

Abstract
Children with biallelic mutations in FANCD1/BRCA2 are at uniquely high risks of leukemia and solid tumors. Preemptive bone marrow transplantation (PE-BMT) has been proposed to avoid the development of leukemia, but empirical study of PE-BMT is unlikely due to the rarity of these children and unknown benefit of PE-BMT. We used survival analysis to estimate the risks of leukemia, and the expected survival if leukemia could be eliminated by curative PE-BMT. We used the results in a decision analysis model to explore the plausibility of PE-BMT for children with variable ages at diagnosis and risks of transplant-related mortality. For example, PE-BMT at one year of age with a 10% risk of transplant-related mortality increased the mean survival by 1.7 years. The greatest benefit was for patients diagnosed between one and three years of age, after which the benefit of PE-BMT decreased with age at diagnosis, and the risk of death from solid tumors constituted a relatively greater burden of mortality. Our methods may be used to model survival for other hematologic disorders with limited empirical data and a pressing need for clinical guidance.
Introduction
Fanconi anemia (FA) is a primarily autosomal recessive inherited bone marrow failure and cancer predisposition syndrome, caused by germline mutations in more than 16 genes involved in the FA/BRCA DNA damage response pathway.1 Patients with FA have an approximately 800-fold increased risk of developing leukemia and an overall risk of more than 50-fold for any malignancy or solid tumor; the cumulative incidence of severe bone marrow failure is more than 50% by age 45 years.2 Characteristic birth defects frequently associated with FA include short stature, café au lait spots, microcephaly, thumb and radial anomalies, and renal structural defects 1. Biallelic mutations in FANCD1/BRCA2 have been reported in rare patients with FA with very early-onset leukemia and solid tumors. Analysis of 27 published cases with these mutations found that the cumulative incidence of any malignancy was 97% by six years of age, and the actuarial risks of leukemia and solid tumor were 79% by 10 years of age and 83% by seven years of age respectively.3
Bone marrow transplantation (BMT) is currently the only treatment that has the potential to restore normal hematologic function in patients with any FA genotype who have bone marrow failure (BMF), myelodysplastic syndrome (MDS) or leukemia.4 The best results were found in recent European experience to involve a matched family donor, patients below age 10 years, transplant without irradiation, and BMF without MDS or leukemia, similar to earlier results from the Center for International Blood and Marrow Transplant Research.5;6 In patients with FA, overall 5-year survival of 80 to 90% may be expected for matched sibling donor transplants, versus 60 to 75% for unrelated donors.5 Survival is lower in patients with hematologic malignancies, in part due to the more frequent use of mismatched or unrelated donors and more intensive marrow suppression.
Patients with FA due to biallelic mutations in FANCD1/BRCA2 have an inordinately high and early risk of leukemia, leading to the suggestion that earlier transplant be considered prior to the development of severe cytopenia.4 There are no published data on the results of such transplants, however. Ten patients with FANCD1/BRCA2 were reported through 2013 who underwent transplant after they had developed leukemia (with one exception who had neutropenia, see Table 1), with only 2 survivors.7–12 Seven died in less than one year after transplant. These patients were not ideal candidates for transplant success, since 8 had acute myeloid leukemia (AML) and one acute lymphoblastic leukemia (ALL);5;13;14 only 3 had matched sibling donors, while 5 had unrelated donors and 2 had partially matched related donors. Cord blood was used in one case, peripheral blood in one, and bone marrow in the others.
Table 1.
Hematopoietic stem cell transplantation in patients with FANCD1/BRCA2 mutations reported in the literature, 2000 to 2013
| ID | Age at hematologic abnormality (yrs) | Reason for BMT | Age at BMT (yrs) | Interval survival post-BMT (yrs) | Donor | Stem Cell Source | Outcome | Age at last report (yrs) | Reference |
|---|---|---|---|---|---|---|---|---|---|
| AP37P | 2 | AML | 2.2 | 1.67 | HLA identical sibling | BM | Dead | 3.87 | 7 |
| 632/1 | 3 | AML | 3.7 | 0.33 | 6/6 URD | BM | Dead | 4 | 8;9 |
| 632/2 | 1.75 | AML | 1.9 | 0.80 | 6/6 MSD | BM | Dead | 2.7 | |
| 800/1 | 0.9 | AML | 1.8 | 0.34 | 6/6 URD | TCD BM | Dead | 2.1 | |
| 800/2 | 0.9 | Neutropenia | 0.8 | 0.17 | 6/6 URD | TCD BM | Dead | 0.97 | |
| 900/1 | 5.2 | ALL | 4.9 | 2.50 | 5/6 Mother | TCD BM | Alive | 7.4 | |
| 984/2 | 4.9 | ALL-AML* | 6.7 | 0.58 | 6/6 URD | UCB | Alive | 7.3 | |
| 2751 | 5.5 | AML | 5.5 | 0.50 | 4/6 and 7/10 Related | PB | Dead | 6 | 10 |
| PT 2 | 1.7 | AML | 1.7 | 0.40 | MUD 8/8 | NA | Dead | 2.1 | 11 |
| Case | 2 | AML | 2 | 0.83 | HLA identical sibling | BM | Dead | 2.8 | 12 |
Abbreviations: ID, identifier; BMT, bone marrow transplant; AML, acute myeloid leukemia; HLA, human leukocyte antigen; BM, bone marrow; URD, Unrelated donor; MSD, matched sibling donor; TCD, T-Cell depleted; ALL, acute lymphoblastic leukemia; UCB, umbilical cord blood; PB, peripheral blood; MUD, matched unrelated donor; NA, not available.
Patient had T-cell ALL at 4.9 yrs of age, went into remission with chemotherapy, and relapsed with AML at 6.3 yrs of age. While being worked up for BMT, a Wilms tumor was found; after nephrectomy, the patient proceeded to transplant at age 6.7 yrs.
Bone marrow transplantation before the development of symptomatic hematologic problems might increase overall survival in patients with FA, particularly those with FANCD1/BRCA2. Healthy transplant recipients (even with FA) would presumably have better tolerance of the complications associated with a BMT, and a successful transplant is anticipated to eliminate the future risk of MDS or leukemia. We have chosen to call this type of transplantation “preemptive (PE-BMT)”15, since it is preventive, rather than “early”, which implies that the outcome being avoided is inevitable, and the only issue is the timing. In reality, only about half of the patients with FANCD1/BRCA2 are predicted to develop leukemia in their lifetime, due to competing risks of solid tumors and death. However, BMT carries non-trivial risks of mortality and morbidity, and exposing an otherwise healthy child to a risky procedure of unknown benefit violates “primum non nocere (first do no harm)”.16 A clinical trial of PE-BMT in those with FANCD1/BRCA2 is not feasible due to the rarity of such patients, and the uncertainty regarding the risks and potential rewards of transplantation in this population.
To address this question, we used statistical methods and decision analysis to explore the utility of PE-BMT in this context. Decision analysis provides a mathematical structure for weighing the risks and benefits of competing strategies. We estimated the overall survival curve if leukemia could be eliminated from the FANCD1/BRCA2 population (actuarial risks) and used the results in a decision analysis to examine the possible benefits of PE-BMT compared with current standard care. We relied on the limited number of published cases with FANCD1/BRCA2 for data on overall survival and development of malignant complications (AML, brain tumors, Wilms tumors, and other solid tumors) prior to BMT.3;10–12;17–19
Methods
Decision model
We developed a Markov decision model from the parental perspective to determine how PE-BMT at one year of age would affect the mean survival of children with FANCD1/BRCA2 compared with a strategy of standard care according to the recent literature reports. Markov, or state-transition, models allow for the recursive simulation of events and risks that change over time, and have been used by others to simulate outcomes of BMT strategies in the presence of uncertainty.20–22 In our Markov model, a cohort of hypothetical cases transition between two mutually exclusive states – Alive and Dead – over multiple cycles (Figure 1). All hypothetical cases begin in the Alive state and transition over time to the Dead state, out of which they cannot transition. Cases receiving standard care have a probability of transitioning to the Dead state that is equal to the age-dependent conditional probability of dying among cases reported in the literature. Children with FANCD1/BRCA2 receiving PE-BMT are at risk of dying from malignancies other than leukemia or from transplant-related mortality (TRM). TRM is the excess mortality attributable to BMT, including risks of infection, graft-versus-host disease (GVHD), and regimen-related toxicity, that is distinct from other causes of death in this population, such as solid tumors. We biased our model in favor of PE-BMT and posited that PE-BMT would not affect the incidence of solid tumors or other causes of death, and that the risk of leukemia would be eliminated. BMT in other FA complementation groups was associated with an increased incidence of head and neck squamous cell carcinoma compared with FA patients who did not have a BMT.23 However, cancer risks associated with more recent conditioning regimens may be lower, and the solid tumors (midline brain, and Wilms tumors) observed in FANCD1/BRCA2 are different from those in other FA groups (head and neck and gynecologic squamous cell carcinomas); the risks may not be increased by BMT or GVHD.
Figure 1. Markov decision model for children with FANCD1/BRCA2.
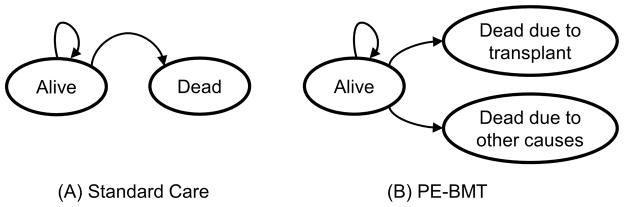
Hypothetical cases transition through the model in 3-month intervals. (A) Beginning in the Alive state, patients receiving Standard Care are at risk of dying from any cause similar to what is expected historically (leukemia or solid tumors). (B) Patients receiving PE-BMT are at risk of dying due to causes related to transplant or from causes other than leukemia, such as solid tumors.
We chose a cycle length of three months, evaluated the decision over 160 cycles, or 40-years duration post-transplant, and we assumed that all events occur half-way through each cycle (“half-cycle correction”).24 We did not adjust the expected survival to reflect decrements in quality of life because we could not find health-state utilities from the parental perspective applicable to our very young population. Hence, we developed a model to provide objective estimates of survival to be used by parents in the context of a family’s values and circumstances. We also did not discount the value of future life-years. We modeled the decision conditioned on survival to ages ranging from 3 months to 10 years to determine at what ages PE-BMT might remain feasible; this analysis informs the decision for patients diagnosed with FA at various ages. All annual probabilities and rates were transformed to the appropriate 3-month equivalents (see Supplement for details.) The decision model was implemented in TreeAge Pro 2014 (TreeAge Software, Inc, Williamstown, MA, http://www.treeage.com).
Data sources
We included the 36 cases of patients with FANCD1/BRCA2 published between the years 2000 through 2013 in our analysis.3;10–12;17–19 We fit several parametric survival curves, including exponential, Weibull, Gompertz, log-normal, and log-logistic functions, to estimate survival from any cause of death, and we used Akaike Information Criterion and visual inspection to assess goodness-of-fit. We estimated the (hypothetical) actuarial survival of children with FANCD1/BRCA2 if leukemia could be eliminated from this population by censoring cases at the development of leukemia and counting death as an event. We calculated the cumulative incidence function in the presence of competing risks25 and the complement of the Kaplan-Meier product-limit estimators26 (“1-KM” estimator) to determine the crude and actuarial risks, respectively, of leukemia and other causes of death. Whereas the crude risk reflects the proportion of patients who experience an event, the actuarial risk represents the hypothetical proportion of patients who would experience an event if the risks of competing events could be removed. All survival analyses were conducted using Stata/SE 13.1 (StataCorp LP, College Station, TX, http://www.stata.com/).
The presumptive advantage of PE-BMT is that transplanting healthy children with FA without leukemia would yield better outcomes than transplanting such children presenting with leukemia, and that by eliminating the risk of leukemia from this population, the children would have greater overall survival. However, due to the particularly early onset of leukemia in these children, an effective policy of preemption would require transplantation during infancy, an age group for which there are no data on TRM for FA. Rather than compare infants with FANCD1/BRCA2 to adolescents with FA in other complementation groups, we consulted a recent publications on BMT in infants with severe combined immunodeficiency or Hurler syndrome who received a BMT within the first year of life (and were without infection at the time of BMT) to estimate a plausible risk of TRM (10%) in otherwise healthy infants (i.e., those without a malignancy).27;28 For our base-case of a one year old patient with FANCD1/BRCA2, we assumed that the cumulative probability of TRM was 10% and that this risk would be spread evenly over the first year following transplantation.
Sensitivity analysis
We varied the decision over a range of ages at BMT and of risks of TRM (Table 2). Long-term survival following BMT in infants and young children with non-malignant disorders such as severe combined immunodeficiency or Hurler syndrome was reported to range from 50% to 95%, with most of the mortality occurring within the first year following BMT.27;28 We thus explored how the decision would change over a wide range of TRM, between 0% and 60% over one year following transplant. We performed one-way and two-way sensitivity analyses, and present threshold values of TRM at which a given strategy has superior mean survival.
Table 2.
Markov model parameters and range tested in sensitivity analysis.
| Model Parameter | Base-case value(s) | Range used in sensitivity analysis | Source |
|---|---|---|---|
| Overall survival | Age-dependent | * | Figure 2a |
| TRM over 1 year | 10% | 0 – 60% | Pai et al27 |
| Survival after PE-BMT | Age-dependent | * | Figure 2c |
| Age at decision | 1 year | 0.25 – 10 years | N/A |
Abbreviations: N/A, not available
Probabilities were not varied in sensitivity analysis.
Results
Survival Analysis
We determined that log-logistic functions provided the best fit for estimating both overall survival and the hypothetical survival if leukemia could be eliminated, from the published FANCD1/BRCA2 population (see Supplement for parameterizations). The median overall survival estimated by the parametric models was 3.5 years of age, and the 10-year overall survival was 8% (Figure 2A). The crude risks of leukemia and death from other causes after treating these as competing risks were 49% (95% CI: 32%–65%) and 48% (95% CI: 30%–63%) by 10 years of age, respectively (Figure 2B). The actuarial risk of leukemia (if the risk of death from other causes could be removed) was 79% (95% CI: 53%–95%) by 10 years of age. If the risk of leukemia could be eliminated in this population, the median survival would be 5 years of age and the 10-year overall survival would be 19% (Figure 2C) due to the high risk of brain and Wilms tumors in this population. The per-year hazard (rate) of death from any cause (mainly leukemia or solid tumors) was largest at four years of age (Figure 2D, dotted line). The hazard of death from causes other than leukemia would peak around five years of age if the risk of leukemia could be removed (solid line).
Figure 2. Survival in patients with FANCD1/BRCA2.

(A) Overall survival. Both non-parametric Kaplan-Meier (solid line) and log-logistic parametric (dotted line) estimators indicate poor survival; less than 10% are expected to survive into their teens. Shaded area indicates 95% confidence intervals. The last survivor was lost to follow-up at age 30. (B) Crude and actuarial risks of leukemia. The actuarial risk of developing leukemia (dotted line) is 79% by age 10. Treating leukemia and death from other causes as competing risks, the crude risks of leukemia (solid line) and death from other causes (dashed line) are 49% and 48% by age 10, respectively. The vertical lines indicate the 95% confidence intervals at those fixed ages. (C) Survival if the risk of leukemia could be eliminated. We censored at the development of leukemia. Both non-parametric Kaplan-Meier (solid line) and parametric log-logistic (dotted line) functions estimated that in the absence of leukemia, 20% would survive until age 10. (D) Annual hazard rates. We plotted the per-year hazard rates for overall survival (dotted line) and survival if leukemia could be eliminated by PE-BMT (solid line). The hazard rate for overall survival peaks between four and five years of age while the hazard rate for survival if leukemia could be eliminated peaks at five years of age.
Base-Case Results and Sensitivity Analyses
For one-year olds receiving standard care, the mean survival was an additional 4.1 years, the median survival was an additional 2.8 years, and overall survival after 5 years and 10 years was 24% and 7%, respectively (Figure 3A, dotted line). Under a base case strategy of PE-BMT at age one year and a 10% risk of TRM over one year (solid line), the mean survival was an additional 5.8 years, the median survival was an additional 3.8 years, and overall survival 5 and 10 years after transplant were 39% and 15%, respectively. Thus PE-BMT at one year of age increased the mean survival by 1.7 years and the median survival by 1.0 years; hence PE-BMT was the dominant strategy under base-case assumptions. Similar analyses are shown in Figures 3B and 3C, suggesting that PE-BMT provides the most survival benefit when performed while the child is less than 3 years of age.
Figure 3. Simulated survival curves for cohorts of children presenting for PE-BMT.
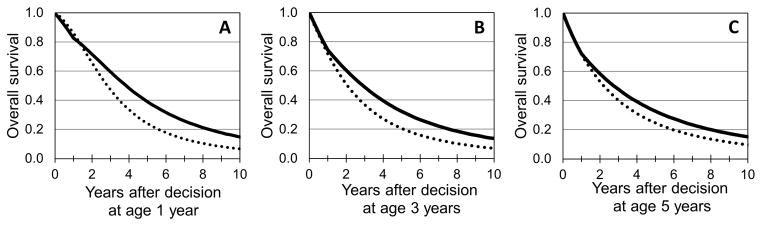
(A) PE-BMT after presentation at one year of age. Simulated survival curve for children receiving PE-BMT at age one year (solid lines) compared with what could be expected if no preemptive action is taken (dotted lines). (B) PE-BMT after presentation at three years of age. (C) PE-BMT after presentation at five years of age. Estimates rely heavily on extrapolation.
As noted above, for children diagnosed at one year of age the expected survival with no intervention is an additional 4.1 years (Figure 4A). PE-BMT would increase the mean survival by 2.3 years if there was no TRM (Figure 4B) or 1.7 years with a 10% risk of TRM, and PE-BMT would remain the superior strategy so long as the risk of TRM over one year was 38% or less. If instead PE-BMT carried a 60% risk of TRM, the expected survival would decrease by 1.3 years. Consider next a child presenting at six months of age. The mean survival expected in the absence of PE-BMT is an additional 4.5 years (Figure 4A). PE-BMT with a 10% risk of TRM would increase the expected survival by 1.7 years (Figure 4B). If the decision was made for a child of 3 years of age, the expected survival would increase by a similar amount, 1.6 years. More generally, PE-BMT remained a viable option at low risks of TRM, but ceased being the superior strategy for children presenting at ages greater than 5 years with risks of TRM greater than 30%. However, the small numbers of published cases of older children and adolescents with FANCD1/BRCA2 limit our confidence in projections made for children presenting for PE-BMT at ages older than three years.
Figure 4. Expected survival of children presenting at ages up to 10 years.
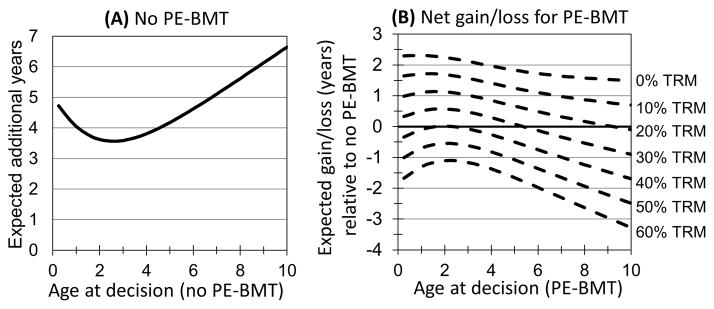
(A) Expected survival of children with no PE-BMT. The expected (mean) survival of children (y-axis) after presenting at age in years (x-axis). If no preemptive action is taken, a child presenting at one year of age is expected to survive an additional four years. (B) The net gain/loss in expected survival from PE-BMT. The additional expected life-years gained or lost (y-axis) by receiving PE-BMT at an age at presentation ranging from birth to 10 years (x-axis) and risk of transplant-related mortality (TRM) ranging from 0% to 60% over one year (dotted lines). PE-BMT at one year of age and a 10% risk of TRM would increase the expected survival by 1.7 years. Estimates for ages greater than three years rely heavily on extrapolation.
Discussion
PE-BMT may increase the mean survival of children with FANCD1/BRCA2 if the procedure is safe enough and the timing is early enough. In our modeling scenarios, ‘safe enough’ in this high risk setting corresponds to 1-year TRM below 30% and early enough is to transplant at less than 5 years of age. The potential benefit of PE-BMT was very sensitive to the risk of TRM, but less sensitive to the age at PE-BMT. Our results depend on the validity of our parametric models for the competing risks of death from leukemia and other causes, which include solid tumors, especially brain and Wilms’ tumors. However, our risk estimates reflect the totality of cases reported through 2013, and our modeled survival curves appear to adequately recapitulate the observed experience.
Although the permissible risk of TRM for PE-BMT is higher than what has been reported in BMT among children with other FA complementation groups 5;13;29 and in infants transplanted for other conditions, 27;28 the expected survival following PE-BMT is still notably short, because of the greater competing risks. Even if there was no risk of TRM, the median survival of this population under standard care would be 5 years of age due to the mortality imposed by the solid tumors (Figure 2C). Improved treatment regimens for the solid tumors associated with FANCD1/BRCA2 are likely to play as big a role in increasing life expectancy as will improvements in survival following BMT.
We did not explicitly model the influence of donor and stem cell source. Among FA patients with other complementation groups who were transplanted for any reason between the years 2000 and 2009, non-relapse mortality after one year was 14% for those with matched sibling donors, versus 24% for those with matched unrelated donors (non-relapse mortality is a proxy for TRM).5 PE-BMT would thus seem a viable option for many with matched sibling donors but for relatively few with matched unrelated donors. Importantly, an individualized risk of TRM for any given patient-donor combination depends on a number of risk factors associated with survival, including stem cell source, donor-recipient gender- or CMV status- mismatch, and conditioning regimen, factors that are not formally included in our model. These factors should be qualitatively assessed as increasing or decreasing the risk for the individual patient at hand versus the population average risk.
Whether parents would elect for their child to undergo PE-BMT is unknown, if the survival benefit offered by PE-BMT is only marginal; if there was no risk of TRM, an optimally-timed preemptive transplant would increase the mean survival by less than 2.5 years. There is also a potential ethical concern about performing a risky procedure on an apparently healthy child. For example, in a study of parental decision making for children with sickle cell disease, 15% of parents stated they would refuse a curative bone marrow transplant even if there was no excess risk of mortality, and only 36% of parents were willing to accept a 15% or greater risk of short-term mortality.30 While children with sickle cell disease do not face the same types of risks as children with FANCD1/BRCA2, the aversion to BMT for sickle cell disease in the absence of symptoms is illustrative of parental aversion to any procedural risk.
The quality of life for patients with FANCD1/BRCA2 as well as their families should also factor into shared decision making. This is complicated since the parents are proxies for their young children, and it is difficult to evaluate using traditional quality of life instruments in this context. Certainly BMT in pediatric populations is associated with decreased quality of life that has both physical and psychosocial bases. Development of GVHD and infection are associated with a worse quality of life.31 In addition, children who receive BMT are kept in protective isolation, both inside and outside the hospital in the months following BMT, which may impact their development at a critical time. Neurocognitive delays were found to be most significant in young patients (ages 0 to 3 years), particularly in those who had received total body irradiation.32 It is difficult to estimate how hospitalization per se impacts the development of children. Even for those who do not undergo BMT, management of solid tumors in this population may also interfere with development and lead to significant other late effects.33
Despite obvious limitations of decision analysis methodology, our results provide an objective quantification of risks vs. benefits that can help parents and physicians decide whether or not to pursue PE-BMT for children with FANCD1/BRCA2. Furthermore, the methods discussed here can be utilized in the context of other disorders for which hematologic malignancy is a major concern.
Supplementary Material
Highlights.
A review of the risks of leukemia and survival in children with FANCD1/BRCA2
An estimate of survival if leukemia could be eliminated
Preemptive transplant may increase survival if transplant-related mortality is low
Acknowledgments
This research was supported in part by the Intramural Research Program of the National Cancer Institute of the National Institutes of Health.
Footnotes
Financial Disclosures: None
Conflicts of Interest: None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Shimamura A, Alter BP. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev. 2010;24:101–122. doi: 10.1016/j.blre.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alter BP, Giri N, Savage SA, et al. Malignancies and survival patterns in the National Cancer Institute inherited bone marrow failure syndromes cohort study. Br J Haematol. 2010;150:179–188. doi: 10.1111/j.1365-2141.2010.08212.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alter BP, Rosenberg PS, Brody LC. Clinical and molecular features associated with biallelic mutations in FANCD1/BRCA2. J Med Genet. 2007;44:1–9. doi: 10.1136/jmg.2006.043257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.MacMillan ML, Wagner JE. Haematopoeitic cell transplantation for Fanconi anaemia - when and how? Br J Haematol. 2010 doi: 10.1111/j.1365-2141.2010.08078.x. [DOI] [PubMed] [Google Scholar]
- 5.Peffault de LR, Porcher R, Dalle JH, et al. Allogeneic hematopoietic stem cell transplantation in Fanconi anemia: the European Group for Blood and Marrow Transplantation experience. Blood. 2013;122:4279–4286. doi: 10.1182/blood-2013-01-479733. [DOI] [PubMed] [Google Scholar]
- 6.Pasquini R, Carreras J, Pasquini MC, et al. HLA-matched sibling hematopoietic stem cell transplantation for fanconi anemia: comparison of irradiation and nonirradiation containing conditioning regimens. Biol Blood Marrow Transplant. 2008;14:1141–1147. doi: 10.1016/j.bbmt.2008.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Howlett NG, Taniguchi T, Olson S, et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science. 2002;297:606–609. doi: 10.1126/science.1073834. [DOI] [PubMed] [Google Scholar]
- 8.Wagner JE, Tolar J, Levran O, et al. Germline Mutations in BRCA2: Shared Genetic Susceptibility to Breast Cancer, Early Onset Leukemia and Fanconi Anemia. Blood. 2004;103:3226–3229. doi: 10.1182/blood-2003-09-3138. [DOI] [PubMed] [Google Scholar]
- 9.MacMillan ML, Auerbach AD, Wagner JE. Risk of malignancy in patients with biallelic BRCA2 mutations. Pediatr Blood & Cancer. 2005;44:539–540. [Google Scholar]
- 10.Chaudhury S, Auerbach AD, Kernan NA, et al. Fludarabine-based cytoreductive regimen and T-cell-depleted grafts from alternative donors for the treatment of high-risk patients with Fanconi anaemia. Br J Haematol. 2008;140:644–655. doi: 10.1111/j.1365-2141.2007.06975.x. [DOI] [PubMed] [Google Scholar]
- 11.Myers K, Davies SM, Harris RE, et al. The clinical phenotype of children with Fanconi anemia caused by biallelic FANCD1/BRCA2 mutations. Pediatr Blood Cancer. 2011;58:462–465. doi: 10.1002/pbc.23168. [DOI] [PubMed] [Google Scholar]
- 12.Yeo CJ, Gilman AL. Interleukin-2-induced Graft-Versus-Leukemia for the Treatment of AML in a BRCA2 Fanconi Anemia Patient. J Pediatr Hematol Oncol. 2013;36:e78–e80. doi: 10.1097/MPH.0b013e31828e5c56. [DOI] [PubMed] [Google Scholar]
- 13.Ayas M, Saber W, Davies SM, et al. Allogeneic hematopoietic cell transplantation for fanconi anemia in patients with pretransplantation cytogenetic abnormalities, myelodysplastic syndrome, or acute leukemia. J Clin Oncol. 2013;31:1669–1676. doi: 10.1200/JCO.2012.45.9719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mitchell R, Wagner JE, Hirsch B, et al. Haematopoietic cell transplantation for acute leukaemia and advanced myelodysplastic syndrome in Fanconi anaemia. Br J Haematol. 2014;164:384–395. doi: 10.1111/bjh.12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alter BP. Fanconi anemia and the development of leukemia. Best Pract Res Clin Haematol. 2014;27:214–221. doi: 10.1016/j.beha.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith CM. Origin and uses of primum non nocere--above all, do no harm! J Clin Pharmacol. 2005;45:371–377. doi: 10.1177/0091270004273680. [DOI] [PubMed] [Google Scholar]
- 17.Dewire MD, Ellison DW, Patay Z, et al. Fanconi anemia and biallelic BRCA2 mutation diagnosed in a young child with an embryonal CNS tumor. Pediatr Blood Cancer. 2009;53:1140–1142. doi: 10.1002/pbc.22139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bodd TL, Van GM, Eiklid K, et al. Fanconi anaemia, BRCA2 and familial considerations - follow up on a previous case report. Acta Paediatr. 2010;99:1741–1743. doi: 10.1111/j.1651-2227.2010.01929.x. [DOI] [PubMed] [Google Scholar]
- 19.Kopic S, Eirich K, Schuster B, et al. Hepatoblastoma in a 4-year-old girl with Fanconi anaemia. Acta Paediatr. 2011;100:780–783. doi: 10.1111/j.1651-2227.2010.02116.x. [DOI] [PubMed] [Google Scholar]
- 20.Beck JR, Pauker SG. The Markov process in medical prognosis. Med Decis Making. 1983;3:419–458. doi: 10.1177/0272989X8300300403. [DOI] [PubMed] [Google Scholar]
- 21.Cutler CS, Lee SJ, Greenberg P, et al. A decision analysis of allogeneic bone marrow transplantation for the myelodysplastic syndromes: delayed transplantation for low-risk myelodysplasia is associated with improved outcome. Blood. 2004;104:579–585. doi: 10.1182/blood-2004-01-0338. [DOI] [PubMed] [Google Scholar]
- 22.Pidala J, Anasetti C, Kharfan-Dabaja MA, et al. Decision analysis of peripheral blood versus bone marrow hematopoietic stem cells for allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2009;15:1415–1421. doi: 10.1016/j.bbmt.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 23.Rosenberg PS, Socie G, Alter BP, Gluckman E. Risk of head and neck squamous cell cancer and death in patients with Fanconi Anemia who did and did not receive transplants. Blood. 2005;105:67–73. doi: 10.1182/blood-2004-04-1652. [DOI] [PubMed] [Google Scholar]
- 24.Briggs A, Sculpher M. An introduction to Markov modelling for economic evaluation. Pharmacoeconomics. 1998;13:397–409. doi: 10.2165/00019053-199813040-00003. [DOI] [PubMed] [Google Scholar]
- 25.Gaynor JJ, Feuer EJ, Tan CC, et al. On the use of cause-specific failure and conditional failure probabilities: Examples from clinical oncology data. J Amer Stat Assoc. 1993;88:400–409. [Google Scholar]
- 26.Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–481. [Google Scholar]
- 27.Pai SY, Logan BR, Griffith LM, et al. Transplantation outcomes for severe combined immunodeficiency, 2000–2009. N Engl J Med. 2014;371:434–446. doi: 10.1056/NEJMoa1401177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boelens JJ, Wynn RF, O’Meara A, et al. Outcomes of hematopoietic stem cell transplantation for Hurler’s syndrome in Europe: a risk factor analysis for graft failure. Bone Marrow Transplant. 2007;40:225–233. doi: 10.1038/sj.bmt.1705718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ayas M, Siddiqui K, Al-Jefri A, et al. Factors affecting the outcome of related allogeneic hematopoietic cell transplantation in patients with Fanconi Anemia. Biol Blood Marrow Transplant. 2014;20:1599–1603. doi: 10.1016/j.bbmt.2014.06.016. [DOI] [PubMed] [Google Scholar]
- 30.Kodish E, Lantos J, Stocking C, et al. Bone marrow transplantation for sickle cell disease. A study of parents’ decisions. N Engl J Med. 1991;325:1349–1353. doi: 10.1056/NEJM199111073251905. [DOI] [PubMed] [Google Scholar]
- 31.Pidala J, Anasetti C, Jim H. Quality of life after allogeneic hematopoietic cell transplantation. Blood. 2009;114:7–19. doi: 10.1182/blood-2008-10-182592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Willard VW, Leung W, Huang Q, Zhang H, Phipps S. Cognitive outcome after pediatric stem-cell transplantation: impact of age and total-body irradiation. J Clin Oncol. 2014;32:3982–3988. doi: 10.1200/JCO.2014.56.2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tanzi EM. Health-related quality of life of hematopoietic stem cell transplant childhood survivors: state of the science. J Pediatr Oncol Nurs. 2011;28:191–202. doi: 10.1177/1043454211408100. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.