

Abstract
Minihepcidins are in vitro and in vivo active mimetics of iron-regulatory hormone hepcidin. They contain various unusual amino acids including: N-substituted, β-homo-, and D-amino acids with their combination depending on particular minihepcidin. In the current study, we sought to limit the use of unusual/more expensive amino acids derivatives by peptide cyclisation. Novel cyclic mimetics of hepcidin were synthesized and tested in vitro and showed activity at low nanomolar concentration. Nonetheless, the most active cyclic compound (mHS17) is approximately ten times less active than the parental minihepcidin PR73. Collectively, our findings suggest that cyclisation is viable approach in the synthesis of hepcidin mimetics.
Keywords: Minihepcidins, Peptides, S-Alkylation of peptides, Iron, Cyclisation
Hepcidin (Figure 1A), a 25 amino acids long peptide hormone, is a key regulator of iron homeostasis in verterbrates1. Its function is mediated through the membrane receptor/iron exporter, ferroportin (Fpn)2. Hepcidin binds to Fpn which in turn causes the internalization of Fpn and its subsequent proteasomal degradation. In consequence, diminished levels of surface Fpn on duodenal enterocytes and hepatic and splenic macrophages restrict systemic iron availability, by decreasing both the absorption of dietary iron in duodenum and the release of recycled iron from macrophages3.
Figure 1.

Comparison of structures of (A) human hepcidin, (B) minihepcidin PR73, (C) inactive disulfide-bond cyclized analog cycloDTHFPI(CIF-CONHCH2CH2S-), and (D) bioactive disulfide-bond cyclized analog cyclo(CDTHFPIC)IF-CONH2.
Hepcidin, ferroportin and related pathways are currently under scrutiny as novel target(s) in the search for new therapeutics for iron disorders, with several leading compounds in advanced stage of development4–6. Hepcidin itself has unfavorable pharmacological properties (t1/2<2.5min)7 and, because of its 4 disulfide bonds, it is notoriously difficult to synthesize8. We developed minihepcidins, rationally designed peptide-based hepcidin mimetics, with potent in vitro and in vivo bioactivity9;10. Minihepcidins are currently under commercial development by Merganser Biotech LLC. Data available to date indicate that minihepcidins could be useful in stand-alone and combination-therapy-regimen(s) for β-thalassemia and polycythemia vera as well as in the treatment of iron-dependent bacterial infections11–14.
Most active minihepcidins contain both natural and unusual amino acids, including N-substituted- and β-homo- amino acids, with the unusual amino acids conferring resistance to proteolysis15–17. The C-terminus of minihepcidins contains a conjugated lipid moiety (C16) that increases plasma half-life and stabilizes the analog(s)18–21. Lipidation may also anchor the peptide in the hydrophobic environment of lipid rafts, increasing its local concentration, and bioactivity21–25. Notably, lipidated retro-inverso minihepcidins were also synthesized and showed potent bioactivity9.
Due to their content of unusual amino acids, certain minihepcidins may be expensive to produce, especially in the large quantities. Additional costs result mainly from the use of β-homo-amino acids (β-homo-L-proline (βhPro) and β-homo-L-phenylalanine (βhPhe)) and the unusual amino acid, 3,3′-diphenyl-L-alanine (Dpa). Therefore we explored a different strategy that would avoid the use of the most expensive amino acid derivatives, produce active hepcidin mimetics, and decrease the cost of synthesis. We chose the peptide cyclisation approach which has a long standing history26. Interestingly, head-to-tail cyclisation of full length hepcidin with and without spacer-residue(s) was recently reported27. However these modifications resulted in inactive derivatives. In our case though, the starting point for modifications was the PR73 minihepcidin (Figure 1B) rather than the full length hormone. Specifically, we employed the previously described S-alkylation method for the bridging of peptide(s) containing two strategically placed Cys residues with either symmetrical bis-halogeno-derivatives28;29 or divinyl sulfone (DVS)30. The use of this particular cyclisation method was prompted both by low cost and wide range of various alkylating analogs available, with an additional benefit of increased rigidity of the bridge that may stabilize the active conformer(s). The availability of various cysteine homologs: (L)Cys, (D)Cys, (L)homoCys, (D)homoCys, (L)Pen, and (D)Pen provides the option of “fine tuning” of selected active derivatives. Moreover, all reactions can be carried out in solution without any protecting groups. Notably, this approach was already applied in peptide drug development31, including phage display32–42 as well as peptide-albumin43;44 and peptide-antibody drug conjugates (ADCs)45.
All linear peptides necessary for this study were synthesized by the solid phase method using CEM Liberty automatic microwave peptide synthesizer (CEM Corporation Inc., Matthews, NC), employing 9-fluorenylmethyloxycarbonyl (Fmoc) chemistry and commercially available amino acid derivatives and reagents (Chem-Impex International, Inc., Wood Dale, IL)46;47. Since minihepcidins contain the biologically important cysteine residue in position 79;10 we used S-tert-butyl protected Cys-derivative (Cys(tBu)) to avoid unwanted interference with S-alkylation of cysteines in these positions.
S-Alkylation/cyclisation of reverse-phase high-performance liquid chromatography (RP-HPLC) purified linear analogs was performed using 2 different protocols, depending on the particular compound’s structure. Generally, for S-alkylation of synthetic linear peptides which are moderately or well soluble in water, we employed the protocol described by Timmerman and co-workers28. These reactions were carried out at ambient temperature in 50 mM ammonium bicarbonate (NH4HCO3) dissolved in a mixture of acetonitrile (ACN) and water 46. In case of hydrophobic peptides (e.g. lipidated analogs) we utilized previously described 1,1,3,3-tetramethylguanidine (TMG) driven reaction of thiol(s) containing compounds with bis-halogeno-derivatives in organic solvent/low molecular weight alcohol (preferably methanol)48, that we adapted to peptides46. Since peptides are rarely well soluble in methanol, we used dimethyl sulfoxide (DMSO) as a solubilizing additive (up to 25%) and excess of TMG (final conc.=0.35%, vol/vol). In addition, we also synthesized bi- and tri-cyclic concatenated derivatives (mHS11-mHS13), in an attempt to benefit from the potential multivalency of binding to multimerized Fpn.
Cyclic crude analogs containing Cys(tBu) residue(s) (position 7) were selectively deprotected using either modified TFMSA protocol or DMSO/TFA oxidation/dimerization followed by reduction with excess of tris(2-carboxyethyl)phosphine hydrochloride (TCEP, 100 eq/30 min)46;49. Notably, an alternative S-tert-butyl deprotection protocol, utilizing 1M HBF4/thioanisole was also described50.
All synthesized cyclic analogs were purified by preparative RP-HPLC and characterized by matrix-assisted laser desorption ionization spectrometry (MALDI-MS) as well as analytical RP-HPLC51 (see Table 1). Structural details for the synthesized analogs are presented in Figures 2, 4 and 5. Notably, for hydrophobic/lipidated analogs, an alternative synthetic protocol was also described52. Generally, cyclisation leading to monocyclic mHS derivatives proceeded efficiently with 1,3-bis(bromomethyl)benzene and 2,6-bis(bromomethyl)pyridine giving superior results. On the other hand, analogs mHS8-10 were particularly difficult to obtain, regardless on used protocol giving estimated yields <5% (based on RP-HPLC purified material).
Table 1.
Analytical and in vitro activity data for mSH analogs.
| Peptide | Composition | MW Calc/Found | RT [min] | EC50 [nM] TREX-hFpn-GFP cells |
|---|---|---|---|---|
| PR73 | C86H133N21O15S | 1733.19/1734.34 | 47.11* | 4.2±0.3 |
| mHS1 | C46H69N15O9S3 | 1072.32/1072.85 | 29.47 | >600 |
| mHS2 | C52H77N17O11S3 | 1212.47/1212.20 | 28.06 | >600 |
| mHS3 | C50H75N15O11S4 | 1190.47/1190.96 | 29.24 | >600 |
| mHS4 | C54H75N15O9S3 | 1174.46/1174.73 | 33.42 | 585.6±71.1 |
| mHS5 | C54H75N15O9S3 | 1174.46/1174.86 | 34.19 | 272.2±18.9 |
| mHS6 | C54H75N15O9S3 | 1174.46/1174.60 | 34.18 | 598.9±114.2 |
| mHS7 | C53H74N16O9S3 | 1175.45/1175.90 | 27.65 | >600 |
| mHS8 | C58H77N15O9S3 | 1224.52/1225.92 | 30.95 | >600 |
| mHS9 | C60H79N15O10S3 | 1266.56/1266.74 | 39.58 | 477.4±76.3 |
| mHS10 | C61H81N15O9S3 | 1264.58/1264.52 | 41.14 | 516.9±131.7 |
| mHS11 | C96H134N28O16S5 | 2096.59/2097.25 | 34.74 | >600 |
| mHS12 | C138H193N41O23S7 | 3018.73/3018.75 | 34.00/34.89 | >600 regioisomers |
| mHS13 | C141H199N41O31S7 | 3188.80/3188.87 | 34.33 | >600 |
| mHS14 | C60H85N17O13S3 | 1348.62/1348.85 | 30.44 | 531.7±36.1 |
| mHS15 | C60H79N15O9S3 | 1250.56/1250.82 | 36.16 | 54.3±2.7 |
| mHS16 | C66H89N17O13S3 | 1424.71/1425.16 | 32.47 | 75.1±0.3 |
| mHS17 | C86H128N18O12S3 | 1702.25/1702.26 | 58.73 | 44.2±6.8 |
| mHS18 | C53H69N13O7S3 | 1096.39/1096.45 | 40.94 | 103.8±13.0 |
| mHS19 | C59H73N13O7S3 | 1172.49/1172.74 | 45.16 | 112.7±10.5 |
| mHS20 | C53H69N13O7S3 | 1096.39/1096.61 | 41.01 | 335.3±41.2 |
| mHS21 | C52H68N12O6S3 | 1053.36/1053.87 | 44.03 | 268.8±29.2 |
| mHS22 | C66H92N16O11S3 | 1381.73/1381.51 | 38.32 | 164.5±14.7 |
| mHS23 | C64H89N15O10S3 | 1324.68/1324.28 | 40.54 | 92.3±19.5 |
| mHS24 | C74H107N15O9S3 | 1446.93/1446.57 | 50.68* | >600 |
| mHS25 | C73H106N14O8S3 | 1403.91/1403.63 | 50.73* | 369.0±35.0 |
| mHS26 | C75H112N14O10S3 | 1465.98/1466.43 | 55.52* | 52.3±10.8 |
| mHS27 | C68H99N13O7S3 | 1306.79/1306.43 | 57.32* | >600 |
| mHS28 | C68H99N13O7S3 | 1306.79/1306.90 | 57.72* | 458.3±87.7 |
Analytical RP-HPLC was performed on a Varian ProStar 210 HPLC system equipped with ProStar 325 Dual Wavelength UV-Vis detector with the wavelengths set at 220 nm and 280 nm (Varian Inc., Palo Alto, CA). Mobile phases consisted of solvent A, 0.1% TFA in water, and solvent B, 0.1% TFA in acetonitrile. Analyses of peptides were performed with an analytical reversed-phase C18 SymmetryShield™ column, 4.6×250 mm, 5 μm (Waters, Milford, MA) or (*) an analytical reversed-phase C4 XBridge™ BEH300 column, 4.6×150 mm, 3.5 μm (Waters, Milford, MA), applying linear gradient of solvent B from 0 to 100% over 100 min (flow rate: 1 ml/min).
Figure 2.
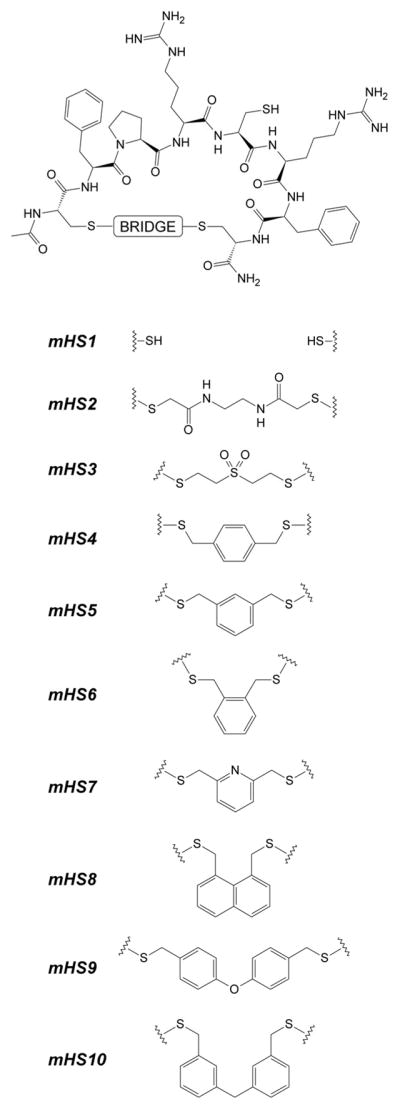
General structure of synthesized analogs mHS1–mHS10.
Figure 4.

Structure of concatenated multi-cyclic derivatives mHS11÷mHS13. For mHS12, examples of 2 regioisomers are depicted.
Figure 5.

Structure of cyclic mHS14÷mHS28 derivatives.
Bicyclic mHS11 was synthesized from its linear analog: Ac-CFPRXRFCFPRXRFC-CONH2 (X=L-Cys(StBu)) using 1,3,5-tris(bromomethyl)benzene as a bridging moiety28. Tricyclic counterparts were synthesized from Ac-CFPRXRFCFPRXRFCFPRXRFC-CONH2 and either commercially available 1,2,4,5-tetrakis(bromomethyl)benzene28 or pentaerythritol tetrakis(2-bromoacetate)46;53. In this case, depending on bridging moiety structure/symmetry, single or multiple products (regioisomers) may be obtained. Single product of S-alkylation is possible if linker used in the process of S-alkylation possesses tetrahedral symmetry (Td) (e.g. pentaerythritol scaffold). As expected, cyclisation reaction with 1,2,4,5-tetrakis(bromomethyl)benzene proceeded with low efficiency, which is most likely due to the generally rigid structure of the final product(s) as well as the fact that such linker may produce theoretically 4 different regioisomers varying in Cys-linker connectivity. Analogous reaction with pentaerythritol tetrakis(2-bromoacetate) gave a single product. Subsequent selective deprotection of the side chains of X-residues (Cys(tBu)→Cys(SH)) in multicyclic analogs using trifluoromethanesulfonic acid (TFMSA) protocol produced the desired analogs mHS11-mHS13. In these harsh conditions the pentaerythritol scaffold remained intact proving that the proposed linker is relatively stable and perhaps require more attention, especially that it efficiently produced tricyclic, sterically hindered compound containing various bulky side chains in the vicinity of S-alkylation sites. Tricyclic analogs obtained via this method would be a logical extension of previously done work on therapeutic bicyclic analogs31 offering potentially even larger diversity, especially if coupled with phage display approach32–42. Notably, the pentaerythritol scaffold was used previously for combinatorial chemistry54 and in the synthesis of polymers and nanomaterials using an atom transfer radical polymerization (ATRP)55;56 but not as an S-alkylating component of peptides’ constraining scaffold(s).
All newly synthesized analogs were tested in vitro57 using a previously described Fpn degradation assay58 with results summarized in Table 1.
Our studies on cyclic analogs were prompted by observation that certain disulfide bridged derivatives of N-terminal portion of hepcidin retain moderate bioactivity59. Specifically, the disulfide –bond cyclized compound cyclo(CDTHFPIC)IF-CONH2 showed EC50=300 nM, while cycloDTHFPI(CIF-CONHCH2CH2S-) was inactive (Figures 1C and 1D respectively), proving that only cyclic analogs with certain structural features are able to bind effectively to Fpn and trigger its degradation. The need for a broader strategy was reemphasized by the inactivity of full length head to tail cyclic hepcidin analogs27. We therefore explored different available linker(s) capable of bridging intramolecular thiols in the prototypical minihepcidin peptide resulting in analogs mHS1-mHS10. The sequence and length of the peptides was chosen based on available data9;58–60. Specifically, we assumed that the hexapeptide FPRCRF, corresponding to the parental sequence FPICIF, is both necessary and sufficient to exert full bioactivity, when its conformation is “properly altered” by cyclisation constrains. This sequence was in turn flanked with Cys residues, to allow for cyclisation based on S-alkylation with bis-thiol-reactive linkers. In addition, Ile → Arg modifications were used to improve solubility and activity59. This sequence is significantly shorter than the hepcidin segment DTHFPICIF from which minihepcidins are derived. Lack of N-terminal dipeptide (DT) has limited effects on bioactivity58, therefore only remaining 3His deletion, which accounts for ~80% loss in activity when combined with the latter (e.g. lack of DTH)58, represents significant challenge and this activity loss can be compensated by proper linker design. Such assumption is plausible since substitution of 3His for fairly similar Phe or (D)His residues has limited effects on overall activity60 and S-alkylation of Cys residue with aromatic linker(s) would produce substituted phenylalanine homolog(s) (see Figure 2). Except aromatic bridging moieties we also tested 2 linear and more flexible linkers afforded by bis-reactive Michael type compound DVS and 2-chloro-N-(2-(2-chloro-acetyloamino)-ethyl)-acetamide, Notably, other thiol(s)-multireactive Michael type compounds, including aromatic derivatives, were also reported32;61.
As shown in Table 1 the most active analog in initial group, mHS5, was produced by bridging with 1,3-bis(bromomethyl)benzene. Interestingly, mHS7, which possesses identical geometry, resulted in inactive analog, suggesting that presence of more hydrophilic/ionizable pyridine group is strongly detrimental in this particular region of molecule. This observation is in line with results from previous mutational and SAR studies that underlined importance of hydrophobic interactions in the vicinity of 4Phe and 9Phe58;60 which are in close proximity to the bridging moiety. The role of hydrophobicity and conformational influence of the linker is additionally validated by results for mHS2 and mHS3 compounds which possess hydrophilic linkers with similar length to that of mHS4 and mHS5 analogs but are inactive. Similarly, analogs mHS4 and mHS6, that closely resemble mHS5 in terms of bridging moiety and its hydrophobicity, are approximately 2 times less active stressing importance of proper spatial arrangements of amino acids side chains for Fpn efficient binding which results from particular linker’s geometry. Based on these results we chose mHS5 and its bridging moiety, 1,3-bis(bromomethyl)benzene, for further studies.
In the next group of concatenated, multicyclic analogs mHS11-mHS13 (Figure 4) we probed whether multivalent cyclic mHS5-derivatives can exert desired bioactivity. Surprisingly all those analogs were inactive. Notably, mHS12 was tested as a mixture of regioisomers, and based on analytical RP-HPLC results there were at least 2 different regioisomers present.
Subsequently we focused on various modifications of mHS5 to determine whether its activity could be improved. Specifically, addition of the Ida-Thr dipeptide at the N-terminus portion of mHS5 is detrimental (analogs mHS5 versus mHS14 and mHS15 versus mHS16), which is unexpected since this fragment corresponds to previously deleted portion (Asp-Thr) of original minihepcidin DTHFPICIF-CONH2, and it was used based on our earlier work on linear minihepcidins10;59. This modification causes ~30–50% loss in activity and its effects may explain negative results for bicyclic mHS11 derivative, which despite being basically mHS5 dimer must have different N- and C-terminal spatial side-chains arrangements due to cyclisation-induced “crowding”, underling once again role of hydrophobic interactions provided by 4Phe and 9Phe. Even the presence of the small acetyl-amino-group in this position is undesirable as analog lacking this particular modification (mHS23) is approximately 2 times more active than AcNH-containing and otherwise identical mHS22 compound. Similarly presence of the substituents at C-terminus (e.g. amide moiety) seems to have negative effects, although they are less pronounced (mHS20 versus mHS21 and mHS24 versus mHS25). Increase of hydrophobicity in 4Phe position (4Phe→Dpa) causes a dramatic increase in potency (5×, mHS5 versus mHS15) which is analogous to the effects observed among its linear counterparts10;59. Since Dpa is considered in this study to be an “expensive” amino acid, we attempted next to develop its viable, less expensive substituent. To this end, we decided to use 3,3-diphenylpropionic- and 3,3,3-triphenylpropionic acids (Dpp and Tpp respectively), which are less expensive des-amino-Dpa homologs. However, this approach requires modification of N-terminal Cys position as both Dpp and Tpp can be only placed as “final” N-terminal residue(s), due to lack of amine group. Based on limited molecular modeling (HyperChem 8.0, Hypercube Inc., Gainesville, FL), we determined that the location and spatial arrangements of Pro residue at mHS5 are such that they permit its substitution for (L)Cys without significant structural change. As a result, we synthesized analogs mHS18 and mHS19 bearing such modifications (e.g. 5Pro→(L)Cys) which showed moderate potency (EC50≈100 nM) and were approximately 2× less potent than initial mHS15 (EC50=54.3±2.7). Notably, there was no apparent effect of hydrophobicity increase on bioactivity as Tpp containing compound (mHS19) showed similar activity to Dpp counterpart (mHS18). The change in absolute Cys configuration ((L)Cys → (D)Cys; mHS18 versus mHS20) resulted in a significant loss of bioactivity (3.2×).
Generally, lipidation improves the bioactivity of cyclic mHS analogs, provided lipid moiety is conjugated via C-terminus of the molecule (mHS17 versus mHS15, mHS24 & mHS25, and mHS26 versus mHS18, mHS27 & mHS28), which agrees with observations for linear minihepcidins59. C16 moiety was introduced either using palmitic acid or iminodiacetic acid mono-n-palmityl amide (IdaNHPal) with spacers afforded by short PEG (1,8-diamino-3,6-dioxaoctane) or 6-aminohexanoic acid (Ahx) respectively. The use of short spacers is necessary for high activity of original linear minihepcidins59 and this principle was also assumed to be important for cyclic counterparts.
The most active cyclic analogs developed in this study are mHS17 (EC50=44.2±6.8 nM) and mHS26 (EC50=52.3±10.8 nM). Despite their bioactivity in the low nanomolar range they are still considerably less potent (10.5× and 12.5× respectively) than parental minihepcidin PR73 (EC50= 4.2±0.3 nM). Nonetheless, they are smaller than PR73, and considerably less expensive to produce, at least based on direct synthesis/materials costs (see Figure 6). Therefore, despite their lower potency, we decided to test whether mHS derivatives can exert desired bioactivity in vivo. We selected lipidated derivatives: mHS17 and mHS26, and “lipid-less” mHS18 as suitable candidates for animal studies62, which were carried out using previously described protocol9;10;58. The peptides were injected subcutaneously and bioactivity was tested solely at 24h post-injection time point, which roughly coincides with maximal activity for linear minihepcidin(s)9;10. Using minihepcidin PR73 as a control, we determined that analogous amounts of mHS peptides (50–200 nmoles) show no desired biological effect (Figure 3B). Since no in vivo activity was observed, we probed plasma stability63–66 of mHS26 and PR73 to determine whether the lack of activity of mHS compounds is due to their proteolytic degradation. In fact, the inactive lipidated analog mHS26 was slightly more resistant to plasma driven degradation (Figure 3C) than the highly bioactive minihepcidin PR73, suggesting that the overall potency rather than stability is the key feature determining in vivo properties/bioactivity of minihepcidins. Such assumption would certainly explain the poor performance of mHS derivatives in in vivo experiments, since animals received similar dosage (50–200 nmoles) with the potency for the most active mHS analogs being at least 10× smaller than of PR73. Cyclisation of mHS peptides certainly improves plasma stability as linear analog mHS1 was rapidly degraded in the same experimental conditions within 6 h.
Figure 6.

Comparison of structures and relative production costs (materials) for analogs PR73, mHS17 and mHS26.
Figure 3.
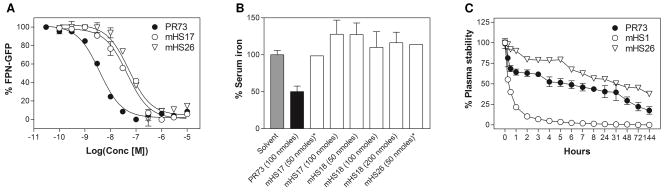
Activity of selected mHS analogs: (A) comparison of dose response curves obtained for analogs PR73 (EC50=4.2±0.3 nM), mHS17 (EC50=44.2±6.8 nM), and mHS26 (EC50=52.3±10.8 nM) in in vitro Fpn degradation assay57, (B) comparison of in vivo activity of selected mHS peptides at 24h time-point62, (C) comparison of plasma stability of PR73 and mHS2666.
In conclusion, a group of small cyclic peptide-based hepcidin agonists was synthesized and tested in vitro and principles of their SAR were established. The most active analogs, mHS17 and mHS26 are considerably cheaper to produce than prototypic, parental minihepcidin PR73, however they show no in vivo potency based on equimolar administration in a mouse model. Nonetheless, in vitro activity of selected mHS peptides is in low nanomolar range suggesting that these novel cyclic analogs may be viable leads for further development of therapeutic hepcidin agonists. Such analogs may be obtained by additional “fine tuning” of mHS properties exploiting commercially available cysteine homologs (e.g. (L)homoCys, (L)β-homoCys, (L)Pen, etc.) or change of the location of the lipidation site (use of lipid-bearing S-alkylating linkers), or both. Significant interest in pharmaceutically-relevant hepcidin agonists certainly warrants further experimentation.
Acknowledgments
This project was partially supported by funds from the Adams and Burnham endowments provided by the Dean’s Office of the David Geffen School of Medicine at UCLA (PR) and NIH grant R01 DK090554 (TG and EN).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Ganz T. Blood. 2003;102:783. doi: 10.1182/blood-2003-03-0672. [DOI] [PubMed] [Google Scholar]
- 2.Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, Kaplan J. Science. 2004;306:2090. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 3.Ganz T, Nemeth E. Biochim Biophys Acta. 2012;1823:1434. doi: 10.1016/j.bbamcr.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fung E, Nemeth E. Haematologica. 2013;98:1667. doi: 10.3324/haematol.2013.084624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Poli M, Asperti M, Ruzzenenti P, Regoni M, Arosio P. Front Pharmacol. 2014;5:86. doi: 10.3389/fphar.2014.00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ruchala P, Nemeth E. Trends Pharmacol Sci. 2014;35:155. doi: 10.1016/j.tips.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xiao JJ, Krzyzanski W, Wang YM, Li H, Rose MJ, Ma M, Wu Y, Hinkle B, Perez-Ruixo JJ. AAPS J. 2010;12:646. doi: 10.1208/s12248-010-9222-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jordan JB, Poppe L, Haniu M, Arvedson T, Syed R, Li V, Kohno H, Kim H, Schnier PD, Harvey TS, Miranda LP, Cheetham J, Sasu BJ. J Biol Chem. 2009;284:24155. doi: 10.1074/jbc.M109.017764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Preza GC, Ruchala P, Pinon R, Ramos E, Qiao B, Peralta MA, Sharma S, Waring A, Ganz T, Nemeth E. J Clin Invest. 2011;121:4880. doi: 10.1172/JCI57693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramos E, Ruchala P, Goodnough JB, Kautz L, Preza GC, Nemeth E, Ganz T. Blood. 2012;120:3829. doi: 10.1182/blood-2012-07-440743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Casu C, Goldberg A, Shah Y, Nemeth E, Ganz T, Gardenghi S, MacDonald B, Rivella S. Blood. 2013;122:431. [Google Scholar]
- 12.Casu C, Oikonomidau R, Nemeth E, Ganz T, MacDonald B, Rivella S. Blood. 2014;124:3231. [Google Scholar]
- 13.Casu C, Oikonomidau R, Shah Y, Nemeth E, Ganz T, MacDonald B, Rivella S. Blood. 2014;124:748. [Google Scholar]
- 14.Arezes J, Jung G, Gabayan V, Valore E, Ruchala P, Gulig PA, Ganz T, Nemeth E, Bulut Y. Cell Host Microbe. 2015;17:47. doi: 10.1016/j.chom.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Biron E, Chatterjee J, Ovadia O, Langenegger D, Brueggen J, Hoyer D, Schmid HA, Jelinek R, Gilon C, Hoffman A, Kessler H. Angew Chem Int Ed Engl. 2008;47:2595. doi: 10.1002/anie.200705797. [DOI] [PubMed] [Google Scholar]
- 16.Frackenpohl J, Arvidsson PI, Schreiber JV, Seebach D. Chembiochem. 2001;2:445. doi: 10.1002/1439-7633(20010601)2:6<445::aid-cbic445>3.3.co;2-i. [DOI] [PubMed] [Google Scholar]
- 17.White TR, Renzelman CM, Rand AC, Rezai T, McEwen CM, Gelev VM, Turner RA, Linington RG, Leung SS, Kalgutkar AS, Bauman JN, Zhang Y, Liras S, Price DA, Mathiowetz AM, Jacobson MP, Lokey RS. Nat Chem Biol. 2011;7:810. doi: 10.1038/nchembio.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ward BP, Ottaway NL, Perez-Tilve D, Ma D, Gelfanov VM, Tschop MH, Dimarchi RD. Mol Metab. 2013;2:468. doi: 10.1016/j.molmet.2013.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosi F, Triola G. Methods Mol Biol. 2013;1047:161. doi: 10.1007/978-1-62703-544-6_12. [DOI] [PubMed] [Google Scholar]
- 20.Zhang L, Bulaj G. Curr Med Chem. 2012;19:1602. doi: 10.2174/092986712799945003. [DOI] [PubMed] [Google Scholar]
- 21.Ingallinella P, Bianchi E, Ladwa NA, Wang YJ, Hrin R, Veneziano M, Bonelli F, Ketas TJ, Moore JP, Miller MD, Pessi A. Proc Natl Acad Sci U S A. 2009;106:5801. doi: 10.1073/pnas.0901007106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Avadisian M, Gunning PT. Mol Biosyst. 2013;9:2179. doi: 10.1039/c3mb70147f. [DOI] [PubMed] [Google Scholar]
- 23.Johannessen L, Remsberg J, Gaponenko V, Adams KM, Barchi JJ, Jr, Tarasov SG, Jiang S, Tarasova NI. Chembiochem. 2011;12:914. doi: 10.1002/cbic.201000563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Augusto MT, Hollmann A, Castanho MA, Porotto M, Pessi A, Santos NC. J Antimicrob Chemother. 2014;69:1286. doi: 10.1093/jac/dkt529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Doyle JR, Harwood BN, Krishnaji ST, Krishnamurthy VM, Lin WE, Fortin JP, Kumar K, Kopin AS. PLoS One. 2014;9:e110502. doi: 10.1371/journal.pone.0110502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kessler H. Angew Chem Int Ed Engl. 1982;21:512. [Google Scholar]
- 27.Clark RJ, Preza GC, Tan CC, van Dijk JW, Fung E, Nemeth E, Ganz T, Craik DJ. Biopolymers. 2013;100:519. doi: 10.1002/bip.22350. [DOI] [PubMed] [Google Scholar]
- 28.Timmerman P, Beld J, Puijk WC, Meloen RH. Chembiochem. 2005;6:821. doi: 10.1002/cbic.200400374. [DOI] [PubMed] [Google Scholar]
- 29.Kemp DS, McNamara M. J Org Chem. 1985;50:5834. [Google Scholar]
- 30.Wilkinson RA, Evans JR, Jacobs JM, Slunaker D, Pincus SH, Pinter A, Parkos CA, Burritt JB, Teintze M. AIDS Res Hum Retroviruses. 2007;23:1416. doi: 10.1089/aid.2007.0027. [DOI] [PubMed] [Google Scholar]
- 31.Timmerman P, Puijk WC, Meloen RH. J Mol Recognit. 2007;20:283. doi: 10.1002/jmr.846. [DOI] [PubMed] [Google Scholar]
- 32.Chen S, Bertoldo D, Angelini A, Pojer F, Heinis C. Angew Chem Int Ed Engl. 2014;53:1602. doi: 10.1002/anie.201309459. [DOI] [PubMed] [Google Scholar]
- 33.Bellotto S, Chen S, Rentero R, I, Wegner HA, Heinis C. J Am Chem Soc. 2014;136:5880. doi: 10.1021/ja501861m. [DOI] [PubMed] [Google Scholar]
- 34.Chen S, Gopalakrishnan R, Schaer T, Marger F, Hovius R, Bertrand D, Pojer F, Heinis C. Nat Chem. 2014;6:1009. doi: 10.1038/nchem.2043. [DOI] [PubMed] [Google Scholar]
- 35.Rentero RI, Sabisz M, Baeriswyl V, Heinis C. Nucleic Acids Res. 2014 doi: 10.1093/nar/gku940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen S, Rentero R, I, Buth SA, Morales-Sanfrutos J, Touati J, Leiman PG, Heinis C. J Am Chem Soc. 2013;135:6562. doi: 10.1021/ja400461h. [DOI] [PubMed] [Google Scholar]
- 37.Angelini A, Cendron L, Chen S, Touati J, Winter G, Zanotti G, Heinis C. ACS Chem Biol. 2012;7:817. doi: 10.1021/cb200478t. [DOI] [PubMed] [Google Scholar]
- 38.Baeriswyl V, Rapley H, Pollaro L, Stace C, Teufel D, Walker E, Chen S, Winter G, Tite J, Heinis C. Chem Med Chem. 2012;7:1173. doi: 10.1002/cmdc.201200071. [DOI] [PubMed] [Google Scholar]
- 39.Baeriswyl V, Heinis C. Protein Eng Des Sel. 2013;26:81. doi: 10.1093/protein/gzs085. [DOI] [PubMed] [Google Scholar]
- 40.Baeriswyl V, Heinis C. Chem Med Chem. 2013;8:377. doi: 10.1002/cmdc.201200513. [DOI] [PubMed] [Google Scholar]
- 41.Baeriswyl V, Calzavarini S, Gerschheimer C, Diderich P, ngelillo-Scherrer A, Heinis C. J Med Chem. 2013;56:3742. doi: 10.1021/jm400236j. [DOI] [PubMed] [Google Scholar]
- 42.Heinis C, Rutherford T, Freund S, Winter G. Nat Chem Biol. 2009;5:502. doi: 10.1038/nchembio.184. [DOI] [PubMed] [Google Scholar]
- 43.Pollaro L, Raghunathan S, Morales-Sanfrutos J, Angelini A, Kontos S, Heinis C. Mol Cancer Ther. 2014 doi: 10.1158/1535-7163.MCT-14-0534. [DOI] [PubMed] [Google Scholar]
- 44.Angelini A, Morales-Sanfrutos J, Diderich P, Chen S, Heinis C. J Med Chem. 2012;55:10187. doi: 10.1021/jm301276e. [DOI] [PubMed] [Google Scholar]
- 45.Angelini A, Diderich P, Morales-Sanfrutos J, Thurnheer S, Hacker D, Menin L, Heinis C. Bioconjug Chem. 2012;23:1856. doi: 10.1021/bc300184m. [DOI] [PubMed] [Google Scholar]
-
46.Peptide synthesis.
Linear peptides were synthesized by the solid phase method using CEM Liberty automatic microwave peptide synthesizer (CEM Corporation Inc., Matthews, NC), applying 9-fluorenylmethyloxycarbonyl (Fmoc) chemistry and commercially available amino acid derivatives and reagents (EMD Biosciences, San Diego, CA and Chem-Impex International, Inc., Wood Dale, IL). Peptides were cleaved from resin using modified reagent K (TFA 94% (v/v); phenol, 2% (w/v); water, 2% (v/v); TIS, 1% (v/v); EDT, 1% (v/v); 2 hours) and precipitated by addition of ice-cold diethyl ether. Subsequently, they were purified by preparative reverse-phase high performance liquid chromatography (RP-HPLC).Synthesis of analogs mHS2-mHS16, mHS22 and mHS23.Cyclic compounds were obtained using modified Timmerman protocol28. Briefly, linear peptides were solubilized in 50% acetonitrile in water containing 50 mM NH4HCO3 (~0.5 mg/mL) and then appropriate bis-functional S-alkylating reagent was added. Reaction(s) were stirred for 18 h and solutions subsequently lyophilized. Obtained solid residues wereselectively deprotected (see below) and purified by preparative RP-HPLC and their purity evaluated by MALDI-MS as well as analytical RP-HPLC. Bis-functional S-alkylating reagents were, for:
mHS2 - 2-Chloro-N-(2-(2-chloro-acetyloamino)-ethyl)-acetamide,
(2) mHS3 - Divinylsulfone, 10 mM conc. of NH4HCO3 was used.
mHS4 - 1,4-Bis(bromomethyl)benzene,
mHS5 - 1,3-Bis(bromomethyl)benzene,
mHS6 - 1,2-Bis(bromomethyl)benzene,
mHS7 - 2,6-Bis(bromomethyl)pyridine,
mHS8 - 1,8-Bis(bromomethyl)naphthalene,
mHS9 - 1-(Chloromethyl)-4-[4-(chloromethyl)phenoxy]benzene,
mHS10 - 1-(Bromomethyl)-3-[3-(bromomethyl)benzyl]benzene,
mHS11 - 1,3,5-Tris(bromomethyl)benzene,
mHS12 - 1,2,4,5-Tetrakis(bromomethyl)benzene,
mHS13 - Pentaerythritol tetrakis(2-bromoacetate),
mHS14-mHS16, mHS22 and mHS23 - 1,3-Bis(bromomethyl)benzene.
- 47.Fields GB, Noble RL. Int J Pept Protein Res. 1990;35:161. doi: 10.1111/j.1399-3011.1990.tb00939.x. [DOI] [PubMed] [Google Scholar]
- 48.Włostowski M, Czarnocka S, Maciejewski P. Tetrahedron Lett. 2010;51:5977. [Google Scholar]
- 49.Cuthbertson A, Indrevoll B. Org Lett. 2003;5:2955. doi: 10.1021/ol035105w. [DOI] [PubMed] [Google Scholar]
- 50.Akaji K, Yoshida M, Tatsumi T, Kimura T, Fujiwara Y, Kiso Y. J Chem Soc Chem Commun. 1990:288. doi: 10.1248/cpb.38.273. [DOI] [PubMed] [Google Scholar]
- 51.Analytical RP-HPLCwas performed on a Varian ProStar 210 HPLC system equipped with ProStar 325 Dual Wavelength UV-Vis detector with the wavelengths set at 220 nm and 280 nm (Varian Inc., Palo Alto, CA). Mobile phases consisted of solvent A, 0.1% TFA in water, and solvent B, 0.1% TFA in acetonitrile. Analyses of peptides were performed with (*) an analytical reversed-phase C4 XBridge™ BEH300 column, 4.6×150 mm, 3.5 μm (Waters, Milford, MA), or an analytical reversed-phase C18 SymmetryShield™ column, 4.6×250 mm, 5 μm (Waters, Milford, MA)applying linear gradient of solvent B from 0 to 100% over 100 min (flow rate: 1 ml/min).
- 52.Micewicz ED, Luong HT, Jung CL, Waring AJ, McBride WH, Ruchala P. Bioorg Med Chem Lett. 2014;24:1452. doi: 10.1016/j.bmcl.2014.02.024. [DOI] [PubMed] [Google Scholar]
- 53.Laliturai J. University of Manchester. 2012. p. 89. [Google Scholar]
- 54.Virta P, Leppanen M, Lonnberg H. J Org Chem. 2004;69:2008. doi: 10.1021/jo035801+. [DOI] [PubMed] [Google Scholar]
- 55.Matyjaszewski K, Tsarevsky NV. Nat Chem. 2009;1:276. doi: 10.1038/nchem.257. [DOI] [PubMed] [Google Scholar]
- 56.Tsarevsky NV, Matyjaszewski K. Chem Rev. 2007;107:2270. doi: 10.1021/cr050947p. [DOI] [PubMed] [Google Scholar]
- 57.In vitro ferroportin (Fpn) degradation assaywas carried out as previously descried58. Briefly, HEK293:TREX-Fpn-GFP, a cell line stably transfected with the human ferroportin-GFP construct under the control of doxycycline-inducible promoter, was plated on poly-D-lysine-coated plates in the presence of 20 μM FAC. Fpn expression was induced with 500 ng/mL doxycycline treatment for 24 hours. Then, doxycycline was washed off, and cells were treated with peptides for 24 hours. Cells were then trypsinized and resuspended at 1 × 106 cells/mL, and the intensity of green fluorescence was analyzed by flow cytometry using FACScan (fluorescence activated cell scanner) Analytic Flow Cytometer (Becton Dickinson, San Jose, CA) with CellQuest version 3.3 software (Becton Dickinson). Cells not induced with doxycycline to express Fpn-GFP were used to establish a gate to exclude background fluorescence. Cells induced with doxycycline, but not treated with any peptides, were used as the positive control. Each peptide treatment was repeated independently 3 to 6 times. The results were expressed as a fraction of the activity of hep25, according to the formula 1 − [(Fx − Fhep25) /(Funtreated − Fhep25)], where F was the mean of the gated green fluorescence and x was the peptide.
- 58.Nemeth E, Preza GC, Jung CL, Kaplan J, Waring AJ, Ganz T. Blood. 2006;107:328. doi: 10.1182/blood-2005-05-2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ganz T, Nemeth E, Preza G, Ruchala PP. 8 435 941 B2. Patent US. 2013:1–28.
- 60.Clark RJ, Tan CC, Preza GC, Nemeth E, Ganz T, Craik DJ. Chem Biol. 2011;18:336. doi: 10.1016/j.chembiol.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen S, Morales-Sanfrutos J, Angelini A, Cutting B, Heinis C. Chembiochem. 2012;13:1032. doi: 10.1002/cbic.201200049. [DOI] [PubMed] [Google Scholar]
- 62.Animal studieswere carried out as previously described9,10,58. Animal studies were approved by the Animal Research Committee at UCLA. Briefly, C57BL/6 mice were obtained from The Jackson Laboratory (Bar Harbor, ME) and were maintained on NIH 31 rodent diet (iron content 336 mg/kg; Harlan Teklad, Indianapolis, IA). Mice were injected subcutaneously either with 100 μL PBS (control) or with 50 or 100 or 200 nmoles peptide in 100 μL PBS. Mice were killed 24 hours later, blood was collected by cardiac puncture, and serum was separated using Microtainer tubes (Becton Dickinson, Franklin Lakes, NJ). Serum iron was determined using a colorimetric assay (Diagnostic Chemicals, Oxford, CT), which was modified for the microplate format so that 50 μL serum was used per measurement. The results were expressed as the percentage of decrease in serum iron when compared with the average value of serum iron levels in PBS-injected mice.
- 63.Eldridge JA, Milewski M, Stinchcomb AL, Crooks PA. Bioorg Med Chem Lett. 2014;24:5212. doi: 10.1016/j.bmcl.2014.09.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kang S, Watanabe M, Jacobs JC, Yamaguchi M, Dahesh S, Nizet V, Leyh TS, Silverman RB. Eur J Med Chem. 2014;90C:448. doi: 10.1016/j.ejmech.2014.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fazio MA, Oliveira VX, Jr, Bulet P, Miranda MT, Daffre S, Miranda A. Biopolymers. 2006;84:205. doi: 10.1002/bip.20396. [DOI] [PubMed] [Google Scholar]
- 66.Plasma stability.Tested analog(s) (10 mM stock solutions in DMSO) were added to freshly prepared mouse plasma (1 μL per 400 μL of plasma, c=25 μM) and incubated at 37 °C. Subsequently small samples of plasma (10 μL) were collected at indicated time-points and immediately diluted with 200 μL of DMSO/ACN mixture (1:1) containing 0.1% of TFA. Then samples were centrifuged at 13000 rpm for 10 min and obtained supernatants analyzed using the Agilent 6460 Triple Quadrupole LC/MS System (Agilent Technologies, Santa Clara, CA), with NM4A-Me peptide as internal standard. Sequence of NM4A-Me: Me-Ida(NHPal)-Phe-Leu-Phe-Arg-Pro-Arg-Asn-amide where Me-Ida(NHPal) is N-methyl-iminodiacetic acid n-palmitylamide.