

Abstract
Streptococcus pyogenes, or group A Streptococcus (GAS), is a pathogen that causes a multitude of human diseases from pharyngitis to severe infections such as toxic shock syndrome and necrotizing fasciitis. One of the primary virulence factors produced by GAS is the peptide toxin streptolysin S (SLS). In addition to its well-recognized role as a cytolysin, recent evidence has indicated that SLS may influence host cell signaling pathways at sublytic concentrations during infection. We employed an antibody array-based approach to comprehensively identify global host cell changes in human epithelial keratinocytes in response to the SLS toxin. We identified key SLS-dependent host responses, including the initiation of specific programmed cell death and inflammatory cascades with concomitant downregulation of Akt-mediated cytoprotection. Significant signaling responses identified by our array analysis were confirmed using biochemical and protein identification methods. To further demonstrate that the observed SLS-dependent host signaling changes were mediated primarily by the secreted toxin, we designed a Transwell infection system in which direct bacterial attachment to host cells was prevented, while secreted factors were allowed access to host cells. The results using this approach were consistent with our direct infection studies and reveal that SLS is a bacterial toxin that does not require bacterial attachment to host cells for activity. In light of these findings, we propose that the production of SLS by GAS during skin infection promotes invasive outcomes by triggering programmed cell death and inflammatory cascades in host cells to breach the keratinocyte barrier for dissemination into deeper tissues.
INTRODUCTION
Streptococcus pyogenes, also known as group A Streptococcus (GAS), is a common colonizer of the skin and mucosal surfaces of humans (1–3). GAS is typically innocuous in these locations or else leads to fairly minor and generally self-limiting infections of the skin or respiratory tract, such as impetigo or pharyngitis (1–3). In cases where an initial infection is left untreated, GAS may cause one of several severe postinfection (p.i.) sequelae, including rheumatic fever or glomerulonephritis (1–3). Furthermore, in rare cases, this exclusively human pathogen breaches the epithelial barrier and invades deeper tissues and blood, resulting in outcomes such as necrotizing fasciitis and Streptococcus toxic shock (1–3). The World Health Organization (WHO) estimates that GAS is responsible for about 18 million cases of severe postinfection sequelae and 700,000 cases of invasive disease each year (2, 4). Combined, GAS infections lead to approximately 500,000 deaths annually (2, 4).
The success of GAS in causing both mild and severe infections is due largely to the myriad of secreted and surface-bound virulence factors expressed by this pathogen. One of the most potent virulence factors produced by GAS is streptolysin S (SLS), a small, ribosomally produced peptide whose mature product is predicted to be 2.7 kDa in size (5–8). SLS is encoded by the streptolysin S-associated gene (sag) cluster. It is produced as a protoxin (SagA) which is subsequently modified by other members of the sag cluster (SagB, SagC, and SagD) to produce the fully functional toxin (6–8). All attempts to purify SLS and elucidate its mature structure have proven unsuccessful due to the unusual nature of its amino acid sequence and the complexity of its posttranslational modifications (5, 8). Although the exact structure of SLS is still unknown, recent studies have indicated that SLS undergoes extensive posttranslational processing that involves the formation of several heterocyclic rings at distinct sites along the length of the peptide (7, 8). These modifications have been shown to be critical for the cytolytic activity of SLS-like peptide toxins (7, 8). The cytolytic activity of SLS has historically been attributed to its ability to induce osmotic deregulation and subsequent lysis through an unknown mechanism, and the toxin has been reported to cause membrane rupture in a variety of eukaryotic cell types and subcellular structures (6, 9–17). The lytic activity of SLS is often measured via red blood cell (RBC) lysis because it is the GAS toxin primarily responsible for the characteristic hemolytic zone that surrounds bacterial colonies on blood agar (18).
Although pore-forming toxins such as SLS have historically been thought of primarily as inducers of lysis and rapid necrotic death in host cells, recent evidence suggests that many of these toxins have more complex roles in influencing host cell signaling at sublytic concentrations during infection. For example, uropathogenic Escherichia coli (UPEC), Aeromonas sp., and Staphylococcus aureus all produce hemolytic toxins that attenuate activation of Akt, a central host signaling kinase that promotes cell survival, growth, and inflammatory responses (19). The Cry5B pore-forming toxin from Bacillus thuringiensis induces host activation of two major stress-associated mitogen-activated protein (MAP) kinase (MAPK) proteins, p38 and Jun N-terminal protein kinase (JNK) (20). Additionally, it has recently been shown that the Panton-Valentine leukocidin (PVL) β-barrel pore-forming toxin produced by S. aureus induces wide-scale inflammatory responses through the nuclear factor kappa B (NF-κB) pathway (21). The streptolysin S toxin has already been identified as a major contributing factor in the successful translocation of group A Streptococcus across the epithelial barrier during the early stages of infection (22). Specifically, SLS was shown to play a role in disruption of intracellular junctions through recruitment of the cysteine protease calpain, which cleaves host proteins such as occludin and E-cadherin (22). SLS has also been implicated in the induction of programmed cell death in macrophages and neutrophils as well as in the inhibition of neutrophil recruitment during infection (16, 17, 23). These recent insights into the additional roles of pore-forming hemolysins highlight the potential contribution of streptolysin S to pathogenesis at the cellular level under conditions that more closely mimic the infection process.
To determine the role of SLS in host cell signaling during infection, we utilized a comprehensive antibody array to identify global changes in host keratinocyte signaling following wild-type (WT) or SLS-deficient GAS infections using an in vitro human epithelial infection model. Using this method, we identified key SLS-dependent host responses, including the activation of the proinflammatory p38 MAPK and nuclear factor kappa B (NF-κB) signaling pathways and the downregulation of prosurvival Akt signaling. Signaling responses identified by array analysis were subsequently confirmed by biochemical and protein identification methods. To determine whether these SLS-dependent host signaling changes required direct bacterium-host cell contact, we designed a Transwell infection system in which direct bacterial attachment was prevented while still allowing secreted factors access to host cells. The results obtained using this approach were consistent with our direct infection studies and answer a longstanding question regarding whether SLS is a toxin that requires bacterial attachment to host cells for activity. These findings suggest that GAS promotes pathogenesis and progression to severe disease by using secreted streptolysin S to induce programmed cell death and inflammatory cascades in infected keratinocytes. To our knowledge, this is the first comprehensive antibody array analysis of a specific virulence factor in a bacterial pathogen. We therefore anticipate that this antibody-based array method and Transwell infection system could be widely applicable to the study of other secreted bacterial toxins and their specific effects on host cell signaling.
MATERIALS AND METHODS
Bacterial cultures.
The GAS M1T1 5448 strains utilized for these experiments included WT GAS, a sagA Δcat SLS-deficient mutant (abbreviated here as the ΔsagA mutant), and a sagA-complemented strain. An illustration of the sag cluster and the hemolytic activity of the strains used in this study may be found in Fig. S1 in the supplemental material. The sagA Δcat GAS strain was generated by the laboratory of Victor Nizet at the University of California, San Diego (6), and the complemented strain was generated in our laboratory by transformation of the ΔsagA mutant with the pDCerm expression vector containing the wild-type sagA gene along with 300 bp directly upstream of sagA to include native promoter sequences (abbreviated here as the ΔsagA+sagA mutant). We estimate that our sagA-complemented strain contains 5 to 10 copies of the pDCerm plasmid. GAS M1T1 5448 strains were grown in Todd-Hewitt broth at 37°C for 16 to 20 h prior to infection of human cells. Supernatant treatments were prepared by growing these strains for 0 to 8 h at 37°C in Dulbecco's modified Eagle's medium (DMEM)–10% fetal bovine serum (FBS). Bacterial cultures were centrifuged (relative centrifugal force [RCF], 2,400) for 10 min to remove the bacteria, and the bacterial supernatants containing SLS were subjected to sterile filtration using a 0.22-μm-pore-size filter prior to use (Millipore).
RBC lysis assay.
Whole sheep blood (BD Biosciences) was washed three times in phosphate-buffered saline (PBS) (Gibco) and diluted to a final concentration of 1:20 (vol/vol) in PBS. Bacterial supernatants (prepared as described above) were combined with the blood/PBS mixture at a ratio of 1:5 (supernatants to blood) and incubated at 37°C overnight. A mixture of 1% Triton–PBS–blood was used as a positive control, representing 100% red blood cell (RBC) lysis, and PBS was used as a negative control for RBC lysis. Experiments were performed in triplicate under each test condition. After overnight incubation, the samples were centrifuged at 275 RCF for 10 min and an aliquot (200 μl) of the supernatant was collected from each sample and transferred to a clear, flat-bottom 96-well plate (Eppendorf) for analysis. A microplate reader was used to determine sample absorbance at 450 nm, a wavelength which allows detection of the heme released from the lysed RBCs. Percent lysis was determined by normalizing absorbance values to the Triton positive control (100% lysis) and to PBS buffer as the negative control.
Keratinocyte culture.
HaCaT human epithelial keratinocytes (24), a kind gift from V. Nizet, were used for these studies. Cells were maintained in DMEM (Life Technologies 11995-073) with 10% heat-inactivated fetal bovine serum (FBS) and incubated at 37°C with 5% CO2. HaCaT cells were maintained in 100-mm-diameter culture dishes (Nunc).
Antibody array.
A Kam-850 antibody microarray kit from Kinexus (Vancouver, BC, Canada) was used for these studies, with protocols adjusted as necessary. HaCaT cells were used for all analyses. Confluent HaCaT cells grown in 100-mm-diameter culture dishes were washed with sterile PBS, and fresh medium (DMEM–10% FBS) was applied prior to GAS infection. Overnight bacterial cultures of GAS were centrifuged and resuspended in fresh Todd-Hewitt broth and their optical densities were normalized. The HaCaT cells were infected with the normalized overnight cultures of wild-type GAS or ΔsagA M1T1 5448 GAS at a multiplicity of infection (MOI) of 10 bacteria per host cell. The infected cells were incubated at 37°C with 5% CO2 for 4 h. At the end of the infection period, media and nonadhered bacteria were aspirated and the cells were washed twice with cold PBS. The cells were then lysed with Kinexus lysis buffer, and the protocol for protein collection, labeling, and antibody array incubation was followed according to the manufacturer's instructions.
Keratinocyte infection. (i) Direct infection conditions.
HaCaT cells were grown to 90% confluence in 6-well tissue culture plates (CytoOne) or in 100-mm-diameter dishes (Nunc). Immediately prior to treatment, the cells were washed with sterile PBS and fresh medium (DMEM–10% FBS) was applied. Overnight bacterial cultures of GAS were centrifuged and resuspended in fresh Todd-Hewitt broth, and their optical densities were normalized. The HaCaT cells were infected with the normalized overnight cultures of wild-type GAS, ΔsagA GAS, or sagA-complemented (ΔsagA+sagA) GAS at an MOI of 10 bacteria per host cell. The infected cells were incubated at 37°C with 5% CO2 for the times indicated for each experiment.
(ii) Transwell infection conditions.
HaCaT cells were grown to 90% confluence in 6-well tissue culture plates (CytoOne). Immediately prior to treatment, the cells were washed with sterile PBS and fresh medium (DMEM–10% FBS) was applied. A sterile 0.4-μm-diameter Transwell insert (Corning) was placed in each well, and fresh cell culture medium was added to the upper chamber according to the manufacturer's instructions. The cells were then treated with normalized overnight cultures of wild-type or mutant GAS at an MOI of 10 by adding the bacteria to the upper chamber of the Transwell system. The infected cells were incubated at 37°C with 5% CO2.
SDS-PAGE and Western blotting.
The protein concentration of each HaCaT sample lysate was determined by the use of a bicinchoninic acid (BCA) assay (Pierce) and bovine serum albumin (BSA) protein standards and normalized prior to loading the samples on a 4% to 15% polyacrylamide gel (Bio-Rad). Samples were transferred to a polyvinylidene difluoride (PVDF) membrane and blocked in 5% BSA–0.1% Tween 20 (Sigma)–Tris-buffered saline (TBS). The membranes were incubated with primary antibodies overnight at 4°C. All primary antibodies were used at a dilution of 1:1,000, with the exceptions of anti-high-mobility group box protein 1 (HMGB1) (1:500) and anti-β-actin (1:2,000). The membranes were washed for 1.5 h in TBS–0.1% Tween 20 and incubated with horseradish peroxidase (HRP)-conjugated secondary antibody at a dilution of 1:5,000 for 1.5 h at room temperature. Membranes were then washed for 1.5 h in TBS–0.1% Tween 20 and incubated with ECL chemiluminescence reagent (Pierce) prior to development on film (Dot Scientific).
Immunofluorescence staining and imaging.
Cells were plated on sterile glass coverslips in 6-well dishes and treated with GAS as described. Following infection, cells were washed in cold PBS and fixed overnight in 4% (wt/vol) paraformaldehyde solution–PBS. Coverslips containing treated cells were then washed in PBS and blocked for 2 h at room temperature in PBS–1% (wt/vol) normal goat serum–2% (vol/vol) Triton–0.5% (vol/vol) Tween 20. The cells were washed with PBS for 1.5 h and incubated with primary antibody at a 1:50 ratio in blocking solution overnight at 4°C. Following incubation with primary antibody, the coverslips were washed for 1.5 h in PBS and then incubated for 2 h at room temperature in secondary antibody (goat anti-rabbit IgG Alexa Fluor 488) using a 1:200 ratio of antibody to blocking solution. Coverslips were washed for 1 h prior to adding DAPI (4′,6-diamidino-2-phenylindole) nuclear stain and rhodamine-phalloidin actin stain at a ratio of 1:1,000 in blocking solution. The coverslips were incubated with nuclear and actin stains for 30 min at room temperature prior to final washes in PBS for 30 min at room temperature. The coverslips were then mounted on glass slides using Fluoromount-G and allowed to set overnight prior to sealing and imaging. Imaging data were collected on a DeltaVision Nikon 90i fluorescence microscope using a standard 20× objective. A Nikon A1R-MP confocal microscope (60× oil objective) was also used to image the localization of HMGB1 following GAS infection. ImageJ software and NIS-Elements Viewer (Nikon) software were used to process the captured images. At least three biological replicates were performed per condition for all immunofluorescence experiments, and at least 3 fields per slide were counted to obtain the reported data. Statistics were determined based on at least 400 cells counted per condition and were pooled from the data from the 3 biological replicates. The graphed data represent averaged values from 3 biological replicates, and error bars indicate standard deviations from the means. Normal distribution was assumed, and significance was determined by analysis of variance (ANOVA) followed by post-ANOVA Dunnett's tests to compare each condition to a corresponding control mean as indicated.
Antibodies and stains.
Antibodies to phospho-Akt Ser473, total Akt, NF-κB p65, IκBα (inhibitory protein of NF-κB), alpha-tubulin, phospho-MAPK p38 (T183 plus T185), total MAPK p38, total heat shock protein 27 (HSP27), phospho-HSP27 (S82), phospho-MEK3/6 (S189 plus S207), total MEK3, total mitogen- and stress-activated protein-serine kinase 1 (MSK1), phospho-MSK1 (S376), total caspase-1, cleaved caspase-1 (p20), and HMGB1 were obtained from Cell Signaling. Antibodies to beta-actin were obtained from AbCam. DAPI nuclear stain was obtained from Cell Signaling, and goat anti-rabbit IgG Alexa Fluor 488 and rhodamine-phalloidin actin stain were obtained from Molecular Probes (Life Technologies).
Ethidium homodimer cell death assay.
HaCaT cells were plated in 24-well tissue culture plates and infected using the conditions described above (MOI = 10). After infection, the cells were washed with sterile PBS. Cells were then covered and incubated at room temperature for 30 min with 4 μM ethidium homodimer 1 (Molecular Probes)–PBS. The level of fluorescence was determined using a plate reader set to 528-nm excitation and 617-nm emission with a cutoff value of 590 nm. The percentage of dead cells was determined by adding 0.1% (wt/vol) Saponin (Sigma) to each well following the initial reading and allowing the plate to incubate for an additional 20 min at room temperature before reading the plate a second time at the same settings. Percent membrane permeabilization values were obtained individually for each well by dividing the initial fluorescence reading value (posttreatment) by the second fluorescence reading value (post-Saponin addition). Experiments were performed in triplicate under each treatment condition, and the average and standard deviation of the values determined under each condition were plotted for comparison. Normal distribution was assumed, and significance was determined by ANOVA, followed by post-ANOVA Dunnett's tests to compare each condition to a corresponding control mean as indicated.
ATP determination assay.
HaCaT keratinocytes were grown to 90% confluence in 6-well dishes and infected with GAS as described above. Lysates were collected on ice immediately following infection and normalized via BCA assay prior to ATP determination. ATP levels were determined using a luminescence-based Molecular Probes ATP Determination kit from Life Technologies. The reported values for each infection condition were normalized to the corresponding uninfected controls. Normal distribution was assumed, and significance was determined by ANOVA, followed by post-ANOVA Dunnett's tests to compare each condition to a corresponding control mean as indicated.
LDH release assay.
HaCaT cells were plated in 24-well tissue culture plates and infected using the conditions described above (MOI = 10). Immediately prior to infection, cells were washed with PBS and supplied with fresh phenol red-free DMEM supplemented with 1% BSA (wt/vol), 2 mM l-glutamine (Gibco), and 1 mM sodium pyruvate (Gibco). Following infection, supernatants were collected and lactate dehydrogenase (LDH) release was evaluated using a cytotoxicity detection kit from Roche. Normal distribution was assumed, and significance was determined by ANOVA, followed by post-ANOVA Dunnett's tests to compare each condition to a corresponding control mean as indicated.
Inhibitor and activator compounds.
The Akt activator compound SC79 was obtained from R&D Systems (25). SB203580, which inhibits p38 MAPK activity, was obtained from Cell Signaling. The NF-κB inhibitor curcumin was obtained from Santa Cruz Biotechnology. Z-VAD-fmk, a general caspase inhibitor, was obtained from R&D Systems. The receptor-interacting protein kinase 1 (RIPK1) kinase inhibitor compound necrostatin-1 was obtained from Sigma-Aldrich. The RIPK3 inhibitor GSK872 was obtained from Calbiochem, and the mixed-lineage kinase domain-like (MLKL) inhibitor necrosulfonamide (NSA) was obtained from Tocris Bioscience. Each of the compounds found to reduce streptolysin S-dependent keratinocyte cytotoxicity (SC79, SB203580, curcumin, necrostatin-1, GSK872, and NSA) was applied to GAS cultures to evaluate bacterial toxicity. All compounds tested were found to have no impact on either growth or viability of GAS (see Fig. S2A to F in the supplemental material), with the exception of curcumin (see Fig. S2C), which affected bacterial growth at early time points (2 h), though this difference was not apparent at later time points (4 h and 6 h). These data confirm that the inhibitors used in this study were not bactericidal in nature.
Caspase-3 and caspase-7 activity assay.
HaCaT cells were plated at a cell density of 13,000 cells per well in a 96-well tissue culture-treated plate (CytoOne) suitable for use on a luminescence plate reader (white, flat bottom). Cells were allowed to attach for 24 h prior to direct infection with WT GAS, ΔsagA GAS, or ΔsagA+sagA GAS at an MOI of 10. Cells were infected for 6 h prior to addition of Caspase-Glo 3/7 reagent (Promega), which contains both cell lysing agents and the luminescent caspase substrate which allows detection of caspase-3 and caspase-7 activity. Luminescence values were determined using a microtiter plate reader. For each biological replicate, each condition was tested in triplicate and the reported values represent the results of one of three independent biological replicates. Normal distribution was assumed, and significance was determined by ANOVA, followed by post-ANOVA Dunnett's tests to compare each condition to a corresponding control mean as indicated.
Statistical analyses.
All statistical analyses were carried out using Graph Pad Prism 6.0. Normal distribution was assumed for all data sets based on the similarity of standard deviations between conditions within each data set and on the absence of outlier values. Significant differences for individual pairs of means were determined by Student's t test, and P values of <0.05 were considered to be significant. For data sets in which means of 3 or more groups were being compared, ANOVA testing was used to determine overall P values. In cases where a significant P value (P < 0.05) was obtained by ANOVA, post hoc Dunnett's tests were carried out to compare the means of all members of a data set to a control mean, which is indicated for each experiment. P values of <0.05 were considered to be significant. Individual P values from t tests and post hoc Dunnett's tests were reported as follows for all data sets: *, P = 0.01 to 0.05; **, P = 0.001 to 0.01; ***, P = 0.0001 to 0.001; ****, P < 0.0001; n.s., not significant.
RESULTS
Streptolysin S induces alterations in keratinocyte survival and inflammatory signaling cascades during GAS infection.
Because the first cells with which GAS typically interacts during skin infection are keratinocytes, we utilized the HaCaT human epithelial keratinocyte cell line to elucidate the effects of streptolysin S on host signal transduction. We conducted an antibody-based array to comprehensively compare the signaling responses of human keratinocytes that were infected with wild-type GAS to the responses of those infected with SLS-deficient GAS. The array we selected contained 850 different antibodies specific for 649 different host signaling proteins (Kinex Kam-850 kit). Optimal infection conditions to evaluate the effects of SLS on host cells were determined by collecting bacterial supernatants from wild-type GAS grown for 0 to 8 h in keratinocyte culture media (DMEM–10% FBS) (Fig. 1A). SLS activity in these supernatants was assessed using a red blood cell lysis assay. Results indicated that wild-type GAS bacteria produce maximal amounts of the SLS toxin during the middle-to-late log phase of bacterial growth (Fig. 1B). To ensure that there were no growth defects in our SLS-deficient and sagA complement strains, we performed a growth curve analysis of these strains and observed growth rates that were identical to the wild-type strain rate (Fig. 1C). To confirm that the hemolytic phenotype was SLS dependent, we collected supernatants from all three strains during late log phase. WT GAS and sagA-complemented GAS strongly induced RBC lysis, while exposure to the ΔsagA mutant did not result in significant lysis (Fig. 1D). On the basis of these data, we evaluated the effects of SLS on keratinocyte signaling by infecting the cells with WT GAS or ΔsagA GAS for 4 h at an MOI of 10, as SLS is robustly produced by GAS under these conditions.
FIG 1.

Active streptolysin S toxin is maximally produced during the mid-to-late log phase of GAS growth. (A) Growth of wild-type GAS in DMEM–10% FBS from 0 h to 8 h. (B) Lytic activity determined via red blood cell (RBC) lysis assay of WT GAS supernatants as a function of bacterial growth. Supernatant samples were taken directly from the cultures used to produce the growth curve shown in the upper left panel. (C) The levels of growth of WT, ΔsagA mutant, and sagA complement (ΔsagA+sagA) GAS strains in DMEM–10% FBS indicate consistent growth for all three strains. (D) An RBC lysis assay was performed using supernatants collected from WT and SLS-mutant cultures during the mid-to-late log phase. The upper and lower limits of RBC lysis were determined by using Triton X-100 detergent as a positive control and PBS as a negative control; percent RBC lysis was determined relative to these values. The results from three replicates are averaged for each condition, and error bars represent standard deviations. Significance was determined by ANOVA (P < 0.0001), and a post hoc Dunnett's test was performed to determine differences in the levels of RBC lysis between treatments. Significance is denoted by asterisks as described in Materials and Methods. Means of data from each condition were compared to the mean level of RBC lysis induced by the ΔsagA mutant.
Following infection, the host cells were lysed and the soluble proteins were analyzed with the Kinex antibody chip. Results from the full array analysis are included in Table S1 in the supplemental material. Calculation of percent error range, z-scores, z-score ratios, and fold changes between the two infection samples (wild-type GAS and ΔsagA GAS) allowed us to identify changes that were likely to be significant. A z-score ratio cutoff value of >|0.95| was used to identify significant candidates (see Table S2 and S3). Proteins whose activity or total levels were substantially up- or downregulated (z-score ratio >|0.95|) under the WT infection condition versus the ΔsagA GAS infection condition were manually sorted into specific signal transduction pathways using DAVID Bioinformatics Software and functional descriptions from the UniProt protein database. Fold changes and z-score ratios between the WT GAS and ΔsagA GAS infections were organized in order of decreasing fold change (see Table S2). Descriptions of the protein candidates identified in the array analysis as well as their known functions are provided in Table S3. Pathways in which the activity levels of numerous signaling proteins were differentially affected in the presence of SLS were selected as candidates for quantitative follow-up experiments. Among some of the most significant differences in protein activation between wild-type and SLS-deficient GAS infections were those observed in proteins involved in the regulation of host cell survival, such as Akt pathway members, as well as stress and inflammatory responses, particularly proteins in the MAPK family and the NF-κB pathway. Other proteins identified from the array included those involved in regulating the host cytoskeleton and cell-cell contact and in DNA damage repair and remodeling, as well as regulators of the cell cycle and translation. From these results, we hypothesized that SLS induces the activation of several stress and inflammatory signaling pathways in keratinocytes to enhance programmed cell death and ultimately contribute to tissue destruction and the progression to severe disease and sepsis.
SLS dampens Akt1/PI3K signaling in epithelial keratinocytes.
The cytotoxic properties of SLS have been well documented in a variety of cell types and subcellular structures in vitro, as well as in vivo using murine skin infection models, but the mechanism of SLS activity is not well understood (6–17, 26). An understanding of the physiological role of this toxin during infection is complicated by reports of its varied effects in different cell types and tissues. SLS has been observed to induce rapid lysis in red blood cells and to contribute to apoptosis or other forms of programmed cell death, such as oncosis, in macrophages and neutrophils (6–17, 26). In murine skin infection models, SLS contributes to the formation of a necrotic lesion at the site of inoculation (6–8). In light of these findings, we hypothesized that SLS may contribute to the regulation of cell death pathways in epithelial keratinocytes. Our antibody array data identified changes in the activity of numerous proteins involved in the regulation of cell growth and survival, and many of the most significant changes occurred in members of the PI3K/Akt1 pathway. Specifically, the antibody array data indicated that phosphorylation at the two sites required for Akt1 activation, T308 and S473, was substantially reduced in wild-type infection compared to infection with the SLS-deficient mutant (see Table S1, S2, and S3 in the supplemental material). Consistent with this observation, the levels of the activated forms of PI3K and PDK1 (positive regulators of Akt activity) were also reduced in WT GAS-infected versus ΔsagA GAS-infected cells (see Table S1, S2, and S3). In contrast, the array results indicated substantially increased levels of the active form of phosphatase and tensin homolog (PTEN), a key inhibitor of the PI3K/Akt1 pathway, in the presence of SLS (see Table S1, S2, and S3). Collectively, these trends suggest that the PI3K/Akt1 pathway is downregulated in host cells in response to SLS.
To confirm the effects of SLS on the PI3K/Akt1 pathway, we infected human keratinocytes with the WT strain, the ΔsagA mutant, and the sagA complement (ΔsagA+sagA) strain and assessed changes in the levels of both total and active (phosphorylated) Akt1. Our results revealed that Akt1 activity is significantly decreased in WT GAS-infected and sagA complementation strain-infected cells, as indicated by loss of phospho-Akt (Fig. 2). Although infection with the ΔsagA mutant induced a modest decrease in the level of phospho-Akt compared to the uninfected control, phospho-Akt levels were significantly lower in infections with SLS-producing strains. As SLS is a secreted toxin, we anticipated that its effects on host cell signaling would occur independently of direct contact between the bacteria and host cells. We therefore utilized a Transwell-based infection assay to further verify that the signaling changes that we observed were due to the activity of the SLS toxin on host cells (see Fig. S3 in the supplemental material). Transwell infection experiments using WT GAS, the ΔsagA mutant, and the sagA-complemented mutant to measure phospho-Akt levels were consistent with our direct infection model, confirming that SLS contributes to the downregulation of this critical survival pathway in human epithelial keratinocytes (Fig. 2). Interestingly, the PI3K/Akt1 pathway has been shown to be downregulated during infection by several other cytolytic bacterial toxins, including the HlyA toxin produced by uropathogenic Escherichia coli (19). Upon activation, one of the major functions of Akt is to inhibit programmed cell death, and lower levels of active Akt in infected host cells could lead to apoptosis or other forms of cell death. We hypothesized, therefore, that the SLS-dependent decrease of the level of phospho-Akt shifts the balance of host cell survival toward the activation of programmed cell death pathways in infected keratinocytes.
FIG 2.
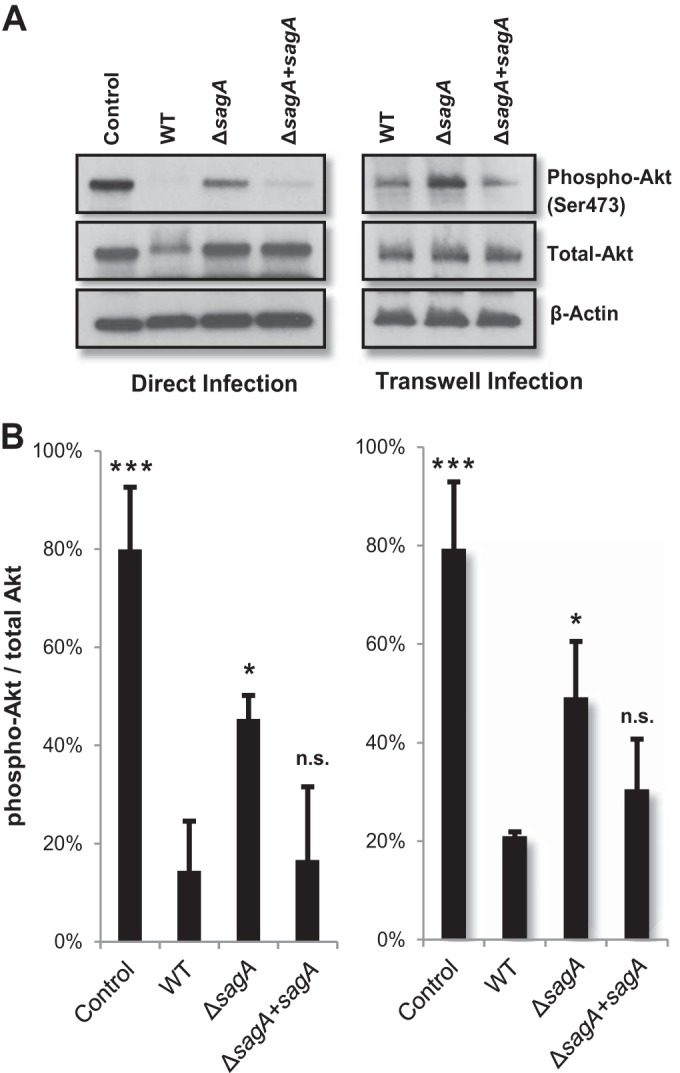
SLS induces loss of phosphorylated Akt in human keratinocytes. (A) HaCaT keratinocytes were infected with GAS directly for 4 h or using the Transwell system for 7 h at an MOI of 10. Levels of phosphorylated Akt in keratinocytes were decreased in the presence of SLS-producing GAS compared to the SLS-deficient mutant. (B) Densitometry data from three independent Western blot analyses showing phospho-Akt/total Akt levels for cells infected with GAS. Error bars represent standard deviations from the means. Similar activity levels were observed for direct infection (left) and infection using the Transwell system (right). Significance was determined by ANOVA (left, P = 0.0003; right, P = 0.0005), and Dunnett's tests were performed post hoc to determine differences in Akt activation compared to wild-type infection for the two infection systems.
SLS accelerates programmed cell death in GAS-infected epithelial keratinocytes.
To determine if SLS-dependent downregulation of the Akt1 cytoprotective cascade leads to enhanced keratinocyte cell death, we measured cytotoxicity using an ethidium homodimer membrane permeabilization assay. At 4 h postinfection, we observed a 10% to 15% loss in viability in infected cells compared to uninfected cells, with only small differences across experimental conditions (Fig. 3A). However, by 6 h postinfection, keratinocyte cell death was dramatically enhanced in the presence of SLS compared to SLS-deficient conditions (Fig. 3A). Similar trends were observed when cells were exposed to GAS using the Transwell infection system, with SLS-producing strains significantly enhancing cell death compared to uninfected cells or cells exposed to the SLS-deficient mutant at 12 h posttreatment (Fig. 3B). Consistent with a loss in keratinocyte membrane integrity as measured by ethidium homodimer assays, exposure to SLS led to enhanced ATP depletion as well as increased LDH release during GAS infection (see Fig. S4 in the supplemental material). These data indicate that SLS is a major contributor to GAS-induced keratinocyte cell death during infection and that direct contact between bacteria and host cells is not required to initiate these events. The slight increase in cell death induced by the ΔsagA mutant compared to the uninfected controls under both the direct infection and Transwell infection conditions is consistent with previous reports of additional contact-dependent and -independent GAS factors, such as streptolysin O (SLO), that contribute to cell death during infection (6, 15, 17, 27–31).
FIG 3.

Keratinocyte viability decreases in the presence of active SLS toxin. GAS-induced cell death was assessed via ethidium homodimer membrane permeabilization assay in HaCaT cells in the presence of WT, SLS-deficient, or sagA-complemented GAS. (A) Keratinocytes were infected with GAS directly for 4 h or 6 h at an MOI of 10. (B) Keratinocytes were exposed to GAS using the Transwell infection system, in which direct contact between bacteria and human cells is prohibited, for 8 h or 12 h at an MOI of 10. In both panels, data from 3 replicates are averaged and error bars represent standard deviations from the means. Significance for each time point was determined by ANOVA (4 h, P = 0.0005; 6 h, P < 0.0001; 8 h, P = 0.0241; 12 h, P < 0.0001). Dunnett's tests were performed to compare means of data from each condition to the wild-type infection data for the corresponding time point.
Having established a role for SLS in enhancing GAS-induced keratinocyte death, we next sought to determine whether Akt signaling contributed to this effect. Prior to GAS infection, HaCaT cells were treated with SC79, an Akt activator (see Fig. S5 in the supplemental material). GAS-induced cell death was then assessed 6 h postinfection via an ethidium homodimer membrane permeabilization assay. Our data demonstrated that activation of Akt could significantly reduce SLS-dependent keratinocyte death during infection (Fig. 4). These data support the hypothesis that the SLS-dependent decrease in phospho-Akt levels contributes to the induction of cell death in GAS-infected keratinocytes.
FIG 4.
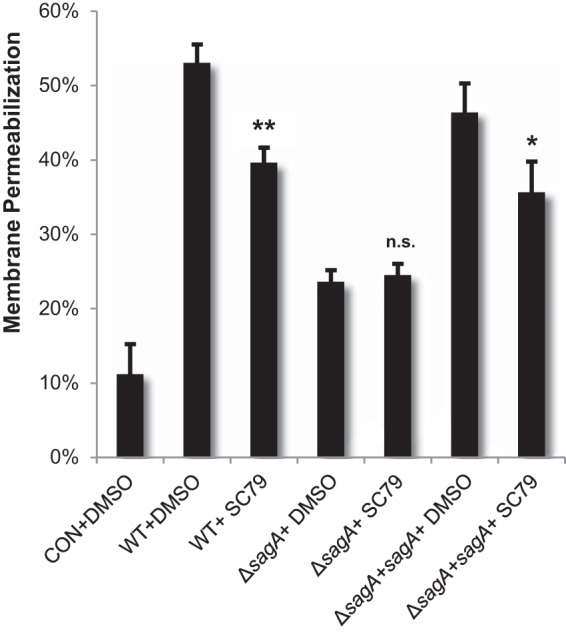
Loss of phospho-Akt contributes to SLS-dependent keratinocyte death. HaCaTs were treated with 8 μg/ml (22 μM) SC79 for 1.5 h prior to infection with GAS. Cells were infected for 6 h, and cell death was detected by ethidium homodimer membrane permeabilization assay. Experiments were performed in triplicate under each condition, and data were averaged; error bars represent standard deviations. Treatment with the Akt activator significantly reduced SLS-induced keratinocyte death, as determined by ANOVA (P < 0.0001) and post hoc Dunnett's testing. Means of vehicle control data from each strain (i.e., WT+DMSO) were compared to data from uninfected control cells (CON+DMSO) and the corresponding pharmacological treatment (i.e., WT+SC79). P values for data from vehicle controls matched to each GAS strain versus treatment with SC79 are indicated.
SLS promotes keratinocyte cell death through activation of the p38 MAPK pathway.
The relationship between the SLS-mediated decrease in phospho-Akt levels and enhanced cell death led us to further investigate the mechanism through which SLS-dependent keratinocyte death is induced during GAS infection. In addition to the downregulation of the Akt1 cytoprotective cascade, the antibody array data indicated a robust SLS-dependent activation of numerous MAPK family members, particularly, members of the p38 MAPK pathway (see Table S1, S2, and S3 in the supplemental material). Interestingly, Akt has been shown to negatively regulate several MAPK kinase kinases (MAP3Ks) upstream of p38 MAPK, including ASK1 and MEKK3 (32, 33). Additionally, the loss of Akt and a consequent increase in p38 signaling have been observed in several cell types in response to a variety of stressors, including several chemotherapeutic agents, adenoviral protein EA1, serum starvation, UV radiation, treatment with tumor necrosis factor alpha (TNF-α), and exposure to ethanol (32, 33). To ascertain whether Akt also plays a role in the negative regulation of p38 signaling during GAS infection, we treated keratinocytes with the Akt activator SC79 or the vehicle control dimethyl sulfoxide (DMSO) at 1.5 h prior to infection. Activation of Akt by SC79 significantly reduced p38 activity (phosphorylation) during GAS infection (Fig. 5). This suggests that the SLS-mediated loss of Akt activity leads to increased activation of the p38 signaling cascade, presumably via Akt-mediated changes in activation of MAP3Ks upstream of p38 (32, 33).
FIG 5.
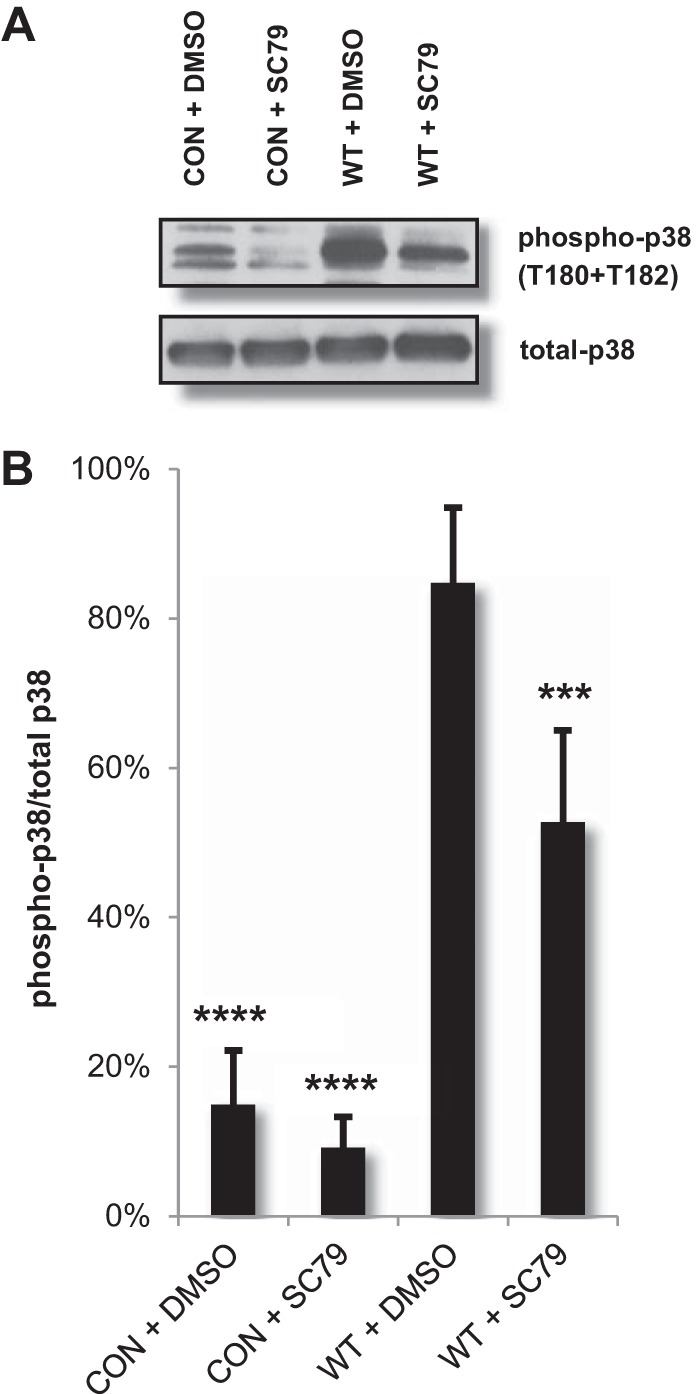
Pharmacological activation of Akt reduces GAS-mediated p38 activation. HaCaTs were treated with 22 μM SC79 (Akt activator) or DMSO for 1.5 h prior to direct infection with GAS at an MOI of 10. Lysates were collected for Western blotting 4 h postinfection. (A) Levels of phosphorylated p38 in keratinocytes were increased in the presence of GAS compared to uninfected cell results, and pretreatment with SC79 significantly reduced p38 activity during infection. (B) Densitometry data from four independent Western blot analyses show phospho-p38/total p38 levels for cells infected with GAS. Error bars indicate standard deviations. ANOVA (P < 0.0001) followed by a Dunnett's test was used to determine significance. The mean data from the experiments performed under each condition were compared to data determined under the WT+DMSO condition.
Because p38 signaling has been shown to play pivotal roles in both programmed cell death and inflammatory cascades in host cells, we hypothesized that SLS-induced activation of this pathway, downstream of phospho-Akt loss, might contribute to SLS-dependent cell death. Activation of p38 signaling has been reported in response to other bacterial toxins, such as pneumolysin from Streptococcus pneumoniae, alpha-hemolysin from Staphylococcus aureus, streptolysin O from GAS, and anthrolysin O from Bacillus anthracis (20, 34–36). To determine whether SLS might also function as an inducer of p38 signaling, we selected several members of the p38 signaling cascade and assessed their activity levels in response to GAS infection. Our results indicated significant SLS-dependent activation of several members of the p38 signaling pathway following both direct and Transwell infection (Fig. 6). Specifically, we observed SLS-dependent activation of MEK3 and MEK6 (Fig. 6A). These kinases have been shown to mutually activate p38 (37). We also observed SLS-dependent phosphorylation of p38 itself (Fig. 6C) and of several of its downstream targets, including heat shock protein 27 (HSP27) (Fig. 6B) and mitogen- and stress-activated protein-serine kinase 1 (MSK1) (Fig. 6D). To determine whether SLS-dependent activation of this pathway was contributing to cell death, we treated keratinocytes with a p38 inhibitor, SB203580, prior to infection with GAS. Inhibition of p38 activity with SB203580 dramatically reduced keratinocyte death during GAS infection in a dose-dependent manner (Fig. 7A). Treatment of HaCaT cells with the p38 inhibitor prior to infection by the ΔsagA mutant had no effect on cell death (Fig. 7B), while it significantly reduced cell death in response to infection by the ΔsagA+sagA complementation strain (Fig. 7C). These data demonstrate that SLS-induced keratinocyte cell death is dependent on the activation of the p38 signaling cascade in host cells.
FIG 6.

SLS enhances activation of the p38 MAPK pathway in infected keratinocytes. HaCaTs were infected with GAS for 4 h via direct infection or for 7 h via the Transwell infection system (at MOI = 10), and lysates were assessed for activation of p38 pathway members. Densitometry analysis of three independent Western blots per target protein was performed to quantify the relative levels of activation of p38 pathway proteins in response to GAS infection using the Transwell or direct infection systems. Averages of data from three biological replicates are shown, with error bars representing standard deviations. Relative levels of activation of proteins are represented as phosphorylated/total protein levels. Statistical significance was determined for each infection condition compared to uninfected cells. Overall P values were determined by ANOVA. (A) Left, P = 0.0027; right, P = 0.0113. (B) Left, P < 0.0001; right, P = 0.0003. (C) Left, P = 0.0004; right, P = 0.0063. (D) Left, 0.0003; right, P = 0.0142. Dunnett's tests were performed post hoc to compare data from each condition to the corresponding uninfected control mean.
FIG 7.

SLS promotes keratinocyte cell death through activation of the p38 MAPK pathway. Keratinocytes were treated with p38 inhibitor SB203580 for 2 h prior to infection with GAS. The inhibitor significantly reduced SLS-dependent keratinocyte death as demonstrated by ethidium homodimer membrane permeabilization assay. These data represent the average results of three biological replicates, with error bars representing standard deviations. Statistical significance was determined for each condition compared to control vehicle-treated cells. Overall P values were determined by ANOVA. (A) P < 0.0001. (B) P < 0.0001. (C) P < 0.0001. Dunnett's tests were performed post hoc to compare each condition to the corresponding vehicle control infection (i.e., WT+DMSO).
SLS-dependent p38 MAPK activation leads to downstream NF-κB signaling.
In addition to regulating the balance of cell survival and cell death, p38 frequently plays an important role in the induction of proinflammatory responses, such as activation of nuclear factor kappa B (NF-κB). Our array data showed an increase in the level of phosphorylated IκB kinase (IKKα), increased RelB (an NF-κB subunit), and increased levels of several NF-κB gene targets, including IκBα (inhibitory protein of NF-κB), COX2 (prostaglandin pathway), several superoxide dismutases (involved in dealing with oxidative stress), heat shock protein 90 (chaperone involved in stress responses), and STAT3 (regulating cell death and cytokine signaling) (see Table S1, S2, and S3 in the supplemental material). Activation of NF-κB signaling has previously been associated with GAS infection, but the virulence factors involved in its activation have not been fully characterized (38–40). To confirm that SLS influences the activation of this inflammatory pathway in epithelial cells, we used immunofluorescence microscopy to visualize localization of the NF-κB p65 subunit during GAS infection of keratinocytes. In its active form, the NF-κB nuclear localization sequences are exposed, leading to its translocation to the nucleus, where it can affect transcription (41). Both direct infection and Transwell infection of HaCaT keratinocytes with WT GAS or the sagA-complemented strain resulted in the robust activation of NF-κB, as seen by a significant increase in nuclear localization (Fig. 8). In contrast, infection with the SLS-deficient mutant resulted in only minimal NF-κB nuclear localization, similar to that seen with the uninfected control (Fig. 8). Consistent with these observations, infection with SLS-producing GAS strains led to a significant loss in total levels of the NF-κB inhibitory protein, IκBα, compared to results seen with the uninfected controls (see Fig. S6 in the supplemental material). Together, these results indicate that SLS is a significant contributor to NF-κB activation during GAS infection of epithelial keratinocytes.
FIG 8.

Streptolysin S enhances proinflammatory signaling through activation of nuclear factor kappa B (NF-κB). HaCaT human keratinocytes were infected directly with GAS at an MOI of 10 for 4 h (A and B) or for 8 h using the Transwell infection system (C and D). The averages of data from three biological replicates are represented for each condition (B and D), with error bars representing standard deviations. Overall P values were determined by ANOVA. (A) P = 0.0013. (B) P < 0.0001. Dunnett's tests were performed to compare each condition to the corresponding uninfected control condition.
We next sought to determine whether SLS-dependent p38 MAPK signaling was initiating NF-κB activation. Pharmacological inhibition of p38 activity with SB203580 during GAS infection significantly reduced SLS-dependent NF-κB nuclear localization, suggesting that p38 is a major upstream contributor to SLS-dependent NF-κB signaling (Fig. 9). To ascertain whether activation of the NF-κB pathway downstream of p38 signaling plays a role in the induction of SLS-mediated keratinocyte death, we treated keratinocytes with the NF-κB inhibitor curcumin prior to GAS infection. Treatment with curcumin significantly reduced SLS-dependent keratinocyte cell death, suggesting that p38-driven activation of the NF-κB pathway plays a role in the induction of programmed cell death in keratinocytes during GAS infection (see Fig. S7 in the supplemental material).
FIG 9.

Streptolysin S enhances activation of nuclear factor kappa B (NF-κB) downstream of p38 activation. HaCaT human keratinocytes were infected directly with GAS at an MOI of 10 for 4 h following a 2-h preincubation with DMSO or p38 activity inhibitor SB203580 at a concentration of 10 μM or 50 μM. (A) Pretreatment with the inhibitor significantly decreased NF-κB nuclear localization in GAS-infected cells. (B) The averages of the results from three biological replicates are graphed for each condition, and error bars represent standard deviations. ANOVA (P < 0.0001) followed by a Dunnett's test was used to determine significance. The mean data from each condition were compared to data from the WT+DMSO condition.
SLS-dependent keratinocyte death occurs primarily through programmed necrosis.
Bacterial pathogens have been observed to induce death in host cells through both caspase-dependent and caspase-independent mechanisms (42–49). To address the dependence of SLS-mediated keratinocyte cell death on the presence of caspases, we treated HaCaT cells with the pan-caspase inhibitor Z-VAD-fmk prior to infection with WT GAS, the ΔsagA mutant, or the sagA-complemented strain. General inhibition of caspases using this inhibitor did not significantly reduce GAS-dependent cell death at 6 h p.i., though the inhibitor did effectively reduce staurosporine-induced membrane permeabilization to control levels (see Fig. S8 and S9 in the supplemental material). These results suggest that GAS-induced keratinocyte cell death occurs primarily through a caspase-independent mechanism. The two primary forms of caspase-dependent cell death are apoptosis and pyroptosis, the latter of which is an inflammatory form of programmed cell death that induces inflammasome activation and plays an important role in the immune response (50, 51). To provide additional evidence that SLS-dependent host cell death does not require caspase activation, we investigated activation of specific caspases associated with apoptosis and pyroptosis during GAS infection. Following infection, we utilized a luminescence-based CaspaseGLO kit (Promega) to determine the activity levels of executioner caspases 3 and 7, which are key mediators of apoptosis. Our results indicated that GAS infection induces a slight increase in the activity of caspases 3 and 7 but that their activity is not SLS dependent, as we detected similar levels across infection conditions by 6 h postinfection (see Fig. S8 in the supplemental material). Since caspases 3 and 7 are activated downstream of both the intrinsic and extrinsic apoptotic cascades, we concluded that SLS-dependent keratinocyte death does not rely on the induction of the classic apoptosis cascade. Similarly, we did not observe SLS-dependent activation (cleavage) of procaspase-1 to caspase-1 in infected keratinocytes from 0 to 6 h p.i. (see Fig. S8). As cleavage of procaspase-1 is a major hallmark of pyroptosis, we concluded that pyroptosis is unlikely to be the primary mechanism of keratinocyte death during GAS infection. These results were consistent with the observation that pan-caspase inhibition did not significantly reduce SLS-dependent keratinocyte death during infection.
As our results indicated that SLS-mediated host cell death does not rely on caspase activation, we hypothesized that SLS was inducing programmed necrosis in GAS-infected keratinocytes. To investigate this hypothesis, we began by evaluating the localization of high-mobility group box protein 1 (HMGB1), a chromatin-binding protein that is present in the nucleus of almost all healthy eukaryotic cells (48, 52, 53). HMGB1 can be used to distinguish apoptosis from necrosis because it is released extracellularly during necrotic death but is sequestered in the nucleus during apoptotic death, even in the case of secondary necrosis (48, 52, 53). Though distinct HMGB1 nuclear localization was clearly apparent under all conditions at 3 h postinfection (data not shown), we observed a notable SLS-dependent reduction in HMGB1 nuclear localization in infected keratinocytes by 6 h p.i. (Fig. 10). These results suggest that SLS induces keratinocyte death through programmed necrosis.
FIG 10.

Release of nuclear HMGB1 from keratinocytes during GAS infection is dependent on the presence of streptolysin S. HaCaT human keratinocytes were infected directly with GAS at an MOI of 10 for 6 h prior to fixation and staining. Confocal microscopy images shown represent one of three independent biological replicates. Actin is stained in red (rhodamine-phalloidin), and HMGB1 is shown in green (goat anti-rabbit IgG Alexa Fluor 488).
One of the best-characterized subtypes of programmed necrosis is necroptosis, which is defined as a proinflammatory cell death mechanism that depends on the formation of a complex between receptor-interacting protein kinase 1 (RIPK1) and RIPK3 (54, 55). This interaction can be inhibited by necrostatin-1, which targets the kinase activity of RIPK1 (54–56). Similarly to other mechanisms of programmed necrosis, necroptosis induces plasma membrane rupture and exhibits a lack of specific apoptotic markers such as chromatin condensation and caspase activation (54, 55). Given the observed SLS-dependent plasma membrane permeabilization and caspase-independent keratinocyte death, we next determined if necroptosis was a possible mechanism for SLS-mediated host cell death. To address this issue, we treated keratinocytes with necrostatin-1 prior to GAS infection. Treatment with necrostatin-1 during WT GAS infection and sagA complementation strain infection significantly inhibited keratinocyte death to levels similar to those seen in control treatments (Fig. 11; see also Fig. S9 in the supplemental material). Furthermore, necrostatin-1-mediated inhibition of keratinocyte death during GAS infection was observed only in the presence of SLS, as the addition of necrostatin-1 did not significantly reduce cell death during SLS-deficient GAS infection (Fig. 11). Consistent with these data, we observed a significant reduction in SLS-dependent keratinocyte death following pharmacological inhibition of RIPK3 and its direct downstream target, mixed-lineage kinase domain-like (MLKL) (see Fig. S10 in the supplemental material). The dependence of SLS-induced host cell death on RIPK1 and RIPK3 kinase activity suggests that SLS may mediate its cytotoxic effects through a necroptosis-like mechanism. We are currently investigating this possibility by assessing whether GAS infection promotes the interaction of RIPK1 and RIPK3 with other host proteins known to be involved in the regulation of programmed necrosis. Further investigation will provide additional details on the signaling cascades that link SLS-dependent modulation of Akt-p38-NF-κB signaling to the induction of RIPK1/RIPK3-dependent programmed necrosis in infected host cells.
FIG 11.

RIPK1 contributes to the activation of programmed cell death in GAS-infected keratinocytes. HaCaTs were incubated with the RIPK1 kinase inhibitor necrostatin-1 for 1 h prior to infection with GAS at an MOI of 10. Cell death was determined by ethidium homodimer membrane permeabilization assay at 6 h postinfection. The averages of results from three biological replicates are shown for each condition, and error bars represent standard deviations. Statistical significance was determined for each condition compared to control vehicle-treated cells. Overall P values were determined by ANOVA. (A) P < 0.0001. (B) P < 0.0001. (C) P < 0.0001. Dunnett's tests were performed post hoc to compare each condition to the corresponding vehicle control infection (i.e., WT+DMSO).
DISCUSSION
In this report, we provide evidence that the mechanism of host cytotoxicity exerted by streptolysin S in GAS-infected keratinocytes involves temporal inactivation of the cytoprotective factor Akt and subsequent activation of the p38 MAPK cascade. Activation of this pathway promotes inflammatory signaling via NF-κB activation and drives SLS-dependent programmed cell death in keratinocytes. We hypothesize that activation of this cascade leads to the production of NF-κB-regulated inflammatory cytokines, such as TNF-α, which may serve as a positive-feedback signal for the initiation of programmed cell death in infected host cells (Fig. 12). Future experiments will aim to identify the specific signaling mediators downstream of NF-κB that ultimately trigger keratinocyte death. Our studies showed that SLS-dependent cell death is caspase independent, induces release of the necrotic marker HMGB1 from the nucleus, and can be reduced through pharmacological inhibition of RIPK1, RIPK3, and MLKL. Together, these findings suggest that SLS-mediated keratinocyte death occurs through programmed necrosis.
FIG 12.

Proposed model summarizing the effect of streptolysin S on keratinocyte signaling during Streptococcus pyogenes infection. During infection, SLS induces osmotic stress, which leads to loss of phospho-Akt and subsequent activation of the p38 MAPK pathway. Activation of p38 leads to NF-κB nuclear localization, which drives the production of inflammatory cytokines. We hypothesize that this inflammatory cytokine production leads to an autocrine signaling loop through cytokine and/or death receptor proteins, ultimately culminating in the induction of programmed cell death in infected keratinocytes. Nec-1, necrostatin-1.
Recent studies have demonstrated that other bacterial pathogens, including Salmonella enterica serovar Typhimurium, Legionella pneumophila, Clostridium septicum, and Clostridium perfringens, can induce programmed necrosis in infected host cells (46–49). Of note, the induction of programmed necrosis in both Clostridium septicum and Clostridium perfringens was found to be dependent on cytolytic toxins produced by these species (48, 49). These findings suggest that, in addition to their well-characterized lytic functions, these and other bacterial cytolytic toxins may be important mediators of host signaling cascades at sublytic concentrations. A study by Goldmann et al. in 2009 provided the first indication that Streptococcus pyogenes cytolysins are involved in the induction of programmed necrosis in infected host cells (17). In that study, infected macrophages were observed to undergo a highly orchestrated form of programmed cell death which was independent of caspase activation and involved loss of mitochondrial membrane potential, induction of reactive oxygen species, and activation of host proteases such as calpains (17). These cytotoxic effects were dependent on the expression of both streptolysin S and streptolysin O (17). It is possible, given our findings, that SLS-mediated programmed necrosis is a major mechanism of host cytotoxicity during group A Streptococcus infection that is common to several cell types, including keratinocytes and macrophages.
The M1T1 5448 strain used in these studies has been observed to induce similar levels of SLS-dependent cell death in HaCaT keratinocytes and in A549 epithelial cells (6). Datta and colleagues have reported that, although the greatest reduction in GAS-mediated keratinocyte cytotoxicity occurs during infection with mutants lacking both SLS and SLO, SLS was determined to serve as the predominant contributor to cytotoxicity in epithelial cells, given the parameters of their study. These results are in line with our observations of a substantial reduction in host cytotoxicity when keratinocytes were infected with the SLS-deficient mutant compared to the wild-type strain or sagA-complemented strain. Of note, we observed a modest but significant increase in cytotoxicity in cells infected with SLS-deficient GAS compared to uninfected cells, and we speculate that SLO, along with other virulence factors, may contribute to this effect. Ruiz et al. investigated the cytotoxic role of SLO during keratinocyte infection using an M6 JRS GAS strain (30). LDH and ethidium homodimer assays were used to assess cytotoxicity in their study. Ruiz et al. reported an SLO-dependent phenotype showing a much higher level of cell death than that observed in our studies as well as in those reported by Datta et al. Given these findings, we speculate that strain-specific differences in SLO expression may account for some of the differences in host cytotoxicity seen during GAS infections (30).
Although SLS has historically been characterized as a simple membrane lysin, our findings indicate that SLS induces specific and precise alterations in host signaling cascades at sublytic concentrations. Our observations echo recent reports that sublytic doses of many other bacterial cytotoxins initiate highly coordinated signaling responses in host cells (19–21, 34–36). Wiles et al. demonstrated that uropathogenic Escherichia coli alpha-hemolysin downregulates the Akt1 cytoprotective cascade in bladder epithelial cells by inducing changes in intracellular calcium levels; this shift leads to the activation of calcium-dependent Akt phosphatases and the ultimate loss of this key prosurvival mediator (19). Staphylococcus aureus alpha-toxin and aerolysin from Aeromonas hydrophila have also been shown to specifically downregulate Akt1 activity during infection (19). Similarly, activation of p38 and NF-κB inflammatory signaling has been observed in response to many other bacterial toxins, including pneumolysin from Streptococcus pneumoniae, alpha-hemolysin and Panton-Valentine leucocidin from Staphylococcus aureus, streptolysin O from S. pyogenes, and anthrolysin O from Bacillus anthracis (20, 21, 34–36). While the induction of inflammatory cascades is a crucial component of the host immune response to bacterial infection, excessive activation of both p38 MAPK and NF-κB can contribute to significant host tissue damage and progression to severe disease.
Many bacterial cytotoxins initiate their effects on host signaling cascades by forming a pore in the host cell membrane (19–21, 34–36). Because of its lytic capabilities, SLS has also historically been described as a pore-forming toxin, despite the lack of definitive evidence to support this hypothesis. Incidentally, recent evidence from our laboratory suggests that SLS may actually target specific ion channels on host cell membranes, leading to membrane rupture as a result of ionic imbalance (D. L. Higashi, N. Biais, D. L. Donahue, J. A. Mayfield, C. R. Tessier, K. Rodriguez, B. L. Ashfeld, J. Luchetti, V. A. Ploplis, F. J. Castellino, and S. W. Lee, submitted for publication). Whether SLS mediates its effects through direct pore formation or by manipulating ion channel function, there is convincing evidence indicating that SLS induces osmotic stress in host cells (6–17, 26). Interestingly, osmotic stress resulting from a variety of stimuli has been shown to trigger loss of Akt signaling and to stimulate the p38 MAPK cascade (19–21, 34–36, 57). The ability to induce osmotic stress in infected host cells, whether directly or through an alternative mechanism, may explain the similarities observed in the host signaling responses to structurally unrelated bacterial cytolysins.
These recent insights regarding bacterial cytotoxin function, coupled with our findings, strengthen the case for reevaluating the physiological role of these toxins in the context of infection. It is possible that the modulation of specific host signaling pathways is a primary function of many cytolytic toxins, such as SLS, under physiologically relevant infection conditions. Should this be the case, it may be possible to supplement current antibiotic-based treatments with highly specific therapies to target the host signaling cascades that are being modulated by toxin-producing pathogens during infection. We anticipate that further investigation into the mechanisms by which SLS enhances programmed cell death and inflammatory signaling in epithelial keratinocytes will provide important insights pertaining to Streptococcus pyogenes pathogenesis and aid in the identification of new therapeutic strategies to combat severe group A Streptococcus disease.
Supplementary Material
ACKNOWLEDGMENTS
We thank our fellow Lee laboratory colleagues, especially Clayton Thomas, who provided valuable insight and expertise that greatly assisted this research.
This work was supported by the Young Innovator Award from the National Institutes of Health, awarded to S.W.L. (NIH-1DP2OD008468-01). R.A.F. is supported by the Albertus Magnus Fellowship provided by Robert C. Boguslaski and by a teaching assistantship through the Eck Institute for Global Health at the University of Notre Dame.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00611-15.
REFERENCES
- 1.Walker MJ, Barnett TC, McArthur JD, Cole JN, Gillen CM, Henningham A, Sriprakash KS, Sanderson-Smith ML, Nizet V. 2014. Disease manifestations and pathogenic mechanisms of group A Streptococcus. Clin Microbiol Rev 27:264–301. doi: 10.1128/CMR.00101-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carapetis JR, Steer AC, Mulholland EK, Weber M. 2005. The global burden of group A streptococcal diseases. Lancet Infect Dis 5:685–694. doi: 10.1016/S1473-3099(05)70267-X. [DOI] [PubMed] [Google Scholar]
- 3.Cunningham MW. 2000. Pathogenesis of group A streptococcal infections. Clin Microbiol Rev 13:470–511. doi: 10.1128/CMR.13.3.470-511.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carapetis JR, Steer AC, Mulholland EK. 2005. Group A streptococcal vaccine development: current status and issues of relevance to less developed countries. World Health Organization Discussion Papers on Child Health. http://whqlibdoc.who.int/hq/2005/WHO_IVB_05.14_eng.pdf. [Google Scholar]
- 5.Molloy EM, Cotter PD, Hill C, Mitchell DA, Ross RP. 2011. Streptolysin S-like virulence factors: the continuing sagA. Nat Rev Microbiol 9:670–681. doi: 10.1038/nrmicro2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Datta V, Myskowski SM, Kwinn LA, Chiem DN, Varki N, Kansal RG, Kotb M, Nizet V. 2005. Mutational analysis of the group A streptococcal operon encoding streptolysin S and its virulence role in invasive infection. Mol Microbiol 56:681–695. doi: 10.1111/j.1365-2958.2005.04583.x. [DOI] [PubMed] [Google Scholar]
- 7.Lee SW, Mitchell DA, Markley AL, Hensler ME, Gonzalez D, Wohlrab A, Dorrestein PC, Nizet V, Dixon JE. 2008. Discovery of a widely distributed toxin biosynthetic gene cluster. Proc Natl Acad Sci U S A 105:5879–5884. doi: 10.1073/pnas.0801338105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mitchell DA, Lee SW, Pence MA, Markley AL, Limm JD, Nizet V, Dixon JE. 2009. Structural and functional dissection of the heterocyclic peptide cytotoxin streptolysin S. J Biol Chem 284:13004–13012. doi: 10.1074/jbc.M900802200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Todd EW. 1938. The differentiation of two distinct serological varieties of streptolysin, streptolysin O and streptolysin S. J Pathol Bacteriol 47:423–445. doi: 10.1002/path.1700470307. [DOI] [Google Scholar]
- 10.Keiser H, Weissmann G, Bernheimer AW. 1964. Studies on lysosomes. IV. Solubilization of enzymes during mitochondrial swelling and disruption of lysosomes by streptolysin S and other hemolytic agents. J Cell Biol 22:101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ofek I, Bergner-Rabinowitz S, Ginsburg I. 1972. Oxygen-stable hemolysins of group A streptococci. 8. Leukotoxic and antiphagocytic effects of streptolysins S and O. Infect Immun 6:459–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bernheimer AW, Schwartz LL. 1964. Lysosomal disruption by bacterial toxins. J Bacteriol 87:1100–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hryniewicz W, Pryjma J. 1977. Effect of streptolysin S on human and mouse T and B lymphocytes. Infect Immun 16:730–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carr A, Sledjeski DD, Podbielski A, Boyle MD, Kreikemeyer B. 2001. Similarities between complement-mediated and streptolysin S-mediated hemolysis. J Biol Chem 276:41790–41796. doi: 10.1074/jbc.M107401200. [DOI] [PubMed] [Google Scholar]
- 15.Sierig G, Cywes C, Wessels MR, Ashbaugh CD. 2003. Cytotoxic effects of streptolysin O and streptolysin S enhance the virulence of poorly encapsulated group A streptococci. Infect Immun 71:446–455. doi: 10.1128/IAI.71.1.446-455.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miyoshi-Akiyama T, Takamatsu D, Koyanagi M, Zhao J, Imanishi K, Uchiyama T. 2005. Cytocidal effect of Streptococcus pyogenes on mouse neutrophils in vivo and the critical role of streptolysin S. J Infect Dis 192:107–116. doi: 10.1086/430617. [DOI] [PubMed] [Google Scholar]
- 17.Goldmann O, Sastalla I, Wos-Oxley M, Rohde M, Medina E. 2009. Streptococcus pyogenes induces oncosis in macrophages through the activation of an inflammatory programmed cell death pathway. Cell Microbiol 11:138–155. doi: 10.1111/j.1462-5822.2008.01245.x. [DOI] [PubMed] [Google Scholar]
- 18.Lancefield RC. 1933. A serological differentiation of human and other groups of hemolytic streptococci. J Exp Med 57:571–595. doi: 10.1084/jem.57.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wiles TJ, Dhakal BK, Eto DS, Mulvey MA. 2008. Inactivation of host Akt/protein kinase B signaling by bacterial pore-forming toxins. Mol Biol Cell 19:1427–1438. doi: 10.1091/mbc.E07-07-0638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kao C-Y, Los FCO, Huffman DL, Wachi S, Kloft N, Husmann M, Karabrahimi V, Schwartz J-L, Bellier A, Ha C, Sagong Y, Fan H, Ghosh P, Hsieh M, Hsu C-S, Chen L, Aroian RV. 2011. Global functional analyses of cellular responses to pore-forming toxins. PLoS Pathog 7:e1001314. doi: 10.1371/journal.ppat.1001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma X, Chang W, Zhang C, Zhou X, Yu F. 2012. Staphylococcal Panton-Valentine leukocidin induces pro-inflammatory cytokine production and nuclear factor-kappa B activation in neutrophils. PLoS One 7:e34970. doi: 10.1371/journal.pone.0034970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sumitomo T, Nakata M, Higashino M, Jin Y, Terao Y, Fujinaga Y, Kawabata S. 2011. Streptolysin S contributes to group A streptococcal translocation across an epithelial barrier. J Biol Chem 286:2750–2761. doi: 10.1074/jbc.M110.171504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin A, Loughman JA, Zinselmeyer BH, Miller MJ, Caparon MG. 2009. Streptolysin S inhibits neutrophil recruitment during the early stages of Streptococcus pyogenes infection. Infect Immun 77:5190–5201. doi: 10.1128/IAI.00420-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. 1988. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol 106:761–771. doi: 10.1083/jcb.106.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jo H, Mondal S, Tan D, Nagata E, Takizawa S, Sharma AK, Hou Q, Shanmugasundaram K, Prasad A, Tung JK, Tejeda AO, Man H, Rigby AC, Luo HR. 2012. Small molecule-induced cytosolic activation of protein kinase Akt rescues ischemia-elicited neuronal death. Proc Natl Acad Sci U S A 109:10581–10586. doi: 10.1073/pnas.1202810109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Voyich JM, Musser JM, DeLeo FR. 2004. Streptococcus pyogenes and human neutrophils: a paradigm for evasion of innate host defense by bacterial pathogens. Microbes Infect 6:1117–1123. doi: 10.1016/j.micinf.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 27.Tsai PJ, Lin YS, Kuo CF, Lei HY, Wu JJ. 1999. Group A Streptococcus induces apoptosis in human epithelial cells. Infect Immun 67:4334–4339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakagawa I, Nakata M, Kawabata S, Hamada S. 2001. Cytochrome c-mediated caspase-9 activation triggers apoptosis in Streptococcus pyogenes-infected epithelial cells. Cell Microbiol 3:395–405. doi: 10.1046/j.1462-5822.2001.00122.x. [DOI] [PubMed] [Google Scholar]
- 29.Aikawa C, Nozawa T, Maruyama F, Tsumoto K, Hamada S, Nakagawa I. 2010. Reactive oxygen species induced by Streptococcus pyogenes invasion trigger apoptotic cell death in infected epithelial cells. Cell Microbiol 12:814–830. doi: 10.1111/j.1462-5822.2010.01435.x. [DOI] [PubMed] [Google Scholar]
- 30.Ruiz N, Wang B, Pentland A, Caparon M. 1998. Streptolysin O and adherence synergistically modulate proinflammatory responses of keratinocytes to group A streptococci. Mol Microbiol 27:337–346. doi: 10.1046/j.1365-2958.1998.00681.x. [DOI] [PubMed] [Google Scholar]
- 31.Timmer AM, Timmer JC, Pence MA, Hsu LC, Ghochani M, Frey TG, Karin M, Salvesen GS, Nizet V. 2009. Streptolysin O promotes group A streptococcus immune evasion by accelerated macrophage apoptosis. J Biol Chem 284:862–871. doi: 10.1074/jbc.M804632200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liao Y, Hung M-C. 2003. Regulation of the activity of p38 mitogen-activated protein kinase by Akt in cancer and adenoviral protein E1A-mediated sensitization to apoptosis. Mol Cell Biol 23:6836–6848. doi: 10.1128/MCB.23.19.6836-6848.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shulga N, Hoek JB, Pastorino JG. 2005. Elevated PTEN levels account for the increased sensitivity of ethanol-exposed cells to tumor necrosis factor-induced cytotoxicity. J Biol Chem 280:9416–9424. doi: 10.1074/jbc.M409505200. [DOI] [PubMed] [Google Scholar]
- 34.Ratner AJ, Hippe KR, Aguilar JL, Bender MH, Nelson AL, Weiser JN. 2006. Epithelial cells are sensitive detectors of bacterial pore-forming toxins. J Biol Chem 281:12994–12998. doi: 10.1074/jbc.M511431200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stassen M, Müller C, Richter C, Neudörfl C, Hültner L, Bhakdi S, Walev I, Schmitt E. 2003. The streptococcal exotoxin streptolysin O activates mast cells to produce tumor necrosis factor alpha by p38 mitogen-activated protein kinase- and protein kinase C-dependent pathways. Infect Immun 71:6171–6177. doi: 10.1128/IAI.71.11.6171-6177.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Porta H, Cancino-Rodezno A, Soberón M, Bravo A. 2011. Role of MAPK p38 in the cellular responses to pore-forming toxins. Peptides 32:601–606. doi: 10.1016/j.peptides.2010.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cowan KJ, Storey KB. 2003. Mitogen-activated protein kinases: new signaling pathways functioning in cellular responses to environmental stress. J Exp Biol 206:1107–1115. doi: 10.1242/jeb.00220. [DOI] [PubMed] [Google Scholar]
- 38.Medina E, Anders D, Chhatwal GS. 2002. Induction of NF-kappaB nuclear translocation in human respiratory epithelial cells by group A streptococci. Microb Pathog 33:307–313. doi: 10.1006/mpat.2002.0532. [DOI] [PubMed] [Google Scholar]
- 39.Tsai P-J, Chen Y-H, Hsueh C-H, Hsieh H-C, Liu Y-H, Wu J-J, Tsou C-C. 2006. Streptococcus pyogenes induces epithelial inflammatory responses through NF-kappaB/MAPK signaling pathways. Microbes Infect 8:1440–1449. doi: 10.1016/j.micinf.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 40.Harder J, Franchi L, Muñoz-Planillo R, Park J-H, Reimer T, Núñez G. 2009. Activation of the Nlrp3 inflammasome by Streptococcus pyogenes requires streptolysin O and NF-kappa B activation but proceeds independently of TLR signaling and P2X7 receptor. J Immunol 183:5823–5829. doi: 10.4049/jimmunol.0900444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Newton K, Dixit VM. 2012. Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol 4:a006049. doi: 10.1101/cshperspect.a006049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.N′Guessan PD, Schmeck B, Ayim A, Hocke AC, Brell B, Hammerschmidt S, Rosseau S, Suttorp N, Hippenstiel S. 2005. Streptococcus pneumoniae R6x induced p38 MAPK and JNK-mediated caspase-dependent apoptosis in human endothelial cells. Thromb Haemost 94:295–303. [DOI] [PubMed] [Google Scholar]
- 43.Stringaris AK, Geisenhainer J, Bergmann F, Balshüsemann C, Lee U, Zysk G, Mitchell TJ, Keller BU, Kuhnt U, Gerber J, Spreer A, Bähr M, Michel U, Nau R. 2002. Neurotoxicity of pneumolysin, a major pneumococcal virulence factor, involves calcium influx and depends on activation of p38 mitogen-activated protein kinase. Neurobiol Dis 11:355–368. doi: 10.1006/nbdi.2002.0561. [DOI] [PubMed] [Google Scholar]
- 44.Kling DE, Tsvang I, Murphy MP, Newburg DS. 2013. Group B Streptococcus induces a caspase-dependent apoptosis in fetal rat lung interstitium. Microb Pathog 61–62:1–10. doi: 10.1016/j.micpath.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krzymińska S, Frąckowiak H, Kaznowski A. 2012. Acinetobacter calcoaceticus-baumannii complex strains induce caspase-dependent and caspase-independent death of human epithelial cells. Curr Microbiol 65:319–329. doi: 10.1007/s00284-012-0159-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Robinson N, McComb S, Mulligan R, Dudani R, Krishnan L, Sad S. 2012. Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat Immunol 13:954–962. doi: 10.1038/ni.2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morinaga Y, Yanagihara K, Nakamura S, Hasegawa H, Seki M, Izumikawa K, Kakeya H, Yamamoto Y, Yamada Y, Kohno S, Kamihira S. 2010. Legionella pneumophila induces cathepsin B-dependent necrotic cell death with releasing high mobility group box1 in macrophages. Respir Res 11:158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kennedy CL, Smith DJ, Lyras D, Chakravorty A, Rood JI. 2009. Programmed cellular necrosis mediated by the pore-forming alpha-toxin from Clostridium septicum. PLoS Pathog 5:e1000516. doi: 10.1371/journal.ppat.1000516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Autheman D, Wyder M, Popoff M, D'Herde K, Christen S, Posthaus H. 2013. Clostridium perfringens beta-toxin induces necrostatin-inhibitable, calpain-dependent necrosis in primary porcine endothelial cells. PLoS One 8:e64644. doi: 10.1371/journal.pone.0064644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bergsbaken T, Fink SL, Cookson BT. 2009. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol 7:99–109. doi: 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Van Cruchten S, Van Den Broeck W. 2002. Morphological and biochemical aspects of apoptosis, oncosis and necrosis. Anat Histol Embryol 31:214–223. doi: 10.1046/j.1439-0264.2002.00398.x. [DOI] [PubMed] [Google Scholar]
- 52.Lotze MT, Tracey KJ. 2005. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol 5:331–342. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- 53.Scaffidi P, Misteli T, Bianchi ME. 2002. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418:191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 54.Kaczmarek A, Vandenabeele P, Krysko DV. 2013. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity 38:209–223. doi: 10.1016/j.immuni.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 55.Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. 2010. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 11:700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 56.Degterev A, Hitomi J, Germscheid M, Ch'en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J. 2008. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol 4:313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen D, Fucini RV, Olson AL, Hemmings BA, Pessin JE. 1999. Osmotic shock inhibits insulin signaling by maintaining Akt/protein kinase B in an inactive dephosphorylated state. Mol Cell Biol 19:4684–4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.