

Abstract
Neuroplasticity involves molecular changes in central nervous system (CNS) synaptic structure and function throughout life. The concept of neural organization allows for synaptic remodeling as a compensatory mechanism to the early pathobiology of Alzheimer’s disease (AD) in an attempt to maintain brain function and cognition during the onset of dementia. The hippocampus, a crucial component of the medial temporal lobe memory circuit, is affected early in AD and displays synaptic and intraneuronal molecular remodeling against a pathological background of extracellular amyloid-beta (Aβ) deposition and intracellular neurofibrillary tangle (NFT) formation in the early stages of AD. Here we discuss human clinical pathological findings supporting the concept that the hippocampus is capable of neural plasticity during mild cognitive impairment (MCI), a prodromal stage of AD and early stage AD.
In 1906, Dr. Alois Alzheimer first described a form of progressive presenile dementia in a female patient named Auguste Deter (Fig. 1), who over time developed memory loss as well as other behavioral sequel and died at the age of 51. Brain autopsy revealed dramatic brain shrinkage and tissue sections stained using silver impregnation procedures demonstrated the presence of senile plaques (SPs) and neurofibrillary tangles (NFTs) (Alzheimer, 1906), now considered the defining lesions of the disease bearing Dr. Alzheimer’s name (Fig. 1). SPs accumulate in the extracellular matrix and contain insoluble fibrils of the amyloid beta (Aβ) fragment, which is cleaved from the larger transmembrane amyloid precursor protein (APP) by successive β cleavage through beta-site APP cleaving enzyme 1 (BACE1) and the γ secretase complex (Haas and Selkoe, 1993; Shoji et al., 1992; Thinakaran and Koo, 2008). NFTs are composed of argyrophilic aggregates of hyperphosphorylated forms of the protein tau (Trojanowski et al., 1993; Yoshiyama et al., 2013). These pathological protein aggregates display a beta-pleated sheet conformation and are thought to interfere with cytoskeletal integrity, which ultimately disrupts synapse and neuronal function. In over 99% of patients, the onset of Alzheimer’s disease (AD) occurs in late adulthood, usually after the age of 65. In the small portion of cases (<1%), the disease has an autosomal dominant pattern of inheritance (“familial AD”), is caused by mutations in one of three different genes (all of which affect amyloid metabolism, including the Amyloid Precursor Protein gene itself) and starts much earlier, with notable mutations in APP, presenilin 1 (PS1), or presenilin 2 (PS2). Interestingly, the original histological slides from Auguste Deter’s autopsy were discovered a few years ago in the basement of a German hospital, and genotyping of these samples showed that she likely was suffering from a familial form of AD (Muller et al., 2013; Rupp et al., 2014), which was consistent with her early age of onset. Late onset AD is the leading cause of dementia in the United States, affecting an estimated 5.2 million people in the United States (Thies and Bleiler, 2013) and is predicted to afflict 13 million people in the USA by 2050.
Figure 1.

(A) Photographs of Dr. Alzheimer and (B) his first patient, Augusta Deter. C. Image of the original postmortem histological slides showing silver impregnated neuritic plaques (D) and a neurofibrillary tangle (E) in the brain of Augusta Deter.
Recent studies have confirmed that AD has a long preclinical stage and some suggest that the disease process begins between 15–20 years prior to emergence of clinical symptoms (Sperling et al., 2014) (Fig. 2). The term mild cognitive impairment (MCI) has been used, synonymous with the term prodromal AD, as describing the intermediate stage between normal brain aging and frank dementia when NFT and Aβ pathology is increased compared to individuals with no cognitive impairment (NCI) (Guillozet et al., 2003; Markesbery, 2010, 2006). The clinical concept of MCI was derived from memory clinics, which attracted milder cases of dementia, as well as longitudinal studies of elderly populations in which subjects were evaluated annually for cognitive status. Many of the subjects with earlier, milder cases of cognitive loss did not exhibit impairment in two cognitive domains, a criterion that was required for an NINDS/ADRDA diagnosis of AD established by McKhann and coworkers (1984). In the 1990’s, such cases were most frequently, but not always, characterized by an amnestic disorder and the term “Mild Cognitive Impairment” (MCI) was popularized by Petersen (1999). While memory disorder clinics reported that amnestic MCI (aMCI) was the most common form of MCI, it was clear that MCI along with other affected single cognitive domains comprised a small, but significant, component of this clinical presentation. Indeed, people with a clinical diagnosis of MCI comprise a heterogeneous cohort of which those who present solely with memory deficits are classified as single domain aMCI, while those who have a deficit in memory as well as other cognitive domains are categorized as multi-domain MCI (mdMCI) (Johnson et al., 2010; Petersen, 2003). Those with aMCI are at a higher risk of developing AD (Johnson et al., 2010; Petersen, 2003). However, some people clinically diagnosed with no cognitive impairment (NCI) or with MCI can exhibit SP and NFT pathology equal to or greater that that seen in mild to moderate AD dementia, challenging the pathologically-based idea that these lesions alone cause dementia (Mufson et al. 1999; Price et al. 2009, Markesbery 2010). Clinical pathological studies indicate that NFTs correlate better than and amyloid plaques with cognition in AD (Bierer et al., 1995; Nelson et al., 2012) However, The “amyloid cascade” hypothesis of AD (Hardy and Higgins GA, 1992) was revised recently to include soluble Aβ oligomers (in addition to insoluble plaques) after multiple studies demonstrated their ability to impair synaptic function and memory (Lesne et al., 2006). Moreover, recent research indicates that there is a dynamic balance between “soluble” oligomeric and “insoluble” fibrillar Aβ pools in the AD brain, with concentrations of soluble Aβ species determining the rate of Aβ aggregation in plaques, and plaque-aggregated Aβ fibrils serving as a reservoir of soluble Aβ in the interstitial fluid surrounding neuronal synapses (Koffie et al., 2009).
Figure 2.

Graphic representation of the putative prolonged trajectory of Alzheimer’s disease cognitive decline throughout Adulthood. Modified from Swerdlow, Neurobiology of Aging, 2007.
Synaptic loss, a hallmark of the AD brain (Scheff et al., 1998, 2003, 2006), correlates with cognitive decline in many vulnerable regions, but not all affected areas (Scheff et al., 2006). In addition, there are deficits in glutamatergic (Hyman, 1987; Palmer et al., 1986; Cross et al., 1987; Hardy et al., 1987; Procter et al., 1987; Cowburn et al., 1988a, b), cholinergic (Davies and Mahoney, 1976; Whitehouse et al, 1982), serotonergic (Mann and Yates 1986), and noradrenergic (Adolfsson et al. 1979; Mann et al. 1983; Zarow et al. 2003; Grudzien et al. 2007; Braak and Del Tredici, 2011; Counts and Mufson, 2010) cortical innervation and as well as an up-regulation of endosomal/lysosomal autophagy molecules, which appear to precede plaque and tangle pathology (Cataldo et al., 1994; Ginsberg et al., 2010a; Nixon, 2005) in cortical innervation in AD. This multiplicity of events supports the view that impairments of multiple processes contribute to the onset of dementia and they are associated with a high degree of inter-individual variability. The mechanism(s) that allow for the preservation of cognitive function in the presence of significant AD neuropathology found at autopsy in individuals without dementia or with MCI is a compelling question (Perez-Nievas et al., 2013).
Dating to the work of Cajal (1901) it has been suggested that the brain is capable of neuroplastic response(s) in the face of changes to the external and internal milieu, aging and disease (see also Jellinger and Attems, 2013; Mesulam, 1999; Scheff and Price, 2001; Scheff et al., 2006). Neuroplasticity may be one of the underlying mechanisms that enable elderly individuals with NCI and MCI with minimal cognitive impairment to function despite the presence of significant AD-like pathology equivalent to someone with AD dementia (Mesulam, 1999; Mufson et al., 2012). The mechanism(s) of this brain resilience to cognitive decline in the presence of abundant pathologies is unclear, but supports the concept of brain or cognitive reserve. According to this concept, regions or circuits within the brain are able to counteract and/or counterbalance age-related alterations or disease pathologies by reorganizing synaptic structure, connections, and ultimately function via a multitude of molecular and cellular pathways (Honer et al., 2012). A primary example of this form of neuroplasticity is found in the hippocampus, a major component of the limbic system that displays neural reorganization after brain injury in animal models of neural damage and in human neurological diseases including epilepsy (Stretton et al., 2014) and AD (DeKosky et al., 2002; Davis et al., 1999). Therefore, the overall goal of this review is to present evidence derived from clinical pathological investigations that the hippocampus, a component of the medial temporal lobe (MTL) memory circuit, displays cellular and structural alterations indicative of neural plasticity during the progression of AD.
Hippocampal Anatomy
The hippocampus is a key component of the medial temporal lobe (MTL) memory circuit, which includes the transentorhinal and entorhinal corties and the subicular complex (Fig. 3). The hippocampus consists of several subdivisions: 1. The dentate gyrus (DG), a tightly packed layer of small granule cells wrapped around the end of the hippocampus proper at the level of the hippocampal fissure, forming a v-shaped wedge. 2. From the DG emerge the components of the Cornu Ammonis: CA4, then CA3, then a very narrow zone termed CA2, and then CA1. The CA fields contain densely packed pyramidal cells (Hyman, 1987; Ramon y Cajal, 1901 Lorente de No, 1934). CA1 then merges with the subiculum, followed by the presubiculum and parasubiculum, then a transition to entorhinal cortex (Brodmann area 28). The term “hippocampus proper” refers to the four CA subfields, while the term “hippocampal formation” subserves the hippocampus proper plus DG and subiculum (Amaral and Lavenex, 2006). The entorhinal cortex contains five layers including a lamina desecans. Layer II/III contains the prominent cell islands consisting of glutamatergic stellate neurons, the axons of which project into the hippocampal dentate gyrus via the perforant pathway (Stranahan and Mattson, 2010). The entorhinal cortex transitions into the transentorhinal cortex (Brodmann area 35) a more traditional six-layered cortex (Taylor and Probst, 2008). The primary projection from the entorhinal to the hippocampus is the perforant pathway (Simonian, 1994; Lorente de Nó, 1934), disruption of which results in defects in learning and memory in animals as well as a profound reactive synaptogenesis response that involves reorganization of glutamatergic neurotransmission signaling (Geddes et al., 1985; Chapman et al., 1999; Masliah et al., 1991; Steward and Vinsant, 1983; Ginsberg, 2005, 2010). Cotman and Lynch (1976) coined the term “reactive synaptogenesis” to describe injury-induced replacement of synapses (Cotman and Lynch, 1976) to differentiate the synapse formation that occurs following injury from that occurring during normal development. This group demonstrated a decline in axon sprouting in aged animals compared to young adult controls, which may be attributed to several different factors such as a reduction in the ability of neurons to synthesize the materials necessary for growth, inability of the target cells to accept new synapses, and the loss of a growth promoting signal or a change in the signal threshold (Cotman, and Scheff, 1979; Scheff et al., 1980). Despite the decline in plasticity with age, the observation that the aged nervous system still retains a significant plasticity response was a major discovery with that was later extended to AD.
Fig. 3.

Photomicrograph of the hippocampal complex immunostained using an antibody against parvalbumin (gold color) and reacted with diaminobenzidine showing the hippocampus, Cornu Ammonis subfields CA1 and CA3, dentate gurus (DG) subiculum (SUB), entorhinal cortex (Brodmann area 28), transentorhinal cortex (area 35), fimbria of the fornix (ff), perfronat pathway (pp), parahippocampal gyrus (Brodmann area 27), presubiculum (area 49) and Schaffer collaterals (SC).
Entorhinal disconnection from the hippocampus occurs in human AD, and is posited to relate to a general failure of synaptic plasticity (Hyman et al., 1990, 1994; Geddes et al., 1986). Hence, the transentorhinal, entorhinal and hippocampal axes are anatomically and functionally connected; this connectivity within the MTL underscores the importance of viewing hippocampal neuroplasticity as a systems-based phenomenon with global circuit-based consequences early in the course of AD.
Hippocampus and Alzheimer’s disease
The hippocampus is one of the earliest brain structures to develop neurodegenerative changes in AD, undergoing profuse NFT accumulation but lesser amyloid pathology deposition in the early stages of AD (Arriagada et al., 1992). AD pathologic lesions and neuronal loss are found in multiple regions within the hippocampal formation as well as in the transentorhinal and entorhinal cortices (Hyman, 1984, 1990; Geddes, 1985; Gomez- Isla et al., 1996; Kordower et al., 2001). Braak and Braak (1991) were the first to characterize the stages of NFT appearance and spread in the AD brain with the earliest changes detected within the MTL. NFTs develop first in the transentorhinal cortex and may spread to the entorhinal cortex (stages I/II), then to the hippocampus (“limbic”, stages III/IV) and then to the medial temporal isocortex (“isocortical”, stages V/VI) (Braak and Braak, 1991; Morris, 2001). By contrast, SPs appear first in association cortex far removed from those areas displaying NFTs, which display patterns that show great inter-individual variation and do not correlate to any significant degree with dementia severity (Mesulam, 1999; Nelson et al., 2012). These observations complicate the simple assumption that amyloid is the driving factor underlying the clinical symptoms of dementia and the formation (s) of NFTs in AD (Hardy and Allsop, 1991) which has since been modified. Interestingly, Dr. Alzheimer wrote “…the plaques are not the cause of senile dementia, but only an accompanying feature of senile involution of the central nervous system” (Alzheimer, 1911). More recently, Mesulam (1999) stated, “It seems as if the Aβ plaques appear at the wrong time and in the wrong places with respect to the clinical dementia and there is little evidence that they cause the NFT”. In light of the continued lack of efficacy of human anti amyloid strategies in AD, these comments may prove to be prescient. By contrast, NFT pathology displays a highly significant correlation with cognitive impairment in AD (Giannakopoulos et. al., 2003) and occurs within the hippocampus very early in the disease processes (Braak and Braak, 1991). However, clinical pathologic data indicate that the hippocampus remains highly malleable despite the abundance of NFT pathology during the onset of AD (Gary et al., 2014).
Hippocampal structural plasticity in MCI and AD
In AD, ultrastructural counts of synapse numbers indicate a reduction in the inner and outer layers of the dentate gyrus (Scheff et al., 1996, 1998), which receives extensive input from the entorhinal cortex (Simonian et al., 1994). Another investigation found a reduction in synapses within the supragranular band below the inner molecular layer in AD (Bertoni-Fredarri et al., 1990). Although decreases in synaptic density are more highly correlated with the degree of cognitive impairment than classic pathological changes related to AD (Terry et al., 1991; Scheff et al., 2006; DeKosky and Scheff, 1990; Scheff and Price, 2003; Sze et al., 1997), very few studies have investigated synaptic contact integrity or evidence for a neuroplastic response in the hippocampus in individuals with MCI or early AD.
A series of studies, which combined unbiased stereology with electron microscopy, failed to demonstrate a significant difference in the total number of synapses within the outer molecular layer of the hippocampus between individuals with aMCI compared to NCI, but there was a significant decrease between early AD and MCI as well as NCI (Scheff et al., 2006). The reduction in synapses in early AD compared to both NCI and MCI did not appear to be associated with a loss of granule cells (West et al., 2004) but likely reflected a loss of afferent innervation from the ipsilateral entorhinal cortex (Hyman et al., 1987; Gomez-Izla et al., 1996; Yasuda et al., 1995; Scharfman and Chao, 2013). Notably, this loss of entorhinal input to the hippocampus has been shown to initiate an extensive sprouting of cholinergic innervation into the molecular layer of the hippocampus (Geddes et al., 1985), where neuritic plaques preferably accumulate, thus leading to a hypothesis that reactive cholinergic sprouting contributes to the pathogenesis of Aβ plaque formation (Geddes et al., 1986). This compensatory structural remodeling within the hippocampus illustrates the neuroplastic capacity of this region to counteract (or contributes to) mounting pathology. By contrast, a subsequent study using the same cohort of cases (Scheff et al., 2006) reported a significant reduction in total synapse number in the striatum radiatum region of the hippocampal CA1 subfield in individuals with MCI compared to NCI (Fig. 4), which correlated with the subject’s Mini-Mental State Exam score (MMSE), but not with NFT Braak stage or apolipoprotein E (ApoE) status, a genetic risk factor for AD (Scheff et al., 2006). Unlike the outer molecular layer, CA1 receives input from the Schaffer collaterals arising from CA3 and not from the glutamatergic neurons of the entorhinal cortex, suggesting differential responses to synapse loss within the hippocampus based upon afferent innervation or chemical phenotype, some of which precipitate synaptic reorganization. A recent report characterized changes in the dendritic branching of the basilar tree of hippocampal CA1 pyramidal neurons. In this study, formalin-fixed tissue autopsy obtained from University of Kentucky Alzheimer’s Disease Center who died with a clinical diagnosis of NCI, MCI or AD was prepared for Golgi impregnation (Fig. 5). Camera lucida drawings of the basilar tree of randomly selected CA1 neurons were analyzed for alterations in dendritic arbor amount, distribution and complexity (Mervis et al., 2013). Quantitation showed a significant increase in dendritic length (18%) and complexity (23%) in CA1 neurons in MCI compared to NCI. Conversely, there was a significant reduction in branch length (−39%) and arbor complexity (−25%) during the progression from MCI to AD (Fig. 5). These findings suggest that the observed increase in CA1 dendritic parameters from NCI to MCI may be another example of a neuroplastic compensatory response to a loss of afferent input early in the course of the disease, which is not maintained as the disease progresses. The role that the reported reduction in total synapse number plays in CA1 neuroplasticity remains unknown. However, these examples of early CA1 neural reorganization may represent a viable window for potential therapeutic strategies aimed at restoring or maintaining hippocampal function during the transition from MCI to AD. Future studies should determine whether alterations in specific synapse subtypes (i.e., perforated vs. non-perforated) are differentially affected and their relation to cognitive decline and brain pathology during the onset of AD. Interestingly, the size of the synaptic contacts was found to be substantially larger in AD cortex compared to non-demented aged controls (Scheff et al., 1990), which was suggested to be part of a compensatory mechanism found in regions of the neocortex and hippocampus (see review Scheff and Price, 2006). These investigators found that as the number of synapses declined in a given region, the size of the residual synapses increased. This synaptic compensatory response was also observed early in the course of the AD (DeKosky and Scheff, 1990).
Figure 4.
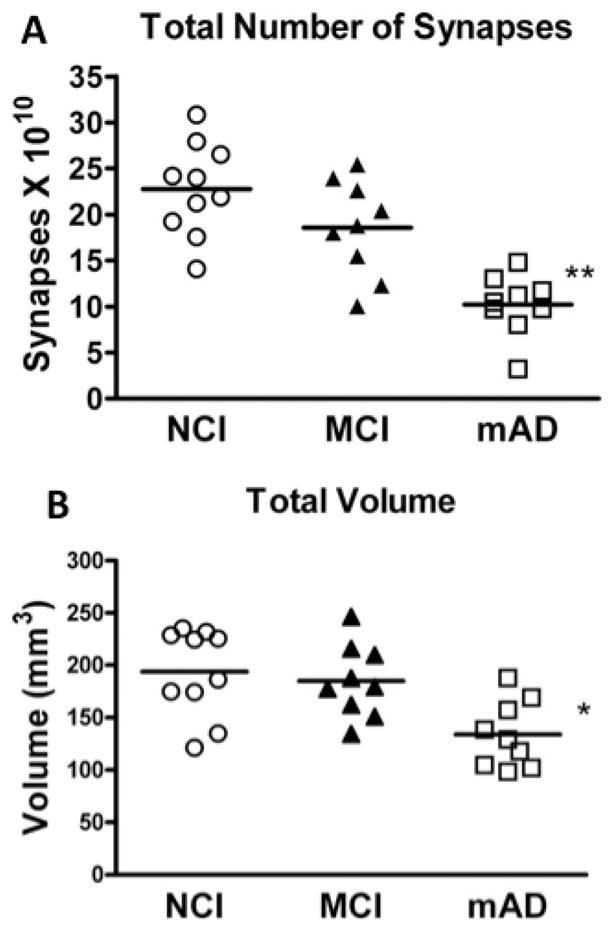
A. Estimates of total number of synapses in stratum radiatum subregion of the hippocampal CA1 across clinical diagnostic groups. ANOVA showed a difference between group means and post-hoc analyses showed a significant reduction in the mild AD (mAD) compared to the no cognitive impairment (NCI) and (B) mild cognitive impairment MCI groups. Post-hoc analysis showed no difference in total synaptic number between the NCI and MCI groups, although the overall group mean was 18% lower in the MCI group. B. Total volume of CA1 stratum radiatum was estimated with the Cavlieri method directly from tissue sections. Individual points represent individual subjects’ scores for each group. Horizontal line indicates group mean. *p< 0.05; **p< 0.01.
Figure 5.

Golgi impregnated hippocampal CA1 pyramidal neurons analyzed for dendritic branching of the basilar tree from cases with a clinical diagnosis of (A) no cognitive impairment, (B) mild cognitive impairment and (C) Alzheimer’s disease. (D) Scholl analysis of the amount, distribution and complexity of the arbor showed a significant increase in these parameters (length, + 18%; complexity, +23%) in MCI CA1 neurons compared to NCI. Conversely, there was a significant reduction in branch length (−39%) and arbor complexity (−25%) in the progression from MCI to AD.
Neuronal structural alterations may not be the only factor(s) contributing to cognitive decline and hippocampal plasticity in MCI. For example, studies have reported significant reductions in synaptic vesicle trafficking-related genes in the AD brain, which interrupt the efficacy of normal synaptic transmission (Yao et al., 2003; Murphy et al., 2003; Kennedy et al., 2005). Counts et al. (2014) examined progressive changes in the expression classes of synaptic gene within single CA1 neurons in subjects who died with a clinical diagnosis of NCI, MCI or moderate AD obtained from the Rush Religious Orders study (RROS). Quantitative analysis revealed a significant downregulation of transcripts encoding regulators of synaptic function including presynaptic vesicle trafficking (e.g., synaptophysin and synaptogyrin), vesicle docking and fusion/release (synaptotagmin and syntaxin 1), and regulators of excitatory postsynaptic function, including PSD-95 and Homer1, in CA1 neurons in MCI and AD compared to NCI (Counts et al., 2014). By constrast, synaptic transcript levels were not significantly different between MCI and AD (Counts et al., 2014), consistent with previous single cell analyses of CA1 neurons in AD (Ginsberg et al., 2000, 2004, 2006; Ginsberg and Che, 2005; Mufson et al., 2006). Downregulation of these markers was associated strongly with poor antemortem cognitive status and AD pathological severity (i.e., Braak stage, NIA-Reagan and CERAD diagnosis) (Counts et al., 2014). Previous biochemical studies of protein levels demonstrated similar findings showing a down regulation of postsynaptic proteins such as PSD-95 and drebrin in the MCI hippocampus compared to NCI subjects, which correlated with poor antemortem cognitive test scores (Counts et al., 2012; Sultana et al., 2010; Hatanpaa et al., 1999). The protein drebrin is a regulator of postsynaptic dendritic spine morphogenesis (Hayashi et al., 1996) and is critical for postsynaptic targeting of PSD-95, which is involved in excitatory postsynaptic plasticity (Takahashi et al., 2003). Interestingly, other groups have found an increase in expression of genes related to mitochondrial bioenergetics, protein homeostasis and the SNARE complex in the MCI hippocampus and entorhinal cortex (Berchtold et al., 2014). A number of synaptic genes showed strong significant correlations most notably in the entorhinal cortex, with fewer in the hippocampus, and these genes were related predominantly to synaptic transmission and synaptic plasticity. Changes in expression of genes that facilitate synaptic excitability and plasticity seem to be associated strongly with worse cognition, and changes in expression of genes that inhibit plasticity were associated positively (somewhat paradoxically) with cognitive scores (Berchtold et al., 2014; Counts et al., 2013). Neurotrophin signaling is also severely affected in MCI and AD, with notable downregulation of cognate neurotrophin receptors TrkA, TrkB, and TrkC in CA1 pyramidal neurons, which also correlates with cognitive decline (MMSE and Global Cognitive Score assessments) (Ginsberg et al., 2010a). These synaptic and neurotrophic alterations suggest that the hippocampus displays greater metabolic demand in MCI and ultimately progressive degeneration, if not maintained. These diverse changes in gene expression indicate that there is a rebalancing of synaptic transmission and plasticity and that synaptic gene transcript and protein levels are expressed differentially and altered in discrete cells and regions in the hippocampus in MCI.
The underlying pathogenic mechanisms that trigger these structural synaptic alterations in the hippocampus during MCI and the onset of AD are unclear, yet several lines of evidence revealed that an aberrant expression of endosomal-lysosomal proteins occurs within hippocampal neurons even before the SP and NFT formation early in AD (Nixon et al., 1992; Cataldo et al., 1994, 1995; Nixon, 2005). Endosomal-lysosomal activity is an essential and dynamic organization of acidified cytoplasmic organelles which perform a variety of functions in neurons such as internalizing nutrients and neurotrophic factors, as well as degrading and recycling receptors (Nixon and Cataldo, 1995; Bishop, 2003). Acid hydrolases such as cathepsins are transported from the Golgi apparatus to late endosomes that contain intracellular material engulfed during autophagy or extracellular material derived from heterophagy (Gordon and Seglen, 1988; Dunn et al., 1990; Gordon et al., 1992). Since endocytic activity is high at nerve terminals and dendrites and is involved in the maintenance of normal synaptic transmission (LaVail and LaVail, 1974; Baas and Heidemann, 1986; Parton and Dotti, 1993), it may also play a role in neuronal plasticity early in disease onset. Ginsberg et al. (2010) reported a significant up-regulation of the endosomal markers rab5 and rab7 in CA1 pyramidal neurons using microarray analysis, real-time quantitative PCR, and immunoblot analyses of regional hippocampal dissections in clinically defined MCI and AD brains, and this up-regulation correlated with cognitive decline as well as with Braak NFT staging (Ginsberg et al., 2010a, 2010b). The reason for this over-activation of endocytic machinery in MCI may be to combat ensuing pathology in vulnerable hippocampal neuronal populations by regulating trophic factor activity in an attempt to compensate for failing endosomal lysosome formation. For example, the early endosome effector rab5 and late endosome effector rab7 regulate nerve growth factor (NGF) signaling (Valdez et al., 2007; Deinhardt et al., 2006; Liu et al., 2007) and up-regulation of rab5 down-regulates the brain-derived neurotrophic factor (BDNF) receptor, TrkB in vitro (Ginsberg et al., 2010a). Both are key players in CNS neuroplasticity responses (Iulita and Cuello, 2014). Since endocytic lysosomal up-regulation occurs prior to SP and NFT formation (Cataldo et al., 1994; 1996), these changes may be a neuroplastic response to even earlier neuronal stresses such as oxidative DNA damage, apoptosis and an increasing failure of endosomal fusion to lysosomes (Lovell and Markesbery, 2007; Wang et al., 2005; Ding et al., 2007; Rodrigues et al., 2012).
Hippocampal Cholinergic Plasticity in MCI and AD
In the early 1970’s a series of articles revealed that lesions of the entorhinal cortex or the perforant pathway, which deprives the hippocampus of its main excitatory glutamatergic input, induces a neuroplasticity response originating from cholinergic medial septal neurons, the major source of cholinergic input to the hippocampus (Mesulam et al., 1983; Mufson et al., 2008), which reinnervates the denervated glutamatergic zones within the molecular layer of the hippocampus in rodents (Cotman et al., 1973; Matthews et al., 1976a, 1976b; Fifkova, 1975; Nadler et al., 1977). The loss of cholinergic input itself most likely plays a pivotal role in the severity of the cognitive and behavioral deficits, especially in the areas of memory and attention and influences the progression of hippocampal neuroplasticity (Mesulam, 2004). Moreover, morphologic studies in postmortem human brain tissues indicate that the cholinergic system has the ability to reorganize during the early phases of AD, including in the hippocampus (DeKosky et al., 2002; Davis et al. 1999).
Hippocampal cholinergic reorganization has been investigated in individuals with AD and MCI. In a study using end stage AD cases, activity for the of the cholinergic lytic enzyme, acetylcholinesterase (AChE), was increased in the outer molecular layer of the dentate gyrus compared to aged controls indicative of septal cholinergic afferent sprouting in humans (Geddes et al., 1985). This study provided evidence that CNS adaptive growth occurs along with the onset of degenerative events in AD. Thirty years later, it was demonstrated that activity levels of choline acetyltransferease (ChAT), the rate-limiting synthetic enzyme for acetylcholine, was increased significantly in the hippocampus of individuals with MCI compared to subjects with NCI or mild-moderate AD (Fig. 6A) (DeKosky et al., 2002; Ikonomovic et al., 2003; Davis et al., 1999). Interestingly, across the three clinical diagnostic groups of NCI, MCI, and mild-moderate AD, increased hippocampal ChAT activity levels correlated with progression of neuritic plaque pathology in entorhinal cortex and hippocampus, supported by the observation that hippocampal ChAT in the MCI group was significantly elevated selectively in the limbic (entorhinal-hippocampal, III/IV) Braak stages (Ikonomovic et al., 2003). These studies also found that ChAT activity in the superior frontal cortex was significantly higher in MCI than in controls, and subjects with mild AD had equal levels to those with NCI (DeKosky et al., 2002). Interestingly, a biochemical up-regulation ChAT enzyme activity was not paralleled by an increase in the density of cholinergic fibers in the same region (Ikonomovic et al., 2007). This lack of an increase in cholinergic axonal innervation of the superior frontal cortex in MCI suggests that structural reorganization of cholinergic profiles is not the mechanism underlying the transient cholinergic plasticity reported in this region. Up-regulation of hippocampal and cortical ChAT activity suggested a chemoplastic response early in the disease process that may maintain cognition and slow the transition from MCI to AD. There was a positive correlation between the up-regulation of hippocampal ChAT activity in MCI and limbic Braak stages (entorhinal-hippocampal, III/IV) (Ikonomovic et al., 2003), suggesting that this elevation is indeed a compensatory response to the entorhinal-hippocampal disconnection syndrome (Fig. 6B, C) (Hyman et al., 1984, 1990; Gomez-Izla et al., 1996). Increases in ChAT activity seen in the hippocampus and superior frontal cortex suggested that cholinergic upregulation is possible in more than one brain area in people with MCI and may be a more general cholinergic response to the onset of AD. Whether other ascending transmitter systems (e.g., noradrenergic and/or dopaminergic, among others) are capable of neuroplasticity during the progression of dementia remains an area of active investigation.
Figure 6.
(A) Choline acetyltransferase (ChAT) activity increased in the hippocampus in MCI and returned to control levels in mild AD. (Band C) Schematic drawings of coronal section of the hippocampus illustrating the loss of innervation to the hippocampus arising from the glutamatergic layer II entorhinal cortex neurons (red) triggering a cholinergic plasticity response (blue) which likely originates from the septal cholinergic projection neurons into the denervated glutamatergic sites in the hippocampus in MCI. Abbreviations: AD: Alzheimer’s disease; CA1, CA2, CA3, CA4- Cornu Ammonis hippocampal subfields; CS- collateral sulcus, DG- dentate gyrus, Ent- entorhinal cortex; f- fornix, gl- granular cell layer of the hippocampus, ml- molecular layer of the hippocampus; NCI- no cognitive impairment, MCI-mild cognitive impairment, pp-perforant pathway, TEC- transentorhinal cortex, Sub-subiculum
Factors underlying hippocampal cholinergic plasticity in individuals with MCI remain to be completely determined. In the case of the hippocampus, it may be triggered by the marked loss of excitatory glutamatergic input into the hippocampus arising from degeneration of entorhinal cortex layer II stellate neurons (Stewart and Scoville, 1976; Klink and Alonso 1997) that occurs in AD and MCI (Gomez-Isla et al., 1996; Kordower et al., 2000). Despite the extensive reduction of the layer II/III stellate neurons within the entorhinal cortex in MCI, alterations to the glutamatergic system in the hippocampus in MCI has received limited investigation. For example, there is a reduction in free glutamate in AD (Hyman et al., 1987), but not in the MCI hippocampus (Rupsingh et al., 2011). Examination of NMDA receptor levels produced conflicting results, with some groups showing a decrease in AD (Greenamyre et al., 1985, 1987) and others showing no change (Geddes et al., 1986; Monaghan et al., 1987; Cowburn et al., 1988b). There is also a reduction in AMPA and mGlu receptor density in AD (Dewar et al., 1991; Ginsberg et al., 2000, 2006). Others report a loss of cortical and hippocampal glutamatergic uptake sites in AD, which were interpreted as nerve terminal loss (Palmer et al., 1986; Cross et al., 1987; Hardy et al., 1987; Procter et al., 1987; Cowburn et al., 1988a, b). These inconsistencies may be due to subregion-selective changes in hippocampal glutamate receptors. An immunohistochemical analyses of glutamate receptor subunit NMDAR1 in the hippocampus reported that AD cases with mild/moderate pathology (Braak I–III) were similar to controls while more severe AD cases (Braak IV–VI) had increased NMDAR1 immunolabeling in the CA fields. In contrast, the dentate gyrus showed a reduction in NMDAR1 labeling particularly within the outer molecular layer the terminal zone of the perforant pathway (Ikonomovic et al., 1999). Further supporting glutamatergic plasticity changes in AD hippocampus, several studies reported that a selective subtype of AMPA glutamate receptor (GluR2) is reduced in AD entorhinal cortex and hippocampus and this downregulation precedes the formation of neurofibrillary pathology (Ikonomovic et al., 1997). Adding to the complexity of glutamatergic changes in AD is the observed reduction in glutamate concentration in AD brain (Ellison et al., 1986; Sasaki et al., 1986; Hyman et al., 1987) that make no distinction between neuronal and metabolic pools, limiting potential interpretations. A clinical pathological study using specific immunological markers of glutamatergic neurons to assess the structural involvement of the glutamatergic system within midfrontal cortex across progressive stages of AD demonstrated a striking pathology-dependent pattern of glutamatergic synaptic remodeling with disease progression (Bell et al., 2007). Subjects with MCI displayed an elevation in glutamatergic presynaptic bouton density, reminiscent to that reported in the hippocampal cholinergic system, which then depletes and is lost with disease progression (DeKosky et al., 2002; Ikonomovic et al., 2003). This increased glutamatergic presynaptic bouton density correlated with improved cognitive performance in the AD group, but not for people with MCI, in which the increase in glutamatergic presynaptic bouton density was paradoxically associated with decreased cognitive ability. The authors suggest two possible explanations: either the upregulation indicates a type of compensatory response intended to counter the effects of preexisting synaptotoxicity, or upregulated terminals are indicative of an uncoordinated aberrant response not indicative of a well- orchestrated synaptic plasticity. Although neither of these interpretations have been supported experimentally, in light of the cholinergic hippocampal and frontal cortex neuroplasticity data and their association with better cognitive performance, the former seems more likely. Taken together, the cholinergic and glutamatergic findings showing an upregulation of neurotransmitter systems lends support to the suggestion that the hippocampal and cortical synapses are indeed more active during MCI than in AD (Goekoop et al., 2006). To this end, the cortical glutamatergic system needs to be more fully investigated in the hippocampus during the onset of AD particularly in the context of therapeutics. In this regard, the use of memantine, an uncompetitive NMDA receptor antagonist and a FDA-approved treatment for moderate to severe AD has been challenged (Reisberg et al., 2006; Danysz and Parsons, 2012).
An unanswered question is why select cholinergic basal forebrain (CBF) neuron subfields respond with a neuroplastic response and others do not in MCI. In this regard, the number of medial septal/diagonal band cholinergic neurons, which are mainly hippocampal projecting, is relatively spared compared to neurons in other CBF nuclei in AD (Mufson et al., 1989; Vogels et al., 1990) in AD and this region does not lose volume in MCI compared to controls (George et al., 2011; Kilimann et al., 2014). The lack of significant degenerative changes in this region may explain its ability to sprout new cholinergic terminals into the denervated hippocampus in response to entorhinal cortex disconnection. In a similar vein, the upregulation of ChAT activity in the frontal cortex may be related to the observation that anterior medial cholinergic neurons located within the substantia innominata, which innervate the frontal cortex (Mesulam et al., 1983; Bierer et al., 1995), are less vulnerable to neuronal degeneration relative to other cholinergic nucleus basalis subfields in AD (Mufson et al., 1989). These CBF neurons are affected to a greater degree than the hippocampal-projecting septal cholinergic neurons in AD (Mufson et al., 1989; Vogels et al., 1990). Maintenance of the neuronal cholinergic phenotype may, at least in part, explain the ability of these neurons to reinnervate the denervated hippocampus and account for the upregulation of ChAT activity in this region early in the disease (DeKosky et al., 2002). The ability to generate new cholinergic profiles is in contrast to that seen in the MCI frontal cortex where despite an upregulation of ChAT activity there is no net increase in cholinergic fibers and varicosities in MCI (Ikonomovic et al., 2007). These studies demonstrate that a biochemical up-regulation of ChAT in MCI frontal cortex does not reflect regional structural reorganization of cholinergic fibers. However, it likely activity compensates for the decrease in cholinergic fiber/axon varicosities found in the AD frontal cortex. Together, these data suggest that two different processes that result in similar chemoplastic responses drive the upregulation of cholinergic activity seen in the MCI hippocampus and frontal cortex. In either case the “drive” for cholinergic reinnervation is strong.
Hippocampal Plasticity and Neurotrophins in MCI and AD
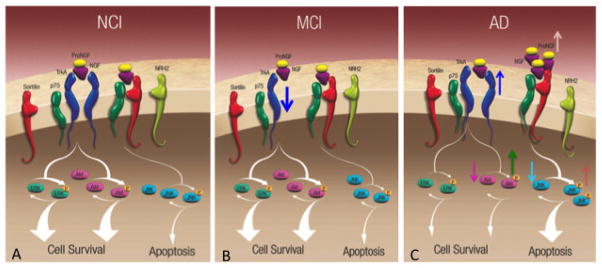
The neurotrophin, Nerve Growth Factor (NGF) is a product of a single gene located on chromosome 1, which codes for two transcripts that give rise to 27 kDa and 35 kDa precursors (Francke et al., 1983; Edwards et al., 1988). NGF is responsible for basal forebrain cholinergic neuron maintenance and survival (Hefti, 1986; Williams et al. 1986); it is produced in the hippocampus and cortex and is retrogradely transported from these regions to the cholinergic neurons within the basal forebrain (Johnson et al., 1987; Seiler and Schwab, 1984). NGF is derived from a precursor protein, proNGF, and is cleaved into a mature form of NGF (Lee et al., 2001). Western blotting revealed that proNGF, not mature NGF, is the predominant form of NGF in the human brain (Fahnestock et al., 2001). NGF binds to two receptors, the cognate NGF tyrosine kinase A (TrkA) receptor and a low affinity p75 pan-neurotrophin receptor (p75NTR) (Ibanez et al., 2002; Chao, 2003; Kaplan and Miller, 2000). NGF binding with TrkA signals downstream survival pathways by activating Akt (Ulrich et al., 1998) while proNGF and p75NTR, together with its co-receptor sortilin (Nykjaer et al., 2004), then activates c-Jun N-terminal protein kinase (JNK) pathways associated with apoptosis (Nykjaer et al., 2005). Since cholinergic basal forebrain neurons located with the medial septal/diagonal band complex are preserved (Mufson 1989; Vogel et al., 1990) and sprout into the molecular layer of the hippocampus in MCI and AD (Geddes et al., 1985; Hyman, 1987), changes in the up- and downstream NGF/proNGF molecular cascade may influence cholinergic plasticity in the hippocampus following perforant path disconnection (Mufson et al., 2012). Despite the ability of the hippocampus to generate replacement of synaptic numbers it is still unclear that the appropriate connections are made and whether the pathologically challenged CNS is able to incorporate an altered circuitry to perform complicated behaviors such as memory and executive functions. Research directed at understanding the effect of CNS plasticity is critical to our understanding of the underlying resilience of the brain during human neurologic disease. The plasticity of the proNGF signaling pathway is particularly important in light of reports that biochemical levels of hippocampal NGF are preserved in MCI and early AD (Mufson et al., 2003).
A recent biochemical study demonstrated that hippocampal proNGF levels increase only in early AD (Mufson et al., 2012), which contrasts to the up-regulation of proNGF seen in both MCI and early stage AD in parietal cortex samples (Peng et al., 2004) obtained from the same set of cases. Western blot analysis revealed that hippocampal TrkA was reduced significantly in MCI compared to NCI and AD. On the other hand, hippocampal p75NTR, sortilin, and its neurotrophin receptor homolog-2 (NRH2) remained stable in the hippocampus (Fig. 7). Interestingly, TrkA was not reduced in MCI cortex, but remained stable in MCI and decreased in early AD (Counts et al., 2004). Hippocampal Akt decreased from NCI to MCI to AD, whereas activated phospho-Akt and the phospho-Akt to Akt ratio were elevated in AD compared to MCI and NCI. Although the precise biological actions of the increase in phospho-Akt remains a challenging question, activated Akt may suppress apoptosis by activating several different anti-apoptotic proteins, suppressing GSK3-mediated apoptotic activities, or by blocking the function of the JNK pathway (Song et al., 2005) late in the disease process. Interestingly, in vivo findings also indicate that p75NTR can also activate Akt via a phosphatidylinositol 3-kinase pathway to facilitate cell survival (Roux and Barker 2002). This phospho-Akt upregulation may be yet another example of hippocampal reorganization, mediating cell survival at a number of levels, depending upon target availability and the requirement for transcriptional or post-transcriptional events to suppress apoptosis within the hippocampus even during the onset of the early stage AD. The role that Akt plays in hippocampal plasticity remains unknown and is an area of active research. Although upstream proNGF receptor binding initiates downstream JNK apoptotic signaling (Mufson et al., 2008), JNK remains stable during the onset of AD and phospho-JNK and the ratio of phospho-JNK to JNK increased significantly in AD compared to NCI and MCI (Mufson et al., 2012). The increase in phospho-JNK may reflect a chronic or accumulative stress process that build during the disease. In the transition from MCI to AD, hippocampal phospho-JNK activation occurs in the face of increased proNGF and phospho-Akt and reduced TrkA, despite no change in amyloid levels (Mufson et al., 2012), which suggests that increasing TrkA and phospho-Akt might offset a shift toward JNK-mediated apoptotic signaling in the AD hippocampus (Fig. 7). Similar to proNGF, it was found that higher hippocampal phospho-JNK levels correlated with lower cognitive test scores, suggesting that pro-apoptotic signaling abnormalities ultimately override the putative compensatory TrkA and AKT-mediated pro-survival cascades as the disease progresses.
Figure 7.

Schematic drawings illustrating changes in the levels of proNGF up- and down stream pathways within the hippocampus during the progression from (A) no cognitive impairment (NCI) to (B) mild cognitive impairment (MCI) to (C) Alzheimer’s disease (AD). Note a down regulation of TrkA (blue arrow) levels in MCI. By contrast, there is an upregulation of TrkA (blue arrow) and proNGF (light brown arrow) in AD. Downstream Akt (dark pink arrow), phopho-Akt (green arrow) increases while JNK is reduced (light blue arrow) and phosphor-JNK (pink arrow) levels increase only in AD. Note that p75NTR, sortilin and neurotrophin receptor homolog-2 (NRH2) remains stable during the progression of AD.
Hippocampal neurogenesis and plasticity
The ability of the CNS to undergo postnatal neurogenesis in the adult brain was once widely assumed not to occur. However, Altman (1963) provided seminal evidence of adult neurogenesis in the granule cells of the dentate gyrus of the hippocampus. Post-natal neurogenesis is now known to occur in at least two brain locations, the subventricular zone of the lateral ventricle (Lois and Alvarez-Buylla, 1993) and the hippocampal subgranular zone (Altman et al., 1965). New neurons, as well as supporting glia, are derived from stem cells residing in these two areas (Doetsch et al., 1999, Johanson et al., 1999). The microenvironment of select hippocampal areas is critical for neurogenesis (Gage et al., 1995). The functional consequence of adult hippocampal neurogenesis is under active investigation. Reduced neurogenesis in the rodent hippocampus results in poorer performance in the Morris water maze, indicating impairment of spatial memory (Rola et al., 2004). On the other hand, neurogenesis reverses memory impairment by altering or repairing dysfunctional neural circuitry (Dash et al., 2001; Akers et al., 2014; Zheng et al., 2013). Hippocampal neurogenesis was increased in AD compared to age-matched individuals (Jin et al., 2004). However, despite this increase in neurogenic markers, there is a significant overall decrease in the number of neurons in the dentate gyrus in AD (West, 1993). There are a few possible explanations for this disconnect. The rate of cell loss may be greater than the rate of formation, especially considering the reduction in neurogenesis with age. Age is a major risk factor for the onset of the behavioral/clinical expression in AD (Gao et al., 1998) and reduces the rate of neurogenesis (Kuhn et al., 1996). Thus, age most likely is a rate-limiting factor in the activation of neurogenesis during the progression of AD. It is also possible that the microenvironment of the AD brain limits either the survivability or differentiation of the newborn neurons, especially in an environment of compromised neurotrophism. The role that neurogenesis plays in the concept of “brain reserve”, which is invoked to explain how people who have extensive brain AD pathology do not exhibit cognitive impairment remains unknown.
Hippocampal Plasticity and AD lesions
It remains unknown whether NFT or SP pathology directly impacts cognitive reserve and therefore the ability to maintain a plastic milieu for protection against dementia onset. Binder and colleagues (Garcia-Sierra et al., 2003; Vana et al. 2011) demonstrated that NFT evolution proceeds from a pretangle to frank tangle formation using site-specific tau antibodies during the progression of AD. During the development of an NFT, the first cellular component to display pretangle material are dendrites and not the perikarya suggesting a protracted NFT developmental phase. Perhaps during this early phase a neuron is still capable of participating in various forms of neural plasticity, including sprouting of new terminals and producing neurotransmitter and neurotrophic receptors. However, it remains to be determined whether appropriate structural and functional integration occurs during neural reorganization within the diseased brain and whether this altered circuitry is sufficiently integrated into the nervous system to perform complicated behavioral and mnemonic tasks. Clinical pathological studies indicate that cholinergic sprouting into the hippocampus occurs during the MCI/prodromal stage of AD when individuals fail to show significant cognitive impairment (DeKosky et al., 2002) despite the presence of tangle pathology in both the hippocampal CA1 sector and entorhinal layer II/III hippocampal projection neurons in MCI and in some aged individuals without cognitive impairment (Mufson et al., 2011, Price et al., 1999, Nelson et al., 2013). These findings suggest that tau dysregulation does not invariably lead to exacerbation of a neurodegenerative phenotype. Perhaps there are tau subclasses that initiate tau loss-of-function or tau gain-of-function mechanisms, which modulate pathogenesis by either exacerbating cellular dysfunction early in the disease or by activating neuronal reorganizational processes, respectively. Thus, the development of subclasses of tau-based therapeutics that specifically target neuroplasticity should be considered as a novel treatment approach aimed at ameliorating or delaying the onset of dementia. If effective, these approaches may prove beneficial for AD as well as other tauopathies.
The role that the key biochemical component of the SP, fibrillar amyloid beta protein, the cleaved product of AβPP processing, plays in brain plasticity is not well understood. However, immunohistochemical investigations of synaptic change in AD revealed that the number of SPs counted in both cortical lamina III and V was significantly greater than in the non-demented controls. However, the number of SPs failed to demonstrate any significant relationship to synaptic numbers in either lamina (Scheff et al., 1990; Scheff and Price, 1993, 2001). Recent data suggests that the amyloid toxic moiety is the oligomeric component of Aβ (Lacor et al. 2007), which damages synapses (Lacor et al., 2007) and results in cognitive impairment (Lesne et al., 2006; Tu et al., 2014). Therefore, further clinical pathological studies are required to determine the relationship between hippocampal plasticity and the oligomeric forms of Aβ as well as other components of proteolytically processed APP, including the C-terminal fragment of APP and other metabolites.
Hippocampal Plasticity and Brain Reserve
Katzman et al., 1988, first introduced the concept of the brain reserve to explain hippocampal structural and biochemical plasticity in the course of AD. Post-mortem examinations in 137 elderly persons revealed a discrepancy between the degree of AD neuropathology and the clinical manifestations at the time of death. Some subjects whose brains had extensive AD pathology had no or very little clinical manifestations of the disease (Katzman et al., 1988). Similar results have since been reported by a number of other investigators (e.g., Mufson et al., 1999; Price et al. 2009, Markesbery et al., 2009; Mufson et al., 2014; Schneider et al., 2009). The seminal paper by Katzman et al. (1988) also reported that these persons had higher brain weights and greater number of neurons as compared to age-matched controls, leading the authors to propose two possible explanations for this phenomenon: these people may have had incipient AD but by some as-yet unknown factor avoided the loss of large numbers of neurons, or alternatively, began with larger brains and more neurons. This led to the concept of a greater brain “reserve”, which might allow for synaptic remodeling as a compensatory mechanism to the early pathobiology of the disease and in turn be able to maintain cognitive abilities in the prodromal stages of the dementia (Mufson et al., 1999; Katzman et al., 1988; Snowdon et al., 1996; Mortimer et al., 1988; DeKosky et al., 2002). It has been suggested that neuroplastic responses are lost as an individual’s dementia progresses (Mesulam, 1999) leading to the consistent observation of a reduction in neuronal viability (Gilmor et al., 1999) and activity in the hippocampus in late-stage AD (DeKosky et al., 2002; Iknonomovic et al., 2003; Davis et al., 1999). An additional phase of plasticity, not evoked until later in the disease, is the development of a neuroplastic response by the inhibitory neuropeptide, galanin (GAL), a G-protein coupled receptor that mediates neurotransmission in the basal forebrain, entorhinal cortex, hippocampus and amygdala (Habert-Ortoli et al., 1994; Smith et al., 1998; Kolakowski et al., 1998; Dutar et al., 1989, Fisone et al., 1987; Coumis et al., 2002; Hartonian et al., 2002; Jhamandas et al., 2002; Mazarati et al., 2000; McDonald et al., 1998; Mufson et al., 2000) and plays an important role in memory and attention (Crawley et al., 1996; Wrenn, 2001) and neuroplasticity (Counts et al., 2003). Basal forebrain GAL immunoreactive fibers hypertrophy and hyperinnervate remaining cholinergic neurons in the medial septal diagonal band complex (Mufson et al., 1993) and nucleus basalis in AD (Bowser et al., 1997; Chan-Palay et al., 1988) but not in MCI (Counts et al., 2006). Single cell gene expression studies showed an upregulation of ChAT expression as well as stable levels of mRNAs encoding select subclasses of protein phosphatase subunits (PP1a and PP1g) in GAL hyperinnervated CBF neurons but downregulation in those not hyperinnervated by GAL in AD (Counts et al., 2008; 2009; 2010). Reduced activity of PP1 and PP2A subunits is implicated in tau hyperphosphorylation, which in turn precipitates NFT pathology and subsequent cytoskeletal destabilization in vulnerable neurons (Mawal-Dewan et al., 2004: Yoshiyama et al., 2013; Wang et al., 2015). Taken together, these observations suggest that GAL remodeling may delay NFT pathology in those remaining non tangle-bearing cholinergic neurons, which project to the hippocampus and cortex in AD (Chan Palay et al., 1988; Mufson et al., 1993; Bowser et al. 1997).
By contrast, there are no reports of a similar GAL fiber plasticity response in the hippocampus during the prodromal phase of AD. However, [125I]h GAL binding experiments using AD tissue have revealed a ~50–100% increase in of binding sites in the hippocampus including the stratum radiatum, pyramidalis, and oriens in the CA1 region, as well as the stratum radiatum in CA3, and the hilus of the dentate gyrus and a ~10–30% increase in the entorhinal cortex in AD (Rodriguez-Puertas et al., 1997). Another in vivo autoradiographic investigation of GAL receptor binding sites revealed a ~3-fold increase in layer II of entorhinal cortex in early compared to late stage AD, where binding levels were increased only slightly over controls (Perez et al., 2002). Both basal forebrain GAL fiber and hippocampal complex receptor plasticity occur on a background of extensive SP and NFT pathology late in the disease process. Since GAL has been suggested to have neuroprotective signaling properties (Wynick et al., 2002, Counts 2009) and prevent NFT formation (Counts et al., 2003), GAL overexpression may be yet another example of a plastic response aimed at maintaining remaining neural function but later in the disease process of AD. The precise role that brain reserve plays in any plasticity response during the onset of AD is still unknown.
Concluding Comments
The hippocampus displays multiple structural, neurochemical, molecular, and cellular alterations during MCI that support its role as a hub for neuroplastic remodeling within the medial temporal lobe. These actions may preserve function of this memory circuit in the face of mounting pathology. Inter-individual differences in the ability of the hippocampus to undergo these compensatory changes may result in the heterogeneity of clinical pathologic findings, supporting the concept of cognitive reserve, whereby certain individuals remain cognitively intact despite the substantive accumulation of AD pathology, while others do not. We suggest that hippocampal neuroplastic pathways provide compelling substrates for therapeutic intervention, wherein mechanisms of brain reserve might be harnessed to modify disease progression in MCI as a molecular switch to counteract or to suppress on select pathogenic pathways that drive the disease.
Highlights.
Hippocampal plasticity in AD
Molecular plasticity in AD
Hippocampal vulnerability in AD
Acknowledgments
This study was supported by grants PO1AG014449, RO1AG043375 and P30AG10161 from the National Institute on Aging, National Institutes of Health. We are indebted to the Catholic nuns, priests, and lay brothers who participated in the Rush Religious Orders Study, the University of Kentucky RADC clinical cohort and to all ADC center participants that provided clinical and pathological data to the various studies discussed in this review.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adolfsson R, Gottfries CG, Roos BE, Winblad B. Changes in the brain catecholamines in patients with dementia of Alzheimer type. Br J Psychiatry. 1979;135:216–223. doi: 10.1192/bjp.135.3.216. [DOI] [PubMed] [Google Scholar]
- Altman J. Audioradiographic investigation of cell proliferation in the brains of rats and cats. Anat Rec. 1963;145:573–591. doi: 10.1002/ar.1091450409. [DOI] [PubMed] [Google Scholar]
- Altman J. Audioradiographic and histological evidence of postnatal neurogenesis in rats. J Comp Neurol. 1965;124:319–335. doi: 10.1002/cne.901240303. [DOI] [PubMed] [Google Scholar]
- Alzheimer A. Über einen eigenartigen schweren Erkrankungsprozeß der Hirnrinde. Neurologisches Centralblatt. 1906;23:1129–1136. [Google Scholar]
- Alzheimer A. Uber eigenartige Krankheitsfaelle des Spaetern Alters, Zeitschrift fuer die gesamte. Neurol Psych. 1911;4:256–286. [Google Scholar]
- Amaral D, Lavenex P. Hippocampal Neuroanatomy. In: Andersen P, Morris R, Amaral D, Bliss T, O’Keefe J, editors. The Hippocampus Book. Oxford University Press; 2006. [Google Scholar]
- Arriagada PV, Growdon JH, Hedley-Whyte ET, et al. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurol. 1992;42:631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- Baas PW, Heideman SR. Microtubule reassembly from nucleating fragments during the regrowth of amputated neurites. J Cell Biol. 1986;104:917–927. doi: 10.1083/jcb.103.3.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berchtold NC, Sabbagh MN, Beach TG, Kim RC, Cribbs DH, Cotman CW. Brain gene expression patterns differentiate mild cognitive impairment from normal aged and alzheimer’s disease. Neurobio Aging. 2014;35:1961–1972. doi: 10.1016/j.neurobiolaging.2014.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertoni-Fredarri C, Fattoretti P, Casoli T, Meier-Ruge W, Ulrich J. Mophological adaptive response of synaptic junctional zones in the human dentate gyrus during aging and Alzheimer’s disease. Brain Res. 1990;517:69–75. doi: 10.1016/0006-8993(90)91009-6. [DOI] [PubMed] [Google Scholar]
- Bierer LM, Hof PR, Purohit DP, Carlin L, Schmeidler J, Davis KL, Perl DP. Neocortical neurofibrillary tangles correlate with dementia severity in Alzheimer’s disease. Arch Neurol. 1995;52:81–88. doi: 10.1001/archneur.1995.00540250089017. [DOI] [PubMed] [Google Scholar]
- Bierer LM, Haroutunian V, Gabriel S, Knott PJ, Carlin LS, Purohit DP, Perl DP, Schmeidler J, Kanof P, Davis KL. Neurochemical correlates of dementia severity in Alzheimer’s disease: relative importance of the cholinergic deficits. J Neurochem. 1995;64:749–760. doi: 10.1046/j.1471-4159.1995.64020749.x. [DOI] [PubMed] [Google Scholar]
- Bishop NE. Dynamics of endosomal sorting. Int Rev Cytol. 2003;232:1–57. doi: 10.1016/s0074-7696(03)32001-7. [DOI] [PubMed] [Google Scholar]
- Bowser R, Kordower JH, Mufson EJ. A confocal microscopic analysis of galaninergic hyperinnervation of cholinergic basal forebrain neurons in Alzheimer’s disease. Brain Pathol. 1997;7:723–730. doi: 10.1111/j.1750-3639.1997.tb01058.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K. The pathological process underlying Alzheimer’s disease in individuals under thirty. Acta Neuropathol. 2011;121:171–181. doi: 10.1007/s00401-010-0789-4. [DOI] [PubMed] [Google Scholar]
- Bell KF, Bennett DA, Cuello AC. Paradoxical upregulation of glutamatergic presynaptic boutons during mild cognitive impairment. J Neurosci. 2007;27:10810–10817. doi: 10.1523/JNEUROSCI.3269-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo AM, Hamilton DJ, Nixon RA. Lysosomal abnormalities in degenerating neurons link neuronal compromise to senile plaque development in Alzheimer disease. Brain Res. 1994;640:68–80. doi: 10.1016/0006-8993(94)91858-9. [DOI] [PubMed] [Google Scholar]
- Cataldo AM, Barnett JL, Berman SA, Li J, Quarless S, Bursztajn S, et al. Gene expression and cellular content of cathepsin D in alzheimer’s disease brain: Evidence for early up-regulation of the endosomal-lysosomal system. Neuron. 1995;14:671–680. doi: 10.1016/0896-6273(95)90324-0. [DOI] [PubMed] [Google Scholar]
- Cataldo AM, Hamilton DJ, Barnett JL, Paskevich PA, Nixon RA. Properties of the endosomal-lysosomal system in the human central nervous system: disturbances mark most neurons in populations at risk to degenerate in Alzheimer’s disease. J Neurosci. 1996;16:186–199. doi: 10.1523/JNEUROSCI.16-01-00186.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo AM, Barnett JL, Pieroni C, Nixon RA. Increased neuronal endocytosis and protease delivery to early endosomes in sporadic Alzheimer’s disease: neuropathologic evidence for a mechanism of increased beta-amyloidogenesis. J Neurosci. 1997;17:6142–6151. doi: 10.1523/JNEUROSCI.17-16-06142.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan-Palay V. Galanin hyperinnervates surviving neurons of the human basal nucleus of Meynert in dementias of Alzheimer’s and Parkinson’s disease: a hypothesis for the role of galanin in accentuating cholinergic dysfunction in dementia. J Comp Neurol. 1988;273:543–557. doi: 10.1002/cne.902730409. [DOI] [PubMed] [Google Scholar]
- Chao MV. Retrograde transport redux. Neuron. 2003;39:1–2. doi: 10.1016/s0896-6273(03)00401-x. [DOI] [PubMed] [Google Scholar]
- Chapman PF, White GL, Jones MW, et al. Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neuro. 1999;2:271–276. doi: 10.1038/6374. [DOI] [PubMed] [Google Scholar]
- Cotman CW, Matthews DA, Taylor D, Lynch G. Synaptic rearrangement in the dentate gyrus: histochemical evidence of adjustments after lesions in immature and adult rats. Proc Natl Acad Sci. 1973;70:3473–3477. doi: 10.1073/pnas.70.12.3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotman CW, Lynch GS. Reactive synaptogenesis in the adult nervous system. In: Barondes SH, editor. Neuronal Recognition. New York: Plenum; 1976. pp. 69–108. [Google Scholar]
- Cotman CW, Scheff SW. Compensatory synapse growth in aged animals after neuronal death. Mech Aging Dey. 1979;9:103–117. doi: 10.1016/0047-6374(79)90124-6. [DOI] [PubMed] [Google Scholar]
- Counts SE, Perez SE, Ginsberg SD, De Lacalle S, Mufson EJ. Galanin in Alzheimer disease. Mol Interv. 2003;3:137–156. doi: 10.1124/mi.3.3.137. [DOI] [PubMed] [Google Scholar]
- Counts SE, Nadeem M, Wuu J, Ginsberg SD, Saragoyi HU, Mufson EJ. Reduction of cortical TrkA but not p75(NTR) protein in early-stage Alzheimer’s disease. Ann Neurol. 2004;56:520–531. doi: 10.1002/ana.20233. [DOI] [PubMed] [Google Scholar]
- Counts SE, Chen EY, Che S, Ikonomovic MD, Wuu J, Ginsberg SD, Dekosky ST, Mufson EJ. Galanin fiber hypertrophy within the cholinergic nucleus basalis during the progression of Alzheimer’s disease. Dement Geriatr Cogn Disord. 2006;21:205–214. doi: 10.1159/000090906. [DOI] [PubMed] [Google Scholar]
- Counts SE, He B, Che S, Ginsberg SD, Mufson EJ. Galanin hyperinnervation upregulates choline acetyltransferase expression in cholinergic basal forebrain neurons in Alzheimer’s disease. Neurodegener Dis. 2008;5:228–231. doi: 10.1159/000113710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counts SE, He B, Che S, Ginsberg SD, Mufson EJ. Galanin fiber hyperinnervation preserves neuroprotective gene expression in cholinergic basal forebrain neurons in Alzheimer’s disease. J Alzheimers Dis. 2009;18:885–896. doi: 10.3233/JAD-2009-1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counts SE, Perez SE, Ginsberg SD, Mufson EJ. Neuroprotective role for galanin in Alzheimer’s disease. EXS. 2010;102:143–162. doi: 10.1007/978-3-0346-0228-0_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counts SE, Mufson EJ. Noradrenaline activation of neurotrophic pathways protects against neuronal amyloid toxicity. J Neurochem. 2010;113:649–660. doi: 10.1111/j.1471-4159.2010.06622.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counts SE, He B, Nadeem M, Wuu J, Mufson EJ. Hippocampal drebrin loss in mild cognitive impairment. Neurodeg Dis. 2011;10:216–219. doi: 10.1159/000333122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counts SE, Alldred MJ, Che S, Ginsberg SD, Mufson EJ. Synaptic gene dysregulation within hippocampal CA1 pyramidal neurons in mild cognitive impairment. Neuropharm. 2014;79:172–179. doi: 10.1016/j.neuropharm.2013.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coumis U, Davies CH. The effects of galanin on long-term synaptic plasticity in the CA1 area of rodent hippocampus. Neurosci. 2002;112:173–182. doi: 10.1016/s0306-4522(02)00007-6. [DOI] [PubMed] [Google Scholar]
- Cowburn R, Hardy J, Roberts P, Briggs R. Presynaptic and postsynaptic glutamatergic function in Alzheimer’s disease. Neurosci Lett. 1988;86:109–113. doi: 10.1016/0304-3940(88)90192-9. [DOI] [PubMed] [Google Scholar]
- Cowburn R, Hardy J, Roberts P, Briggs R. Regional distribution of pre- and postsynaptic glutamatergic function in Alzheimer’s disease. Brain Res. 1988b;452:403–407. doi: 10.1016/0006-8993(88)90048-0. [DOI] [PubMed] [Google Scholar]
- Crawley JN. Minireview. Galanin-acetylcholine interactions: relevance to memory and Alzheimer’s disease. Life Sci. 1996;58:2185–2199. doi: 10.1016/0024-3205(96)00093-8. [DOI] [PubMed] [Google Scholar]
- Cross AJ, Slater P, Candy JM, Perry EK, Perry RH. Glutamate deficits in Alzheimer’s disease. J Neurosurg Psychiatry. 1987;50:357–358. [Google Scholar]
- Cruts M, Theuns J, Van Broeckhoven C. Locus-specific mutation databases for neurodegenerative brain diseases. Human Mut. 2012;33:1340–1344. doi: 10.1002/humu.22117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danysz W, Parsons CG. Alzheimer’s disease, beta-amyloid, glutamate, NMDA receptors and memantine--searching for the connections. Br J Pharmacol. 2012;167:324–352. doi: 10.1111/j.1476-5381.2012.02057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash PK, Mach SA, Moore AN. Enhanced neurogenesis in the rodent hippocampus following traumatic brain injury. J Neurosci Res. 2001;63:313–319. doi: 10.1002/1097-4547(20010215)63:4<313::AID-JNR1025>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Davies P, Maloney AJ. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet. 1976;2:1403. doi: 10.1016/s0140-6736(76)91936-x. [DOI] [PubMed] [Google Scholar]
- Davis KL, Mohs RC, Marin D, et al. Cholinergic markers in elderly patients with early signs of Alzheimer disease. JAMA. 1999;281:1401–1406. doi: 10.1001/jama.281.15.1401. [DOI] [PubMed] [Google Scholar]
- Deinhardt K, Salinas S, Verastegui C, Watson R, Worth D, Hanrahan S, Bucci C, Schiavo G. Rab5 and rab7 control endocytic sorting along the axonal retrograde transport pathway. Neuron. 2006;52:293–305. doi: 10.1016/j.neuron.2006.08.018. [DOI] [PubMed] [Google Scholar]
- DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol. 1990;27:457–464. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- DeKosky ST, Ikonomovic MD, Styren SD, et al. Upregulation of choline acetyltransferase activity in hippocampus and frontal cortex of elderly subjects with mild cognitive impairment. Ann Neurol. 2002;51:145–155. doi: 10.1002/ana.10069. [DOI] [PubMed] [Google Scholar]
- Dewar D, Chalmers DT, Graham DI, McCulloch J. Glutamate metabotropic and AMPA binding sites are reduced in Alzheimer’s disease: an autoradiographic study of the hippocampus. Brain Res. 1991;553:58–64. doi: 10.1016/0006-8993(91)90230-s. [DOI] [PubMed] [Google Scholar]
- Ding Q, Dimayuga E, Keller JN. Oxidative damage, protein synthesis, and protein degradation in Alzheimer’s disease. Curr Alzheimer Res. 2007;4:73–79. doi: 10.2174/156720507779939788. [DOI] [PubMed] [Google Scholar]
- Doetsch F, Caille I, Lim DA, Garcia-Verdugo JM, Alvarez-Buylla A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell. 1999;97:703–716. doi: 10.1016/s0092-8674(00)80783-7. [DOI] [PubMed] [Google Scholar]
- Dunn WA., Jr Studies on the mechanisms of autophagy: maturation of the autophagic vacuole. J Cell Biol. 1990;110:1935–1945. doi: 10.1083/jcb.110.6.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutar P, Lamour Y, Nicoll RA. Galanin blocks the slow cholinergic EPSP in CA1 pyramidal neurons from ventral hippocampus. Eur J Pharmacol. 1989;164:355–360. doi: 10.1016/0014-2999(89)90477-9. [DOI] [PubMed] [Google Scholar]
- Edwards RH, Selby MJ, Mobley WC, Weinrich SL, Hruby DE, Rutter WJ. Processing and secretion of nerve growth factor: expression in mammalian cells with a vaccinia vector. Mol Cell Biol. 1988;8:2456–2464. doi: 10.1128/mcb.8.6.2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison DW, Beal MF, Mazurek MF, Bird ED, Martin JB. A postmortem study of amino acid neurotransmitters in Alzheimer’s disease. Ann Neurol. 1986;20:616–621. doi: 10.1002/ana.410200510. [DOI] [PubMed] [Google Scholar]
- Fahnestock M, Mchalski B, Xu B, Coughlin MD. The precursor pro-nerve growth factor is the predominant form of nerve growth factor in brain and is increased in Alzheimer’s disease. Mol Cell Neurosci. 2001;18:210–220. doi: 10.1006/mcne.2001.1016. [DOI] [PubMed] [Google Scholar]
- Fifkova E. Two types of terminal degeneration in the molecular layer of the dentate fascia following lesions of the entorhinal cortex. Brain Res. 1975;96:169–175. doi: 10.1016/0006-8993(75)90592-2. [DOI] [PubMed] [Google Scholar]
- Fisone G, Wu CF, Consolo S, Nordstrom O, Brynne N, Bartfai T, Melander T, Hokfelt T. Galanin inhibits acetylcholine release in the ventral hippocampus of the rat: histochemical, autoradiographic, in vivo, and in vitro studies. Proc Natl Acad Sci. 1987;84:7339–7343. doi: 10.1073/pnas.84.20.7339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke U, de Martinville B, Coussens L, Ullrich A. The human gene for the beta subunit of nerve growth factor is located on the proximal short arm of chromosome 1. Science. 1983;222:1248–1251. doi: 10.1126/science.6648531. [DOI] [PubMed] [Google Scholar]
- Gage FH, Coates PW, Palmer TD, Kuhn HG, Fisher LJ, Suhonen JO, Peterson DA, Suhr ST, Ray J. Survival and differentiation of adult neuronal progenitor cells transplanted to the adult brain. Proc Natl Acad Sci USA. 1995;92:11879–11883. doi: 10.1073/pnas.92.25.11879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S, Hendrie HC, Hall KS, Hui S. The relationships between age, sex, and the incidence of dementia and Alzheimer disease: a meta-analysis. Arch Gen Psychiatry. 1998;55:809–815. doi: 10.1001/archpsyc.55.9.809. [DOI] [PubMed] [Google Scholar]
- Garcia-Sierra F, Ghoshal N, Quinn B, Berry RW, Binder LI. Conformational changes and truncation of tau protein during tangle evolution in Alzheimer’s disease. J Alzheimers Dis. 2003;5:65–77. doi: 10.3233/jad-2003-5201. [DOI] [PubMed] [Google Scholar]
- Geddes JW, Monaghan DT, Cotman CW, Lott IT, Kim RC, Chiu HC. Plasticity of hippocampal circuitry in Alzheimer’s disease. Science. 1985;230:1179–1181. doi: 10.1126/science.4071042. [DOI] [PubMed] [Google Scholar]
- Geddes JW, Chang-Chui H, Cooper SM, Lott IT, Cotman CW. Density and distribution of NMDA receptors in the human hippocampus in Alzheimer’s disease. Brain Res. 1986;399:156–161. doi: 10.1016/0006-8993(86)90611-6. [DOI] [PubMed] [Google Scholar]
- Geddes JW, Anderson KJ, Cotman CW. Senike plaques as aberrant sprout-stimulating structures. Exp Neurol. 1986;94:767–776. doi: 10.1016/0014-4886(86)90254-2. [DOI] [PubMed] [Google Scholar]
- Geddes JW, Cotman CW. Plasticity in hippocampal excitatory amino acid receptors in Alzheimer’s disease. Neurosci Res. 3:672–678. doi: 10.1016/0168-0102(86)90062-3. [DOI] [PubMed] [Google Scholar]
- George S, Mufson EJ, Leurgans S, Shah RC, Ferrari C, Detoledo-Morrell L. MRI-based volumetric measurement of the substantia innominata in amnestic MCI and mild AD. Neurobiol Aging. 2011;32:1756–1764. doi: 10.1016/j.neurobiolaging.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannakopoulos P, Herrmann FR, Bussiere T, Bouras C, Kovari E, Perl DP, Morrison JH, Gold G, Hof PR. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology. 2003;60:1495–1500. doi: 10.1212/01.wnl.0000063311.58879.01. [DOI] [PubMed] [Google Scholar]
- Gilmor ML, Erickson JD, Varoqui H, et al. Preservation of nucleus basalis neurons containing choline acetyltransferase and the vesicular acetylcholine transporter in the elderly with mild cognitive impairment and early Alzheimer’s disease. J Comp Neurol. 1999;411:693–704. [PubMed] [Google Scholar]
- Ginsberg SD, Alldred MJ, Counts SE, et al. Microarray analysis of hippocampal CA1 neurons implicates early endosomal dysfunction during Alzheimer’s disease progression. Biol Psychiatry. 2010a;68:885–893. doi: 10.1016/j.biopsych.2010.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg SD, Mufson EJ, Counts SE, Wuu J, Alldred MJ, Nixon RA, Che S. Regional selectivity of rab5 and rab7 protein upregulation in mild cognitive impairment and Alzheimer’s disease. J Alzheimers Dis. 2010b;22:631–639. doi: 10.3233/JAD-2010-101080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg SD. Glutamatergic neurotransmission expression profiling in the mouse hippocampus after perforant-path transection. Am J Geriatr Psychiatry. 2005;13:1052–1061. doi: 10.1176/appi.ajgp.13.12.1052. [DOI] [PubMed] [Google Scholar]
- Ginsberg SD, Hemby SE, Lee VMY, Eberwine JH, Trojanowski JQ. Expression profile of transcripts in Alzheimer’s disease neurofibrillary tangle-bearing CA1 neurons identifies new potential mediators of neurodegeneration. Ann Neurol. 2000;48:77–87. [PubMed] [Google Scholar]
- Ginsberg SD, Elarova I, Ruben M, Tan F, Counts SE, Eberwine JH, Trojanowski JQ, Hemby SE, Mufson EJ, Che S. Single cell gene expression analysis: implications for neurodegenerative and neuropsychiatric disorders. Neurochem Res. 2004;29:1053–1064. doi: 10.1023/b:nere.0000023593.77052.f7. [DOI] [PubMed] [Google Scholar]
- Ginsberg SD, Che S. Expression profile analysis within the human hippocampus: comparison of CA1 and CA3 pyramidal neurons. J Comp Neurol. 2005;487:107–118. doi: 10.1002/cne.20535. [DOI] [PubMed] [Google Scholar]
- Ginsberg SD, Che S, Counts SE, Mufson EJ. Single cell gene expression profiling in Alzheimer’s disease. NeuroRx. 2006;3:302–318. doi: 10.1016/j.nurx.2006.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg SD. Alterations in discrete glutamate receptor subunits in adult mouse dentate gyrus granule cells following perforant path transection. Anal Bioanal Chem. 2010;397:3349–3358. doi: 10.1007/s00216-010-3826-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goekoop R, Scheltens P, Barkhof F, Rombouts S. Cholinergic challenge in Alzheimer patients and mild cognitive impairment differentially affects hippocampal activation--a pharmacological fMRI study. Brain. 2006;129:141–157. doi: 10.1093/brain/awh671. [DOI] [PubMed] [Google Scholar]
- Gomez-Isla T, Price JL, McKeel DW, Morris JC, Growdon JH, Hyman BT. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J Neurosci. 1996;16:4491–4500. doi: 10.1523/JNEUROSCI.16-14-04491.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon PB, Hoyvik H, Seglen PO. Prelysosomal and lysosomal connections between autophagy and endocytosis. Biochem J. 1992;283:361–369. doi: 10.1042/bj2830361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon PB, Seglen PO. Prelysosomal convergence of autophagic and endocytic pathways. Biochem Biophys Res Commun. 1988;151:40–47. doi: 10.1016/0006-291x(88)90556-6. [DOI] [PubMed] [Google Scholar]
- Greenamyre JT, Penney JB, Young AB, D’Amato CJ, Hicks SP, Shoulson I. Alterations in L-glutamate binding in Alzheimer’s and Huntington’s diseases. Science. 1985;227:1496–1499. doi: 10.1126/science.2858129. [DOI] [PubMed] [Google Scholar]
- Greenamyre JT, Penney JB, D’Amato CJ, Young AB. Dementia of the Alzheimer’s Type: Changes in Hippocampal L-[3H]Glutamate Binding. J Neurochem. 1987;48:543–551. doi: 10.1111/j.1471-4159.1987.tb04127.x. [DOI] [PubMed] [Google Scholar]
- Grudzien A, Shaw P, Weintraub S, Bigio E, Mash DC, Mesulam MM. Locus coeruleus neurofibrillary degeneration in aging, mild cognitive impairment and early Alzheimer’s disease. Neurobiol Aging. 2007;28:327–335. doi: 10.1016/j.neurobiolaging.2006.02.007. [DOI] [PubMed] [Google Scholar]
- Guillozet AL, Weintraub S, Mash DC, Mesulam MM. Neurofibrillary tangles, amyloid, and memory in aging and mild cognitive impairment. Arch Neurol. 2003;60:729–736. doi: 10.1001/archneur.60.5.729. [DOI] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Cellular processing of beta-amyloid precursor protein and the genesis of amyloid beta-peptide. Cell. 1993;75:1039–1042. doi: 10.1016/0092-8674(93)90312-e. [DOI] [PubMed] [Google Scholar]
- Habert-Ortoli E, Amiranoff B, Loquet I, Laburthe M, Mayaux JF. Molecular cloning of a functional human galanin receptor. Proc Natl Acad Sci. 1994;91:9780–9783. doi: 10.1073/pnas.91.21.9780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Cowburn R, Barton A, Reynolds G, Lofdahl E, O’Carroll A, Wester P, Winblad B. Region-specific loss of glutamate innervation in Alzheimer’s disease. Neurosci Lett. 1987;73:77–80. doi: 10.1016/0304-3940(87)90034-6. [DOI] [PubMed] [Google Scholar]
- Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci. 1991;12:383–388. doi: 10.1016/0165-6147(91)90609-v. [DOI] [PubMed] [Google Scholar]
- Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- Hartonian I, Mufson EJ, de Lacalle S. Long-term plastic changes in galanin innervation in the rat basal forebrain. Neurosci. 2002;115:787–795. doi: 10.1016/s0306-4522(02)00453-0. [DOI] [PubMed] [Google Scholar]
- Hatanpaa K, Isaacs KR, Shirao T, Brady DR, Rapoport SI. Loss of proteins regulating synaptic plasticity in normal aging of the human brain and in Alzheimer disease. J Neuropathol Exp Neurol. 1999;58:637–643. doi: 10.1097/00005072-199906000-00008. [DOI] [PubMed] [Google Scholar]
- Hefti F. Nerve growth factor promotes survival of septal cholinergic neurons after fimbral transections. J Neurosci. 1986;6:2155–2162. doi: 10.1523/JNEUROSCI.06-08-02155.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honer WG1, Barr AM, Sawada K, Thornton AE, Morris MC, Leurgans SE, Schneider JA, et al. Cognitive reserve, presynaptic proteins and dementia in the elderly. Transl Psy. 2012;2:1–8. doi: 10.1038/tp.2012.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman BT, Van Hoesen GW, Damasio AR, Barnes CL. Alzheimer’s disease: cell-specific pathology isolates the hippocampal formation. Science. 1984;225:1168– 1170. doi: 10.1126/science.6474172. [DOI] [PubMed] [Google Scholar]
- Hyman BT, Van Hoesen GW, Kromer LJ, Damasio AR. Perforant pathway changes and the memory impairment of Alzheimer’s disease. Ann Neurol. 1986;20:472–481. doi: 10.1002/ana.410200406. [DOI] [PubMed] [Google Scholar]
- Hyman BT, Van Hoesen GW, Damasio AR. Alzheimer’s disease: Glutamate depletion in the hippocampal perforant pathway zone. Ann Neurol. 1987;22:37–40. doi: 10.1002/ana.410220110. [DOI] [PubMed] [Google Scholar]
- Hyman BT, Kromer BA, Van Hoesen GW. Reinnervation of the hippocampus perforant pathway zone in Alzheimer’s disease. Ann Neurol. 1987;21:259–267. doi: 10.1002/ana.410210307. [DOI] [PubMed] [Google Scholar]
- Hyman BT, Van Hoesen GW, Damasio AR. Memory-related neural systems in Alzheimer’s disease: an anatomic study. Neurology. 1990;40:1721–1730. doi: 10.1212/wnl.40.11.1721. [DOI] [PubMed] [Google Scholar]
- Ibanez CF. Jekyll-Hyde neurotrophins: the story of proNGF. Trends Neurosci. 2002;25:284–286. doi: 10.1016/s0166-2236(02)02169-0. [DOI] [PubMed] [Google Scholar]
- Ikonomovic MD, Mizukami K, Davies P, Hamilton R, Sheffield R, Armstrong DM. The loss of GluR2(3) immunoreactivity precedes neurofibrillary tangle formation in the entorhinal cortex and hippocampus of Alzheimer brains. J Neuropathol Exp Neurol. 1997;56:1018–1027. doi: 10.1097/00005072-199709000-00007. [DOI] [PubMed] [Google Scholar]
- Ikonomovic MD, Mizukami K, Warde D, Sheffield R, Hamilton R, Wenthold RJ, Armstrong DM. Distribution of glutamate receptor subunit NMDAR1 in the hippocampus of normal elderly and patients with Alzheimer’s disease. Exp Neurol. 1999;160:194–204. doi: 10.1006/exnr.1999.7196. [DOI] [PubMed] [Google Scholar]
- Ikonomovic MD, Mufson EJ, Wuu J, et al. Cholinergic plasticity in hippocampus of individuals with mild cognitive impairment: correlation with Alzheimer’s neuropathology. J Alzheimers Dis. 2003;5:39–48. doi: 10.3233/jad-2003-5106. [DOI] [PubMed] [Google Scholar]
- Ikonomovic MD, Abrahamson EE, Isanski BA, Wuu J, Mufson EJ, DeKosky ST. Superior frontal cortex cholinergic axon density in mild cognitive impairment and early Alzheimer disease. Arch of Neurol. 2007;64:1312–1317. doi: 10.1001/archneur.64.9.1312. [DOI] [PubMed] [Google Scholar]
- Iulita MF, Do CS, Ower AK, Fortress AM, Aguilar LF, Hanna M, Wisniewski T, Granholm AC, Buhusi M, Busciglio J, Cuello AC. Nerve growth factor metabolic dysfunction in Down’s syndrome brains. Brain. 2014;137:860–872. doi: 10.1093/brain/awt372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger KA, Attems J. Neuropathological approaches to cerebral aging and neuroplasticity. Dialogues in Clinical Neuroscience. 2013;15:29–43. doi: 10.31887/DCNS.2013.15.1/kjellinger. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhamandas JH, Harris KH, MacTavish D, Jassar BS. Novel excitatory actions of galanin on rat cholinergic basal forebrain neurons: implications for its role in Alzheimer’s disease. J Neurophysiol. 2002;87:696–704. doi: 10.1152/jn.00416.2001. [DOI] [PubMed] [Google Scholar]
- Jin K, Peel AL, Mao XO, Xie L, Cottrell BA, Henshall DC, Greenberg DA. Increased hippocampal neurogenesis in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2004;101:343–347. doi: 10.1073/pnas.2634794100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson CB, Svensson M, Wallstedt L, Janson AM, Frisen J. Neural stem cells in the adult brain. Exp Cell Res. 1999;253:733–736. doi: 10.1006/excr.1999.4678. [DOI] [PubMed] [Google Scholar]
- Johnson JK, Pa J, Boxer AL, et al. Baseline predictors of clinical progression among patients with dysexecutive mild cognitive impairment. Dement Geriatr Cogn Disord. 2010;30:344–351. doi: 10.1159/000318836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan DR, Miller FD. Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol. 2000;10:381–391. doi: 10.1016/s0959-4388(00)00092-1. [DOI] [PubMed] [Google Scholar]
- Katzman R, Terry R, DeTeresa R, et al. Clinical, pathological and neurochemical changes in dementia: a subgroup with preserved mental status and numerous neocortical plaques. Ann Neurol. 1988;23:138–144. doi: 10.1002/ana.410230206. [DOI] [PubMed] [Google Scholar]
- Kennedy MB, Beale HC, Carlisle HJ, Washburn LR. Integration of biochemical signalling in spines. Nat Rev Neurosci. 2005;6:423–434. doi: 10.1038/nrn1685. [DOI] [PubMed] [Google Scholar]
- Kilimann I, Grothe M, Heinsen H, Alho EJ, Grinberg L, Amaro E, et al. Subregional basal forebrain atrophy in Alzheimer’s disease: a multicenter study. J Alzheimers Dis. 2014;40:687–700. doi: 10.3233/JAD-132345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klink R, Alonso A. Morphological characteristics of layer II projection neurons in the rat medial entorhinal cortex. Hippocampus. 1997;7:571–583. doi: 10.1002/(SICI)1098-1063(1997)7:5<571::AID-HIPO12>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Koffie RM, Meyer-Luehmann M, Hashimoto T, et al. Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci U S A. 2009;106:4012–4017. doi: 10.1073/pnas.0811698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolakowski LF, O’Neill GP, Howard AD, Broussard SR, Sullivan KA, Feighner SD, Sawzdargo M, Nguyen T, Kargman S, Shiao LL, et al. Molecular characterization and expression of cloned human galanin receptors GALR2 and GALR3. J Neurochem. 1998;71:2239– 2251. doi: 10.1046/j.1471-4159.1998.71062239.x. [DOI] [PubMed] [Google Scholar]
- Kordower JH, Chu Y, Stebbins GT, et al. Loss and atrophy of layer II entorhinal cortex neurons in elderly people with mild cognitive impairment. Ann Neurol. 2001;49:202–213. [PubMed] [Google Scholar]
- Kuhn HG, Gage FH. Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J Neurosci. 1996;16(6):2027–2033. doi: 10.1523/JNEUROSCI.16-06-02027.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J Neurosci. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaVail JH, LaVail MM. Retrograde axonal transport in the central nervous system. Science. 1974;176:1416–1417. doi: 10.1126/science.176.4042.1416. [DOI] [PubMed] [Google Scholar]
- Lee R, Kermani P, Teng KK, Hempstead BL. Regulation of cell survival by secreted proneurotrophins. Science. 2001;294:1945–1948. doi: 10.1126/science.1065057. [DOI] [PubMed] [Google Scholar]
- Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Liu J, Lamb D, Chou MM, Liu YJ, Li G. Nerve growth factor-mediated neurite out growth via regulation of Rab5. Mol Biol Cell. 2007;18:1375–1384. doi: 10.1091/mbc.E06-08-0725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lois C, Alvarez-Buylla A. Proliferating subventricular zone cells in the adult mammalian forebrain can differentiate into neurons and glia. Proc Natl Acad Sci USA. 1993;90:2074–2077. doi: 10.1073/pnas.90.5.2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorente de Nó R. Studies on the structure of the cerebral cortex. II. Continuation of the study of the Ammonic system. J Psychol Neurol. 1934;46:113–177. [Google Scholar]
- Lovell MA, Markesbery WA. Oxidative damage in mild cognitive impairment and early Alzheimer’s disease. J Neurosci Res. 2007;85:3036–3040. doi: 10.1002/jnr.21346. [DOI] [PubMed] [Google Scholar]
- Mann DM, Yates PO, Hawkes J. The pathology of the human locus ceruleus. Clin Neuropathol. 1983;2:1–7. [PubMed] [Google Scholar]
- Mann DM, Yates PO. Neurotransmitter deficits in Alzheimer’s disease and in other dementing disorders. Hum Neurobiol. 1986;5:147–158. [PubMed] [Google Scholar]
- Markesbery WR. Neuropathologic alterations in mild cognitive impairment: a review. J Alzheimer’s Dis. 2010;19:221. doi: 10.3233/JAD-2010-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markesbery WR, Schmitt FA, Kryscio RJ, Davis DG, Smith CD, Wekstein DR. Neuropathologic substrate of mild cognitive impairment. Arch Neurol. 2006;63:38–46. doi: 10.1001/archneur.63.1.38. [DOI] [PubMed] [Google Scholar]
- Masliah E, Fagan AM, Terry RD, DeTeresa R, Mallory M, Gage FH. Reactive synaptogenesis assessed by synaptophysin immunoreactivity is associated with GAP-43 in the dentate gyrus of the adult rat. Exp Neurol. 1991;113:131–142. doi: 10.1016/0014-4886(91)90169-d. [DOI] [PubMed] [Google Scholar]
- Matthews DA, Cotman C, Lynch G. An electron microscopic study of lesion- induced synaptogenesis in the dentate gyrus of the adult rat. I. Magnitude and time course of degeneration. Brain Res. 1976a;115:1–21. doi: 10.1016/0006-8993(76)90819-2. [DOI] [PubMed] [Google Scholar]
- Matthews DA, Cotman C, Lynch G. An electron microscopic study of lesion- induced synaptogenesis in the dentate gyrus of the adult rat. II. Reappearance of morphologically normal synaptic contacts. Brain Res. 1976b;115:23–41. doi: 10.1016/0006-8993(76)90820-9. [DOI] [PubMed] [Google Scholar]
- Mawal-Dewan M, Henley J, Van de Voorde A, Trojanowski JQ, Lee VMY. The phosphorylation state of tau in the developing rat brain is regulated by phosphoprotein phosphatases. J Biol Chem. 1994;269:30981–30987. [PubMed] [Google Scholar]
- Mazarati AM, Hohmann JG, Bacon A, Liu H, Sankar R, Steiner RA, Wynick D, Wasterlain CG. Modulation of hippocampal excitability and seizures by galanin. J Neurosci. 2000;20:6276–6281. doi: 10.1523/JNEUROSCI.20-16-06276.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald MP, Gleason TC, Robinson JK, Crawley JN. Galanin inhibits performance on rodent memory tasks. Ann N Y Acad Sci. 1998;863:305–322. doi: 10.1111/j.1749-6632.1998.tb10704.x. [DOI] [PubMed] [Google Scholar]
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, et al. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- Mervis RF, Patel CJ, Foley SK, Zajd S, Aradi M, Solanki P, Walha GS, Scheff SE, Mufson EJ. Hippocampal CA1 dendritic neuroplasticity in mild cognitive impairment and Alzheimer’s disease. Neurosci Abstr 2013 [Google Scholar]
- Mesulam MM, Mufson EJ, Levey AI, Wainer BH. Cholinergic innervation of cortex by the basal forebrain: cytochemistry and cortical connections of the septal area, diagonal band nuclei, nucleus basalis (substantia innominata), and hypothalamus in the rhesus monkey. J Comp Neurol. 1983;214:170–197. doi: 10.1002/cne.902140206. [DOI] [PubMed] [Google Scholar]
- Mesulam MM. Neuroplasticity failure in Alzheimer’s disease: Bridging the gap between plaques and tangles. Neuron. 1999;24:521. doi: 10.1016/s0896-6273(00)81109-5. [DOI] [PubMed] [Google Scholar]
- Mesulam M, Shaw P, Mash D, Weintraub S. Cholinergic nucleus basalis tauopathy emerges early in the aging-MCI-AD continuum. Ann Neurol. 2004;55:815–828. doi: 10.1002/ana.20100. [DOI] [PubMed] [Google Scholar]
- Monaghan DT, Geddes JW, Yao D, Chung C, Cotman CW. [3H]TCP binding sites in Alzheimer’s disease. Neurosci Lett. 1987;73:197–200. doi: 10.1016/0304-3940(87)90017-6. [DOI] [PubMed] [Google Scholar]
- Morris JC, Price AL. Pathologic correlates of nondemented aging, mild cognitive impairment, and early-stage Alzheimer’s disease. J Mol Neurosci. 2001;17:101–118. doi: 10.1385/jmn:17:2:101. [DOI] [PubMed] [Google Scholar]
- Morris GP, Clark IA, Vissel Bryce B. Inconsistencies and Controversies Surrounding the Amyloid Hypothesis of Alzheimer’s Disease. Acta Neuropathologica Communications. 2014;2:135. doi: 10.1186/s40478-014-0135-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortimer JA. Etiology of dementia of Alzheimer’s type. Vol. 1. New York: Wiley; 1988. Do psychosocial risk factors contribute to Alzheimer’s disease? pp. 39–52. [Google Scholar]
- Mufson EJ, Bothwell M, Hersh LB, Kordower JH. Nerve growth factor receptor immunoreactive profiles in the normal, aged human basal forebrain: colocalization with cholinergic neurons. J Comp Neurol. 1989b;285:196–217. doi: 10.1002/cne.902850204. [DOI] [PubMed] [Google Scholar]
- Mufson EJ, Cochran E, Benzing W, Kordower JH. Galaninergic innervation of the cholinergic vertical limb of the diagonal band (Ch2) and bed nucleus of the stria terminalis in aging, Alzheimer’s disease and Down’s syndrome. Dementia. 1993;4:237–250. doi: 10.1159/000107329. [DOI] [PubMed] [Google Scholar]
- Mufson EJ, Chen EY, Cochran EJ, et al. Entorhinal cortex beta-amyloid load in individuals with mild cognitive impairment. Exp Neurol. 1999;158:469–490. doi: 10.1006/exnr.1999.7086. [DOI] [PubMed] [Google Scholar]
- Mufson EJ, Deecher DC, Basile M, Izenwasse S, Mash DC. Galanin receptor plasticity within the nucleus basalis in early and late Alzheimer’s disease: an in vitro autoradiographic analysis. Neuropharm. 2000;39:1404–1412. doi: 10.1016/s0028-3908(00)00011-3. [DOI] [PubMed] [Google Scholar]
- Mufson EJ, Ginsberg SD, Ikonomovic MD, DeKosky ST. Human cholinergic basal forebrain: chemoanatomy and neurologic dysfunction. J Chem Neuroanat. 2003;26(4):233–242. doi: 10.1016/s0891-0618(03)00068-1. [DOI] [PubMed] [Google Scholar]
- Mufson EJ, Ikonomovic MD, Styren SD, Counts SE, Wuu J, Leurgans S, Bennett DA, Cochran EJ, DeKosky ST. Preservation of brain nerve growth factor in mild cognitive impairment and Alzheimer disease. Arch Neurol. 2003;60:1143–1148. doi: 10.1001/archneur.60.8.1143. [DOI] [PubMed] [Google Scholar]
- Mufson EJ, Counts SE, Che S, Ginsberg SD. Neuronal gene expression profiling: uncovering the molecular biology of neurodegenerative disease. Prog Brain Res. 2006;158:197–223. doi: 10.1016/S0079-6123(06)58010-0. [DOI] [PubMed] [Google Scholar]
- Mufson EJ, Counts SE, Perez SE, Ginsberg SD. Cholinergic system during the progression of Alzheimer’s disease: therapeutic implications. Expert Rev Neurother. 2008;8:1703–1718. doi: 10.1586/14737175.8.11.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mufson EJ, He B, Nadeem M, Perez SE, Counts SE, Leurgans S, Fritz J, Lah J, Ginsberg SD, Wuu J, Scheff SW. Hippocampal proNGF signaling pathways and β-amyloid levels in mild cognitive impairment and Alzheimer disease. J Neuropathol Exp Neurol. 2012;71:1018–1029. doi: 10.1097/NEN.0b013e318272caab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mufson EJ, Binder L, Counts SE, DeKosky ST, de Toledo-Morrell L, et al. Mild cognitive impairment: pathology and mechanisms. Acta Neuropathol. 2012;123:13–30. doi: 10.1007/s00401-011-0884-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mufson EJ, He B, Nadeem M, Perez SE, Counts SE, Leurgans S, Fritz J, Lah J, Ginsberg SD, Wuu J, Scheff SW. Hippocampal proNGF signaling pathways and β-amyloid levels in mild cognitive impairment and Alzheimer’s disease. J Neuropathol Exp Neurol. 2012;71:1018–1029. doi: 10.1097/NEN.0b013e318272caab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mufson EJ, Ward S, Binder L. Prefibrillar tau oligomers in mild cognitive impairment and Alzheimer’s disease. Neurodegener Dis. 2014;13:151–153. doi: 10.1159/000353687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller U, Winter P, Graeber MB. A presenilin 1 mutation in the first case of Alzheimer’s disease. Lancet Neurol. 2013;12:129–130. doi: 10.1016/S1474-4422(12)70307-1. [DOI] [PubMed] [Google Scholar]
- Murphy TH. Activity-dependent synapse development: changing the rules. Nat Neurosci. 2003;6:9–11. doi: 10.1038/nn0103-9. [DOI] [PubMed] [Google Scholar]
- Nadler JV, Cotman CW, Lynch G. Histochemical evidence of altered development of cholinergic fibers in the rat dentate gyrus following lesions. I. Time course after complete unilateral entorhinal lesion at various ages. J Comp Neurol. 1977;171:561–587. doi: 10.1002/cne.901710409. [DOI] [PubMed] [Google Scholar]
- Nelson PT1, Alafuzoff I, Bigio EH, Bouras C, Braak H, Cairns NJ, Castellani RJ, Crain BJ, et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. 2012;71:362–381. doi: 10.1097/NEN.0b013e31825018f7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA. Endosome function and dysfunction in Alzheimer’s disease and other neurodegenerative diseases. Neurobiol Aging. 2005;26:373–382. doi: 10.1016/j.neurobiolaging.2004.09.018. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Cataldo AM. The endosomal-lysosomal system of neurons: new roles. Trends Neurosci. 1995;18:489–496. doi: 10.1016/0166-2236(95)92772-i. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Cataldo AM, Paskevich PA, Hamilton DJ, Wheelock TR, Kanaley-Andrews L. The lysosomal system in neurons. Involvement at multiple stages of Alzheimer’s disease pathogenesis. Ann N Y Acad Sci. 1992;674:65–88. doi: 10.1111/j.1749-6632.1992.tb27478.x. [DOI] [PubMed] [Google Scholar]
- Nykjaer A, Lee R, Teng KK, Jansen P, Madsen P, Nielsen MS, Jacobsen C, Kliemannel M, Schwarz E, Willnow TE, Hempstead BL, Petersen CM. Sortilin is essential for proNGF-induced neuronal cell death. Nature. 2004;427:843–848. doi: 10.1038/nature02319. [DOI] [PubMed] [Google Scholar]
- Nykjaer A, Willnow TE, Petersen CM. p75NTR—live or let die. Curr Opin Neurobiol. 2005;15:49–57. doi: 10.1016/j.conb.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Palmer AM, Procter AW, Stratmann GC, Bowen DM. Excitatory amino acid-releasing and cholinergic neurones in Alzheimer’s disease. Neurosci Lett. 1986;66:199–204. doi: 10.1016/0304-3940(86)90190-4. [DOI] [PubMed] [Google Scholar]
- Parton RG, Dotti CG. Cell biology of neuronal endocytosis. J Neurosci Res. 1993;36:1–9. doi: 10.1002/jnr.490360102. [DOI] [PubMed] [Google Scholar]
- Peng S, Wuu J, Mufson EJ, Fahnestock M. Increased proNGF levels in subjects with mild cognitive impairment and mild Alzheimer’s disease. J Neuropathol Exp Neurol. 2004;63:641–649. doi: 10.1093/jnen/63.6.641. [DOI] [PubMed] [Google Scholar]
- Perez S, Basile M, Mash DC, Mufson EJ. Galanin receptor over-expression within the amygdala in early Alzheimer’s disease: an in vitro autoradiographic analysis. J Chem Neuroanat. 2002;24:109–116. doi: 10.1016/s0891-0618(02)00034-0. [DOI] [PubMed] [Google Scholar]
- Perez-Nievas BG, Stein TD, Tai HC, Dols-Icardo O, Scotton TC, Barroeta-Espar I, Fernandez-Carballo L, de Munain EL, Perez J, Marquie M, Serrano-Pozo A, Frosch MP, Lowe V, Parisi JE, Petersen RC, Ikonomovic MD, López OL, Klunk W, Hyman BT, Gómez-Isla T. Dissecting phenotypic traits linked to human resilience to Alzheimer’s pathology. Brain. 2013;136:2510–2526. doi: 10.1093/brain/awt171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen RC, Smith GE, Ivnik RJ, Kokmen E, Tangalos EG. Memory function in very early Alzheimer’s disease. Neurol. 1994;44:867–872. doi: 10.1212/wnl.44.5.867. [DOI] [PubMed] [Google Scholar]
- Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, Kokmen E. Mild cognitive impairment: clinical characterization and outcome. Arch Neurol. 1999;56:303–308. doi: 10.1001/archneur.56.3.303. [DOI] [PubMed] [Google Scholar]
- Petersen RC. Mild cognitive impairment clinical trials. Nat Rev Drug Discov. 2003;2:646–653. doi: 10.1038/nrd1155. [DOI] [PubMed] [Google Scholar]
- Price JL, McKeel DW, Jr, Buckles VD, et al. Neuropathology of nondemented aging:presumptive evidence for preclinical Alzheimer disease. Neurobiol Aging. 2009;30:1026–1036. doi: 10.1016/j.neurobiolaging.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Procter AW, Palmer AM, Francis PT, Lowe SL, Neary D, Murphy E, Doshi R, Bowen DM. Evidence of Glutamatergic Denervation and Possible Abnormal Metabolism in Alzheimer’s Disease. J Neurochem. 1988;50:790–802. doi: 10.1111/j.1471-4159.1988.tb02983.x. [DOI] [PubMed] [Google Scholar]
- Rajendran L, Annaert W. Membrane trafficking pathways in Alzheimer’s disease. Traffic. 2012;13:759–770. doi: 10.1111/j.1600-0854.2012.01332.x. [DOI] [PubMed] [Google Scholar]
- Ramon y Cajal S. On a special ganglion of the spheno-occipital cortex. Trabajos del Laboratorio de lnvestigaciones Biologicas de la universidad de Madrid. 1901:TOMO 1. [Google Scholar]
- Reisberg B, Doody R, Stoffler A, Schmitt F, Ferris S, Mobius HJ. A 24-week open-label extension study of memantine in moderate to severe Alzheimer disease. Arch Neurol. 2006;63:49–54. doi: 10.1001/archneur.63.1.49. [DOI] [PubMed] [Google Scholar]
- Rodrigues R, Smith MA, Wang X, Perry G, Lee HG, Zhu X, Petersen RB. Molecular neuropathogenesis of Alzheimer’s disease: an interaction model stressing the central role of oxidative stress. Future Neurol. 2012;7:287–305. doi: 10.2217/fnl.12.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Puertas R, Nilsson S, Pascual J, Pazos A, Hokfelt T. 125I-galanin binding sites in Alzheimer’s disease: increases in hippocampal subfields and a decrease in the caudate nucleus. J Neurochem. 1998;68:1106–1113. doi: 10.1046/j.1471-4159.1997.68031106.x. [DOI] [PubMed] [Google Scholar]
- Rola R, Raber J, Rizk A, Otsuke S, VandenBerg SR, Morhardt DR, Fike JR. Radiation-induced impairment of hippocampal neurogenesis is associated with cognitive deficits in young mice. Exp Neurol. 2004;188:316–330. doi: 10.1016/j.expneurol.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Roux PP, Barker PA. Neurotrophin signaling through the p75 neutrophin receptor. Prog Neurobiol. 2002;67:203–233. doi: 10.1016/s0301-0082(02)00016-3. [DOI] [PubMed] [Google Scholar]
- Rupp C, Beyreuther K, Maurer K, Kins S. A presenilin 1 mutation in the first case of Alzheimer’s disease: Revisited. Alzheimers Dement. 2014;10:869–72. doi: 10.1016/j.jalz.2014.06.005. [DOI] [PubMed] [Google Scholar]
- Rupsingh R, Borrie M, Smith M, Wells JL, Bartha R. Reduced hippocampal glutamate in Alzheimer disease. Neurobiol Aging. 2011;32:802–810. doi: 10.1016/j.neurobiolaging.2009.05.002. [DOI] [PubMed] [Google Scholar]
- Sasaki H, Muramoto O, Kanazawa I, Kosaka K, Iizuka R. Regional distribution of amino acid transmitters in postmortem brains of presenile and senile dementia of Alzheimer type. Ann Neurol. 1986;19:263–269. doi: 10.1002/ana.410190307. [DOI] [PubMed] [Google Scholar]
- Scharfman HE, Chao MV. The entorhinal cortex and neurotrophin signaling in Alzheimer’s disease and other disorders. Cogn Neurosci. 2013;4:123–35. doi: 10.1080/17588928.2013.826184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheff SW, Benardo LS, Cotman CW. Decline of reactive fiber growth in the dentate gyrus of aged rats compared to young adult rats following entorhinal cortex removal. Brain Res. 1980;199:21–38. doi: 10.1016/0006-8993(80)90227-9. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Price DA. Synapse loss in the temporal lobe in Alzheimer’s disease. Ann Neurol. 1993;33:190–199. doi: 10.1002/ana.410330209. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Sparks L, Price DA. Quantitative assessment of synaptic density in the entorhinal cortex in Alzheimer’s disease. Ann Neurol. 1996;34:356–361. doi: 10.1002/ana.410340309. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Price DA. Synaptic density in the inner molecular layer of the hippocampal dentate gyrus in alzheimer disease. J Neuropath Exp Neurol. 1998;57:1146–53. doi: 10.1097/00005072-199812000-00006. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Price DA. Alzheimer’s disease-related synapse loss in the cingulate cortex. J Alzheimers Dis. 2001;3:495–505. doi: 10.3233/jad-2001-3509. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Price DA. Synaptic pathology in Alzheimer’s disease: a review of ultrastructural studies. Neurobiol Aging. 2003;24:1029–1046. doi: 10.1016/j.neurobiolaging.2003.08.002. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Price DA, Schmitt FA, Mufson EJ. Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol Aging. 2006;27:1372–1384. doi: 10.1016/j.neurobiolaging.2005.09.012. [DOI] [PubMed] [Google Scholar]
- Schneider JA, Aggarwal NT, Barnes L, Boyle P, Bennett DA. The neuropathology of older persons with and without dementia from community versus clinic cohorts. J Alz Dis. 2009;18:691–701. doi: 10.3233/JAD-2009-1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoji M, Golde TE, Ghiso J, Cheung TT, Estus S, Shaffer LM, Cai XD, McKay DM, Tintner R, Frangione B, et al. Production of the Alzheimer amyloid beta protein by normal proteolytic processing. Science. 1992;258:126–129. doi: 10.1126/science.1439760. [DOI] [PubMed] [Google Scholar]
- Simonian NA, Rebeck GW, Hyman BT. Functional integrity of neural systems related to memory in Alzheimer’s disease. Prog Brain Res. 1994;100:245–254. doi: 10.1016/s0079-6123(08)60791-8. [DOI] [PubMed] [Google Scholar]
- Smith KE, Walker MW, Artymyshyn R, Bard J, Borowsky B, Tamm JA, Yao WJ, et al. Cloned human and rat galanin GALR3 receptors. Pharmacology and activation of G-protein inwardly rectifying K+ channels. J Biol Chem. 1998;273:23321–23326. doi: 10.1074/jbc.273.36.23321. [DOI] [PubMed] [Google Scholar]
- Snowdon DA, Gross MD, Butler SM. Antioxidants and reduced functional capacity in the elderly: findings from the Nun Study. J Gerontol A Biol Sci Med Sci. 1996;51:M10–M16. doi: 10.1093/gerona/51a.1.m10. [DOI] [PubMed] [Google Scholar]
- Song G, Ouyang G, Bao S. The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med. 2005;9:59–71. doi: 10.1111/j.1582-4934.2005.tb00337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling R, Mormino E, Johnson K. The evolution of preclinical Alzheimer’s disease: Implications for prevention trials. Neuron. 2014;84:608–622. doi: 10.1016/j.neuron.2014.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward O, Scoville SA. Cells of origin of entorhinal cortical afferents to the hippocampus and fascia dentate of the rat. J Comp Neurol. 1976;169:347–370. doi: 10.1002/cne.901690306. [DOI] [PubMed] [Google Scholar]
- Steward O, Vinsant SL. The process of reinnervation in the dentate gyrus of adult rats: a quantitative electron microscopic analysis of terminal proliferation and reactive synaptogenesis. J Comp Neurol. 1983;214:370–386. [Google Scholar]
- Stretton J, Sidhu MK, Winston GP, Bartlett P, McEvoy AW, Symms MR, Koepp MJ, et al. Working memory network plasticity after anterior temporal lobe resection: a longitudinal functional magnetic resonance imaging study. Brain. 2014;137:1439–53. doi: 10.1093/brain/awu061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranahan AM, Mattson MP. Selective vulnerability of neurons in layer II of the entorhinal cortex during aging and Alzheimer’s disease. Neur Plast. 2010;2010:1–8. doi: 10.1155/2010/108190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultana R, Banks WA, Butterfield DA. Decreased levels of PSD95 and two associated proteins and increased levels of BCl2 and caspase 3 in hippocampus from subjects with amnestic mild cognitive impairment: insights into their potential roles for loss of synapses and memory, accumulation of Abeta, and neurodegeneration in a prodromal stage of Alzheimer’s disease. J Neurosci Res. 2010;88:469–477. doi: 10.1002/jnr.22227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sze CI, Troncoso JC, Kawas C, Mouton P, Price DL, Martin LJ. Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer disease. J Neuropathol Exp Neurol. 1997;56:933–944. doi: 10.1097/00005072-199708000-00011. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Sekino Y, Tanaka S, Mizui T, Kishi S, Shirao T. Drebrin-dependent actin clustering in dendritic filopodia governs synaptic targeting of postsynaptic density-95 and dendritic spine morphogenesis. J Neurosci. 2003;23:6586–6595. doi: 10.1523/JNEUROSCI.23-16-06586.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor KI, Probst A. Anatomic localization of the transentorhinal region of the entorhinal cortex. Neurobiol Aging. 2008;29:1591–1596. doi: 10.1016/j.neurobiolaging.2007.03.024. [DOI] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, et al. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Thies W, Bleiler L. 2013 Alzheimer’s disease facts and figures. Alzheimer’s & Dementia: The Journal of the Alzheimer’s Association. 2013;9:208–245. doi: 10.1016/j.jalz.2013.02.003. [DOI] [PubMed] [Google Scholar]
- Thinakaran G, Koo EH. Amyloid precursor protein trafficking, processing, and function. J Biol Chem. 2008;283:29615–29619. doi: 10.1074/jbc.R800019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trojanowski JQ. The Molecular and Genetic Basis of Neurological Disease. J Neuropath Exp Neurol. 1993;52:550. [Google Scholar]
- Tu S, Okamoto S, Lipton SA, Xu H. Oligomeric Aβ-induced synaptic dysfunction in Alzheimer ‘s disease. Molec Neurodegen. 2014;9:48. doi: 10.1186/1750-1326-9-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich E, Duwel A, Kauffmann-Zeh A, Lyon D, Rudkin B, Evan G, Martin-Zanca D. Specific TrkA survival signals interfere with different apoptotic pathways. Oncogene. 1998;16:825–832. doi: 10.1038/sj.onc.1201842. [DOI] [PubMed] [Google Scholar]
- Vana L, Kanaan NM, Ugwu IC, Wuu J, Mufson EM, Lester I. Progression of tau pathology in cholinergic basal forebrain neurons in mild cognitive impairment and Alzheimer’s disease. Am J Pathol. 2011;179:2533–2550. doi: 10.1016/j.ajpath.2011.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Xiong S, Xie C, Markesbery WR, Lovell MA. Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer’s disease. J Neurochem. 2005;93:953–962. doi: 10.1111/j.1471-4159.2005.03053.x. [DOI] [PubMed] [Google Scholar]
- Wang Y, Yang R, Gu J, Yin X, Jin N, Xie S, Wang Y, Chang H, Qian W, Shi J, Iqbal K, et al. Cross talk between PI3K-AKT-GSK-3β and PP2A pathways determines tau hyperphosphorylation. Neurobiol Aging. 2015;36:188–200. doi: 10.1016/j.neurobiolaging.2014.07.035. [DOI] [PubMed] [Google Scholar]
- West M. Regionally specific loss of neurons in the aging human hippocampus. Neurobiol Aging. 1993;14:287–293. doi: 10.1016/0197-4580(93)90113-p. [DOI] [PubMed] [Google Scholar]
- West MJ, Kawas CH, Stewart WF, Rudow GL, Troncoso JC. Hippocampal neurons in pre-clinical Alzheimer’s disease. Neurobio Aging. 2004;25:1205–1212. doi: 10.1016/j.neurobiolaging.2003.12.005. [DOI] [PubMed] [Google Scholar]
- Whitehouse PJ, Price DL, Struble RG, Clark AW, Coyle JT, Delong MR. Alzheimer’s disease and senile dementia. Loss of neurons in the basal forebrain. Science. 1982;215:1237–1239. doi: 10.1126/science.7058341. [DOI] [PubMed] [Google Scholar]
- Williams LR, Varon S, Peterson GM, Wictorin K, Fischer W, Bjorklund A, Gage FH. Continuous infusion of nerve growth factor prevents basal forebrain neuronal death after fimbria fornix transection. Proc Natl Acad Sci USA. 1986;83:9231–9325. doi: 10.1073/pnas.83.23.9231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrenn CC, Crawley JN. Pharmacological evidence supporting a role for galanin in cognition and affect. Prog Neuropsychopharmacol Biol Psy. 2001;25:283–299. doi: 10.1016/s0278-5846(00)00156-1. [DOI] [PubMed] [Google Scholar]
- Wynick D, Bacon A. Targeted disruption of galanin: new insights from knock-out studies. Neuropep. 2002;36:132–144. doi: 10.1054/npep.2002.0888. [DOI] [PubMed] [Google Scholar]
- Yao PJ, Zhu M, Pyun EI, Brooks AI, Therianos S, Meyers VE, et al. Defects in expression of genes related to synaptic vesicle trafficking in frontal cortex of Alzheimer’s disease. Neurobio Dis. 2003;12:97–109. doi: 10.1016/s0969-9961(02)00009-8. [DOI] [PubMed] [Google Scholar]
- Yasuda RP, Ikonomovic MD, Sheffield R, Rubin RT, Wolfe BB, Armstrong DM. Reduction of AMPA-selective glutamate receptor subunits in the entorhinal cortex of patients with Alzheimer’s disease pathology: a biochemical study. Brain Res. 1995;678:161–167. doi: 10.1016/0006-8993(95)00178-s. [DOI] [PubMed] [Google Scholar]
- Yoshiyama Y, Lee VM, Trojanowski JQ. J Therapeutic strategies for tau mediated neurodegeneration. Neurol Neurosurg Psychiatry. 2013;84:784–795. doi: 10.1136/jnnp-2012-303144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdez G, Philippidou P, Rosenbaum J, Akmentin W, Shao Y, Halegoua S. Trk- signaling endosomes are generated by Rac-dependent macroendocytosis. Proc Natl Acad Sci USA. 2007;104:12270–12275. doi: 10.1073/pnas.0702819104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vana L, Kanaan NM, Ugwu IC, Wuu J, Mufson EJ, Binder LI. Progression of tau pathology in cholinergic basal forebrain neurons in mild cognitive impairment and Alzheimer’s disease. Am J Pathol. 2011;179:2533–2550. doi: 10.1016/j.ajpath.2011.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogels OJ, Broere CA, ter Laak HJ, ten Donkelaar HJ, Nieuwenhuys R, Schulte BP. Cell loss and shrinkage in the nucleus basalis Meynert complex in Alzheimer’s disease. Neurobiol Aging. 1990;11:3–13. doi: 10.1016/0197-4580(90)90056-6. [DOI] [PubMed] [Google Scholar]
- Zarow C, Lyness SA, Mortimer JA, Chui HC. Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer’ and Parkinson disease. Arch Neurol. 2003;60:337–341. doi: 10.1001/archneur.60.3.337. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Thompson R, Zhang H, Xu H. APP processing in Alzheimer’s disease. Mol Brain. 2011;4:3. doi: 10.1186/1756-6606-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, ZhuGe Q, Zhong M, Chen G, Shao B, Wang H, Mao X, Xie L, Jin K. Neurogenesis in adult human brain after traumatic brain injury. J Neurotrauma. 2013;30:1872–1880. doi: 10.1089/neu.2010.1579. [DOI] [PMC free article] [PubMed] [Google Scholar]