

Summary
Host defense against viruses and intracellular parasites depends on effector CD8+ T cells whose optimal clonal expansion, differentiation, and memory properties require signals from CD4+ T cells. Here we addressed the role of dendritic cell (DC) subsets in initial activation of the two T cell types and their co-operation. Surprisingly, initial priming of CD4+ and CD8+ T cells was spatially segregated within the lymph node and occurred on different DC with temporally distinct patterns of antigen-presentation via MHCI vs. MHCII molecules. DC that co-present antigen via both MHC molecules were detected at a later stage; these XCR1+-DC are the critical platform involved in CD4+ T cell augmentation of CD8+ T cell responses. These findings delineate the complex choreography of cellular interactions underlying effective cell-mediated anti-viral responses, with implications for basic DC subset biology as well as for translational application to the development of vaccines that evoke optimal T cell immunity.
Introduction
The induction of an adaptive immune response requires the interaction of several lymphoid and myeloid cell types. For the generation of cytotoxic T lymphocytes (CTL), initial activation of naïve CD8+ T cells occurs via antigen-presenting cells (APC) that engage the antigen-specific T cell receptor (TCR) and other stimulatory surface receptors of these lymphocytes (Curtsinger and Mescher, 2010). The critical MHCI molecules involved in TCR recognition by CD8+ T cells can be loaded with antigenic determinants by a direct antigen-presentation pathway involving cytosolic proteins or by a cross-presentation pathway, which is fueled by extracellular proteins (Kurts et al., 2010). The latter is believed to play an essential role for pathogens that do not directly infect professional APC.
A second conventional T cell, the CD4+ helper T cell, is activated via antigen-presenting MHCII molecules. In distinction to the ligands involved in activation of CD8+ T cells, antigenic peptides presented by MHCII molecules are typically derived from extracellular proteins or intracellular proteins that are recycled from the cell surface (Germain, 1994). These CD4+ T cells provide crucial soluble and membrane-associated signals to antigen-specific B lymphocytes, leading to effective adaptive humoral immunity (Crotty, 2014). As with B cells and humoral responses, CD4+ T cells also provide molecular “help” to CTL, optimizing cellular immune responses by enhancing CD8+ T cell clonal expansion, differentiation and survival (Castellino and Germain, 2006).
Although the functional parallel is clear, a conceptual problem in comparing CD4+ T cell help for humoral vs. cellular responses in mouse models is that the interaction between CD4+ and CD8+ T cells cannot be direct, based on TCR engagement, as mouse CD8+ T cells do not express the necessary MHCII molecules to provide ligands for the CD4+ T cell TCR. This paradox was resolved by experiments showing that dendritic cells (DC) serve as a platform to mediate communication between CD4+ and CD8+ T cells (Mitchison and O'Malley, 1987; Ridge et al., 1998). Both T cell subsets must interact with the same DC in an antigen- and TCR-dependent manner, meaning that the “platform” DC must present antigen to CD4+ and CD8+ T cells via both the MHCI and MHCII pathways, respectively (Bennett et al., 1997; Cassell and Forman, 1987).
Given that naïve lymphocytes specific for a specific foreign antigen are rare, it has been argued that the likelihood of a (simultaneous, random) three-cell encounter is too low to be effective at driving the responses in question (Bevan, 2004). This argument has been weakened by experiments showing that i) a DC that had interacted with a CD4+ T cell could help a CD8+ T cell even after the CD4+ T cell was removed, removing the need for contemporaneous three cell clustering (Ridge et al., 1998) and ii) DC-CD4+ T cell interactions lead to the production of the chemokines CCL3/4 that attract CD8+ T cells via CCR5 to the licensed DC optimizing rare cell contacts (Castellino et al., 2006).
The same intravital imaging methods that revealed such chemokine-mediated guidance also showed that upon encounter with antigen-laden DC, T cells arrest and initiate long-term interactions lasting for several hours (16-20h) (Bousso and Robey, 2003; Miller et al., 2002; Stoll et al., 2002). This means that both CD4+ and CD8+ T cells would be substantially delayed in finding a common DC even with chemokine guidance. Furthermore, the past decade has seen an increasingly detailed parsing of dendritic cells into distinct subsets with specific localizations within secondary lymphoid tissues (Gerner et al., 2012; Kissenpfennig et al., 2005) as well as the emergence of strong evidence for preferential presentation of antigen via MHCI and MHCII by different DC types (den Haan et al., 2000; Dudziak et al., 2007; Schnorrer et al., 2006). Together, the dynamic considerations and the complexity of DC biology raise the crucial issue of when, where and on which DC do CD4+ and CD8+ T cells become activated and communicate?
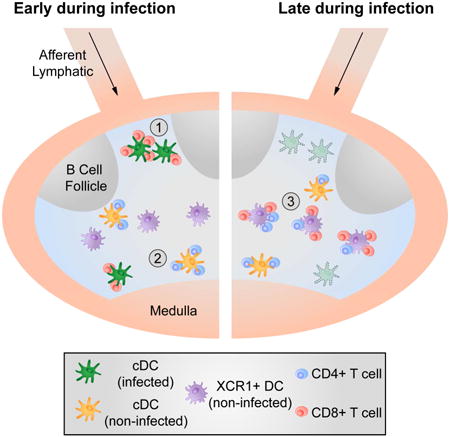
In light of these unresolved questions, the present study aimed to elucidate the spatial and temporal events that occur during CD4+ T cell augmentation of CD8+ T cells responses (‘help’) and to reveal the location and identity of the DC subset(s) that serve(s) as the communication platform for CD4+ and CD8+ T cells. Surprisingly, we found that early post-infection, antigen-specific activation of CD4+ and CD8+ T cells is spatially separated and mediated via non-infected and infected DC, respectively. XCR1+ (CD8α+) DC, which have been described to play a central role in CD8+ T cell priming, appeared to be dispensable for this initial activation of CD8+ T cells. After the triggering of both T cell subsets, CD4+ and CD8+ T cells translocated to a specific area in the paracortex, where they interacted with a third, non-infected XCR1+ DC population, defining the platform for delivery of help. These findings delineate the complex choreography of cellular interactions underlying effective cell-mediated anti-viral responses with parallels to the spatiotemporal events involved in delivery of CD4+ T cell help during humoral immune responses.
Results
Direct priming of CD8+ T cells does not require XCR1+ DC
To understand how CD8+ T cells and CD4+ T cells interact with DC during the induction of robust cell-mediated immune responses, we used a model system of vaccinia virus (VV) infection that supports both direct and cross-presentation pathways and elicits a CD4+ help-dependent CD8+ T cell response (Norbury et al., 2001; Wiesel et al., 2010). To allow for a time-resolved analysis of the cellular events, we initially carried out our experiments with the replication-deficient variant MVA (modified vaccinia virus Ankara). This attenuated virus produces a single round of infection with full expression of early and late viral antigens (Drexler et al., 2004). Four hours after i.v. infection of mice with MVA-GFP we could detect infected DC based on GFP expression. Phenotypic analysis of infected DC revealed a comparable infection rate among CD8α+ and CD11b+ DC in the spleen (Figure 1A). After infection with a recombinant virus that also expresses the ovalbumin-derived SIINFEKL determinant (MVA-NP-S-GFP), the infected DC also presented virally-expressed antigens via MHCI as quantified by antibody staining with clone 25.D1, which recognizes SIINFEKL bound to the mouse MHCI molecule H-2Kb. To test whether such directly infected DC could drive CD8+ T cell proliferation, we infected Kbm1 animals (Kbm1 = mutant Kb unable to bind SIINFEKL) with MVA-OVA and MVA-OVA-Kb, respectively. In this experimental set-up only MVA-OVA-Kb infected DC were able to present antigen and promote proliferation of OT-I cells (CD8+ ovalbumin-specific, TCR transgenic T cells) while non-infected or MVA-OVA infected DC were unable to stimulate OT-I cell proliferation in culture (Figure 1B). This demonstrates that directly infected DC present viral antigens and induce proliferation of antigen-specific CD8+ T cells ex vivo. In vivo we could detect direct interactions between MVA-OVA-GFP infected DC and transferred OT-I lymphocytes shortly after infection using intravital 2-photon microscopy (IVM) and analysis of stained LN sections (Movie S1 and Figure 1C). Arrested T cells were typically seen at the subcapsular sinus (SCS) and formed clusters in the interfollicular area and the cortical ridge as previously reported (Hickman et al., 2008; Kastenmuller et al., 2013). Because both DC and macrophages populate the area in which we see clusters of OT-I T cells early after infection, we depleted macrophages using clodronate liposomes or used additional DC-specific reporter animals (Figures S1A-C) and analyzed clusters shortly after MVA-OVA-GFP infection. These data also indicated that DC are the predominant cellular targets of early antigen recognition by the OT-I T cells.
Figure 1. Direct priming of CD8+ T cells does not require XCR1+ DC.
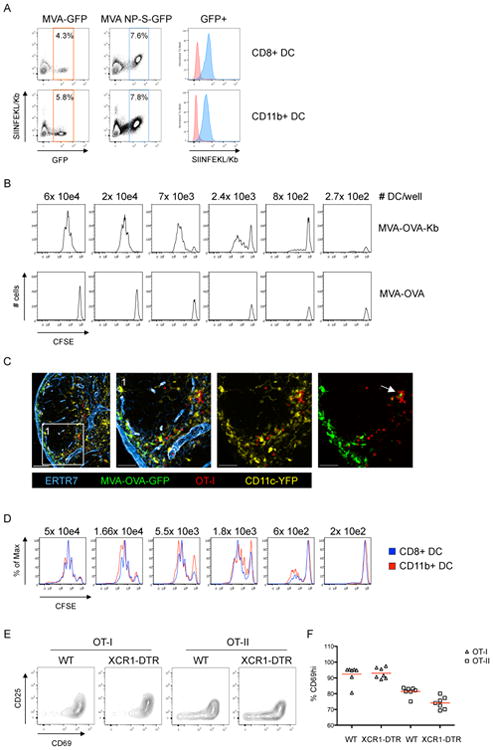
(A) Analysis of splenic DC after i.v. infection with MVA-GFP or MVA-NP-SIINFEKL-GFP (8h p.i.). (B) Analysis of OT-I proliferation after ex vivo coincubation with isolated splenic DC from Kbm1 mice infected with MVA-OVA or MVA-OVA-Kb (8h pi). (C) Immunofluorescent (IF) images of a dLN showing clustering/interaction between transferred OT-I cells and infected (GFP-expressing) DC (MVA-OVA-GFP; f.p.; 8h p.i.). (D) Analysis of OT-I proliferation after coincubation with DC subsets sorted ex vivo (MVA-OVA; i.v.; 8h p.i.). (E/F) Activation marker (CD25/CD69) upregulation on transferred OT-I and OT-II cells in the popliteal LN 12h after f.p. infection (MVA-OVA). Representative plots (E) and analysis (F) are shown comparing DTX-treated WT and XCR1-DTR animals. Data are representative of three (n=3) (A-D) and two (n=4) (E/F) independent experiments. ** = p≤0.01, ns = non-significant, scale bars 100/50 μm. See also Figure S1 and Movie S1.
To elucidate whether different infected DC subsets have a differential capacity to stimulate OT-I cells, we infected mice i.v., sorted splenic CD11b+ or CD8α+ DC 8h later and co-cultured the sorted cells with CFSE-labeled OT-I cells. At 72h post co-culture we consistently observed similar proliferation of OT-I cells after co-incubation with either DC subset (Figure 1D, S1D). CD8α+ DC substantially overlap with the XCR1+ DC subpopulation (Becker et al., 2014). Therefore, to further test whether CD8α+ DC are required for activation of OT-I cells in vivo, we transferred OT-I cells into WT or XCR1-DTR animals (Yamazaki et al., 2013), treated them with Diphtheria toxin (DTX) to deplete the XCR1+ DC, infected the animals with MVA-OVA in the footpad (f.p.), and then analyzed the expression of the early activation markers CD69 and CD25 on OT-I cells in the draining lymph node (dLN) 12h later. We found that the early activation of OT-I cells was unaltered in the absence of XCR1+ DC (Figure 1E/F). In contrast we found a small but consistent reduction in the early activation of OT-II cells (CD4+) also specific for OVA but presented by MHCII molecules (Figure 1E/F). In summary we conclude that MVA infects various DC subsets in vivo that express and present antigens to CD8+ T cells leading to activation and T cell proliferation. For this initial activation XCR1+(CD8α+) expressing DC appear to be dispensable.
Early activation of CD4+ and CD8+ T cells occurs on spatially distinct DC
Having established that antigen-specific CD8+ T cells are initially activated by infected DC, we wished to clarify whether CD4+ T cells can provide cognate help via such infected, CD8+ T cell engaged DC. To this end, we transferred OT-I and OT-II cells into mice, infected the animals with MVA-OVA and observed the migratory behavior of the transferred T cells in situ using IVM. As expected, we could readily detect arrested OT-I cells clustering around infected DC. Surprisingly, however OT-II cells did not co-arrest with their CD8+ T cell counterparts (Movie S2 and Figure 2A). Instead, they migrated similarly to polyclonal CD4+ control T cells at around 10 μm/min (Figure 2B). Later after infection (8-12h) we were unable to detect OT-I/OT-II co-clusters using IVM (data not shown) although kinetic experiments with isolated cells recovered from these animals revealed that the majority of the OT-II cells were activated (CD69hi) (Figure 2C) and therefore were likely to have engaged on antigen-rich APC by this time point. These findings suggested that OT-II cells might be activated in deeper areas of the LN that are not typically visualized using IVM. Therefore we analyzed frozen LN sections to identify the location OT-I and OT-II cell co-clusters. In line with our IVM data, we found that OT-II cells did not accumulate and cluster in the SCS area in contrast to OT-I cells (Figure 2D), which were found in proximity to MVA-infected (GFP-expressing) cells. OT-II cells did not cocluster substantially with OT-I, showing only random colocalization with OT-I at frequencies similar to OVA antigen-unspecific polyclonal CD4+ T cells (Figure S2A/B). To further assess this spatial separation of CD4+ and CD8+ T cells during initial antigen-dependent priming after viral infection, we analyzed spleen sections at similar time points. Again we found a segregation of antigen-specific CD4+ and CD8+ T cells. OT-I cells were localized at the marginal zone in proximity to infected APC (Figure S2C/D). In contrast, OT-II cells remained in the white pulp, where they clustered and were activated by non-infected (GFP-negative) APC (Figure S2C/D).
Figure 2. Priming of CD4+ and CD8+ T cells occurs on spatially distinct DC.
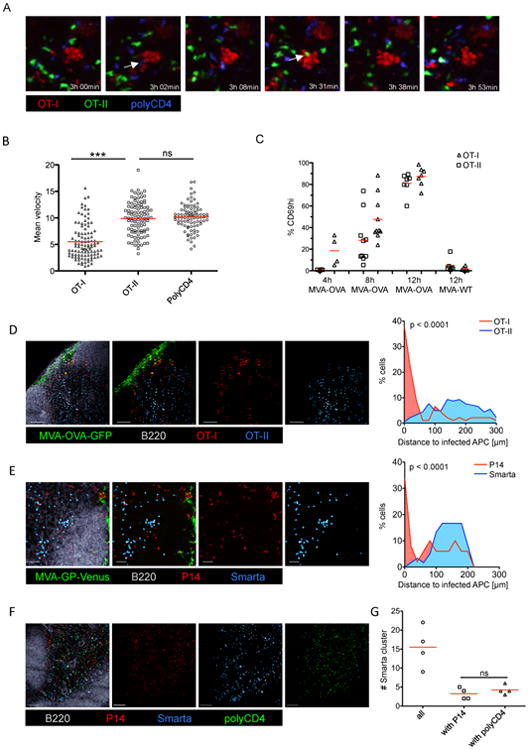
(A) Images from IVM of the popliteal LN 3-4h after MVA-OVA f.p. infection. OT-I, OT-II and control cells (polyclonal CD4+ T cells) were transferred 24h prior to infection. White arrows indicate brief interaction between clustered OT-I and OT-II or control cells (see also Movie S2). (B) Analysis of the mean velocity of transferred T cells using the data shown in Movie S2, (red bars indicate mean values). (C) Analysis of CD69 upregulation on transferred OT-I and OT-II cells in the dLN at different time-points after f.p. infection (MVA-OVA/MVA WT). (D/E/F) IF images of a dLN showing the localization of (D) transferred OT-I and OT-II cells (MVA-OVA-GFP), (E) transferred P14 and Smarta cells (MVA-GP-Venus), (F) transferred P14, Smarta and polyclonal CD4+ T cells (MVA-GP) 10h p.i. (G) Quantification of cluster abundance from four experiments as in F.
Data are representative of at least two independent experiments (A/B; n=10), (C/G; n=4-8; pooled data) (D-F; n=10). *** = p≤0.001, ns = non-significant, scale bars (D/E/F) 100 μm, See also Figure S2 and Movie S2.
To determine if the segregation of OT-I and OT-II cells during activation is a phenomenon related to their particular T cell receptors (TCR), we generated MVA-GP-Venus and analyzed a different TCR transgenic T cell pair specific for the Lymphocytic choriomeningitis virus (LCMV) glycoprotein (GP) (Smarta/CD4+, P14/CD8+). Similar to our previous results we found accumulation of antigen-specific CD8+ T cells (P14) around MVA-GP-Venus-infected DC while antigen-specific CD4+ T cells (Smarta) accumulated in the paracortex (Figure 2E and S2E). Smarta cells formed homogenous clusters in the paracortex of dLN that were only randomly intermixed with P14 cells, similar to non-specific control cells (Figure 2F/G). To further evaluate whether the observed separated activation of antigen-specific CD4+ vs. CD8+ T cells is a more general feature of initial activation we examined two additional experimental systems. As a first approach we used recombinant Adenovirus infections and found that OT-I cells translocated to the SCS and IFA to interact with directly infected APC, while OT-II cells remained in the paracortex where they were activated in an antigen-specific manner (Figure S2F-I). Second, we immunized mice with soluble OVA protein and LPS as adjuvant. Again we found that activation of OT-I and OT-II cells is predominantly separated (Figure S2J-M, Movie S3). In summary, we conclude that the initial activation of antigen-specific CD4+ and CD8+ T cells is segregated and involves distinct DC in different locations within the lymph node or spleen.
Identification of DC that present viral antigen via both MHCI and MHCII later after infection
These findings were surprising in light of previous reports demonstrating that CD4+ help for CD8+ T cells occurs on a single DC co-presenting MHCI and MHCII antigens (Bennett et al., 1997; Cassell and Forman, 1987) and our prior studies showing how chemokines guide T cells to DC co-presenting MHCI and MHCII ligands (Castellino et al., 2006). One way to reconcile the present observations with these prior findings is to postulate that the licensing and/or the delivery of help occurs later during the course of infection. To examine this possibility, we transferred OT-I, OT-II, and polyclonal CD4+ T cells into mice that had been infected for 30h and analyzed the location of these transferred cells in LN sections 8h after transfer (Figure 3A). With this experimental set-up, we could readily detect OT-I and OT-II cell co-clusters, while polyclonal CD4+ T cells showed an unbiased distribution (Figure 3B). When systematically comparing cellular positioning in the LN early (10h) vs. late (38h) after infection we found marked differences for OT-I cells and modest differences for OT-II cells (Figure 3C). This reflects the predominant activation of OT-I at the SCS/IFA early after infection (10h) vs. the presence of antigen-bearing DC in the paracortex at later time-points (38h). These paracortical DC were able to present antigen to and activate both OT-I and OT-II T cells, as indicated by the OT-I and OT-II T cells expressing activation markers in co-clusters surrounding such DC (Figure 3D). Thus, later during infection a common DC, positioned in the peripheral paracortex, presents antigen able to productively engage the TCR of both CD4+ and CD8+ T cells. Use of two experimental approaches to block DC migration (site removal, lymph vessel obliteration) revealed that migratory DC were not required for the formation of OT-I/OT-II cell co-clusters during the late phase of infection (Figure S3A/B). Additionally, Batf3 KO animals that lack migratory CD103+ DC but only a fraction of LN resident CD8α+ (XCR1+) DC (Edelson et al., 2010) showed mixed OT-I/OT-II cell co-clusters, confirming that CD103+ migratory DC were dispensable for the formation of such clusters (Figure S3B). It is important to note that the presence of clusters consisting of three different cell types (OT-I/OT-II/DC) does not necessarily mean that those ternary interactions occur or are necessary for delivery of help under physiological conditions as in these experiments an artificially high number of precursor T cells was used to facilitate detection of the co-presenting DC.
Figure 3. CD4+ and CD8+ T cells co-cluster later during infection.
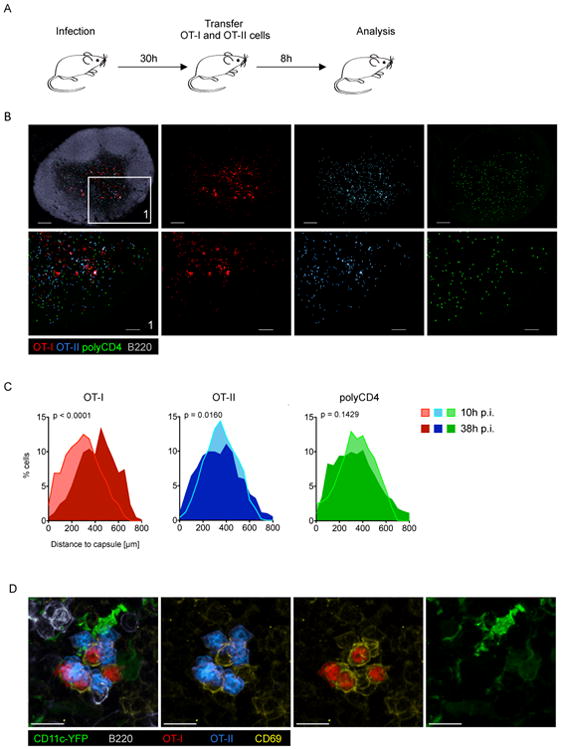
(A) Schematic of the experimental set-up to reveal antigen-bearing cells later in the course of infection (MVA-OVA). (B) IF images of the dLN showing the localisation of OT-I, OT-II and control cells (polyclonal CD4 T cells). (C) Histograms showing cellular localisation 10h (see Figure 2) or 38h p.i. (see experimental set-up shown in 3A). (D) IF image of the dLN showing a mixed OT-I/OT-II cell cluster and activation status (CD69). Data are representative of 10 (B/D; n=20) or three (C; n=3) independent experiments. Scale bars (B) 200 μm/100 μm, (D) 10 μm. See also Figure S3.
Non-infected cross-presenting XCR1+ DC are the information-transmission platform for CD4+ and CD8+ T cells
Given that XCR1+ DC were dispensable for early CD4+ and CD8+ T cell activation (Figure 1) but play a central role in immunogenic CTL priming (Shortman and Heath, 2010), we hypothesized that these DC might be involved in the CD4+ and CD8+ T cell co-clustering we observed above. To investigate this hypothesis, we first attempted to identify which DC subset presents antigen to OT-I cells at a late stage of infection. CD11b+ and CD8α+ DC were sorted 30h after infection and co-cultured with CFSE-labeled OT-I cells. In contrast to the results obtained early (8h) after infection (Figure 1D), at this later time point exclusively CD8α+ (XCR1+) DC induced OT-I cell proliferation (Figure 4A, S4A/B). To test whether this is due to the known propensity of this DC subset to cross-present antigen, we infected C57BL/6 and Kbm1 mice with MVA-OVA-Kb, sorted the CD8α+ DC subset 30h later and co-incubated these DC with CFSE-labeled OT-I cells (Figure 4B). If direct antigen-presentation was still occurring, directly infected Kbm1 DC should still be able to drive OT-I cell proliferation due to virally driven Kb expression (Figure 1B). However, this was not the case, supporting the notion that later during infection, at least when using replication incompetent viruses, cross-presentation becomes the dominant pathway for MHCI loading with viral antigens.
Figure 4. Non-infected cross-presenting XCR1+ DC are the information-transmission platform for CD4+ and CD8+ T cells.
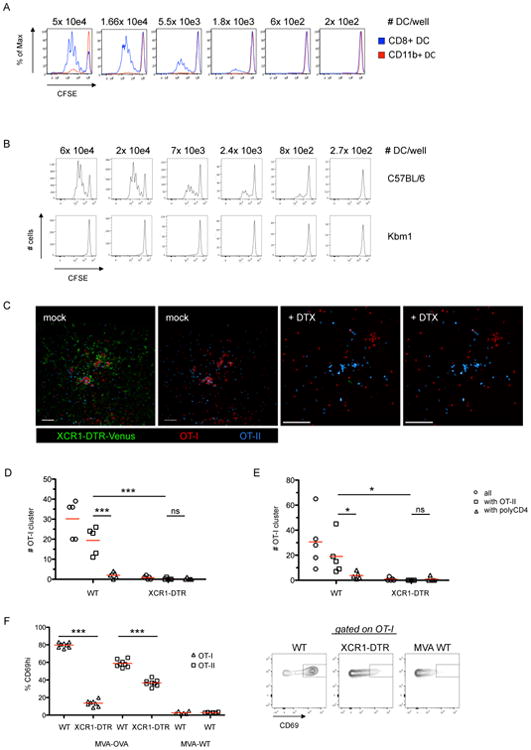
(A) Proliferation of CFSE labeled OT-I cells after ex vivo coincubation with sorted splenic DC subsets (MVA-OVA; i.v.; 30h p.i.). (B) Proliferation of CFSE labeled OT-I cells after ex vivo coincubation with isolated splenic DC from WT or Kbm1 mice 30h p.i. (MVA-OVA-Kb; i.v.). (C) Images of dLN using the experimental set-up as in Figure 3A. XCR1-DTR-Venus mice were treated with PBS or DTX. (D/E) T cell cluster abundance in the presence or absence of XCR1 DC using a (D) semi-automated or (E) fully automated analysis. (F) Analysis and representative plots of CD69 expression on OT-I/OT-II cells that were transferred 28h post infection (MVA-OVA/MVA WT; f.p.) and analyzed 12h later in the dLN. Data are representative of three independent experiments (n=8). *** = p≤0.001, scale bars 50 μm. See also Figure S4.
We then examined whether XCR1+ DC also served as the platform that communicates with both antigen-specific CD4+ and CD8+ T cells in vivo. We infected XCR1-DTR-Venus mice, treated them with DTX or PBS and transferred OT-I and OT-II cells into these animals 30h post infection and DTX treatment. XCR1+ DC were detected in the middle of mixed OT-I/OT-II cell co-clusters in PBS-treated animals 8h after T cell transfer (Figure 4C). The XCR1+ DC were not broadly distributed throughout the LN as in the steady state (Figure S4C), but rather formed aggregates that were intermixed with the co-clustered T cells (Figure 4C/S4D). DTX depletion of XCR1+ DC led to a loss of mixed OT-I/OT-II cell co-clusters, while leaving distinct OT-II and separate, rare OT-I cell clusters (Figure 4C-E). We next quantified the requirement for XCR1+ DC in the stimulation of OT-I and OT-II cells at this late phase post-infection. Twelve hours after T cell transfer we harvested the dLN and analyzed OT-I and OT-II cells for CD69 expression using flow cytometry. In the absence of XCR1+ DC, the fraction of activated OT-I cells dropped from 80% in WT to 15% in XCR1-DTR mice (Figure 4F). The activation of OT-II cells was modestly reduced from 60% to 40% if XCR1+ DC were absent. Together, these data indicate that cross-presenting XCR1+ DC serve as a platform for interaction (simultaneously or sequentially) with both antigen-specific CD4+ and CD8+ T cells at late times after infection.
XCR1+ DC are also critical for communication between CD4+ and CD8+ T cells during productive VV infection
We next examined whether these findings applied to events following infection with replication competent vaccinia virus (VV) that does not require antigen cross-presentation for T cell priming due to ongoing infection of DC (Xu et al., 2010). First, we addressed whether initial CD4+ and CD8+ T cells priming is also spatially separated during VV infection. To this end, we transferred OT-I and OT-II cells into WT animals and infected them with VV-OVA. IVM 10h p.i. confirmed the near absolute separation of arrested OT-I and OT-II cells, with the former forming clusters near the LN capsule, the latter in deeper areas of the LN (Figure 5A and Movie S4). Similar to MVA-OVA infection, VV-OVA infection induced the accumulation of OT-I cells at the SCS where they interacted with infected DC (Figure 5B and S5A/B) (Hickman et al., 2011). In contrast, OT-II cells remained in the paracortical areas of the LN and did not co-cluster with OT-I (Figure 5B and S5C/D).
Figure 5. XCR1+ DC are the information transfer platform for CD4+ and CD8+ T cells during VV infection.
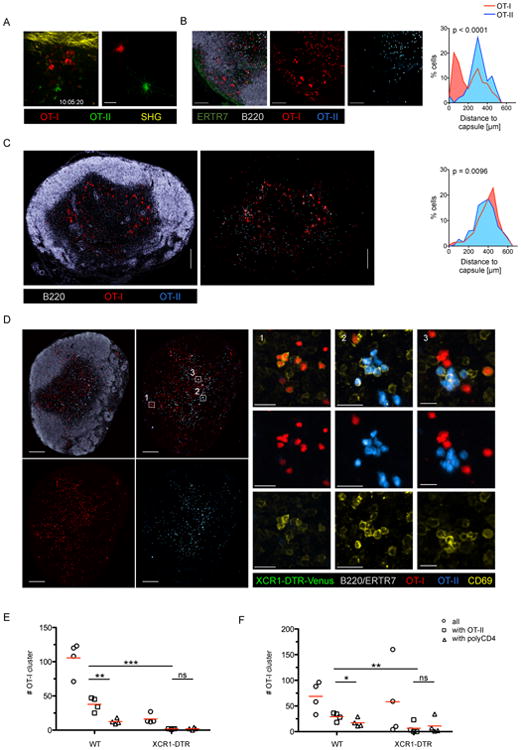
(A) IF image and translated tracks from IVM of the popliteal LN 10-11h after VV-OVA infection (f.p.). See also Movie S3. (B) IF images of a dLN showing the localization and cluster formation of transferred OT-I and OT-II cells (VV-OVA; f.p.; 10h p.i.). (C/D) IF images of the dLN of a XCR-DTR-Venus mice treated with PBS (C) or DTX (D) showing the localization of labeled OT-I and OT-II cells following the experimental set-up shown in Figure 3A using VV-OVA. (E/F) T cell cluster abundance in the presence or absence of XCR1 DC using a (D) semi-automated or (E) fully automated analysis. Data are representative of three independent experiments (n=4). Scale bars (A) 50 μm, (B) 100μm, (C) 200 μm, (D) 200 μm/20 μm. See also Figure S5 and Movie S4.
Next, we addressed whether late co-clustering of CD8+ and CD4+ T cells occurs after VV infection. Naïve OT-I and OT-II cells were transferred into WT mice 30h post infection and the dLN were examined by fluorescent microscopy 8h later. Central sagittal sections showed that mixed T cell clusters consisted of activated (CD69+) cells in the peripheral paracortex rather than in the deep paracortical central region (Figure 5C and S5E). Using VV-infected XCR1-DTR-Venus mice as recipients we found that mixed OT-I/OT-II cell co-clusters were organized around XCR1+ DC at this later time-point (Figure S5F). Depletion of XCR1+ DC led to a loss of mixed OT-I/OT-II cell clusters, confirming that XCR1+ DC are the predominant population involved in co-presentation of MHCI and MHCII determinants at this later stage, making them likely platforms for the delivery of help (Figure 5D-F). In the absence of XCR1+ DC OT-II cell clusters were still present in the paracortex, typically in proximity to the medullary area. OT-I cell clusters were also present albeit in lower frequency, and importantly, were separated from OT-II cells (Figure 5E/F). Persistent OT-I clusters in the absence of XCR1+ DC reflect ongoing VV replication and continued infection of LN resident DC. Such clusters were largely absent after infection with the replication-deficient in MVA (Figure 3C/D), most likely due to the absence of infected DC at this time point.
Localization of endogenous activated CD8+ T cells during VV infection
We next turned to an assessment of whether endogenous T cells are activated at similar anatomical sites as transferred TCR transgenic cells and whether both CD4+ and CD8+ T cells similarly seek out XCR1+ DC later during infection. Labeled OT-I cells were transferred to WT recipients and the mice infected with VV-OVA. 8h later we examined whether activated (CD69hi) endogenous T cells were part of activated OT-I cell clusters. CD69hi non-transgenic cells were found in close proximity to activated OT-I cells, arguing for an activation of endogenous cells by the same DC (Figure 6A). To analyze the location of activated T cells later during infection when CD69 expression is downregulated, we employed IFNγ (YFP) reporter animals. As expected, YFP positive cells were not seen on fixed LN sections from naïve mice (Figure S6A). In contrast, 38h p.i. we found YFP positive cells in the dLN that consisted of CD4+, CD8+ and double negative (CD3+) T cells as well as NK cells (Figure 6B/C). CD8+ T cells showed an increased cellular volume (Figure 6D) and the highest YFP expression (Figure 6E) as measured by forward scatter signal and the mean fluorescent intensity (MFI).
Figure 6. Localization of endogenous activated CD8+ T cells during VV infection.
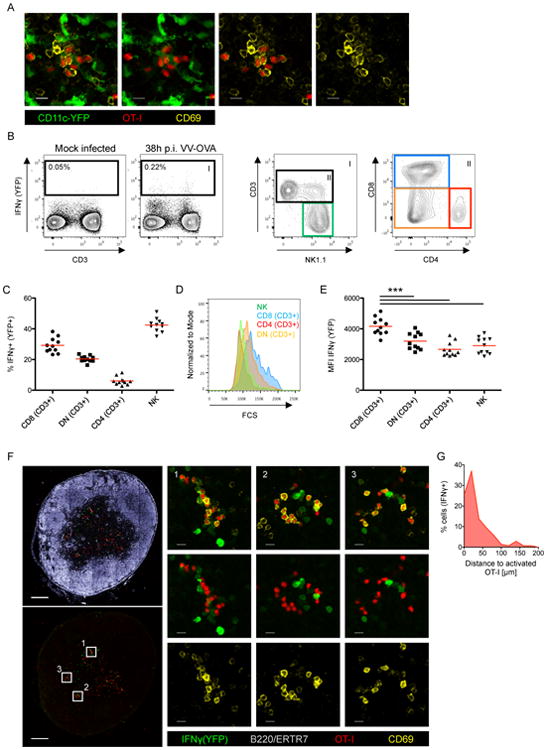
(A) IF Images of a LN showing co-localization of transferred OT-I cells and endogenous (non-OT-I) CD69hi cells (VV-OVA; f.p.; 10h p.i.). (B/C/D/E) Analysis of IFNγ(YFP+) reporter animals (VV-OVA; 36h p.i.). (B) Graphs show the gating strategy, (C) the cellular distribution, (D) the size distribution and (E) the mean fluorescence intensity (MFI) of the YFP signal of IFNγ+(YFP+) cells (red bars indicates mean values). (F) IF images showing the localization of YFP expressing cells 36h after infection of IFNγ (YFP+) reporter animals (VV-OVA; f.p.). OT-I cells were transferred 8h before analysis. (G) Histogram shows the distance between IFNγ+(YFP+) cells and activated (CD69hi) OT-I cells. Data are representative of two independent experiments (n=8) (C/E/G) shows pooled data. *** = p≤0.001, scale bar (A) 10 μm, (F) 200 μm/10 μm. See also Figure S6.
Using immunofluorescent analysis of LN sections from IFNγ reporter animals 40h after infection (VV-OVA), we detected YFP+ cells at the SCS and in the paracortex. To examine whether the YFP+ cells represent recently activated T cells, we blocked the entry of newly arriving naïve T cells to the dLN using CD62L antibodies 14h post infection. Under this condition, we found bright YFP+ CD8+ cells in the paracortex (Figure S6B). Dim YFP+ cells positioned in the paracortex were CD4+, CD8+ or double negative (Figure S6C). In contrast, YFP+ NK cells were also dim but positioned at the SCS area rather than the paracortex (Figure S6B/D). Even at a 100-fold higher dose of VV-OVA (108) we did not observe cytokine-mediated activation of endogenous CD44+ CD8+ T cells, arguing that the identified enlarged, paracortical YFP+ CD8+ T cells reflect previously activated antigen-specific T cells (Figure S6E). To determine whether the location of such endogenous activated CD8+ T cells corresponds to areas of the dLN in which we expect CD4+ T cell help to be delivered, we transferred OT-I cells as described in Figure 3A. Endogenous YFP+ bright cells in the paracortex were CD69 negative or low and typically adjacent to clustered, transferred OT-I cells (Figure 6F). Examination of the distance between bright YFP+ cells in the paracortex and the transferred activated OT-I cells revealed that the majority of those cells were closer than 20μm (Figure 6G). These several observations are consistent with the view that the sequential model in which initial T cell priming occurs on distinct DC subsets in different LN regions and subsequently, information exchange occurs on XCR1+ DC as platform that co-presents antigen via both MHCI and MHCII molecules, applies not just to TCR transgenic models, but to polyclonal anti-viral responses as well.
VV-specific T cells activated in the absence of XCR1+ DC are ‘helpless’
If the XCR1+ DC platform is dispensable for early CTL activation but important for later differentiation and survival of the activated CTL, XCR1+ DC depletion should negatively impact proliferation, effector differentiation and memory CTL function, equivalent to the situation of CTL priming in the absence of CD4+ T cell help (Wiesel and Oxenius, 2012). To test this prediction, we first analyzed the CD8+ T cell immune response on d8 after VV infection in the presence or absence of CD4+ T cells. We found a significant reduction in the total number of antigen-specific CD8+ T cells in the spleen on d8 after infection (Figure 7A). A similar level of reduction in the immunodominant (B8R) CD8+ Tcell response was also seen after depletion of XCR1+ DC (Figure 7B). Combined CD4+ T cell and XCR1+ DC depletion showed no additional reduction as compared to CD4+ T cell depletion alone, arguing that help is delivered via XCR1+ DC (Figure 7C). To see if the observed reduction of the B8R-specific CD8+ T cell response upon XCR1+ DC depletion reflected the lack of help delivered via this DC population and not an unrelated function independent of antigen presentation to CD4+ T cells, we analyzed mixed BM (bone marrow) chimeric mice. MHCII KO × XCR1-DTR BM chimeric mice (50/50) were generated and infected with VV-OVA 8 weeks after reconstitution (Figure S7A/B). In these animals application of DTX results in a 50% depletion of XCR1+ DC with the remaining XCR1+ DC lacking expression of MHCII. Such MHCII-deficient DC cannot serve as a platform for delivery of help (Figure 7D). The depletion of XCR1+ MHCII+ DC led to a significant reduction in the anti-viral B8R-specific CD8+ T cell response on d8 post priming (Figure 7E).
Figure 7. VV-specific T cells activated in the absence of XCR1+ DC are ‘helpless’.
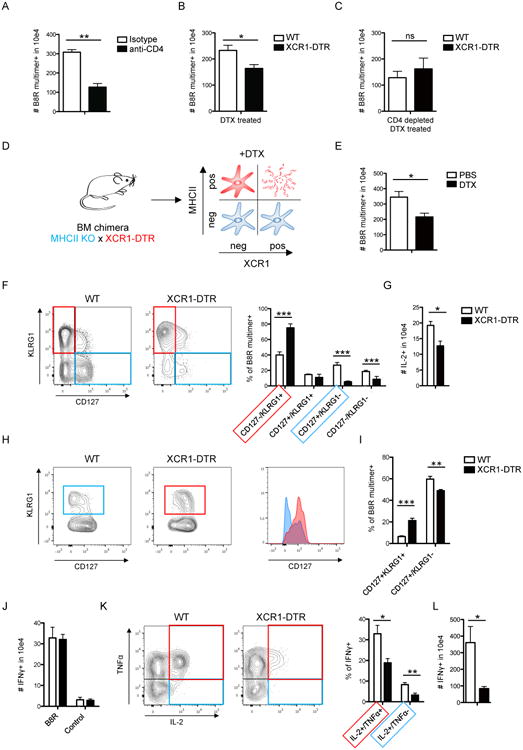
(A-C) Analysis of the total numbers of B8R multimer-specific splenic CD8+ T cells 8 days after VV-OVA infection (i.p.). Comparison of (A) isotype vs. CD4 depleted mice, (B) WT vs. XCR1-DTR mice treated with DTX and (C) WT vs. XCR1-DTR mice treated with DTX and anti-CD4 antibodies. (D) Schematic of DC composition in bone-marrow (bm) chimeric animals (MHCII KO × XCR1-DTR → WT). (E) Antiviral CD8+ immune response is shown comparing DTX vs. PBS treated bm chimeric animals on d8 (VV-OVA i.p.). (F/G) Analysis of B8R specific immune responses d8 p.i. (VV-OVA i.p.), comparing WT vs. XCR1-DTR animals treated with DTX, showing (F) the phenotype of B8R multimer-specific CD8+ T cells and (G) the amount of IL-2 producing CD8+ T cells after peptide (B8R) stimulation for 5h. (H-L) Analysis of the immune response 60 p.i. (VV-OVA i.p.), comparing WT vs. XCR1-DTR animals treated with DTX. (H) Phenotype and (I) relative distribution of B8R multimer-specific memory subsets. (J) Absolute numbers of IFNγ-producing CD8+ T cells, (K) relative distribution of polyfunctional CD8+ T cells (gated on IFNγ+) after peptide (B8R) stimulation. (L) Recall response d5 after Lm-B8R challenge. Graph shows total numbers of IFNγ-producing CD8+ T cells after peptide (B8R) stimulation. Data are representative of three or two (L) independent experiments (n=4). The graphs show mean +SEM. * = p≤0.05, ** = p≤0.01, *** = p≤0.001, ns = “non significant”. See also Figures S7.
We further examined whether the absence of XCR1+ DC impacts CD8+ T cell differentiation to an effector or memory state (Janssen et al., 2003; Shedlock and Shen, 2003; Sun and Bevan, 2003). We found a striking shift towards terminally differentiated effector cells (CD127-/KLRG1+) and a relative loss of memory precursors (CD127+/KLRG1-) if XCR1+ DC were depleted (Figure 7F), along with a significant reduction in the capacity of the activated CD8+ T cells to produce IL-2 (Figure 7G). A similar loss in IL-2 producing cells was also observed when starting the depletion after infection (Figure S7C-E). In contrast, the capacity of antigen-specific CD8+ T cells to produce IFNγ appeared to be unaltered when comparing these conditions (Figure S7F). Next we analyzed the memory response in mice that were previously infected with VV-OVA in the presence or absence of XCR1+ DC. Interestingly, we detected only a small but significant reduction in the total numbers of B8R multimer-specific CD8+ T cell in the memory phase if XCR1+ DC were absent during priming (Figure S7G). However, analysis of memory subsets on d60 post prime showed a significant increase in KLRG1+ B8R multimer-specific memory T cells if XCR1+ DC were depleted during priming (Figure 7H/I). This memory subset was characterized by prominent CD127 expression typically seen in classical central memory T cells (CD127hi/KLRG1-). The antigen-specific CD8+ memory T cells had a full capacity to produce IFNγ if XCR1+ DC were absent during priming (Figure 7J). Yet, these memory CD8+ T cells had a profound defect in IL-2 production (Figure 7K), which was characterized by a reduction of polyfunctional T cells (IFNγ+ TNFα+ IL-2+) (Figure 7K) and a reduced amount of IL-2 production on a single cell level as measured by the MFI (Figure S7H). Finally to test the capacity of the memory cells, generated in the absence of presence of XCR1+ DC to undergo optimal secondary expansion, we rechallenged such mice with L. monocytogenes expressing the B8R peptide (Lm-B8R). 5 days after challenge we found that mice that lacked XCR1+ DC during the priming with VV-OVA failed to mount a robust recall response against LM-B8R (Figure 7L), providing a physiological relevance of XCR1+ DC as a critical platform for delivery of cognate helper signals from CD4+ T cells.
Discussion
Here we report the spatio-temporal dynamics of CD4+ and CD8+ T cells early after viral infection and the role of distinct DC subpopulations in both activation of and communication between CD4+ and CD8+ T cells during this crucial phase of the adaptive immune response. Our data reveal a complex choreography during the initiation of cell-mediated immunity, the specific features of which help clarify what seem to be contradictory observations in the literature. We find that (i) initial activation of CD4+ and CD8+ T cells is spatially separated and involves distinct DC; (ii) CD8+ T cells are first activated by infected DC and CD4+ T cells by non-infected DC; (iii) later during infection a third DC population (XCR1+ DC) presents antigen to both CD4+ and CD8+ T cells; and (iv) these XCR1+ DC are a platform for communication between CD4+ and CD8+ T cells, shaping the differentiation of the latter and modulating memory programming even in situations in which cross-presentation per se is not required.
The finding that early CD4+ and CD8+ T cell activation post-infection is separated and orchestrated at distinct anatomical localizations was surprising. This feature may have been missed in previous studies because of an exclusive focus on the dynamic behavior of CD8+ T cells (Hickman et al., 2011; Hickman et al., 2008; Kastenmuller et al., 2013) or the use of peptide-pulsed DC when co-analysis of both CD4+ and CD8+ behavior was studied (Beuneu et al., 2006; Castellino et al., 2006). Several reports previously showed a propensity of distinct DC subsets to present via either MHCI or MHCII molecules when using protein antigens (den Haan et al., 2000; Dudziak et al., 2007; Schnorrer et al., 2006). Factors that regulate such differential antigen-presentation among DC subsets have been described (Dudziak et al., 2007; Vander Lugt et al., 2014) and numerous viral immune evasion proteins that interfere with antigen-presentation have been identified (Alcami and Koszinowski, 2000). However, the profound spatial segregation of CD4+ vs. CD8+ T cell activation early after infection requires additional investigation to more fully understand the basis for this phenomenon and its relevance to the acute and memory phases of immunity. Given the preferential localization of DC subsets with subregions of the LN (Gerner et al., 2012; Kissenpfennig et al., 2005), these new findings suggest a complex combination of intrinsic DC biology and pathogen-associated effects on antigen presentation and localization of DC subsets will greatly affect the nature of the ensuing cell-mediated response.
It is unknown how many naïve CD8+ T cells actually require signals derived from CD4+ T cells to mount a robust and functional memory CD8+ T cell response. In any given naïve mouse repertoire of a few hundred CD8+ T cells specific for a foreign antigen, it might just be (the proliferative progeny of) a few initially activated T cells that receive functional ‘help’ from CD4+ T cells. This quantitative issue places some limits on the interpretation of our results. Because we cannot directly visualize the specific subset of ‘helped’ CD8+ T cells as they receive the necessary molecular signals, we cannot formally exclude the possibility that the relevant memory CD8+ T cell pool is formed by the offspring of a few T cells that do not correspond to the bulk behavior of the cells we quantify. Specifically, it is possible that a minor, but biologically relevant population of naïve CD8+ T cells encounters a cross-presenting (helped) XCR1+ DC first rather than undergoing initial activation on a directly infected non-licensed DC as in our proposed model.
Nonetheless, several lines of reasoning support the notion that CD4+ T cell help is primarily delivered to activated rather than naïve CD8+ T cells. First, although CCR5 expression can occur in a TCR-independent manner, optimal upregulation of this chemokine receptor occurs upon antigen activation, giving the T cells the capacity to follow chemokine signals to the licensed DC (Castellino et al., 2006). Second, naïve T cells interact for many hours with DC that present high potency foreign antigens. During this period (defined as Phase II by (Mempel et al., 2004), the DC-engaged CD8+ T cells would not be able to search for the optimal (licensed) DC. After these long-lasting interactions, activated T cells enter a third phase that is characterized by short interactions with DC (Mempel et al., 2004). To date the biological relevance of this third phase has remained elusive. Our model assigns it a potential specific biological function, namely the search for licensed DC. Third, besides CD8+ T cells, CD4+ T cells also require pre-activation in order to express the molecules that are required to deliver help, in particular CD40 ligand (Bennett et al., 1998; Ridge et al., 1998; Schoenberger et al., 1998). Finally, our new model reveals a close similarity between the cellular events that occur during CD4+ help for B cells and for CD8+ T cells. B cells and CD8+ T cells are activated separately from CD4+ T cell helpers at different anatomical locations before they come together for signal exchange (McHeyzer-Williams et al., 2006).
Besides the implication that pre-activated rather than naïve lymphocytes deliver/receive help, LN-resident XCR1+ DC have been identified as the critical platform on which such signals are transmitted. Interestingly, migratory DC seemed dispensable for initial CD8+ T cell activation or for provision of help. Vaccinia virus particles directly disseminate to the dLN after local intradermal infection of the skin (Lin et al., 2013), in contrast to Herpes simplex virus that requires migratory DC to shuttle antigen to the dLN (Bedoui et al., 2009). With the latter virus, migratory DC are required to hand off antigen to LN-resident DC (Allan et al., 2006). This hand-off is in line with our work showing the crucial requirement of LN-resident XCR1+ DC rather than migratory DC to act as a critical platform to provide help to CD8+ T cells.
The XCR1+ DC subset has primarily received attention due to its capacity to cross-present antigens (Shortman and Heath, 2010). Our experiments have uncovered an important additional function by using a model that does not require cross-presentation for CD8+ T cell priming (Xu et al., 2010). In this situation the absence of XCR1+ DC had a small effect on the primary immune response, compared to the absence of CD4+ T cell help. However, we found a profound role of XCR1+ DC on the differentiation of CD8+ T cells and the functionality of the resulting memory T cells, which largely lacked the ability to produce the IL-2 needed for an optimal recall response (Feau et al., 2011). This function of XCR1+ DC is optimized by signals from CD4+ helper T cells, consistent with the equivalent reduction of the CD8+ T cell response after depletion of XCR1+ DC or of CD4+ T cells (Figure 7A-C).
The observations reported here provide a new level of understanding of the complex cell-cell interactions that underlie effective cell-mediated immune responses. Two distinct conventional T cell subsets (CD4+ and CD8+ αβ T cells) and a variety of different DC subtypes (CD11b+, XCR1+ and possibly others) operate in a staged, dynamic process to provide both early effectors and memory cells that later support host defense upon re-infection. In the context of previous findings on the contribution of chemokine signaling to optimization of communication involving T cell subsets and DC (Castellino et al., 2006; Hickman et al., 2011; Hugues et al., 2007), and evidence for phased changes in T cell migratory dynamics after viral infection (Mempel et al., 2004), we are now able to draw an increasing complete picture of how this limb of the adaptive immune system operates to enable rare cells to generate robust acute and memory responses. The new evidence for distinct roles of DC subsets in primary activation of CD4+ vs. CD8+ T cells and as a platform for their communication also provides guidance for how to best direct vaccine components to drive specific aspects of immunity.
Experimental Procedures
Animals
Mice were purchased from Jackson or Janvier Labs or maintained at in-house facilities. All mice were maintained in specific pathogen free conditions at an Association for Assessment and Accreditation of Laboratory Animal Care-accredited animal facility. All procedures were approved by the NIAID Animal Care and Use Committee (National Institutes of Health, Bethesda, MD) and the North Rhine-Westphalia State Environment Agency (LUA NRW), respectively. For details on mouse strains see supplementary information.
Viruses, bacteria, and infections
107-108 IU recombinant MVA, 106 - 107 PFU VV-OVA, 2× 107 PFU Ad-OVA-GFP or 5× 103 CFU LM-B8R were diluted in PBS and injected in the footpad (foothock (Kamala, 2007)), i.v. or i.p.
Adoptive T cell transfer
OT-I, OT-II, P14, Smarta or polyclonal control CD4+ T cells were sorted using a MACS CD4 or CD8 negative selection kit (Miltenyi) combined with biotinylated anti-CD44 (IM7, BD Biosciences). 2-4× 106 cells were transferred i.v.
In vitro proliferation assay
OT-I cells were isolated and labelled with CFSE 5 μM (Invitrogen), followed by an ex vivo 72h co-incubation with isolated splenic DC or LN-derived DC.
Isolation of DC and cell sorting
Spleens or LNs were harvested and digested with Collagenase/DNAse for 30min followed by a DC enrichment step using MACS CD11c positive selection kit (Miltenyi) and sorting based on CD11c, MHCII, CD8 and CD11b staining using a FACSAria (BD Biosciences) cell sorter. Cellular purity was >95%.
Flow Cytometry
For analysis LN and spleens were harvested and single cell suspensions were generated. For details on antibodies see supplementary information.
Immunofluorescence Staining
PLP-fixed, frozen tissues were were cut, stained, mounted and acquired on a 710 confocal microscope (Carl Zeiss Microimaging). For details on antibodies see supplementary information.
Intravital two-photon imaging
Mice were anesthetized, popliteal LNs were exposed, and intravital microscopy was performed using a protocol modified from a previous report (Kastenmuller et al., 2013). Raw imaging data were processed and analyzed with Imaris (Bitplane). For details see supplementary information.
Analysis of imaging data
Images were systematically analyzed using a semi-automated (Imaris/Bitplane) and a fully-automated approach. For details see supplementary information.
Statistical Analysis
Student t test (two-tailed) and Mann-Whitney test were used for the statistical analysis of differences between two groups with normal and non-normal distribution.
Supplementary Material
Acknowledgments
We would like to thank S. Ebbinghaus and S. Rathmann for technical assisstance; C. Kurts for scientific discussions and critically reading the manuscript; G. Sutter for help with generation of rec. MVA's; J. Bennink, R.M. Kedl, P. Knolle and D.H. Busch for kindly providing VV-OVA, LM-B8R, AdOVA and MHCI multimers, respectively. This research was supported by the Intramural Research Program, NIAID, NIH. W.K. and N.G. are members of the DFG Excellence Cluster ImmunoSensation in Bonn, Germany and are supported by grant SFB670/SFB704. W.K. is supported by NRW-Rückkehrerprogramm of the German state of Northrhine-Westfalia. T. K. was supported by the Kishimoto Foundation, grants from The Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT) and Japan Society for the Promotion of Science. F.K. was supported by the Human Frontier Science Program (HFSP RGY007/2011) and the Einstein Foundation Berlin.
Footnotes
Author contributions: S.E. and A.G. planned and performed experiments and analyzed data, M.Y.G., K.K. and H.H. analyzed data and designed experiments, F.K. developed analysis software and analyzed imaging data, T.K. and N.G. were involved in study design. R.N.G and W.K. conceptually designed the study, analyzed the data and wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alcami A, Koszinowski UH. Viral mechanisms of immune evasion. Trends Microbiol. 2000;8:410–418. doi: 10.1016/S0966-842X(00)01830-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan RS, Waithman J, Bedoui S, Jones CM, Villadangos JA, Zhan Y, Lew AM, Shortman K, Heath WR, Carbone FR. Migratory dendritic cells transfer antigen to a lymph node-resident dendritic cell population for efficient CTL priming. Immunity. 2006;25:153–162. doi: 10.1016/j.immuni.2006.04.017. [DOI] [PubMed] [Google Scholar]
- Becker M, Guttler S, Bachem A, Hartung E, Mora A, Jakel A, Hutloff A, Henn V, Mages HW, Gurka S, et al. Ontogenic, Phenotypic, and Functional Characterization of XCR1(+) Dendritic Cells Leads to a Consistent Classification of Intestinal Dendritic Cells Based on the Expression of XCR1 and SIRPalpha. Front Immunol. 2014;5:326. doi: 10.3389/fimmu.2014.00326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedoui S, Whitney PG, Waithman J, Eidsmo L, Wakim L, Caminschi I, Allan RS, Wojtasiak M, Shortman K, Carbone FR, et al. Cross-presentation of viral and self antigens by skin-derived CD103+ dendritic cells. Nat Immunol. 2009;10:488–495. doi: 10.1038/ni.1724. [DOI] [PubMed] [Google Scholar]
- Bennett SR, Carbone FR, Karamalis F, Flavell RA, Miller JF, Heath WR. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature. 1998;393:478–480. doi: 10.1038/30996. [DOI] [PubMed] [Google Scholar]
- Bennett SR, Carbone FR, Karamalis F, Miller JF, Heath WR. Induction of a CD8+ cytotoxic T lymphocyte response by cross-priming requires cognate CD4+ T cell help. J Exp Med. 1997;186:65–70. doi: 10.1084/jem.186.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuneu H, Garcia Z, Bousso P. Cutting edge: cognate CD4 help promotes recruitment of antigen-specific CD8 T cells around dendritic cells. J Immunol. 2006;177:1406–1410. doi: 10.4049/jimmunol.177.3.1406. [DOI] [PubMed] [Google Scholar]
- Bevan MJ. Helping the CD8+ T-cell response. Nature Reviews Immunology. 2004;4:595–602. doi: 10.1038/nri1413. [DOI] [PubMed] [Google Scholar]
- Bousso P, Robey E. Dynamics of CD8+ T cell priming by dendritic cells in intact lymph nodes. Nat Immunol. 2003;4:579–585. doi: 10.1038/ni928. [DOI] [PubMed] [Google Scholar]
- Cassell D, Forman J. Linked recognition of helper and cytotoxic antigenic determinants for the generation of cytotoxic T lymphocytes. Ann N Y Acad Sci. 1987;532:51–60. doi: 10.1111/j.1749-6632.1988.tb36325.x. [DOI] [PubMed] [Google Scholar]
- Castellino F, Germain RN. Cooperation between CD4+ and CD8+ T cells: When, Where, and How? Annual Review of Immunology. 2006;24:519–540. doi: 10.1146/annurev.immunol.23.021704.115825. [DOI] [PubMed] [Google Scholar]
- Castellino F, Huang AY, Altan-Bonnet G, Stoll S, Scheinecker C, Germain RN. Chemokines enhance immunity by guiding naive CD8+ T cells to sites of CD4+ T cell-dendritic cell interaction. Nature. 2006;440:890–895. doi: 10.1038/nature04651. [DOI] [PubMed] [Google Scholar]
- Crotty S. T Follicular Helper Cell Differentiation, Function, and Roles in Disease. Immunity. 2014;41:529–542. doi: 10.1016/j.immuni.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtsinger JM, Mescher MF. Inflammatory cytokines as a third signal for T cell activation. Curr Opin Immunol. 2010;22:333–340. doi: 10.1016/j.coi.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Haan JM, Lehar SM, Bevan MJ. CD8(+) but not CD8(-) dendritic cells cross-prime cytotoxic T cells in vivo. J Exp Med. 2000;192:1685–1696. doi: 10.1084/jem.192.12.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drexler I, Staib C, Sutter G. Modified vaccinia virus Ankara as antigen delivery system: how can we best use its potential? Curr Opin Biotechnol. 2004;15:506–512. doi: 10.1016/j.copbio.2004.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudziak D, Kamphorst AO, Heidkamp GF, Buchholz VR, Trumpfheller C, Yamazaki S, Cheong C, Liu K, Lee HW, Park CG, et al. Differential Antigen Processing by Dendritic Cell Subsets in Vivo. Science. 2007;315:107–111. doi: 10.1126/science.1136080. [DOI] [PubMed] [Google Scholar]
- Edelson BT, Kc W, Juang R, Kohyama M, Benoit LA, Klekotka PA, Moon C, Albring JC, Ise W, Michael DG, et al. Peripheral CD103+ dendritic cells form a unified subset developmentally related to CD8alpha+ conventional dendritic cells. J Exp Med. 2010;207:823–836. doi: 10.1084/jem.20091627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feau S, Arens R, Togher S, Schoenberger SP. Autocrine IL-2 is required for secondary population expansion of CD8(+) memory T cells. Nat Immunol. 2011;12:908–913. doi: 10.1038/ni.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain RN. MHC-dependent antigen processing and peptide presentation: providing ligands for T lymphocyte activation. Cell. 1994;76:287–299. doi: 10.1016/0092-8674(94)90336-0. [DOI] [PubMed] [Google Scholar]
- Gerner MY, Kastenmuller W, Ifrim I, Kabat J, Germain RN. Histo-cytometry: a method for highly multiplex quantitative tissue imaging analysis applied to dendritic cell subset microanatomy in lymph nodes. Immunity. 2012;37:364–376. doi: 10.1016/j.immuni.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman HD, Li L, Reynoso GV, Rubin EJ, Skon CN, Mays JW, Gibbs J, Schwartz O, Bennink JR, Yewdell JW. Chemokines control naive CD8+ T cell selection of optimal lymph node antigen presenting cells. J Exp Med. 2011;208:2511–2524. doi: 10.1084/jem.20102545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman HD, Takeda K, Skon CN, Murray FR, Hensley SE, Loomis J, Barber GN, Bennink JR, Yewdell JW. Direct priming of antiviral CD8+ T cells in the peripheral interfollicular region of lymph nodes. Nat Immunol. 2008;9:155–165. doi: 10.1038/ni1557. [DOI] [PubMed] [Google Scholar]
- Hugues S, Scholer A, Boissonnas A, Nussbaum A, Combadiere C, Amigorena S, Fetler L. Dynamic imaging of chemokine-dependent CD8+ T cell help for CD8+ T cell responses. Nat Immunol. 2007;8:921–930. doi: 10.1038/ni1495. [DOI] [PubMed] [Google Scholar]
- Janssen EM, Lemmens EE, Wolfe T, Christen U, von Herrath MG, Schoenberger SP. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature. 2003;421:852–856. doi: 10.1038/nature01441. [DOI] [PubMed] [Google Scholar]
- Kamala T. Hock immunization: a humane alternative to mouse footpad injections. J Immunol Methods. 2007;328:204–214. doi: 10.1016/j.jim.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastenmuller W, Brandes M, Wang Z, Herz J, Egen JG, Germain RN. Peripheral Prepositioning and Local CXCL9 Chemokine-Mediated Guidance Orchestrate Rapid Memory CD8+ T Cell Responses in the Lymph Node. Immunity. 2013;38:502–513. doi: 10.1016/j.immuni.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissenpfennig A, Henri S, Dubois B, Laplace-Builhe C, Perrin P, Romani N, Tripp CH, Douillard P, Leserman L, Kaiserlian D, et al. Dynamics and function of Langerhans cells in vivo: dermal dendritic cells colonize lymph node areas distinct from slower migrating Langerhans cells. Immunity. 2005;22:643–654. doi: 10.1016/j.immuni.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Kurts C, Robinson BW, Knolle PA. Cross-priming in health and disease. Nat Rev Immunol. 2010;10:403–414. doi: 10.1038/nri2780. [DOI] [PubMed] [Google Scholar]
- Lin LC, Flesch IE, Tscharke DC. Immunodomination during peripheral vaccinia virus infection. PLoS Pathog. 2013;9:e1003329. doi: 10.1371/journal.ppat.1003329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHeyzer-Williams LJ, Malherbe LP, McHeyzer-Williams MG. Checkpoints in memory B-cell evolution. Immunol Rev. 2006;211:255–268. doi: 10.1111/j.0105-2896.2006.00397.x. [DOI] [PubMed] [Google Scholar]
- Mempel TR, Henrickson SE, Von Andrian UH. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature. 2004;427:154–159. doi: 10.1038/nature02238. [DOI] [PubMed] [Google Scholar]
- Miller MJ, Wei SH, Parker I, Cahalan MD. Two-photon imaging of lymphocyte motility and antigen response in intact lymph node. Science. 2002;296:1869–1873. doi: 10.1126/science.1070051. [DOI] [PubMed] [Google Scholar]
- Mitchison NA, O'Malley C. Three-cell-type clusters of T cells with antigen-presenting cells best explain the epitope linkage and noncognate requirements of the in vivo cytolytic response. Eur J Immunol. 1987;17:1579–1583. doi: 10.1002/eji.1830171109. [DOI] [PubMed] [Google Scholar]
- Norbury CC, Princiotta MF, Bacik I, Brutkiewicz RR, Wood P, Elliott T, Bennink JR, Yewdell JW. Multiple antigen-specific processing pathways for activating naive CD8+ T cells in vivo. J Immunol. 2001;166:4355–4362. doi: 10.4049/jimmunol.166.7.4355. [DOI] [PubMed] [Google Scholar]
- Ridge JP, Di Rosa F, Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature. 1998;393:474–478. doi: 10.1038/30989. [DOI] [PubMed] [Google Scholar]
- Schnorrer P, Behrens GM, Wilson NS, Pooley JL, Smith CM, El-Sukkari D, Davey G, Kupresanin F, Li M, Maraskovsky E, et al. The dominant role of CD8+ dendritic cells in cross-presentation is not dictated by antigen capture. Proc Natl Acad Sci U S A. 2006;103:10729–10734. doi: 10.1073/pnas.0601956103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenberger SPS, Toes RER, van der Voort EIE, Offringa RR, Melief CJC. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature. 1998;393:480–483. doi: 10.1038/31002. [DOI] [PubMed] [Google Scholar]
- Shedlock DJ, Shen H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science. 2003;300:337–339. doi: 10.1126/science.1082305. [DOI] [PubMed] [Google Scholar]
- Shortman K, Heath WR. The CD8+ dendritic cell subset. Immunol Rev. 2010;234:18–31. doi: 10.1111/j.0105-2896.2009.00870.x. [DOI] [PubMed] [Google Scholar]
- Stoll S, Delon J, Brotz TM, Germain RN. Dynamic imaging of T cell-dendritic cell interactions in lymph nodes. Science. 2002;296:1873–1876. doi: 10.1126/science.1071065. [DOI] [PubMed] [Google Scholar]
- Sun JC, Bevan MJ. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science (New York, NY) 2003;300:339–342. doi: 10.1126/science.1083317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Lugt B, Khan AA, Hackney JA, Agrawal S, Lesch J, Zhou M, Lee WP, Park S, Xu M, DeVoss J, et al. Transcriptional programming of dendritic cells for enhanced MHC class II antigen presentation. Nat Immunol. 2014;15:161–167. doi: 10.1038/ni.2795. [DOI] [PubMed] [Google Scholar]
- Wiesel M, Joller N, Ehlert AK, Crouse J, Sporri R, Bachmann MF, Oxenius A. Th cells act via two synergistic pathways to promote antiviral CD8+ T cell responses. J Immunol. 2010;185:5188–5197. doi: 10.4049/jimmunol.1001990. [DOI] [PubMed] [Google Scholar]
- Wiesel M, Oxenius A. From crucial to negligible: Functional CD8+T-cell responses and their dependence on CD4+T-cell help. Eur J Immunol. 2012;42:1080–1088. doi: 10.1002/eji.201142205. [DOI] [PubMed] [Google Scholar]
- Xu RH, Remakus S, Ma X, Roscoe F, Sigal LJ. Direct presentation is sufficient for an efficient anti-viral CD8+ T cell response. PLoS Pathog. 2010;6:e1000768. doi: 10.1371/journal.ppat.1000768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki C, Sugiyama M, Ohta T, Hemmi H, Hamada E, Sasaki I, Fukuda Y, Yano T, Nobuoka M, Hirashima T, et al. Critical roles of a dendritic cell subset expressing a chemokine receptor, XCR1. J Immunol. 2013;190:6071–6082. doi: 10.4049/jimmunol.1202798. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.