in the different groups, we examined the feces of the cohoused mice as well as mice from Taconic Biosciences, reportedly supplied as sfb positive, for the presence of sfb by PCR. Initially sfb-negative mice sharing a cage with sfb-positive mice became positive for sfb, as were the mice from the supplier known to carry sfb (see Fig. S1 in the supplemental material). Stool samples from mice from The Jackson Laboratory, reportedly negative for sfb, were confirmed by PCR to be sfb negative.
FIG 2.
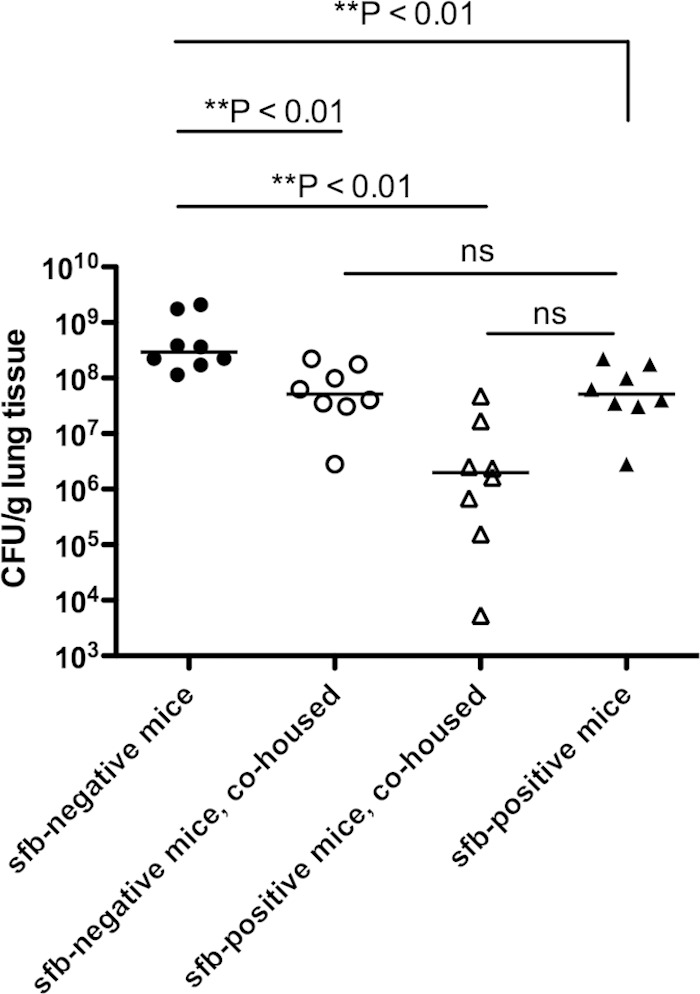
sfb-negative mice acquire increased resistance to S. aureus pneumonia after cohousing with sfb-positive mice. Female sfb-negative C57BL/6 mice and age-matched, female, sfb-positive C57BL/6 mice were housed together in isolated cages for 2 weeks prior to bacterial challenge. All of the cohoused mice were sfb positive at the time of bacterial challenge. Eight additional sfb-negative or sfb-positive mice were not cohoused for the same period and remained sfb negative or sfb positive, respectively. After 2 weeks, mice were challenged with 5 × 108 CFU of MRSA i.n. and sacrificed after 18 h for determination of the number of bacterial CFU per gram of lung tissue. Symbols represent individual animals, and horizontal bars represent the medians. P values were determined by the Kruskal-Wallis test (overall ANOVA; P < 0.01), and P values depict Dunn's multiple pairwise comparisons for nonparametric distributions. ns, not significant.
Protective effect of sfb gut colonization on susceptibility to S. aureus pneumonia.
To determine whether the difference in susceptibility to S. aureus pneumonia was due to the presence of sfb in the murine intestine, we administered sfb-containing stool (obtained from initially germfree mice that had been monocolonized with sfb [23]) or PBS via gavage to 4-week-old sfb-negative mice 2 weeks prior to challenge with S. aureus. sfb was detected by PCR in the stools of mice 2 weeks after the gavage with sfb. The PBS-gavaged mice were negative for sfb. When challenged with S. aureus, mice that had acquired sfb were more resistant to infection, as demonstrated by significantly lower bacterial counts in lungs (4.9-fold difference) (Fig. 3A) and a trend (P = 0.08) toward lower bacterial counts in spleens (11-fold difference) (Fig. 3B). When challenged with a higher bacterial inoculum in a survival assay, initially sfb-negative mice that had become sfb positive were also at much lower risk of succumbing to MRSA lung infection than continuously sfb-negative mice. All sfb-negative mice died by 36 h, while 70% of mice rendered sfb positive survived (Fig. 3C).
FIG 3.
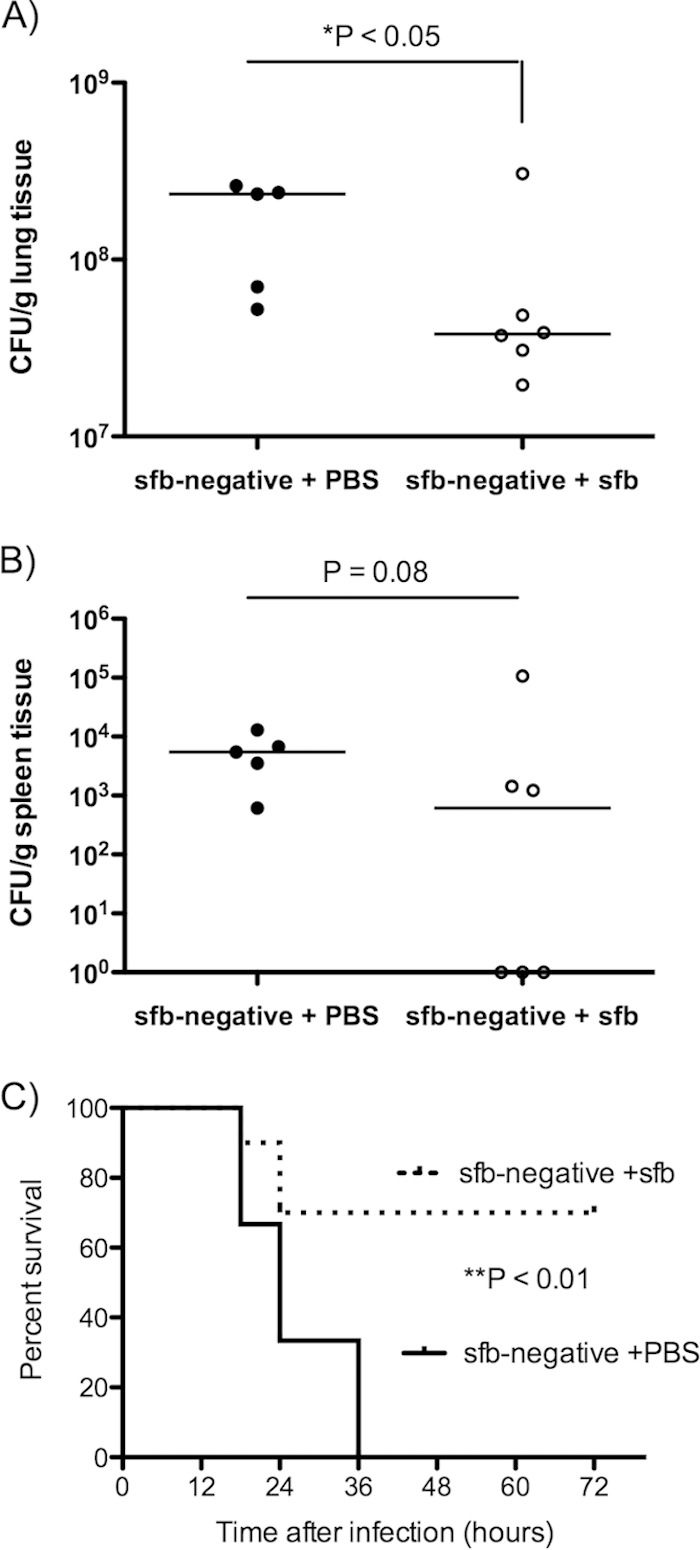
sfb-negative mice acquire increased resistance to S. aureus pneumonia after colonization with sfb in the GI tract. sfb-negative C57BL/6 mice were gavaged with either PBS (sfb-negative + PBS; n = 5) or sfb-containing stool (sfb-negative + sfb; n = 6) 2 weeks prior to intranasal challenge with MRSA strain LAC at a dose of 5 × 108 CFU (each circle represents an individual animal) (A and B) or 2 × 109 CFU (for a survival experiment) (n = 10 in each group) (C). The data show numbers of bacterial CFU per gram of lung tissue (A) or spleen tissue (B) at 18 h postinfection. (C) Kaplan-Meier survival curves. Symbols represent individual animals, and horizontal bars represent the medians. P values were determined by the Mann-Whitney U test (A and B) or by the log rank test (C).
sfb gut colonization is associated with differential pulmonary type 17 immunity.
We measured levels of type 17-associated cytokines in the BALF after S. aureus challenge and tested whether acquisition of sfb via gut colonization alters type 17 immunity in the lung by initially comparing sfb-negative mice that became sfb positive, either after cohousing with sfb-positive Taconic mice or after gavage with sfb-containing stool. As shown in Fig. 4A to D, we found that BALF levels of IL-22, one of the main Th17 effector cytokines, differed significantly between groups of mice with and without sfb colonization. Eight hours after intranasal challenge with MRSA, both sfb-negative and sfb-positive mice had similar IL-22 levels (Fig. 4A), but by 18 h postinfection, all sfb-positive mice (whether initially sfb positive or rendered sfb positive after cohousing with sfb-positive mice or after sfb gavage) had significantly higher IL-22 levels than those of sfb-negative mice (Fig. 4B to D).
FIG 4.



Type 17 immunity differs in the lungs of sfb-negative and sfb-positive mice. Eight and 18 h after infection with MRSA, mice were euthanized and BALF was obtained for cytokine analysis. Bars represent the means, and error bars represent standard deviations (SD). P values were determined by the Mann-Whitney U test for 2-group comparisons or by the Kruskal-Wallis test with Dunn's multiple-comparison test. (A) BALF IL-22 level at 8 h postinfection; (B) BALF IL-22 level at 18 h postinfection; (C) BALF IL-22 level at 18 h postinfection, after cohousing; (D) BALF IL-22 level at 18 h postinfection in Jackson (sfb-negative) mice that acquired sfb after gavage 2 weeks prior to infection; (E) BALF IL-17 level at 8 h postinfection; (F) BALF IL-17 level at 18 h postinfection; (G) BALF IL-17 level at 18 h postinfection, after cohousing; (H) BALF IL-17 level at 18 h postinfection in Jackson mice that acquired sfb after gavage 2 weeks prior to infection; (I) BALF IL-6 level at 8 h postinfection; (J) BALF IL-6 level at 18 h postinfection; (K and L) Ly6G-positive cells (neutrophils) in BALF 18 h after infection of cohoused mice (K) or sfb-colonized Jackson mice (L). (M to P) Mouse lung sections were stained with anti-RORγt antibody (upper left squares; blue), anti-IL-22 (upper middle squares; red), and anti-CD3e (upper right squares; green) and examined by confocal microscopy. Lower left squares depict phase-contrast microscopy, and lower right squares show merged confocal images. (M) Lung tissue sections of an sfb-positive mouse, showing many cells positive for RORγt, IL-22, and CD3 (whitish-yellow cells in merged confocal image), likely representing Th17 or Th22 cells, as well as RORγt- and IL-22-positive but CD3-negative cells (purple), possibly representing ILC3s. (N) Lung tissue sections of a previously sfb-negative mouse after acquisition of sfb 2 weeks prior by nasogastric lavage, also showing both RORγt+ IL-22+ CD3+ and RORγt+ IL-22+ CD3− cell populations, but in smaller quantities overall, and very few CD3-positive cells. (O) Lung tissue sections of an sfb-negative mouse showing even fewer RORγt+ or IL-22+ cells, despite a denser cell infiltrate, and no RORγt+ IL-22+ CD3− cells. Isotype control stains for panels M to O are shown in Fig. S2a to c in the supplemental material. (P) Numbers of dually IL-22- and TCRβ-positive cells in the BALF of mice 8 h after infection, as determined by intracellular staining by flow cytometry. There were significantly more IL-22- and TCRβ-positive cells in sfb-positive than in sfb-negative mice.
We did not observe significant differences in levels of IL-17, another key cytokine in Th17 immunity, between any groups of mice (Fig. 4E to H; see Fig. S2b and S2c in the supplemental material), except for a small increase in IL-17 in the BALF of initially sfb-negative mice after they became sfb positive through cohousing (Fig. 4G), indicating that this cytokine likely does not play a very important role in S. aureus pneumonia, at least not at the time points we examined.
Consistent with the known role of IL-6 in promoting IL-22 expression (24), we also found that sfb-negative mice had significantly less IL-6 in BALF at an early time point (8 h) after challenge with S. aureus (Fig. 4I). At 18 h, sfb-negative mice had significantly more IL-6 in BALF (Fig. 4J), possibly due to the very high bacterial levels driving IL-6 production in the sfb-negative mice at this time point, while the sfb-positive mice were beginning to clear the infection.
Th17 immunity leads to neutrophil attraction (25) and production of antimicrobial peptides (11). sfb-positive mice (both initially colonized mice and initially sfb-negative mice after cohousing with sfb-positive mice or after gavage with sfb) had larger numbers of Ly6G-positive cells (neutrophils) in BALF (Fig. 4K and L), supporting their propensity toward Th17 pulmonary responses.
In order to identify the types of cells capable of producing IL-22 in the lungs after infection with MRSA, we stained lung tissue sections of infected sfb-positive mice for RORγt, IL-22, and CD3e (Fig. 4M to O). We found two predominant populations of cells that were positive for IL-22. One group of cells was positive for RORγt, IL-22, and CD3 and likely represents Th17 or Th22 cells or γδ T cells. Another population of cells was positive for RORγt and IL-22 but negative for CD3 and possibly represents innate lymphoid cells of type 3 (ILC3s). Although these images are not meant to quantify the exact numbers of cells present in lung tissues of sfb-negative and sfb-positive mice, IL-22-producing cells were qualitatively more prevalent in sfb-positive mice (Fig. 4M) than in sfb-negative mice that recently acquired sfb (Fig. 4N) and sfb-negative mice (Fig. 4O). For control stains of images, please see Fig. S2 in the supplemental material.
Since studies by other groups showed that γδ T cells are the primary source of IL-17 in a similar murine model of S. aureus pneumonia (26), and since we observed no difference in IL-17 levels, we predicted that γδ T cells were not likely to be involved in the sfb-induced responses, and we focused instead on αβ T cells (Th17 cells). Therefore, we quantified the numbers of IL-22-producing Th17 cells by intracellular staining with flow cytometry, using cells isolated from BALF 8 h after infection, and we found significantly larger numbers of IL-22+ TCRβ+ cells in sfb-positive than sfb-negative mice (Fig. 4P).
IL-22 is critical for host defense against severe S. aureus pneumonia.
To further define the role of IL-22 in S. aureus pneumonia, we tested whether antibody-mediated neutralization of IL-22 or administration of recombinant IL-22 affected the clearance of S. aureus from the murine lung. sfb-positive mice given a monoclonal antibody to IL-22 intranasally 4 h prior to infection with S. aureus subsequently exhibited a nearly 2-log increase in bacterial burdens in the lungs compared to mice receiving control IgG (Fig. 5A), indicating a significantly higher susceptibility to infection in the absence of IL-22.
FIG 5.

Neutralization of IL-22 leads to more severe S. aureus pneumonia, while exogenous IL-22 administration increases resistance of sfb-negative mice to infection. C57BL/6 mice from Taconic Biosciences (sfb positive) received a monoclonal anti-IL-22 antibody or control IgG 4 h prior to challenge with 5 × 108 CFU of MRSA intranasally. (A) Numbers of bacterial CFU per gram of lung tissue at 18 h postinfection. (B and C) C57BL/6 mice from The Jackson Laboratory (sfb negative) received exogenous recombinant IL-22 or the control protein BSA during challenge with 5 × 108 CFU of MRSA LAC intranasally. Numbers of bacterial CFU per gram of lung (B) or spleen (C) were determined at 18 h postinfection. Symbols represent individual animals, and horizontal bars represent the medians. P values were determined by the Mann-Whitney U test for 2-group comparisons or by the Kruskal-Wallis test with Dunn's multiple-comparison test.
Alternatively, administration of recombinant IL-22 intranasally simultaneously with challenge with S. aureus protected sfb-negative mice from development of severe pneumonia, as indicated by significantly lower bacterial burdens in the lungs and spleens (Fig. 5B and C). Note that intranasal administration of IL-22 led to an about 60-fold increase in BALF IL-22 concentrations (see Fig. S3a in the supplemental material). Exogenous administration of anti-IL-22 antibody did not lead to significant measurable differences in BALF IL-22 and IL-17 concentrations (see Fig. S3b and c, respectively). Exogenous rIL-22 administration to sfb-positive mice simultaneously with challenge with MRSA strain LAC had no significant protective effect on susceptibility to MRSA pneumonia (see Fig. S4), suggesting that there is no additional benefit of adding IL-22 (at least at this dose) in an sfb-positive setting.
DISCUSSION
Understanding the immune responses important for host defense against S. aureus pneumonia, which is a leading cause of death worldwide, is critical for the development of vaccines and immunotherapies. In this study, we examined whether the GI microbiota, particularly the presence of sfb, has an effect on the susceptibility of mice to S. aureus pneumonia. Our findings indicate that sfb-positive mice are more resistant than sfb-negative mice when challenged in an S. aureus pneumonia model and that this resistance is correlated with a type 17 innate immune response. sfb-negative mice were much more susceptible to S. aureus pneumonia, with higher bacterial burdens in the lungs and spleens and higher mortality after challenge with S. aureus. Cohousing sfb-negative mice with sfb-positive mice for 14 days, and thereby transferring sfb, and likely other GI microbiota, significantly decreased the susceptibility to S. aureus pneumonia and increased mouse survival. Furthermore, we demonstrated that sfb accounts for most of this change in phenotype by showing that transfer of sfb to sfb-negative mice via orogastric lavage increased the resistance to S. aureus lung infection. Overall, a strong case is made for a critical role of sfb-driven type 17 immunity in the ability to clear S. aureus from infected lungs and, in particular, a role for IL-22, whose production is closely linked to the presence of sfb microbiota in the GI tract. Thus, even though the lung does not contain many GI organisms, these clearly influence innate immunity in this tissue, indicating a far-reaching role of GI microbiota-driven immune effectors in tissues beyond the GI tract.
While the results implicate the sfb component of the GI microbiota in the susceptibility of C57BL/6 mice to S. aureus lung infection, there may still have been some genetic variability contributing as a cofactor in the experiments comparing mice from different vendors (i.e., The Jackson Laboratory and Taconic Biosciences). The inbred mouse line C57BL/6 is widely used in animal models, and often, little attention is paid to the breeding facility from which mice are purchased for experiments. Although all C57BL/6 mouse strains originated from the same ancestor, separation in different laboratories over multiple decades and generations has resulted in the emergence of genetic and phenotypic differences. A recent study compared in detail the genomic and phenotypic differences of the C57BL/6J (established at The Jackson Laboratory in 1948) and C57BL/6N (passed on to the National Institutes of Health [NIH] in 1951 and later transferred to Taconic Biosciences in 1991) strains, and it found 34 single nucleotide polymorphisms (SNPs), 2 indels, and 15 structural variants, as well as a range of phenotypic differences (27), including physiologic, metabolic, and behavioral differences. Simon et al. found that male C57BL/6N mice compared to male C57BL/6J mice showed enhanced resistance to Listeria monocytogenes and an increased proinflammatory response (represented by increased plasma levels of IL-6, interferon-inducible protein 10 [IP-10], and chemokine ligand 2 [CCL2]) (27). Whether this difference was due to a genetic or environmental factor (e.g., the GI tract microbiota) is not known. Nonetheless, it is possible that some of these genetic differences also contributed to the innate immune response of C57BL/6 mice to S. aureus in our experiments comparing mice from different sources. However, genetic variation is very unlikely to play a role in our experiments comparing mice from the same source with and without sfb gavage.
The alteration of susceptibility to S. aureus pneumonia in our study is in accordance with a study by Fagundes et al., who demonstrated that colonization by indigenous microbiota alters the way that a host reacts to infectious stimuli through activation of TLR-dependent pathways (28). They showed that germfree mice were more susceptible to Klebsiella pneumoniae pneumonia and that priming germfree mice with lipopolysaccharide (LPS) or other TLR agonists increased their inflammatory responsiveness and led to better bacterial clearance and mouse survival (28). Two studies recently demonstrated that the GI tract microbiota also regulates immune defenses in the respiratory tract against influenza virus infection via the TLR7 pathway (29) or inflammasome activation (30). Gut microbiota are likely important not only in infections in the lung, as microbiota modulate tumoral immune surveillance in this tissue via the Th17 pathway as well (31). In Cheng et al.'s study, mice treated with antibiotics had increased susceptibility to lung tumor development, which could be rescued by administration of γδT17 cells or IL-17 (31). It is generally well described that mice without a microbiota have a vastly immature immune system, which likely contributes to differences in susceptibility to infection. Our work is unique in regard to examining a specific commensal organism affecting susceptibility to a nonenteric infection.
The Th17 pathway has been shown to play an important role in mucosal host defense against several respiratory pathogens, including Klebsiella pneumoniae (8), Pseudomonas aeruginosa (32), Chlamydia pneumoniae (33, 34), Mycoplasma pneumoniae (35), and Mycobacterium tuberculosis (36; reviewed in reference 5). Type 17 immunity has been described to be important in defense against S. aureus pneumonia in several other studies, although some with conflicting results (9–11). The S. aureus virulence factor α-hemolysin leads to increased transcription of host cytokine and chemokine genes, including that encoding the p19 subunit of IL-23, producing a prominent cellular Th17 response (37). Mice lacking the IL-17 receptor or IL-22 or which are coinfected with influenza A virus and thereby have an inhibited Th17 immunity display impaired clearance of S. aureus compared to wild-type or influenza virus-free mice (10). These results contrast with those of Choi et al., who reported that IL-17−/− and IL-22−/− mice showed no difference in MRSA clearance compared to wild-type mice (9). Whether these conflicting results are related to variable GI tract microbiota among mouse strains, to different bacterial challenge strains, or to other experimental variables is unclear.
IL-22 is a member of the IL-10 family of immune mediators that is expressed by several types of lymphocytes, including those of the innate and adaptive immune systems (38). It has multiple functions, among which is modulation of tissue responses during inflammation and antimicrobial molecule production (i.e., beta-defensin, Reg3γ, and lipocalin 2) (39–41). IL-22 has been shown to mediate the early host defense against bacterial pathogens (41) and appears to prevent bacterial dissemination in the GI tract, e.g., after infection with Citrobacter rodentium (41) or Salmonella enterica serotype Typhimurium (39). In the lung, IL-22 has been shown to mediate the mucosal host defense against Klebsiella pneumoniae pneumonia (8), and IL-22-expressing γδ T cells were protective in a model of Bacillus subtilis-induced pulmonary fibrosis (42).
Given our observation that increased resistance to S. aureus pneumonia was associated with higher IL-22 levels, we tested its role directly and found that antibody-mediated neutralization of IL-22 led to an increased susceptibility to S. aureus pneumonia in sfb-positive mice. Importantly, we also found that exogenous administration of IL-22 to sfb-negative mice rescued their defense against S. aureus pneumonia to a resistance level similar to that of sfb-positive mice, which further supports the protective role of IL-22 in S. aureus pneumonia. These findings suggest that therapeutic administration of recombinant IL-22 is a potential new approach to treatment of S. aureus pneumonia.
We did not observe significant differences in levels of IL-17, another key cytokine in Th17 immunity, between any groups of mice, except for a slight increase in MRSA-infected mice after sfb acquisition through cohousing, indicating that this cytokine likely does not play a very important role in S. aureus pneumonia, at least not at the time points we examined. It is possible that modulation of the pulmonary immune response by the enteric microbiota affects only specific cytokines. We observed at least two populations of cells capable of producing IL-22 in the lung after challenge with MRSA. The first population comprised cells positive for IL-22, CD3, and RORγt, likely representing Th17 or Th22 cells (43), while the second group was negative for CD3 but positive for IL-22 and RORγt, possibly representing ILC3s (44).
Our data also highlight the importance of choosing an appropriate control mouse strain for these types of experiments and might explain the conflicting results seen in prior studies. Another explanation for reported conflicting results might relate to the finding that different pathogens can induce different cytokine production patterns and effector functions in Th17 cells (45).
Protective responses to S. aureus pneumonia were associated with an increase in IL-6, a Th17-associated cytokine, at an early time point. Previous studies have shown that IL-6 is a pleiotropic cytokine involved in the cross talk between immune cells and stromal cells, activates the STAT3 signaling axis, is one of the main regulators of Th17 cell differentiation (46), and promotes IL-22 expression (24). IL-6-deficient mice showed an impaired defense against pneumococcal pneumonia (47). Interestingly, IL-6 and IL-1b levels are elevated in mice orally treated with antibiotics resulting in commensal depletion compared to those in untreated mice and are associated with more severe illness (21). This is in accordance with our findings that sfb-negative mice had lower IL-6 levels than sfb-positive mice 8 h after infection and that initially sfb-negative mice had markedly increased IL-6 expression after acquisition of sfb. sfb-negative mice in our study had higher IL-6 levels at 18 h postinfection, possibly reflecting a delayed response or a response due to the continuing high levels of S. aureus in the lungs. IL-6 produced by macrophages during MRSA infection stimulates the pulmonary epithelium via STAT3 signaling to produce the antimicrobial protein Reg3γ, which has bacteriostatic and bactericidal effects against MRSA (9). Further studies are needed to examine the link between the GI microbiota and the production of certain antimicrobial proteins in the lung.
We found an increased susceptibility of sfb-negative mice to S. aureus lung infection and could demonstrate that this was related to IL-22 production. In human disease, infection with S. aureus, and particularly MRSA, can rapidly progress toward a fatal infection over a short period, indicating the importance of the innate immune response to infection. We do not know at this time whether humans who get these invasive S. aureus pneumonias have or lack organisms in the GI microbiota that predispose them to increased susceptibility, but as progress in understanding the interaction of the microbiota and the immune system continues, it might be possible to ensure that individuals carry organisms that can maximize their innate resistance to infection. Our findings also suggest that exogenous IL-22 can be used as a therapy for S. aureus pneumonia, a disease process in desperate need of improved therapies.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the National Institutes of Health (NICHD grant K12HD047349-07 to S. Gauguet, grant K08AI108690 to N. K. Surana, grant R01 EY022054 to M. Gadjeva, and grant R01 HL092515 to G. P. Priebe) and by National Institute of Allergy and Infectious Diseases (NIAID) grants AI46706 and AI057159, a component of award number U54 AI057159 (to G. B. Pier).
We are greatly thankful to James Lederer for performing the BALF cytokine Luminex assays, to Roderick Bronson for interpreting the histological images, and to Michael Coyne for helpful advice.
We have no conflicts of interest relevant to this article.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00037-15.
REFERENCES
- 1.Dantes R, Mu Y, Belflower R, Aragon D, Dumyati G, Harrison LH, Lessa FC, Lynfield R, Nadle J, Petit S, Ray SM, Schaffner W, Townes J, Fridkin S, Emerging Infections Program-Active Bacterial Core Surveillance MRSA Surveillance Investigators . 2013. National burden of invasive methicillin-resistant Staphylococcus aureus infections, United States, 2011. JAMA Intern Med 173:1970–1978. doi: 10.1001/jamainternmed.2013.10423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rubinstein E, Kollef MH, Nathwani D. 2008. Pneumonia caused by methicillin-resistant Staphylococcus aureus. Clin Infect Dis 46(Suppl 5):S378–S385. doi: 10.1086/533594. [DOI] [PubMed] [Google Scholar]
- 3.Vardakas KZ, Matthaiou DK, Falagas ME. 2009. Incidence, characteristics and outcomes of patients with severe community acquired-MRSA pneumonia. Eur Respir J 34:1148–1158. doi: 10.1183/09031936.00041009. [DOI] [PubMed] [Google Scholar]
- 4.Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, Harrison LH, Lynfield R, Dumyati G, Townes JM, Craig AS, Zell ER, Fosheim GE, McDougal LK, Carey RB, Fridkin SK, Active Bacterial Core Surveillance MRSA Investigators . 2007. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 298:1763–1771. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- 5.Tsai HC, Velichko S, Hung LY, Wu R. 2013. IL-17A and Th17 cells in lung inflammation: an update on the role of Th17 cell differentiation and IL-17R signaling in host defense against infection. Clin Dev Immunol 2013:267971. doi: 10.1155/2013/267971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Beaucoudrey L, Puel A, Filipe-Santos O, Cobat A, Ghandil P, Chrabieh M, Feinberg J, von Bernuth H, Samarina A, Janniere L, Fieschi C, Stephan JL, Boileau C, Lyonnet S, Jondeau G, Cormier-Daire V, Le Merrer M, Hoarau C, Lebranchu Y, Lortholary O, Chandesris MO, Tron F, Gambineri E, Bianchi L, Rodriguez-Gallego C, Zitnik SE, Vasconcelos J, Guedes M, Vitor AB, Marodi L, Chapel H, Reid B, Roifman C, Nadal D, Reichenbach J, Caragol I, Garty BZ, Dogu F, Camcioglu Y, Gulle S, Sanal O, Fischer A, Abel L, Stockinger B, Picard C, Casanova JL. 2008. Mutations in STAT3 and IL12RB1 impair the development of human IL-17-producing T cells. J Exp Med 205:1543–1550. doi: 10.1084/jem.20080321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Minegishi Y, Saito M, Nagasawa M, Takada H, Hara T, Tsuchiya S, Agematsu K, Yamada M, Kawamura N, Ariga T, Tsuge I, Karasuyama H. 2009. Molecular explanation for the contradiction between systemic Th17 defect and localized bacterial infection in hyper-IgE syndrome. J Exp Med 206:1291–1301. doi: 10.1084/jem.20082767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aujla SJ, Chan YR, Zheng M, Fei M, Askew DJ, Pociask DA, Reinhart TA, McAllister F, Edeal J, Gaus K, Husain S, Kreindler JL, Dubin PJ, Pilewski JM, Myerburg MM, Mason CA, Iwakura Y, Kolls JK. 2008. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med 14:275–281. doi: 10.1038/nm1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choi SM, McAleer JP, Zheng M, Pociask DA, Kaplan MH, Qin S, Reinhart TA, Kolls JK. 2013. Innate Stat3-mediated induction of the antimicrobial protein Reg3gamma is required for host defense against MRSA pneumonia. J Exp Med 210:551–561. doi: 10.1084/jem.20120260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kudva A, Scheller EV, Robinson KM, Crowe CR, Choi SM, Slight SR, Khader SA, Dubin PJ, Enelow RI, Kolls JK, Alcorn JF. 2011. Influenza A inhibits Th17-mediated host defense against bacterial pneumonia in mice. J Immunol 186:1666–1674. doi: 10.4049/jimmunol.1002194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robinson KM, McHugh KJ, Mandalapu S, Clay ME, Lee B, Scheller EV, Enelow RI, Chan YR, Kolls JK, Alcorn JF. 2014. Influenza A virus exacerbates Staphylococcus aureus pneumonia in mice by attenuating antimicrobial peptide production. J Infect Dis 209:865–875. doi: 10.1093/infdis/jit527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Priebe GP, Walsh RL, Cederroth TA, Kamei A, Coutinho-Sledge YS, Goldberg JB, Pier GB. 2008. IL-17 is a critical component of vaccine-induced protection against lung infection by lipopolysaccharide-heterologous strains of Pseudomonas aeruginosa. J Immunol 181:4965–4975. doi: 10.4049/jimmunol.181.7.4965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gopal R, Rangel-Moreno J, Slight S, Lin Y, Nawar HF, Fallert Junecko BA, Reinhart TA, Kolls J, Randall TD, Connell TD, Khader SA. 2013. Interleukin-17-dependent CXCL13 mediates mucosal vaccine-induced immunity against tuberculosis. Mucosal Immunol 6:972–984. doi: 10.1038/mi.2012.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu W, Huang J, Duan B, Traficante DC, Hong H, Risech M, Lory S, Priebe GP. 2012. Th17-stimulating protein vaccines confer protection against Pseudomonas aeruginosa pneumonia. Am J Respir Crit Care Med 186:420–427. doi: 10.1164/rccm.201202-0182OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chewning JH, Weaver CT. 2014. Development and survival of Th17 cells within the intestines: the influence of microbiome- and diet-derived signals. J Immunol 193:4769–4777. doi: 10.4049/jimmunol.1401835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clarke TB. 2014. Early innate immunity to bacterial infection in the lung is regulated systemically by the commensal microbiota via Nod-like receptor ligands. Infect Immun 82:4596–4606. doi: 10.1128/IAI.02212-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, Tanoue T, Imaoka A, Itoh K, Takeda K, Umesaki Y, Honda K, Littman DR. 2009. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139:485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ivanov II, Frutos RL, Manel N, Yoshinaga K, Rifkin DB, Sartor RB, Finlay BB, Littman DR. 2008. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe 4:337–349. doi: 10.1016/j.chom.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ivanov II, Littman DR. 2010. Segmented filamentous bacteria take the stage. Mucosal Immunol 3:209–212. doi: 10.1038/mi.2010.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cooke KR, Hill GR, Gerbitz A, Kobzik L, Martin TR, Crawford JM, Brewer JP, Ferrara JL. 2000. Hyporesponsiveness of donor cells to lipopolysaccharide stimulation reduces the severity of experimental idiopathic pneumonia syndrome: potential role for a gut-lung axis of inflammation. J Immunol 165:6612–6619. doi: 10.4049/jimmunol.165.11.6612. [DOI] [PubMed] [Google Scholar]
- 21.Chen LW, Chen PH, Hsu CM. 2011. Commensal microflora contribute to host defense against Escherichia coli pneumonia through Toll-like receptors. Shock 36:67–75. doi: 10.1097/SHK.0b013e3182184ee7. [DOI] [PubMed] [Google Scholar]
- 22.Fox AC, McConnell KW, Yoseph BP, Breed E, Liang Z, Clark AT, O'Donnell D, Zee-Cheng B, Jung E, Dominguez JA, Dunne WM, Burd EM, Coopersmith CM. 2012. The endogenous bacteria alter gut epithelial apoptosis and decrease mortality following Pseudomonas aeruginosa pneumonia. Shock 38:508–514. doi: 10.1097/SHK.0b013e31826e47e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Umesaki Y, Okada Y, Matsumoto S, Imaoka A, Setoyama H. 1995. Segmented filamentous bacteria are indigenous intestinal bacteria that activate intraepithelial lymphocytes and induce MHC class II molecules and fucosyl asialo GM1 glycolipids on the small intestinal epithelial cells in the ex-germ-free mouse. Microbiol Immunol 39:555–562. doi: 10.1111/j.1348-0421.1995.tb02242.x. [DOI] [PubMed] [Google Scholar]
- 24.Li L, Shi QG, Lin F, Liang YG, Sun LJ, Mu JS, Wang YG, Su HB, Xu B, Ji CC, Huang HH, Li K, Wang HF. 2014. Cytokine IL-6 is required in Citrobacter rodentium infection-induced intestinal Th17 responses and promotes IL-22 expression in inflammatory bowel disease. Mol Med Rep 9:831–836. doi: 10.3892/mmr.2014.1898. [DOI] [PubMed] [Google Scholar]
- 25.Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, Oliver P, Huang W, Zhang P, Zhang J, Shellito JE, Bagby GJ, Nelson S, Charrier K, Peschon JJ, Kolls JK. 2001. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med 194:519–527. doi: 10.1084/jem.194.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng P, Liu T, Zhou WY, Zhuang Y, Peng LS, Zhang JY, Yin ZN, Mao XH, Guo G, Shi Y, Zou QM. 2012. Role of gamma-delta T cells in host response against Staphylococcus aureus-induced pneumonia. BMC Immunol 13:38. doi: 10.1186/1471-2172-13-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Simon MM, Greenaway S, White JK, Fuchs H, Gailus-Durner V, Wells S, Sorg T, Wong K, Bedu E, Cartwright EJ, Dacquin R, Djebali S, Estabel J, Graw J, Ingham NJ, Jackson IJ, Lengeling A, Mandillo S, Marvel J, Meziane H, Preitner F, Puk O, Roux M, Adams DJ, Atkins S, Ayadi A, Becker L, Blake A, Brooker D, Cater H, Champy MF, Combe R, Danecek P, di Fenza A, Gates H, Gerdin AK, Golini E, Hancock JM, Hans W, Holter SM, Hough T, Jurdic P, Keane TM, Morgan H, Muller W, Neff F, Nicholson G, Pasche B, Roberson LA, Rozman J, Sanderson M, Santos L, Selloum M, Shannon C, Southwell A, Tocchini-Valentini GP, Vancollie VE, Westerberg H, Wurst W, Zi M, Yalcin B, Ramirez-Solis R, Steel KP, Mallon AM, de Angelis MH, Herault Y, Brown SD. 2013. A comparative phenotypic and genomic analysis of C57BL/6J and C57BL/6N mouse strains. Genome Biol 14:R82. doi: 10.1186/gb-2013-14-7-r82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fagundes CT, Amaral FA, Vieira AT, Soares AC, Pinho V, Nicoli JR, Vieira LQ, Teixeira MM, Souza DG. 2012. Transient TLR activation restores inflammatory response and ability to control pulmonary bacterial infection in germfree mice. J Immunol 188:1411–1420. doi: 10.4049/jimmunol.1101682. [DOI] [PubMed] [Google Scholar]
- 29.Wu S, Jiang ZY, Sun YF, Yu B, Chen J, Dai CQ, Wu XL, Tang XL, Chen XY. 2013. Microbiota regulates the TLR7 signaling pathway against respiratory tract influenza A virus infection. Curr Microbiol 67:414–422. doi: 10.1007/s00284-013-0380-z. [DOI] [PubMed] [Google Scholar]
- 30.Ichinohe T, Pang IK, Kumamoto Y, Peaper DR, Ho JH, Murray TS, Iwasaki A. 2011. Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc Natl Acad Sci U S A 108:5354–5359. doi: 10.1073/pnas.1019378108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheng M, Qian L, Shen G, Bian G, Xu T, Xu W, Shen G, Hu S. 2014. Microbiota modulate tumoral immune surveillance in lung through a gammadeltaT17 immune cell-dependent mechanism. Cancer Res 74:4030–4041. doi: 10.1158/0008-5472.CAN-13-2462. [DOI] [PubMed] [Google Scholar]
- 32.Liu J, Feng Y, Yang K, Li Q, Ye L, Han L, Wan H. 2011. Early production of IL-17 protects against acute pulmonary Pseudomonas aeruginosa infection in mice. FEMS Immunol Med Microbiol 61:179–188. doi: 10.1111/j.1574-695X.2010.00764.x. [DOI] [PubMed] [Google Scholar]
- 33.Wolf K, Plano GV, Fields KA. 2009. A protein secreted by the respiratory pathogen Chlamydia pneumoniae impairs IL-17 signalling via interaction with human Act1. Cell Microbiol 11:769–779. doi: 10.1111/j.1462-5822.2009.01290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou X, Chen Q, Moore J, Kolls JK, Halperin S, Wang J. 2009. Critical role of the interleukin-17/interleukin-17 receptor axis in regulating host susceptibility to respiratory infection with Chlamydia species. Infect Immun 77:5059–5070. doi: 10.1128/IAI.00403-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu Q, Martin RJ, Rino JG, Breed R, Torres RM, Chu HW. 2007. IL-23-dependent IL-17 production is essential in neutrophil recruitment and activity in mouse lung defense against respiratory Mycoplasma pneumoniae infection. Microbes Infect 9:78–86. doi: 10.1016/j.micinf.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lockhart E, Green AM, Flynn JL. 2006. IL-17 production is dominated by gammadelta T cells rather than CD4 T cells during Mycobacterium tuberculosis infection. J Immunol 177:4662–4669. doi: 10.4049/jimmunol.177.7.4662. [DOI] [PubMed] [Google Scholar]
- 37.Frank KM, Zhou T, Moreno-Vinasco L, Hollett B, Garcia JG, Bubeck Wardenburg J. 2012. Host response signature to Staphylococcus aureus alpha-hemolysin implicates pulmonary Th17 response. Infect Immun 80:3161–3169. doi: 10.1128/IAI.00191-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zenewicz LA, Flavell RA. 2011. Recent advances in IL-22 biology. Int Immunol 23:159–163. doi: 10.1093/intimm/dxr001. [DOI] [PubMed] [Google Scholar]
- 39.Raffatellu M, George MD, Akiyama Y, Hornsby MJ, Nuccio SP, Paixao TA, Butler BP, Chu H, Santos RL, Berger T, Mak TW, Tsolis RM, Bevins CL, Solnick JV, Dandekar S, Baumler AJ. 2009. Lipocalin-2 resistance confers an advantage to Salmonella enterica serotype Typhimurium for growth and survival in the inflamed intestine. Cell Host Microbe 5:476–486. doi: 10.1016/j.chom.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R. 2004. IL-22 increases the innate immunity of tissues. Immunity 21:241–254. doi: 10.1016/j.immuni.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 41.Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, Abbas AR, Modrusan Z, Ghilardi N, de Sauvage FJ, Ouyang W. 2008. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med 14:282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- 42.Simonian PL, Wehrmann F, Roark CL, Born WK, O'Brien RL, Fontenot AP. 2010. Gammadelta T cells protect against lung fibrosis via IL-22. J Exp Med 207:2239–2253. doi: 10.1084/jem.20100061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eyerich S, Eyerich K, Pennino D, Carbone T, Nasorri F, Pallotta S, Cianfarani F, Odorisio T, Traidl-Hoffmann C, Behrendt H, Durham SR, Schmidt-Weber CB, Cavani A. 2009. Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J Clin Invest 119:3573–3585. doi: 10.1172/JCI40202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Van Maele L, Carnoy C, Cayet D, Ivanov S, Porte R, Deruy E, Chabalgoity JA, Renauld JC, Eberl G, Benecke AG, Trottein F, Faveeuw C, Sirard JC. 2014. Activation of type 3 innate lymphoid cells and interleukin 22 secretion in the lungs during Streptococcus pneumoniae infection. J Infect Dis 210:493–503. doi: 10.1093/infdis/jiu106. [DOI] [PubMed] [Google Scholar]
- 45.Zielinski CE, Mele F, Aschenbrenner D, Jarrossay D, Ronchi F, Gattorno M, Monticelli S, Lanzavecchia A, Sallusto F. 2012. Pathogen-induced human TH17 cells produce IFN-gamma or IL-10 and are regulated by IL-1beta. Nature 484:514–518. doi: 10.1038/nature10957. [DOI] [PubMed] [Google Scholar]
- 46.Camporeale A, Poli V. 2012. IL-6, IL-17 and STAT3: a holy trinity in auto-immunity? Front Biosci 17:2306–2326. doi: 10.2741/4054. [DOI] [PubMed] [Google Scholar]
- 47.van der Poll T, Keogh CV, Guirao X, Buurman WA, Kopf M, Lowry SF. 1997. Interleukin-6 gene-deficient mice show impaired defense against pneumococcal pneumonia. J Infect Dis 176:439–444. doi: 10.1086/514062. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.