

Abstract
Legionella pneumophila is a bacterial pathogen that thrives in alveolar macrophages, causing a severe pneumonia. The virulence of L. pneumophila depends on its Dot/Icm type IV secretion system (T4SS), which delivers more than 300 effector proteins into the host, where they rewire cellular signaling to establish a replication-permissive niche, the Legionella-containing vacuole (LCV). Biogenesis of the LCV requires substantial redirection of vesicle trafficking and remodeling of intracellular membranes. In order to achieve this, several T4SS effectors target regulators of membrane trafficking, while others resemble lipases. Here, we characterized LpdA, a phospholipase D effector, which was previously proposed to modulate the lipid composition of the LCV. We found that ectopically expressed LpdA was targeted to the plasma membrane and Rab4- and Rab14-containing vesicles. Subcellular targeting of LpdA required a C-terminal motif, which is posttranslationally modified by S-palmitoylation. Substrate specificity assays showed that LpdA hydrolyzed phosphatidylinositol, -inositol-3- and -4-phosphate, and phosphatidylglycerol to phosphatidic acid (PA) in vitro. In HeLa cells, LpdA generated PA at vesicles and the plasma membrane. Imaging of different phosphatidylinositol phosphate (PIP) and organelle markers revealed that while LpdA did not impact on membrane association of various PIP probes, it triggered fragmentation of the Golgi apparatus. Importantly, although LpdA is translocated inefficiently into cultured cells, an L. pneumophila ΔlpdA mutant displayed reduced replication in murine lungs, suggesting that it is a virulence factor contributing to L. pneumophila infection in vivo.
INTRODUCTION
Legionella pneumophila is a bacterial pathogen which infects and replicates in protozoa, as well as in human alveolar macrophages and lung epithelial cells, causing a severe pneumonia called Legionnaires' disease (1). Several secretion systems allow L. pneumophila to export proteins into its environment and interact with host cells (1, 2). The Legionella type I and type II secretion systems (T1SS and T2SS) have been shown to contribute to L. pneumophila virulence (3, 4); however, only the Dot/Icm type IV secretion system (T4SS) is essential for intracellular replication and pathogenesis (5, 6). The Dot/Icm T4SS delivers more than 300 effector proteins into the host cell, where they manipulate cell signaling (7, 8). This enables the bacteria to evade phagolysosomal degradation and instead establish a replication-permissive endoplasmic reticulum (ER)-derived niche, the Legionella-containing vacuole (LCV) (9–11).
The biogenesis of the LCV is a multistep process and involves substantial remodeling of the membrane of the nascent Legionella phagosome (12, 13). Consequently, a large number of effectors secreted by Legionella are membrane-associated proteins which target cellular regulators of membrane trafficking, such as Rab GTPases (14, 15), or directly exploit and manipulate host lipids (16–18). To achieve membrane association, Legionella effectors highjack at least two common host cell mechanisms. Several effectors contain a classical C-terminal CAAX-box motif which is recognized by prenyltransferases and posttranslationally modified with a geranylgeranyl (20-carbon) or a farnesyl (15-carbon) lipid anchor (19, 20). After the lipid transfer, Ras-converting enzyme 1 (RCE-1) removes the amino acid residues after the modified cysteine and isoprenyl cysteine carboxyl methyltransferase (IcmT) methylates the new C terminus. Together, these irreversible modifications increase the hydrophobicity and membrane affinity of the effectors, facilitating their membrane association. Legionella ensures the efficient modification of effectors by recruiting host proteins, such as farnesyl transferase, RCE-1, and IcmT to the LCV in a T4SS-dependent manner (20).
Alternatively to using a covalently attached lipid anchor, several Legionella effectors associate with membranes by binding phosphatidylinositol phosphates (PIPs) (16, 21), negatively charged glycerophospholipids which contain a mono- or polyphosphorylated myo-inositol ring (22). PIPs are versatile signaling molecules, and specific enrichment of one subspecies gives identity to organelle membranes, leading to selective recruitment of PIP-binding proteins. L. pneumophila actively modulates the PIP composition of membranes. The effectors SidF and SidP are PIP phosphatases, which hydrolyze the D3 phosphate of different 3-phosphorylated inositol headgroups (23, 24). This conversion of PIP species is important for the transition of the phosphatidylinositol-3-phosphate (PI3P)-rich early Legionella-containing phagosome to the mature phosphatidylinositol-4-phosphate (PI4P)-rich LCV. In addition, VipD removes PI3P from early endosomes, contributing to the decoupling of the LCV from the normal phagosomal maturation pathway (25).
VipD belongs to the large family of phospholipases, which hydrolyze either the carboxyl ester (subfamilies A and B), the glycerol-oriented (subfamily C), or the alcohol-oriented (subfamily D) phosphodiester bonds of phospholipid substrates. L. pneumophila encodes at least 15 putative phospholipase A (PLA) enzymes (18, 25), but other than VipD and PlaB, the latter of which was implicated in virulence in vivo (26), their functions in infection are not well defined. Additional effectors belonging to the other phospholipase subfamilies exist. The effector PlcC/CegC1 is a novel Zn2+-dependent phospholipase C (PLC) and, together with two homologues, PlcA and PlcB, contributes to virulence in the Galleria mellonella infection model (27). The effector LpdA was first recognized in a bioinformatic search for effectors with potential function in phospholipid biosynthesis due to its homology with eukaryotic PLDs (28). Eukaryotic PLDs use mainly phosphatidylcholine (PC) but can also accept other phospholipid substrates, to generate the free lipid headgroup and the important signaling molecule phosphatidic acid (PA) (29). LpdA localizes to the LCV and perturbs PA distribution upon ectopic expression in cells, suggesting that it might modulate PA levels on the LCV (28). However, important features of LpdA such as the substrate specificity and the consequences of its activity on host cell physiology remain unknown. In this study, we characterized the activity of LpdA, investigated its mode of membrane association, and determined its effect on host cells in vitro and during mouse infection in vivo to further develop our understanding of its function in L. pneumophila infection.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
The bacterial strains used in the present study are listed in Table 1. Escherichia coli strains were cultured using Luria-Bertani broth or agar. L. pneumophila 130b (AA100) was grown at 37°C on buffered charcoal yeast extract (CYE) plates or in N-(2-acetamido)-2-aminoethanesulfonic acid buffered yeast extract (AYE) broth (30, 31). If required, Legionella media were supplemented with 25 μg of kanamycin/ml and 6 μg of chloramphenicol/ml. All chemicals and reagents were obtained from Sigma-Aldrich, Ltd., unless indicated otherwise.
TABLE 1.
Bacterial strains used in the study
| Strain | Serogroup/genotypea | Source or reference(s) |
|---|---|---|
| Legionella pneumophila | ||
| 130b (AA100, ATCC BAA-74) | O1; clinical isolate | 30, 31 |
| 130b ΔdotA | dotA gene disrupted with a Kanr cassette | 32 |
| 130b ΔlpdA (ICC1080) | lpdA (lpw_19211) gene disrupted with a Kanr cassette | This study |
| Escherichia coli | ||
| BL21(DE3)Star | Novagen | |
| Top10 | Invitrogen |
Kanr, kanamycin resistance.
Cell culture and transfection.
Human alveolar epithelial A549 cells, cervical epithelial HeLa cells, and HEK293E cells were maintained at 37°C in Dulbecco modified Eagle medium supplemented with 10% fetal bovine serum, GlutaMAX (Invitrogen), and nonessential amino acids in a humidified atmosphere of 5% CO2. THP-1 human monocytic and RAW 264.7 murine macrophage-like cells were cultured in RPMI 1640 supplemented with 10% fetal bovine serum and GlutaMAX (Invitrogen). THP-1 cells were differentiated by the addition of 50 ng of phorbol 12-myristate 13-acetate/ml for 72 h. A549, HeLa, THP-1, and RAW 264.7 cells were obtained from the American Type Culture Collection (ATCC) and subjected to minimal passaging. HEK293E cells were kindly provided by Avinash R. Shenoy (Imperial College, London, United Kingdom). The transfection of HeLa and A549 cells with eukaryotic expression plasmids was performed according to the manufacturer's instructions using GeneJuice (Novagen) or jetPRIME (Polyplus Transfection) reagent, respectively.
Plasmid and strain construction.
All plasmids, primers, and restriction enzymes (NEB, Inc.) used and created in this study are described in Tables 2, 3, and 4. Gene sequences were extracted from the draft genome sequence of L. pneumophila strain 130b (32; see also ftp://ftp.sanger.ac.uk/pub/pathogens/Legionella/pneumophila/130b/) or obtained by sequence homology search using NCBI BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Unless indicated otherwise, chromosomal DNA of L. pneumophila 130b was used as the PCR template. The mutagenesis of the catalytic lysine residues of LpdA was performed sequentially with a QuikChange II site-directed mutagenesis kit (Agilent Technologies) and pRK5 Myc LpdA (pICC1565) as the starting template. The mutated gene was then subcloned in other plasmids. The expression plasmid for ER-targeted mCherry protein was obtained by introducing a C-terminal KDEL ER-targeting and an N-terminal calreticulin ER retention signal into pmCherry-C1 (Clontech) by reverse PCR. Standard molecular biology techniques were used, and the identities of all plasmids were confirmed by sequencing. Plasmids were transformed into L. pneumophila by electroporation (33).
TABLE 2.
Plasmids used and created in this study
| Plasmid and derivative(s) | Description/function | Expressed protein | Primer sequence (5′–3′) | Restriction site | Source or reference |
|---|---|---|---|---|---|
| pRK5 | Expression of proteins with N-terminal Myc tag in mammalian cells | Clontech | |||
| pICC1565 | Myc-LpdA | CGCACTGAGGATCCATGCTCCAATTTTTTGAGAAAGGT | BamHI | This study | |
| GCGTCGCAGAATTCTCAAGAACAAACACAACAGGTTC | EcoRI | ||||
| pICC1566 | Myc-LpdAΔC5aa | CGCACTGAGGATCCATGCTCCAATTTTTTGAGAAAGGT | BamHI | This study | |
| GCGTCGCAGAATTCTCAGGTTCTTTTAGGCGATATC | EcoRI | ||||
| pICC1585 | Myc-SidC PI4P | GTCATAGGATCCAAATATTCCTCCAAGCCATTATTGG | BamHI | This study | |
| GATACCCTGCAGCTATTTCTTTATAACTCCCGTGTAC | PstI | ||||
| pICC1575 | Myc-LpdA K165R | As for pICC1565, with pICC1574 as a template | This study | ||
| pICC1578 | Myc-LpdA K376R | As for pICC1565, with pICC1577 as a template | This study | ||
| pICC1581 | Myc-LpdA KK165/376RR | As for pICC1565, with pICC1580 as a template | This study | ||
| pICC1341 | pRK5-HA; expression of proteins with N-terminal HA tag in mammalian cells | 21 | |||
| pICC1567 | HA-LpdA | As for pICC1565 | This study | ||
| pICC1568 | HA-LpdAΔC5aa | As for pICC1566 | This study | ||
| pICC1569 | HA-LpdA C440F | CGCACTGAGGATCCATGCTCCAATTTTTTGAGAAAGGT | BamHI | This study | |
| GCGTCGCAGAATTCTCAAGAACAAACAAAACAGGTTC | EcoRI | ||||
| pICC1574 | HA-LpdA K165R | See Table 3 | This study | ||
| pICC1577 | HA-LpdA K376R | See Table 3 | This study | ||
| pICC1580 | HA-LpdA KK165/376RR | See Table 3 | This study | ||
| pEGFP-N1 | Expression of proteins with C-terminal GFP tag in mammalian cells | Clontech | |||
| pICC1586 | hs PLCδ1-PH-GFP | ATGCGCTCGAGATGGACTCGGGCCGGG | XhoI | This study | |
| AGCATGGATCCTCGATGTTGAGCTCCTTCAGGAAG | BamHI | ||||
| pICC562 | pMMB207c-4HA; vector for the expression of proteins with four N-terminal HA tags in L. pneumophila | 67 | |||
| pICC1570 | 4HA-LpdA | TCGCACTGAGGTACCATGCTCCAATTTTTTGAGAAAGGT | KpnI | This study | |
| TCGCATCTAGATCAAGAACAAACACAACAGGTTC | XbaI | ||||
| pICC1576 | 4HA-LpdA KK165/376RR | As for pICC1570, with pICC1580 as a template | This study | ||
| pXDC61 | Expression of proteins with N-terminal β-lactamase TEM1-tag in L. pneumophila | 68 | |||
| pICC1571 | TEM1-LpdA | As for pICC1570 | This study | ||
| pICC1579 | TEM1-LpdA KK165/376RR | As for pICC1570, with pICC1580 as a template | This study | ||
| pXDC31 | Expression of GFP in L. pneumophila | 40 | |||
| pET28a(+) | Expression of a His6 tag fusion protein in E. coli | Novagen | |||
| pICC1572 | His-LpdA | CGCACTGAGGATCCATGCTCCAATTTTTTGAGAAAGGT | BamHI | This study | |
| GCGTCGCAGAATTCTCAAGAACAAACACAACAGGTTC | EcoRI | ||||
| pICC1573 | LpdA-His | CATGACCATGGGACTCCAATTTTTTGAGAAAGGT | NcoI | This study | |
| TCATGCTCGAGAGAACAAACACAACAGGTTC | XhoI | ||||
| pICC1582 | LpdA KK165/376RR-His | As for pICC1573, with pICC1580 as a template | This study | ||
| pMXs-IRES-Puro | pMXs-IP; vector for viral transduction of mammalian cells | Clontech | |||
| pICC1583 | GFPSPO reporter | CTAGGGATCCGCCACCATGGTGAGCAAGG | This study | ||
| GCATGAATTCTTAACTAGTCTTAGTGGCGTCATC | |||||
| pm-Cherry-C1 | Expression of proteins with mCherry tag in mammalian cells | Clontech | |||
| pICC1584 | pmCherryER | ER-targeted mCherry | See Table 3 | This study |
TABLE 3.
Primers used for site-directed mutagenesis and reverse PCRs in this study
| Description/function | Primer sequence (5′–3′) | Source |
|---|---|---|
| Introduction of a K165R mutation into LpdA by site-directed mutagenesis PCR | ATAATTCGATTGCGTCCAACCATAATAGAGCCATCTGGATTG | This study |
| CAATCCAGATGGCTCTATTATGGTTGGACGCAATCGAATTAT | ||
| Introduction of a K376R mutation into LpdA by site-directed mutagenesis PCR | GAAAAAAGAGGTTATTCATATGAGATTTATGTCAATTGATGGGCAGG | This study |
| CCTGCCCATCAATTGACATAAATCTCATATGAATAACCTCTTTTTTC | ||
| Fusion of a calreticulin ER retention signal to mCherry by reverse PCR | GAGCAGCAGCGGCACGGATAGCAGCATGGTGGCGACCGGTAGGG-CCTTCTTGGCCTAGCAGT | This study |
| AGCAGTGAGCAAGGGCGAGGAG | ||
| Fusion of a KDEL ER targeting signal to mCherry by reverse PCR | GAGCTGTAACTGATCATAATCAGCCATACC | This study |
| ATCCTTTCTAGATCCGGTGGATCCC |
TABLE 4.
Primers used for the construction of the L. pneumophila ΔlpdA strain ICC1080
| Function | Primer sequence (5′–3′) | Source |
|---|---|---|
| Amplification of the 5′-flanking region | CATAATGACAGGTAAGGCTTC | This study |
| CAGTGGGTACCGATTCACTCATAAATTAATTTTACCTAAT | ||
| Amplification of the 3′-flanking region | GCGACTCTAGATTTTAGGCTGTTTTGATCAAGCC | This study |
| ATCTCAATTACAGGAAATGCAAAC | ||
| Sequencing of the lpdA genomic region | CTTGCCATGTTCTGCTATATATTTC | This study |
| GTGATATTGTATGCACTTCCATC |
To create the L. pneumophila 130b ΔlpdA mutant strain, a PCR deletion cassette consisting of the kanamycin resistance (Kanr) cassette from pSB315 (34) embedded in the 3′ and 5′ chromosomal flanking regions of the lpdA gene was produced and transformed into L. pneumophila 130b by natural transformation (35). Transformants were selected by plating on CYE agar containing 25 μg of kanamycin/ml. Replacement of the lpdA gene with the Kanr cassette and the integrity of the chromosomal flanking regions was confirmed by sequencing of the genomic region.
Generation of stable A549 cell lines by transduction.
Viral transductions were performed as described previously (36). Briefly, HEK293E cells were transfected with pICC1583 and the pCMV-VSV-G envelope and pCMV-MMLV-gag-pol packaging plasmids using Lipofectamine 2000 (Life Technologies); the medium was replaced after 24 h, and the cell supernatant containing virions was harvested at 48 h posttransfection. The supernatant was passed through a 0.45-μm-pore-size filter and added to A549 cells. After 24 h, the cells were washed with Dulbecco's phosphate-buffered saline (D-PBS) and incubated with growth medium supplemented with 1.5 μg of puromycin (Gibco)/ml. After 1 week of selection, the expression of the green fluorescent protein (GFP) reporters throughout the cell population was confirmed by immunofluorescence (IF), and cells were frozen or further passaged without selection.
Lipase activity assay.
The substrate specificity of recombinant His6-tagged LpdA was assessed as described previously (27). Since solubility tests showed that His6-tagged LpdA was poorly soluble, assays were carried out with bacterial lysates. For the generation of lysates, strains were grown until achieving exponential growth, and expression was induced by addition of 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) for 4 h. Expression of the recombinant proteins with the expected apparent molecular mass (52 kDa) was confirmed by SDS-PAGE. Subsequently, pelleted bacteria were suspended in 1/20 of the original culture volume of 10 mg of lysozyme/ml and 1 μl of Triton X-100/ml in 40 mM Tris-HCl (pH 7.5, 25°C) at 37°C for 30 min. After repeated passage through a 26-gauge needle, the lysates were resuspended in one-half of the original culture volume of 40 mM Tris-HCl (pH 7.5, 25°C). Then, 0.2 mM lipid substrates—dipalmitoyl-phosphatidylcholine (PC), 99% purity; dipalmitoyl-phosphatidylglycerol (PG), 99% purity; dipalmitoyl-phosphatidylethanolamine (PE), 99% purity; dipalmitoyl-phosphatidylserine (PS), 99% purity; or 0.25 mM PI and PIPs (phosphatidylinositol diC16 [PIC16], phosphatidylinositol 3-phosphate diC16 [PI3P], phosphatidylinositol 4-phosphate diC16 [PI4P], phosphatidylinositol 5-phosphate diC16 [PI5P], phosphatidylinositol 3,4-bisphosphate diC16 [PI3,4P], phosphatidylinositol 3,5-bisphosphate diC16 [PI3,5P], phosphatidylinositol 4,5-bisphosphate diC16 [PI4,5P], and phosphatidylinositol 3,4,5-trisphosphate diC16 ([PI3,4,5P])—were incubated with buffer (40 mM Tris-HCl [pH 7.5, 25°C]) or lysates of BL21 or BL21 expressing LpdA-His6 or an LpdA KK165/376RR-His6 inactive mutant in a 1:1 ratio. After 4 to 5 h of incubation at 37°C with agitation, the lipids were extracted (37) and analyzed by thin-layer chromatography (TLC) using chloroform-methanol-water (65:25:4 [vol/vol/vol]) as the running phase and pure lipid substrates and phosphatidic acid (1,2-dimyristoyl-sn-glycero-3-phosphate monosodium salt [PA]) as references. The TLC plate was developed using 0.2% naphthol blue-black. PI, PA, and PIPs were purchased from Echelon Biosciences; all other lipid substrates were purchased from Avanti Polar Lipids, Inc., or Sigma-Aldrich.
Metabolic labeling and isolation of palmitoylated proteins.
HeLa cells (one well/condition in six-well plates) were transfected with expression plasmids for LpdA or its variants, grown overnight, and then incubated with 25 μM 17-octadecynoic acid (ODYA; Cambridge Biosciences). After 7 h, the medium was removed, and the cells were washed with PBS and scraped into 100 μl of lysis buffer containing 1% Triton X-100, 0.1% SDS, and complete EDTA-free protease inhibitor (Roche). After 15 min of incubation, large debris was removed by centrifugation at 20,000 × g for 10 min at 4°C, and the protein concentration in the supernatant was measured. Click-chemistry was then performed on equal amounts of protein. Briefly, 1 mM CuSO4, 1 mM Tris(2-carboxyethyl)phosphine hydrochloride (TCEP), 100 μM Tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA), and a 100 μM concentration of a trifunctional azido-TAMRA-PEG-biotin (AzTB) capture reagent (38, 39) were added to the clarified lysate, and the mixture was incubated for 45 min at room temperature. The reaction was stopped by adding 10 mM EDTA, and the proteins were precipitated by the addition of 1 ml of chilled methanol and centrifugation (20,000 × g, 30 min, 4°C). The pellet was washed three times with 1 ml of methanol, air dried, and subsequently resuspended in 100 μl of PBS (0.2% SDS, 1 mM EDTA). A 10-μl portion of the solution was kept as an input control. The biotinylated proteins were isolated using Dynabeads MyOne streptavidin C1 (Invitrogen; 50 μl of slurry per mg of protein), washed three times with at least 0.5 ml of PBS (0.2% SDS), and subsequently boiled in 50 μl of 1× SDS-gel sample buffer. Equal volumes of supernatant were subjected to SDS-PAGE and Western blotting. LpdA and control proteins were labeled with mouse anti-HA.11 (clone 16B12; MMs-101P, Cambridge Bioscience), mouse anti-Myc tag (catalog no. 05-724; Millipore), rabbit anti-calnexin (SPA-860F, Stressgen), and anti-tubulin (T6199; Sigma) antibodies in combination with appropriate horseradish peroxidase-labeled secondary antibodies (Jackson ImmunoResearch). Labeled proteins were visualized with a EZ-ECL detection reagent (Geneflow) and a Fuji Las3000 imager.
Infection of eukaryotic cells and intracellular growth assay.
L. pneumophila strains were cultured for infection as described previously (32). The bacteria were suspended in cell culture media, supplemented with chloramphenicol (6 μg/ml) and 1 mM IPTG if required, and added to A549 cells at a multiplicity of infection (MOI) of 15 to 100. To synchronize infection, samples were centrifuged for 5 min at 600 × g.
For growth assays, individual wells of a black 96-well plate with translucent bottoms were seeded with 8.5 × 104 THP-1 cells. After differentiation for 72 h, the medium was replaced with 100 μl of growth medium supplemented with 6 μg of chloramphenicol/ml and 1 mM IPTG. The cells were infected with L. pneumophila strains containing the pXDC31 GFP expression plasmid (40) at an MOI of 0.1 to 0.5 in triplicate. At 1 h postinfection, the medium was replaced with one containing 100 μg of gentamicin/ml, and the cells were incubated for a further 1 h and then washed three times with PBS. The infected cells were incubated in 100 μl of supplemented growth medium without phenol red, and the GFP fluorescence was recorded over 92 h in a FLUOstar Omega plate reader (BMG Labtech) equipped with an atmospheric control unit. Readings were taken every 2 h. Fluorescence measurements were corrected for the background fluorescence signal of uninfected cells, and each curve was normalized to a zero starting value.
β-Lactamase (TEM1) translocation assay.
The translocation of LpdA was tested as described previously (32). Briefly, RAW 264.7 cells were infected with L. pneumophila 130b expressing TEM1 fusions of LpdA, an inactive mutant of LpdA, or control proteins; then, after 1 h of incubation and washing, 20 μl of freshly prepared CCF2-AM β-lactamase substrate (LiveBLAzer FRET-B/G loading kit; Invitrogen) was added, and the fluorescence emission at 450 and 520 nm was measured from the bottom using a Fluostar Optima plate reader at 3 h postinfection. The translocation rate was expressed as the fold increase in the 450/520-nm emission ratio of each infected cell sample versus the emission ratio of uninfected cells.
Immunofluorescence microscopy.
Cells on coverslips were fixed with 3.2% paraformaldehyde (PFA), washed with D-PBS, incubated with 50 mM ammonium chloride, and washed and permeabilized with 0.1% Triton X-100. After blocking with PBS containing 2% (wt/vol) bovine serum albumin and 2% natural donkey serum, the samples were stained sequentially with primary and secondary antibodies and mounted using ProLong antifade reagent (Invitrogen). The primary antibodies (including catalog numbers) used were as follows: mouse anti-HA.11 (clone 16B12; MMs-101P; Cambridge Bioscience), mouse anti-Myc tag (05-724, Millipore), rabbit anti-Legionella lipopolysaccharide (PA1-7227; Affinity BioReagents), rabbit anti-Giantin (ab24586; Abcam), anti-c-Myc–fluorescein isothiocyanate conjugate (F2047; Sigma), and anti-HA-TRITC (H9037; Sigma). The secondary antibodies were donkey anti-rabbit or anti-mouse IgG Alexa Fluor 488 and donkey anti-rabbit or anti-mouse IgG rhodamine red X (RRX) (all from Jackson ImmunoResearch). The DNA was visualized with Hoechst 33342 dye. To visualize mitochondria, transfected cells were treated with MitoTracker Red (Life Technologies) prior to fixation. Samples were analyzed using an Axio Z1 imager microscope. Typically, 50 to 100 cells were assessed per condition, and at least two independent biological repeats were carried out per experimental series. Representative images were deconvoluted and processed using AxioVision software (Carl Zeiss).
Infection of mice.
All mouse procedures were approved by the University of Melbourne Animal Ethics Committee and carried out as described previously (41). Briefly, groups of mice were inoculated separately with ∼2.5 × 106 CFU of each L. pneumophila 130b strain under investigation. For consistency, L. pneumophila 130b wild-type (WT) or ΔlpdA mutant strains both carried the empty p4HA (pICC562) plasmid. At 72 h postinoculation, the mice were sacrificed, and their lung tissues were extracted. The tissue was homogenized, and complete host cell lysis was ensured by incubation in 0.1% saponin for 15 min at 37°C. Serial dilutions of the homogenate were plated onto plain CYE agar plates to determine the number of viable bacteria.
RESULTS
L. pneumophila infection causes drastic changes to the host cell PA distribution.
LpdA is conserved across the most commonly used sequenced prototype L. pneumophila isolates (130b [32], Philadelphia [42], Corby [43], Alcoy [44], Lens, Paris [45]; 97 to 98% identity; see Fig. S1 in the supplemental material). In order to investigate whether LpdA affects cellular distribution of PA, we generated an A549 lung epithelial cell line stably expressing a PA biosensor consisting of the PA-binding domain of the yeast protein Spo20 fused to GFP (A549-SPO) (46). In unchallenged cells GFP-Spo20 exclusively localizes to the nucleus (Fig. 1A), but upon stimulation of production it is rapidly recruited to sites where PA is generated (28, 46). A549-SPO cells were infected with L. pneumophila wild type, ΔlpdA mutant, or ΔlpdA mutant complemented with a plasmid encoding 4HA-tagged LpdA. Infection with the T4SS-deficient L. pneumophila ΔdotA strain was used as a control. IF microscopy at 6 h postinfection revealed that wild-type L. pneumophila but not the ΔdotA mutant induced redistribution of GFP-Spo20 from the nucleus, resulting in a diffuse signal throughout the cells (Fig. 1A). A similar redistribution of GFP-Spo20 was observed as early as 2 h, and remained after 24 h, postinfection (data not shown). The quantification of this effect by visual counting showed that at 6 h postinfection ca. 80% of cells infected with wild-type L. pneumophila had no nuclear GFP-Spo20 signal (Fig. 1B). In contrast, at the same time, >80% of the cells infected with the L. pneumophila ΔdotA mutant retained a clear nuclear GFP-Spo20 signal. Analysis and quantification of the PA redistribution in the cells infected with either the L. pneumophila ΔlpdA mutant or the complemented strain showed that both induced migration of GFP-Spo20 from the nucleus with the same kinetics and to the same extent as wild-type L. pneumophila (Fig. 1B and data not shown). These results show that upon infection, L. pneumophila induces rapid T4SS-dependent, but LpdA-independent, changes to the PA levels of host cells. The lack of obvious LpdA-dependent PA generation was not due to impaired translocation of the effector by L. pneumophila 130b into host cells, as TEM1-β lactamase translocation assays confirmed delivery of LpdA, and an inactive LpdA KK165/376RR mutant in which two catalytically important lysine residues were mutated (28), into host cells (see Fig. S2 in the supplemental material). This suggests that any impact of LpdA during infection might be subtle and masked by other effectors.
FIG 1.
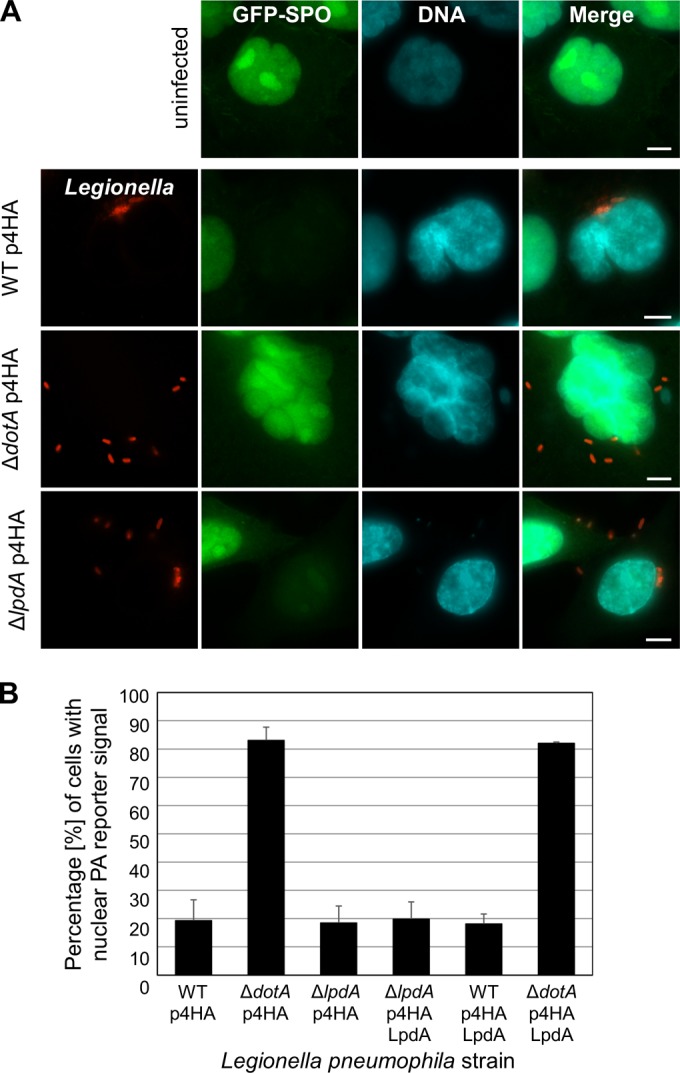
L. pneumophila infection causes drastic changes to the cellular PA distribution. A549 cells stably expressing the PA reporter GFP-SPO were left uninfected or infected for 6 h with an L. pneumophila 130b wild type (WT), ΔdotA mutant, or ΔlpdA mutant carrying the empty p4HA or p4HA LpdA plasmid and immunostained using an anti-L. pneumophila antibody (red). DNA was visualized with Hoechst DNA dye (blue). (A) Samples were examined by IF microscopy. Images are representative of at least three independent experiments. Scale bar, 5 μm. (B) Quantification of the effect of infection on the cellular distribution of the GFP-SPO PA reporter by IF counting. Error bars represent the mean standard deviations of three independent experiments.
LpdA is targeted to membranes by posttranslational palmitoylation.
Since the PA reporter assay (Fig. 1) suggested that the function of LpdA during infection is masked by redundant effectors, we continued our study using ectopic expression of hemagglutinin (HA)- or Myc-tagged LpdA or inactive mutants. In HeLa cells (Fig. 2A) and A549 lung epithelial cells (data not shown), HA-LpdA (Fig. 2A) or Myc-LpdA (data not shown) localized to small vesicular structures, which were distributed throughout the cell and, to a lesser extent, to the plasma membrane. Since similar distribution patterns for LpdA were observed in both cell lines, further experiments were carried out with HeLa cells because of their higher and more reproducible transfection efficiency. Cotransfection of these constructs with GFP-Spo20 showed redistribution of the reporter from the nucleus to vesicular structures and plasma membrane sites, which were enriched in LpdA. The catalytically inactive mutant LpdA KK165/376RR failed to induce the redistribution of GFP-Spo20, confirming that LpdA generates PA at the plasma membrane and vesicular structures (Fig. 2B).
FIG 2.

Ectopically expressed LpdA localizes to the plasma membrane and small vesicles, where it generates PA. IF images of HeLa cells transfected with pRK5 HA, pRK5 HA-LpdA WT, or LpdA KK165/376RR (KKRR) mutant alone (A) or together with the GFP-SPO PA reporter (B). The results are representative of at least three independent experiments. Scale bar, 10 μm.
Bioinformatic sequence analysis of LpdA using CSS-Palm 2.0 (47) revealed that the five C-terminal amino acid residues of LpdA contain three cysteines, which represent a potential site for posttranslational S-palmitoylation (Fig. 3A). Two of these cysteines are conserved across LpdA homologues of the most commonly used L. pneumophila isolates (see Fig. S1 in the supplemental material). In order to determine the role of these residues for the membrane association of LpdA, we created a truncation mutant (LpdAΔC5aa) and visualized its localization upon ectopic expression by IF. To examine the role of a nonconserved cysteine in the localization of LpdA, a C440F mutant corresponding to the amino acid replacement found in other LpdA homologues was included. Although full-length LpdA and LpdA C440F were indistinguishable and localized to vesicles and the plasma membrane, the LpdAΔ5aa mutant showed a diffuse cytoplasmic distribution (Fig. 3B). Notably, similarly to the wild-type effector, LpdAΔ5aa induced depletion of the GFP-Spo20 reporter from the nucleus (see Fig. S3 in the supplemental material), indicating that the truncated protein has lipase activity and that the loss of membrane association is not due to misfolding.
FIG 3.

Posttranslational palmitoylation targets LpdA to membranes. (A) Putative C-terminal palmitoylation site of LpdA. The three cysteines predicted to be modified with a palmitic acid moiety are underlined. (B) HA-LpdA, HA-LpdA C440F, and HA-LpdAΔC5aa were ectopically expressed in HeLa cells and immunostained, and their localization was analyzed by IF microscopy. Shown are representative images of at least three independent experiments. Scale bar, 10 μm. (C) Metabolic labeling and protein isolation strategy. HeLa cells expressing LpdA were metabolically labeled with the palmitic acid analogue ODYA. After cell lysis, a biotin moiety and a fluorophore were ligated using click chemistry. Posttranslationally modified proteins were isolated using streptavidin pulldown (PD) and analyzed by Western blotting (WB). (D) Representative result of the WB analysis of PD and input fractions using HA-conjugated, calnexin (endogenous metabolic labeling control), and tubulin (separation control) antibodies.
To elucidate whether the C-terminal membrane association motif of LpdA is palmitoylated, we performed bio-orthogonal metabolic labeling with a lipid reporter, an emerging strategy used to profile protein lipidation (48). HeLa cells expressing HA-LpdA or HA-LpdAΔC5aa were incubated with growth medium supplemented with 17-octadecynoic acid (ODYA), a palmitic acid analogue that is readily incorporated into proteins by palmitoyltransferases (Fig. 3C) (49). After cell lysis, the trifunctionalized AzTB capture reagent (38, 39) was ligated to the ODYA moiety in a Cu(I)-catalyzed cycloaddition, allowing the isolation and analysis of modified proteins by streptavidin pulldown (PD), Western blotting (WB) (Fig. 3D), and in-gel fluorescence detection (not shown). Anti-tubulin Western blotting confirmed equal concentrations of the PD input lysates and the effective elimination of unmodified proteins and cytosolic content from the isolated PD fractions. The detection of similar amounts of the endogenous, palmitoylated protein calnexin (Fig. 3D) (49) in all ODYA-treated samples, but not the unlabeled control sample, validated comparable and specific metabolic labeling and PD of modified proteins. Anti-HA WB revealed weak background binding of HA-LpdA in the absence of—and to a similar extent, HA-LpdAΔC5aa in the presence of—ODYA to the beads. Importantly, compared to HA-LpdAΔC5aa and the controls, HA-LpdA was enriched in the isolated PD fraction (Fig. 3D). This showed that LpdA is palmitoylated in the host cell and that the last five residues are critical for this modification.
LpdA localizes to Rab4- and Rab14-containing vesicles.
To determine which subcellular membranes LpdA targets, we examined the colocalization of LpdA with a panel of marker proteins for different cellular membranes (GFP-Rab1, -Rab2, -Rab4, -Rab5, -Rab6, -Rab7, -Rab10, -Rab11, -Rab14, and -LC3) and the mitochondria. The vesicular structures formed by LpdA colocalized with GFP-Rab4- and GFP-Rab14-positive membranes (Fig. 4, insets). The association was lost when the C-terminal palmitoylation motif of LpdA was removed (see Fig. S4 in the supplemental material). Sporadic association of LpdA with GFP-Rab7- and -Rab5-positive late and early endosomes was also observed (see Fig. S5A in the supplemental material). Notably, expression of LpdA did not seem to change the distribution of Rab4- or Rab14-positive vesicles or early and late endosomes compared to cells transfected with the LpdA inactive mutant (data not shown), suggesting that LpdA localizes to, but does not compromise, these transport vesicles. No colocalization with the mitochondria or GFP-Rab1, -Rab2, -Rab6, -Rab10, -Rab11 or the autophagosome marker GFP-LC3 was observed (see Fig. S5A to C in the supplemental material). It is noteworthy that Rab1, Rab2, and Rab6, which are usually found at the Golgi apparatus, showed in some LpdA-expressing cells a more diffuse or vesicular distribution rather than a compact perinuclear localization (data not shown). Our data show that vesicular LpdA mainly localizes to GFP-Rab4- and -Rab14-positive vesicles, which are involved in recycling transport processes between the plasma membrane and endosomes and endosomes and the Golgi apparatus.
FIG 4.

Vesicular LpdA localizes to Rab4 (A)- and Rab14 (B)-containing vesicles. HeLa cells were cotransfected with pRK5 HA or pRK5 HA-LpdA and plasmids for the expression of marker proteins of different cellular compartments and processed for IF microscopy. Rab4/Rab14- and LpdA-positive vesicles are highlighted with arrows in the magnified insets. Similar results were observed in at least two independent experiments. Scale bar, 10 μm.
LpdA hydrolyzes phosphatidylinositol, phosphatidylinositol-3- and phosphatidylinositol-4-phosphate, and phosphatidylglycerol.
The plasma membrane and Rab4- and Rab14-positive vesicles, which are targeted by LpdA, are made up of several different lipid species (50). In order to define the substrate specificity of LpdA, we performed activity assays with a selection of purified lipids. We first attempted to purify recombinant His6-tagged LpdA and the inactive mutant LpdA KK165/376RR. However, as the recombinant proteins were insoluble, PLD activity was determined using E. coli lysates containing wild-type or mutant LpdA. Bacterial lysates were incubated with phosphatidylcholine (PC), phosphatidylserine (PS), phosphatidylethanolamine (PE), and phosphatidylglycerol (PG) for 5 h (Fig. 5A) or phosphatidylinositiol and phosphatidylinositiol-monophosphates (Fig. 5B) and -polyphosphates (data not shown) for 4 h, and the samples were analyzed by TLC as described previously (27). Incubation of PG but not PC (Fig. 5A), PE, or PS (data not shown) with lysates containing wild-type LpdA revealed PA generation. Furthermore, wild-type LpdA efficiently released PA from PI, PI3P, and PI4P but not PI5P or polyphosphorylated PIPs (Fig. 5B). Importantly, substrate turnover did not occur in incubations with lysates of bacteria containing the empty plasmid or expressing the LpdA KK165/376RR mutant. These results confirm that LpdA has PLD activity, specifically releasing PA from PI, PI3P, PI4P, and PG in vitro.
FIG 5.
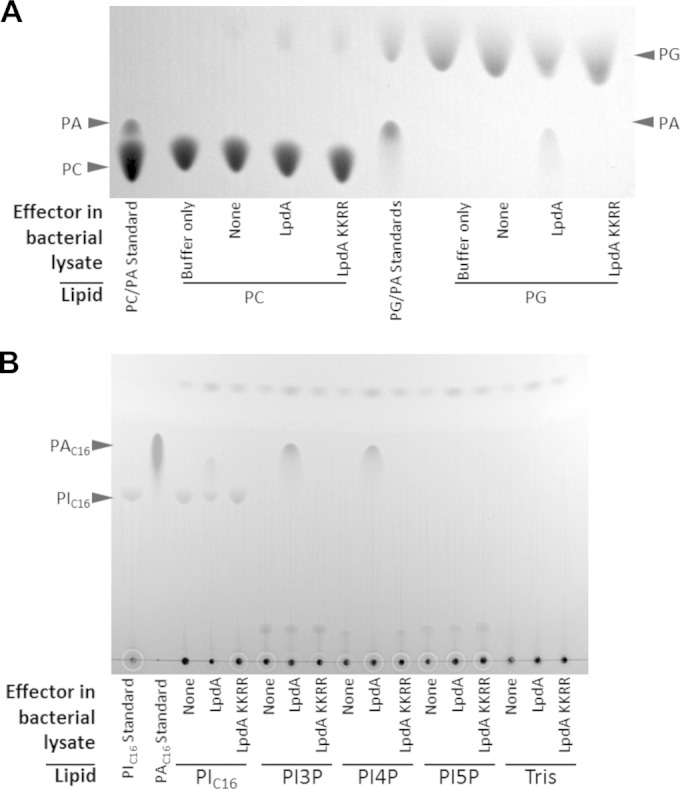
LpdA catalyzes the conversion of PG, PI, PI3P, and PI4P to PA. Lysates of E. coli with or without expression of His6-tagged LpdA or LpdA KK165/376RR (KKRR) or a buffer control were incubated with 0.2 mM PC or PG for 5 h at 37°C (A) or 0.25 mM PIC16 or monophosphorylated PIC16 for 4 h at 37°C (B). Lipids were extracted and analyzed by TLC. Corresponding pure PC, PG, PIC16, and PA standards were included. Similar results were obtained in at least two independent experiments.
LpdA induces disruption of the Golgi apparatus.
PI3P and PI4P are important determinants of the maturation of phagolysosomes, Golgi function, and LCV biogenesis (16, 51, 52). Since LpdA can hydrolyze these PIPs in vitro, we analyzed whether LpdA affects the distribution of PIP-binding reporter proteins in the cell. We coexpressed the active lipase or the inactive mutant with the PI3P and PI4P reporters 2×FYVE-GFP (Fig. 6A) or Myc-SidC PIP4 (Fig. 6B), respectively (21). 2×FYVE-GFP displayed strong association with LpdA-containing vesicles (Fig. 6A); however, its distributions were similar in mock-, LpdA- (Fig. 6A), or LpdA KK165/376RR (data not shown)-transfected cells. The Myc-SidC PIP4 reporter localized to perinuclear structures which were reminiscent of the Golgi and to very small vesicles throughout mock-transfected cells (Fig. 6B). Ectopically expressed LpdA did not colocalize with the PI4P reporter or induce its loss from membranes; however, in LpdA-expressing cells, the perinuclear Golgi body-like SidC PI4P structures seemed to have fragmented into small and medium sized vesicles, showing that LpdA's activity impacts PI4P-containing compartments.
FIG 6.

LpdA induces rearrangement of PI4P-containing compartments. HeLa cells were cotransfected with pRK5 HA or pRK5 HA-LpdA and a plasmid expressing the PI3P-binding protein FYVE fused to GFP (A) or the Myc-tagged PI4P binding domain of SidC (B), stained, and analyzed IF microscopy. Vesicles in which LpdA and the PI3P marker colocalize are highlighted with arrows in the magnified inset in panel A. Similar results were observed in at least three independent experiments. Scale bar, 5 μm.
In order to elucidate whether the action of LpdA has consequences for the integrity of the Golgi apparatus, we immunostained LpdA-transfected cells for the Golgi marker protein Giantin. This revealed that LpdA did not colocalize with the Golgi marker (Fig. 7A); however, in cells expressing the wild-type LpdA, but not in cells expressing the catalytically inactive mutant, the normal compact, tight stacks of Golgi cisternae had disappeared and, instead, disintegration into separated stacks or even small, vesicle-like fragments was apparent. At the same time, LpdA did not localize to or affect the integrity of the ER, indicating that the fragmentation of the Golgi apparatus was not due to action of the effector at the ER, which could interfere with early steps of the secretory pathway (see Fig. S6 in the supplemental material). Quantification of the Golgi damage revealed that ca. 90% of the LpdA KK165/376RR- or GFP-expressing cells, but only ca. 30% of the LpdA-expressing cells, retained an intact Golgi apparatus (Fig. 7B). Notably, the Golgi disruption induced by the LpdA mutant lacking the C-terminal palmitoylation motif was significantly higher (*, P = 0.0243, two-tailed, unpaired t test) than for the wild-type enzyme, suggesting that spatial control can restrict the activity of LpdA. IF analysis of infected A549 cells revealed a T4S-dependent, but LpdA-independent, disruption of the Golgi apparatus into ministacks and vesicles as early as 2 h 30 min postinfection (see Fig. S7 in the supplemental material), indicating that several effectors are involved in triggering the loss of integrity. Taken together, our data indicate that, although LpdA does not directly associate with Golgi membranes, its activity can impact on the integrity of the Golgi apparatus, most likely by perturbing PA levels and the lipid and protein transport by Rab14-positive vesicles between plasma membrane, endosomes, and the Golgi.
FIG 7.

LpdA activity in the cell disrupts the Golgi apparatus. HeLa cells were transfected with pRK5 HA-LpdA WT, pRK5 HA-LpdA KK165/376RR (KKRR), pRK5 HA-LpdAΔ5aa, or pRK5 HA-GFP and immunostained for HA and the Golgi protein Giantin. Samples were imaged (A), and the integrity of the Golgi apparatus was assessed in 100 transfected cells (B). Full and dotted arrows in the panel A inset image highlight Giantin-positive fragments and LpdA-containing vesicles, respectively, which do not colocalize. The images are representative of at least three independent experiments. Scale bar, 10 μm. In panel B, the error bars represent the mean standard deviations of three independent experiments. Significance was analyzed using a two-tailed, unpaired t test.
LpdA contributes to the virulence of L. pneumophila in A/J mice.
The infection data (Fig. 1 and see Fig. S7 in the supplemental material) suggested that the activity of LpdA in cells might be masked by functionally redundant effectors. In line with this, an L. pneumophila LpdA-null mutant was previously reported to show no growth defect in the environmental host Acanthamoeba castellanii (28). Similarly, we found that the L. pneumophila 130b ΔlpdA mutant was not attenuated for growth in THP-1 macrophages (see Fig. S8 in the supplemental material). We reasoned, therefore, that the contribution of LpdA to virulence might only become apparent under the highly selective pressure encountered in the mammalian lung. In order to determine whether LpdA has a role in virulence, we infected A/J mice with L. pneumophila 130b, ΔlpdA mutant, or ΔlpdA mutant complemented with a plasmid, allowing expression of 4HA-tagged LpdA, and examined the bacterial growth in the murine lung after 72 h. The L. pneumophila ΔlpdA mutant showed a reduced bacterial load in murine lungs compared to L. pneumophila wild-type and ΔlpdA 4HA-LpdA strains in two independent experiments (Fig. 8). This difference between the ΔlpdA mutant and the complemented strain was statistically significant in both experiments (Fig. 8, P = 0.0041 and P = 0.0355; Mann-Whitney U test); however, due to a higher variation in the number of recovered wild-type bacteria, it was only significant between the ΔlpdA mutant and wild type once (Fig. 8A, P = 0.0025, Mann-Whitney U test). There was no statistically significant difference between the wild-type and the complemented strain. In order to test the role of the PLD activity of LpdA for its function in the murine infection model, an L. pneumophila ΔlpdA strain expressing the 4HA-LpdA KK165/376RR inactive mutant was included. This experiment showed that in contrast to the expression of wild-type 4HA-LpdA, the inactive LpdA mutant did not restore full virulence of the ΔlpdA mutant (Fig. 8B, P = 0.0482, Mann-Whitney U test). Taken together, our data indicate that, although functionally redundant effectors seem to exist, LpdA contributes to the optimal survival of L. pneumophila in vivo and that its lipase activity is required to fulfill this function.
FIG 8.

LpdA contributes to the fitness of L. pneumophila 130b during pulmonary infections of A/J mice. The results of two independent experiments (A and B) are shown. A/J mice were infected with L. pneumophila wild-type (WT) and ΔlpdA mutant strains by intranasal inoculation. At 72 h postinfection, the animals were sacrificed and the CFU in the lungs determined by plating. (A) Both the wild type and the complemented ΔlpdA strain grew significantly (P = 0.0025 and P = 0.0041, respectively [Mann-Whitney U test]) better than the mutant strain in the lungs of mice. (B) The ΔlpdA strain and the ΔlpdA strain expressing the inactive LpdA KK165/376RR (KKRR) mutant are significantly (P = 0.0355 and P = 0.0482, respectively [Mann-Whitney U test]) attenuated compared to the complemented L. pneumophila ΔlpdA p4HA LpdA strain.
DISCUSSION
L. pneumophila uses several effectors, such as SidF, LecE, and LegS2, which have enzymatic activities that directly modulate membrane lipid composition and metabolism (23, 28, 53). Moreover, many other effectors share homology with eukaryotic lipases (18). The high abundance of lipid-converting enzymes indicates that subversion of host cell lipids is crucial for L. pneumophila infection; however, our knowledge about the biology of most of these effectors remains limited. Here, we report that LpdA is an L. pneumophila effector that exploits host cell mediated posttranslational S-palmitoylation as membrane-targeting mechanism. To our knowledge, LpdA is the first palmitolylated L. pneumophila effector, although other T4SS effectors have previously been shown to exploit posttranslational prenylation for membrane targeting (19, 20). Bacterial type III secretion system effectors, such as Salmonella SspH2 and SseI or Pseudomonas AvrPphB, as well as several viruses, have been shown to use posttranslational palmitoylation for subcellular targeting (54–56), suggesting that its exploitation is an ancient strategy in the coevolution of pathogens and hosts.
LpdA is a lipolytic effector which has been proposed to be a PLD modulating phospholipid biosynthesis on the LCV (28). Our study revealed that LpdA confers a moderate advantage for survival and replication in the lungs of A/J mice to wild-type L. pneumophila and the complemented strain compared to the ΔlpdA mutant. Importantly, our data indicate that this requires the catalytic activity of LpdA. The difference between the ΔlpdA and complemented ΔlpdA strains was more consistent across experiments than the one between ΔlpdA and wild-type L. pneumophila, since the latter showed larger variation in bacterial load compared to the complemented mutant. The increased variation could be due to higher heterogeneity of LpdA expression in the wild-type population. Notably, hardly any L. pneumophila effector null mutants, and not even strains lacking up to one-third of the known effectors, are attenuated in growth in most L. pneumophila cell infection models (57). An L. pneumophila JR32 ΔlpdA mutant was previously reported to replicate as well as the wild-type strain in amoebae (28). Similarly, we found that the L. pneumophila 130b ΔlpdA strain was not attenuated for replication in THP-1 macrophages. In the murine lung, the bacteria are confronted with the diverse antibacterial mechanisms of macrophages and neutrophils, creating a highly selective environment in which the impact of individual effectors, such as LpdA, might become more apparent than in monoculture infections.
To gain a deeper understanding of the function of LpdA on a molecular level, we performed in vitro substrate specificity assays. These showed that LpdA has PLD activity toward PI and its derivatives, PI3P and PI4P, which all are important signaling molecules. PI3P is also hydrolyzed by the PLA effector VipD, which leads to loss of the PI3P-binding proteins from the membrane (25). Ectopic coexpression of LpdA with PI3P and PI4P reporter proteins did not cause any loss of the reporters from the membrane, suggesting that LpdA does not hydrolyze these two lipids or that the amounts which are removed are too small to impact on membrane anchoring of the reporter proteins.
In vitro, LpdA also hydrolyzed PG, which is a minor lipid constituent of eukaryotic membranes but can amount to up to 20% of bacterial membranes. Overexpression of LpdA could therefore have adverse effects on the integrity of the bacteria and causes unspecific release of LpdA. However, using the inactive LpdA KK165/376RR mutant, we demonstrated that the translocation of LpdA occurs independently of its activity but requires a functional T4SS. In eukaryotic cells, PG is mainly found in mitochondrial membranes. However, neither our experiments nor the work of Viner et al. (28) provided any indication that LpdA is targeted to mitochondria or affects their integrity. In infected cells, LpdA localizes to the LCV (28). The exact lipid composition of the LCV is unknown. However, since L. pneumophila releases outer membrane vesicles (OMVs), which can fuse with the LCV membrane (58), it is likely that the LCV membrane also contains PG of bacterial origin, which could serve as the substrate for LpdA. OMVs released by L. pneumophila have been shown to interfere with phagosome-lysosome maturation and fusion of endosomes with the LCV (58), which is desirable for the bacteria at early stages of the infection. However, the maturation into the replication-permissive LCV relies on fusion of ER-derived vesicles with the LCV, for which the transition to a more ER-like lipid composition could be favorable. LpdA could function in this adjustment process.
Our data show that ectopic expression of LpdA induces drastic changes to cellular PA levels and has a detrimental effect on the integrity of the Golgi apparatus. Ectopic-expression levels of proteins might be substantially higher than during infection and therefore cause unspecific alterations of the cell physiology; however, our data show that L. pneumophila infection induces drastic redistribution of a PA reporter in host cells, as well as a rapid dispersal of the Golgi body, a phenomenon which was also reported earlier by Rothmeier et al. (59). Both phenotypes were T4S dependent, but not affected by deletion of LpdA, suggesting that LpdA has a small or very specific, tightly controlled role and that other redundant effectors mask its effect. A large number of effectors that were shown to target host cell mediators of the secretory pathway (60) could contribute to the Golgi disruption. Additional lipases encoded in Legionella genomes and effectors such as LecE, which activates host cell PA phosphatase, leading to the conversion of PA into diacylglycerol (28), could contribute to generation and turnover of PA, explaining why no accumulation of our PA reporter on any specific cellular membrane or the LCV was detected. In light of the involvement of redundant effectors during infection, only ectopic expression could be used to unravel the contribution of LpdA to these phenotypes in infection.
Using ectopic expression, the data show that the lipase activity of LpdA is required for PA generation and the disruption of the Golgi apparatus. PA is an important signaling molecule and was implicated in the regulation of membrane tubulation and vesicle formation at recycling endosomes and the Golgi apparatus (61–63). In particular, the structure and function of the Golgi apparatus was shown to be sensitive to disturbance of the cellular PA homeostasis. The inhibition of phosphatidic acid synthesis was previously shown to disrupt the Golgi structure (64). Robust activation of human PLD, which generates PA and regulates membrane trafficking at various cellular sites, including the Golgi structure, is required for induction of Golgi fragmentation by the drug ilimaquinone (65, 66).
LpdA triggers Golgi disruption without localizing to the organelle, which suggests that changes in lipid composition and PA levels in distant membranes sites adversely affect membrane transport processes to and/or the lipid composition of the Golgi apparatus, which result in its breakdown. The main PA generation sites are the plasma membrane and Rab4- and Rab14-containing vesicles, to which LpdA localizes; however, the observation that LpdA lacking the palmitoylation motif, which does not specifically localize to these sites anymore, induces even more pronounced Golgi disruption, leads to the hypothesis that LpdA-mediated perturbation of the cellular PA homeostasis rather than the modulation of a specific vesicular trafficking pathway can cause this effect.
Taken together, we established here that the effector LpdA is a PLD that can hydrolyze several lipid substrates to modulate cellular PA levels and contributes to virulence of L. pneumophila in the murine lung. We discovered that Legionella exploits host cell-mediated posttranslational S-palmitoylation to exert spatial control over LpdA's activity in the cell. Moreover, our work revealed that L. pneumophila infection induces rapid disruption of the Golgi apparatus, as well as dramatic changes in host cell PA levels, with unknown benefits for the infection and involvement of unidentified effectors besides LpdA, warranting future research in the interdependence of these phenotypes and the role of phospholipase effectors in L. pneumophila pathogenesis.
Supplementary Material
ACKNOWLEDGMENTS
We thank Miguel Seabra (Imperial College [IC], London, United Kingdom) for the Rab GTPase expression plasmids, Nicolas Vitale (Institute of Cellular and Integrative Neurosciences, University of Strasbourg, Strasbourg, France) for the GFP-SPO reporter plasmids, Xavier Charpentier for the pXDC31 plasmid (Université Lyon 1, Lyon, France), Xin Li (IC) for the PLCδ1 template DNA, Avinash Shenoy (IC) for providing reagents for the creation of stable cell lines, and Julian Rayner and Matthew Jones (Wellcome Trust Sanger Institute, Cambridge, United Kingdom) for initial advice on the analysis of palmitoylation.
This study was supported by grants from the Medical Research Council UK, German Research Foundation (DFG) grants DFG FL 359/6-1, 6-2, and grants to E.L.H. from the Australian National Health and Medical Research Council (APP606788).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00785-15.
REFERENCES
- 1.Newton HJ, Ang DK, van Driel IR, Hartland EL. 2010. Molecular pathogenesis of infections caused by Legionella pneumophila. Clin Microbiol Rev 23:274–298. doi: 10.1128/CMR.00052-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allombert J, Fuche F, Michard C, Doublet P. 2013. Molecular mimicry and original biochemical strategies for the biogenesis of a Legionella pneumophila replicative niche in phagocytic cells. Microbes Infect 15:981–988. doi: 10.1016/j.micinf.2013.09.007. [DOI] [PubMed] [Google Scholar]
- 3.Fuche F, Vianney A, Andrea C, Doublet P, Gilbert C. 2015. Functional type 1 secretion system involved in Legionella pneumophila virulence. J Bacteriol 197:563–571. doi: 10.1128/JB.02164-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rossier O, Starkenburg SR, Cianciotto NP. 2004. Legionella pneumophila type II protein secretion promotes virulence in the A/J mouse model of Legionnaires' disease pneumonia. Infect Immun 72:310–321. doi: 10.1128/IAI.72.1.310-321.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Segal G, Purcell M, Shuman HA. 1998. Host cell killing and bacterial conjugation require overlapping sets of genes within a 22-kb region of the Legionella pneumophila genome. Proc Natl Acad Sci U S A 95:1669–1674. doi: 10.1073/pnas.95.4.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vogel JP, Andrews HL, Wong SK, Isberg RR. 1998. Conjugative transfer by the virulence system of Legionella pneumophila. Science 279:873–876. doi: 10.1126/science.279.5352.873. [DOI] [PubMed] [Google Scholar]
- 7.Lifshitz Z, Burstein D, Peeri M, Zusman T, Schwartz K, Shuman HA, Pupko T, Segal G. 2013. Computational modeling and experimental validation of the Legionella and Coxiella virulence-related type IVB secretion signal. Proc Natl Acad Sci U S A 110:E707–E715. doi: 10.1073/pnas.1215278110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu W, Banga S, Tan Y, Zheng C, Stephenson R, Gately J, Luo ZQ. 2011. Comprehensive identification of protein substrates of the Dot/Icm type IV transporter of Legionella pneumophila. PLoS One 6:e17638. doi: 10.1371/journal.pone.0017638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Horwitz MA. 1983. Formation of a novel phagosome by the Legionnaires' disease bacterium (Legionella pneumophila) in human monocytes. J Exp Med 158:1319–1331. doi: 10.1084/jem.158.4.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kagan JC, Roy CR. 2002. Legionella phagosomes intercept vesicular traffic from endoplasmic reticulum exit sites. Nat Cell Biol 4:945–954. doi: 10.1038/ncb883. [DOI] [PubMed] [Google Scholar]
- 11.Swanson MS, Isberg RR. 1995. Association of Legionella pneumophila with the macrophage endoplasmic reticulum. Infect Immun 63:3609–3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prashar A, Terebiznik MR. 2015. Legionella pneumophila: homeward bound away from the phagosome. Curr Opin Microbiol 23:86–93. doi: 10.1016/j.mib.2014.11.008. [DOI] [PubMed] [Google Scholar]
- 13.So EC, Mattheis C, Tate EW, Frankel G, Schroeder GN. 2015. Creating a customized intracellular niche: subversion of host cell signaling by Legionella type IV secretion system effectors. Can J Microbiol 6:1–19. [DOI] [PubMed] [Google Scholar]
- 14.Goody RS, Itzen A. 2013. Modulation of small GTPases by Legionella. Curr Top Microbiol Immunol 376:117–133. doi: 10.1007/82_2013_340. [DOI] [PubMed] [Google Scholar]
- 15.Mousnier A, Schroeder GN, Stoneham CA, So EC, Garnett JA, Yu L, Matthews SJ, Choudhary JS, Hartland EL, Frankel G. 2014. A new method to determine in vivo interactomes reveals binding of the Legionella pneumophila effector PieE to multiple Rab GTPases. mBio 5:e01148-14. doi: 10.1128/mBio.01148-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haneburger I, Hilbi H. 2013. Phosphoinositide lipids and the Legionella pathogen vacuole. Curr Top Microbiol Immunol 376:155–173. doi: 10.1007/82_2013_341. [DOI] [PubMed] [Google Scholar]
- 17.Ivanov SS, Roy C. 2013. Host lipidation: a mechanism for spatial regulation of Legionella effectors. Curr Top Microbiol Immunol 376:135–154. doi: 10.1007/82_2013_344. [DOI] [PubMed] [Google Scholar]
- 18.Lang C, Flieger A. 2011. Characterisation of Legionella pneumophila phospholipases and their impact on host cells. Eur J Cell Biol 90:903–912. doi: 10.1016/j.ejcb.2010.12.003. [DOI] [PubMed] [Google Scholar]
- 19.Ivanov SS, Charron G, Hang HC, Roy CR. 2010. Lipidation by the host prenyltransferase machinery facilitates membrane localization of Legionella pneumophila effector proteins. J Biol Chem 285:34686–34698. doi: 10.1074/jbc.M110.170746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Price CT, Al-Quadan T, Santic M, Jones SC, Abu Kwaik Y. 2010. Exploitation of conserved eukaryotic host cell farnesylation machinery by an F-box effector of Legionella pneumophila. J Exp Med 207:1713–1726. doi: 10.1084/jem.20100771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harding CR, Mattheis C, Mousnier A, Oates CV, Hartland EL, Frankel G, Schroeder GN. 2013. LtpD is a novel Legionella pneumophila effector that binds phosphatidylinositol 3-phosphate and inositol monophosphatase IMPA1. Infect Immun 81:4261–4270. doi: 10.1128/IAI.01054-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Di Paolo G, De Camilli P. 2006. Phosphoinositides in cell regulation and membrane dynamics. Nature 443:651–657. doi: 10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- 23.Hsu F, Zhu W, Brennan L, Tao L, Luo ZQ, Mao Y. 2012. Structural basis for substrate recognition by a unique Legionella phosphoinositide phosphatase. Proc Natl Acad Sci U S A 109:13567–13572. doi: 10.1073/pnas.1207903109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Toulabi L, Wu X, Cheng Y, Mao Y. 2013. Identification and structural characterization of a Legionella phosphoinositide phosphatase. J Biol Chem 288:24518–24527. doi: 10.1074/jbc.M113.474239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gaspar AH, Machner MP. 2014. VipD is a Rab5-activated phospholipase A1 that protects Legionella pneumophila from endosomal fusion. Proc Natl Acad Sci U S A 111:4560–4565. doi: 10.1073/pnas.1316376111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schunder E, Adam P, Higa F, Remer KA, Lorenz U, Bender J, Schulz T, Flieger A, Steinert M, Heuner K. 2010. Phospholipase PlaB is a new virulence factor of Legionella pneumophila. Int J Med Microbiol 300:313–323. doi: 10.1016/j.ijmm.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 27.Aurass P, Schlegel M, Metwally O, Harding CR, Schroeder GN, Frankel G, Flieger A. 2013. The Legionella pneumophila Dot/Icm-secreted effector PlcC/CegC1 together with PlcA and PlcB promotes virulence and belongs to a novel zinc metallophospholipase C family present in bacteria and fungi. J Biol Chem 288:11080–11092. doi: 10.1074/jbc.M112.426049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Viner R, Chetrit D, Ehrlich M, Segal G. 2012. Identification of two Legionella pneumophila effectors that manipulate host phospholipids biosynthesis. PLoS Pathog 8:e1002988. doi: 10.1371/journal.ppat.1002988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jenkins GM, Frohman MA. 2005. Phospholipase D: a lipid centric review. Cell Mol Life Sci 62:2305–2316. doi: 10.1007/s00018-005-5195-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Edelstein PH. 1986. Control of Legionella in hospitals. J Hosp Infect 8:109–115. doi: 10.1016/0195-6701(86)90037-X. [DOI] [PubMed] [Google Scholar]
- 31.Engleberg NC, Drutz DJ, Eisenstein BI. 1984. Cloning and expression of Legionella pneumophila antigens in Escherichia coli. Infect Immun 44:222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schroeder GN, Petty NK, Mousnier A, Harding CR, Vogrin AJ, Wee B, Fry NK, Harrison TG, Newton HJ, Thomson NR, Beatson SA, Dougan G, Hartland EL, Frankel G. 2010. Legionella pneumophila strain 130b possesses a unique combination of type IV secretion systems and novel Dot/Icm secretion system effector proteins. J Bacteriol 192:6001–6016. doi: 10.1128/JB.00778-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen DQ, Huang SS, Lu YJ. 2006. Efficient transformation of Legionella pneumophila by high-voltage electroporation. Microbiol Res 161:246–251. doi: 10.1016/j.micres.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 34.Galan JE, Ginocchio C, Costeas P. 1992. Molecular and functional characterization of the Salmonella invasion gene invA: homology of InvA to members of a new protein family. J Bacteriol 174:4338–4349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stone BJ, Kwaik YA. 1999. Natural competence for DNA transformation by Legionella pneumophila and its association with expression of type IV pili. J Bacteriol 181:1395–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shenoy AR, Wellington DA, Kumar P, Kassa H, Booth CJ, Cresswell P, MacMicking JD. 2012. GBP5 promotes NLRP3 inflammasome assembly and immunity in mammals. Science 336:481–485. doi: 10.1126/science.1217141. [DOI] [PubMed] [Google Scholar]
- 37.Bligh EG, Dyer WJ. 1959. A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 38.Heal WP, Jovanovic B, Bessin S, Wright MH, Magee AI, Tate EW. 2011. Bio-orthogonal chemical tagging of protein cholesterylation in living cells. Chem Commun 47:4081–4083. doi: 10.1039/c0cc04710d. [DOI] [PubMed] [Google Scholar]
- 39.Heal WP, Wright MH, Thinon E, Tate EW. 2012. Multifunctional protein labeling via enzymatic N-terminal tagging and elaboration by click chemistry. Nat Protoc 7:105–117. doi: 10.1038/nprot.2011.425. [DOI] [PubMed] [Google Scholar]
- 40.Hovel-Miner G, Pampou S, Faucher SP, Clarke M, Morozova I, Morozov P, Russo JJ, Shuman HA, Kalachikov S. 2009. SigmaS controls multiple pathways associated with intracellular multiplication of Legionella pneumophila. J Bacteriol 191:2461–2473. doi: 10.1128/JB.01578-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harding CR, Stoneham CA, Schuelein R, Newton H, Oates CV, Hartland EL, Schroeder GN, Frankel G. 2013. The Dot/Icm effector SdhA is necessary for virulence of Legionella pneumophila in Galleria mellonella and A/J mice. Infect Immun 81:2598–2605. doi: 10.1128/IAI.00296-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chien M, Morozova I, Shi S, Sheng H, Chen J, Gomez SM, Asamani G, Hill K, Nuara J, Feder M, Rineer J, Greenberg JJ, Steshenko V, Park SH, Zhao B, Teplitskaya E, Edwards JR, Pampou S, Georghiou A, Chou IC, Iannuccilli W, Ulz ME, Kim DH, Geringer-Sameth A, Goldsberry C, Morozov P, Fischer SG, Segal G, Qu X, Rzhetsky A, Zhang P, Cayanis E, De Jong PJ, Ju J, Kalachikov S, Shuman HA, Russo JJ. 2004. The genomic sequence of the accidental pathogen Legionella pneumophila. Science 305:1966–1968. doi: 10.1126/science.1099776. [DOI] [PubMed] [Google Scholar]
- 43.Steinert M, Heuner K, Buchrieser C, Albert-Weissenberger C, Glockner G. 2007. Legionella pathogenicity: genome structure, regulatory networks and the host cell response. Int J Med Microbiol 297:577–587. doi: 10.1016/j.ijmm.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 44.D'Auria G, Jimenez-Hernandez N, Peris-Bondia F, Moya A, Latorre A. 2010. Legionella pneumophila pangenome reveals strain-specific virulence factors. BMC Genomics 11:181. doi: 10.1186/1471-2164-11-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cazalet C, Rusniok C, Bruggemann H, Zidane N, Magnier A, Ma L, Tichit M, Jarraud S, Bouchier C, Vandenesch F, Kunst F, Etienne J, Glaser P, Buchrieser C. 2004. Evidence in the Legionella pneumophila genome for exploitation of host cell functions and high genome plasticity. Nat Genet 36:1165–1173. doi: 10.1038/ng1447. [DOI] [PubMed] [Google Scholar]
- 46.Kassas N, Tryoen-Toth P, Corrotte M, Thahouly T, Bader MF, Grant NJ, Vitale N. 2012. Genetically encoded probes for phosphatidic acid. Methods Cell Biol 108:445–459. doi: 10.1016/B978-0-12-386487-1.00020-1. [DOI] [PubMed] [Google Scholar]
- 47.Ren J, Wen L, Gao X, Jin C, Xue Y, Yao X. 2008. CSS-Palm 2.0: an updated software for palmitoylation sites prediction. Protein Eng Des Sel 21:639–644. doi: 10.1093/protein/gzn039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tate EW, Kalesh KA, Lanyon-Hogg T, Storck EM, Thinon E. 2015. Global profiling of protein lipidation using chemical proteomic technologies. Curr Opin Chem Biol 24C:48–57. doi: 10.1016/j.cbpa.2014.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Martin BR, Cravatt BF. 2009. Large-scale profiling of protein palmitoylation in mammalian cells. Nat Methods 6:135–138. doi: 10.1038/nmeth.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van Meer G, Voelker DR, Feigenson GW. 2008. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol 9:112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.De Matteis MA, Wilson C, D'Angelo G. 2013. Phosphatidylinositol-4-phosphate: the Golgi and beyond. Bioessays 35:612–622. doi: 10.1002/bies.201200180. [DOI] [PubMed] [Google Scholar]
- 52.Jeschke A, Zehethofer N, Lindner B, Krupp J, Schwudke D, Haneburger I, Jovic M, Backer JM, Balla T, Hilbi H, Haas A. 2015. Phosphatidylinositol 4-phosphate and phosphatidylinositol 3-phosphate regulate phagolysosome biogenesis. Proc Natl Acad Sci U S A 112:4636–4641. doi: 10.1073/pnas.1423456112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Degtyar E, Zusman T, Ehrlich M, Segal G. 2009. A Legionella effector acquired from protozoa is involved in sphingolipids metabolism and is targeted to the host cell mitochondria. Cell Microbiol 11:1219–1235. doi: 10.1111/j.1462-5822.2009.01328.x. [DOI] [PubMed] [Google Scholar]
- 54.Blanc M, Blaskovic S, van der Goot FG. 2013. Palmitoylation, pathogens and their host. Biochem Soc Trans 41:84–88. doi: 10.1042/BST20120337. [DOI] [PubMed] [Google Scholar]
- 55.Dowen RH, Engel JL, Shao F, Ecker JR, Dixon JE. 2009. A family of bacterial cysteine protease type III effectors utilizes acylation-dependent and -independent strategies to localize to plasma membranes. J Biol Chem 284:15867–15879. doi: 10.1074/jbc.M900519200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hicks SW, Charron G, Hang HC, Galan JE. 2011. Subcellular targeting of Salmonella virulence proteins by host-mediated S-palmitoylation. Cell Host Microbe 10:9–20. doi: 10.1016/j.chom.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.O'Connor TJ, Adepoju Y, Boyd D, Isberg RR. 2011. Minimization of the Legionella pneumophila genome reveals chromosomal regions involved in host range expansion. Proc Natl Acad Sci U S A 108:14733–14740. doi: 10.1073/pnas.1111678108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fernandez-Moreira E, Helbig JH, Swanson MS. 2006. Membrane vesicles shed by Legionella pneumophila inhibit fusion of phagosomes with lysosomes. Infect Immun 74:3285–3295. doi: 10.1128/IAI.01382-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rothmeier E, Pfaffinger G, Hoffmann C, Harrison CF, Grabmayr H, Repnik U, Hannemann M, Wolke S, Bausch A, Griffiths G, Muller-Taubenberger A, Itzen A, Hilbi H. 2013. Activation of Ran GTPase by a Legionella effector promotes microtubule polymerization, pathogen vacuole motility, and infection. PLoS Pathog 9:e1003598. doi: 10.1371/journal.ppat.1003598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hilbi H, Haas A. 2012. Secretive bacterial pathogens and the secretory pathway. Traffic 13:1187–1197. doi: 10.1111/j.1600-0854.2012.01344.x. [DOI] [PubMed] [Google Scholar]
- 61.Giridharan SS, Cai B, Vitale N, Naslavsky N, Caplan S. 2013. Cooperation of MICAL-L1, syndapin2, and phosphatidic acid in tubular recycling endosome biogenesis. Mol Biol Cell 24:1776–1790. doi: 10.1091/mbc.E13-01-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stamnes M, Schiavo G, Stenbeck G, Sollner TH, Rothman JE. 1998. ADP-ribosylation factor and phosphatidic acid levels in Golgi membranes during budding of coatomer-coated vesicles. Proc Natl Acad Sci U S A 95:13676–13680. doi: 10.1073/pnas.95.23.13676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang J-S, Gad H, Lee SY, Mironov A, Zhang L, Beznoussenko GV, Valente C, Turacchio G, Bonsra AN, Du G, Baldanzi G, Graziani A, Bourgoin S, Frohman MA, Luini A, Hsu VW. 2008. A role for phosphatidic acid in COPI vesicle fission yields insights into Golgi maintenance. Nat Cell Biol 10:1146–1153. doi: 10.1038/ncb1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Siddhanta A, Backer JM, Shields D. 2000. Inhibition of phosphatidic acid synthesis alters the structure of the Golgi apparatus and inhibits secretion in endocrine cells. J Biol Chem 275:12023–12031. doi: 10.1074/jbc.275.16.12023. [DOI] [PubMed] [Google Scholar]
- 65.Riebeling C, Morris AJ, Shields D. 2009. Phospholipase D in the Golgi apparatus. Biochim Biophys Acta 1791:876–880. doi: 10.1016/j.bbalip.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sonoda H, Okada T, Jahangeer S, Nakamura S. 2007. Requirement of phospholipase D for ilimaquinone-induced Golgi membrane fragmentation. J Biol Chem 282:34085–34092. doi: 10.1074/jbc.M705593200. [DOI] [PubMed] [Google Scholar]
- 67.Dolezal P, Aili M, Tong J, Jiang JH, Marobbio CM, Lee SF, Schuelein R, Belluzzo S, Binova E, Mousnier A, Frankel G, Giannuzzi G, Palmieri F, Gabriel K, Naderer T, Hartland EL, Lithgow T. 2012. Legionella pneumophila secretes a mitochondrial carrier protein during infection. PLoS Pathog 8:e1002459. doi: 10.1371/journal.ppat.1002459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.de Felipe KS, Glover RT, Charpentier X, Anderson OR, Reyes M, Pericone CD, Shuman HA. 2008. Legionella eukaryotic-like type IV substrates interfere with organelle trafficking. PLoS Pathog 4:e1000117. doi: 10.1371/journal.ppat.1000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.