

Significance
Peptidoglycan (PG) is a unique and essential cross-linked, bag-like macromolecule that completely encases the cytoplasmic membrane and confers shape and rigidity to a bacterial cell. Therefore, bacterial cell growth is tightly coupled to PG expansion, requiring the coordinate activity of hydrolases that cleave the cross-links and synthases that catalyze the cross-link formation. This study highlights the importance of cross-link cleavage and its regulation in PG biogenesis by demonstrating the modulation of a cross-link–specific PG hydrolytic enzyme, MepS, by a previously unidentified degradation system consisting of an outer membrane lipoprotein, NlpI and a periplasmic protease, Prc. These studies facilitate better understanding of bacterial cell wall synthesis, which is a target of several antimicrobial therapeutic agents.
Keywords: bacterial morphogenesis, peptidoglycan, regulated proteolysis, MepS, NlpI-Prc
Abstract
Bacterial growth and morphogenesis are intimately coupled to expansion of peptidoglycan (PG), an extensively cross-linked macromolecule that forms a protective mesh-like sacculus around the cytoplasmic membrane. Growth of the PG sacculus is a dynamic event requiring the concerted action of hydrolases that cleave the cross-links for insertion of new material and synthases that catalyze cross-link formation; however, the factors that regulate PG expansion during bacterial growth are poorly understood. Here, we show that the PG hydrolase MepS (formerly Spr), which is specific to cleavage of cross-links during PG expansion in Escherichia coli, is modulated by proteolysis. Using combined genetic, molecular, and biochemical approaches, we demonstrate that MepS is rapidly degraded by a proteolytic system comprising an outer membrane lipoprotein of unknown function, NlpI, and a periplasmic protease, Prc (or Tsp). In summary, our results indicate that the NlpI–Prc system contributes to growth and enlargement of the PG sacculus by modulating the cellular levels of the cross-link–cleaving hydrolase MepS. Overall, this study signifies the importance of PG cross-link cleavage and its regulation in bacterial cell wall biogenesis.
Peptidoglycan (PG or murein) is a unique and essential constituent of eubacterial cell walls, thus making it an excellent target for several antimicrobial agents. It is a single, large, extensively cross-linked macromolecule that forms a mesh-like sacculus protecting cells against intracellular turgor pressure in addition to conferring cell shape. Structurally, the PG sacculus is made up of linear glycan strands cross-linked to each other by short peptide chains forming a continuous layer around the cytoplasmic membrane. The glycan strands are made up of alternating N-acetyl muramic acid (NAM) and N-acetyl glucosamine (NAG) disaccharide units in which NAM is covalently attached to a peptide chain containing 2- to 5-amino acid residues, with the pentapeptide consisting of l-alanine (ala)−d-glutamic acid (glu)−meso-diaminopimelic acid (mDAP)−d-ala−d-ala. Normally, d-ala of one peptide chain is cross-linked to mDAP of another peptide chain of an adjacent glycan strand, resulting in an extensively cross-linked single- or multilayered sacculus (1).
Because the murein sacculus totally encircles the cytoplasmic membrane, growth of a cell is tightly coupled to expansion of PG. Growth of the PG sacculus is a dynamic and coordinated event requiring concerted action both of murein hydrolases that facilitate cleavage of cross-links for the insertion of nascent murein material and of synthases that catalyze cross-link formation between adjacent glycan strands (Fig. 1) (2, 3).
Fig. 1.

Schematic of PG sacculus expansion. (A) Depiction of PG sacculus enlargement during growth of a bacterial cell. New murein strands (colored red) are incorporated into the preexisting murein strands (colored blue) to expand the PG sacculus (2, 3). (B) A model showing cleavage and resynthesis of cross-links for successful insertion of new glycan strands for expansion of the PG sacculus (8). The green bars indicate peptide stems whereas the small dark red bars represent the cross-bridges between the peptide stems of adjacent glycan strands.
Escherichia coli encodes multiple PG synthases that catalyze the formation of d-ala−mDAP cross-links in the PG sacculus. The class I enzymes (PBP1a and PBP1b, encoded by mrcA and mrcB, respectively) are bifunctional and possess both glycosyl transferase (GT) and transpeptidase (TP) activities whereas class II enzymes (PBP2 and PBP3, encoded by mrdA and ftsI, respectively) are monofunctional and possess only the TP activity (4, 5). The TP activity of PBP1a and PBP1b is activated, respectively, by their cognate lipoprotein cofactors LpoA and LpoB, located in the outer membrane (6, 7).
Given that the interconnecting peptide bridges in the PG sacculus need to be cleaved for the insertion of new murein material, hydrolytic enzymes with such activity are expected to be critical for PG expansion and thus for bacterial viability (2, 3, 8). Essential cross-link–specific hydrolases have recently been identified in both Gram-positive and -negative bacteria (9–13). E. coli possesses several hydrolytic enzymes specific for d-ala−mDAP cross-links (termed d,d-endopeptidases based on their specificity) (14); of these peptidases, Spr, YebA, and YdhO (renamed MepS, MepM, and MepH, respectively, in which mep stands for murein endopeptidase) are critical for growth under normal physiological conditions because a mutant lacking all these three endopeptidases is unable to incorporate new murein and exhibits rapid lysis (9).
Although cross-link cleavage is fundamental for PG growth, it is obvious that such cleavage needs to be stringently regulated at the spatiotemporal level to avoid lethal breakage and rupture of the PG sacculus. In addition, to maintain the continuum and integrity of PG, cleavage must be tightly coupled to cross-link formation, but the underlying mechanisms are not known.
To understand how this potentially lethal hydrolytic activity is controlled in the cell, here, we examined the regulation of the d,d-endopeptidase MepS. We found that MepS is highly abundant in the exponential phase of growth, with levels falling sharply at the onset of the stationary phase. Using combined genetic, molecular, and biochemical approaches, we demonstrate that MepS is degraded rapidly by a previously unknown proteolytic system comprising a tetratricopeptide repeat (TPR)-containing outer membrane (OM) lipoprotein, NlpI, and a periplasmic protease, Prc. In summary, we show that the NlpI–Prc system regulates PG synthesis by altering the levels of MepS, a hydrolase that breaks the cross-links for insertion of new murein material during growth of the PG sacculus.
Results
Growth Phase Specificity of PG Hydrolase MepS.
MepS is an OM lipoprotein, belonging to the NlpC/P60 superfamily of peptidases, that contributes to the essential murein hydrolytic activity in E. coli (9). Because this observation reflected a vital role for MepS in growth and enlargement of the PG sacculus, we examined the level of cellular MepS during various phases of growth, by Western analysis using a functional C-terminal 3XFlag-tagged derivative at the native chromosomal locus (Table S1). Fig. 2 shows that the level of MepS is high throughout the exponential phase of growth, with levels falling steeply at the onset of the stationary phase, with further gradual decrease as cells progress into the late stationary phase.
Table S1.
Bacterial strains used in this study
| Strain | Genotype* | Source† |
| BL21 (λDE3) | ompT rB−mB− (PlacUV5::T7gene1) | Novagen |
| BW25113 | lacIq rrnB3 ∆lacZ4787 ∆(araBAD)567 ∆(rhaBAD)568 hsdR514 | (37) |
| BW27783 | BW25113 ∆(araFGH) Φ(∆araEp PCP8−araE) | (38) |
| MG1655 | rph1 ilvG rfb-50 | Laboratory collection |
| MR801‡ | MG1655 mepS-Flag-Kan | MG1655X P1(DY378-mepS-Flag-Kan) |
| MR802 | MR801 (KanS) | MR801/pCP20 |
| MR803 | MR802 ΔnlpI::Kan | MR802 X P1 (ΔnlpI::Kan) |
| MR804 | MR802 Δprc::Kan | MR802 X P1(Δprc::Kan) |
| MR805 | MR802 ΔnlpI::frt | MR803/pCP20 |
| MR806 | MR805Δprc::Kan | MR805 X P1(Δprc::Kan) |
| MR807 | MG1655 ΔphoA::frt ΔmepS::frt | MG1655 ΔphoA ΔmepS::Kan/pCP20 |
| MR808 | MR807 ΔnlpI::Kan | MR807 X P1 (ΔnlpI::Kan) |
| MR809 | MR807 Δprc::Kan | MR807 X P1(Δprc::Kan) |
| MR810 | MG1655 ΔmepS::frt | MG1655 ΔmepS::Kan/pCP20 |
| MR811 | MG1655 ΔnlpI::frt | MG1655 ΔnlpI::Kan/pCP20 |
| MR812 | MG1655 Δprc::frt | MG1655 ΔnlpI::Kan/pCP20 |
| MR813 | MR810 ΔnlpI::frt | MR810 ΔnlpI::Kan/pCP20 |
| MR814 | MR810 Δprc::Kan | MR810 X P1(Δprc::Kan) |
| MR815 | MR811 Δprc::Kan | MR811 X P1(Δprc::Kan) |
| MR816 | BW27783 ΔmepM::frt | BW27783 ΔmepM::Kan/pCP20 |
| MR818‡ | MR810 prc-HA-Cam | MR810 X P1(prc-HA-Cam) |
| MR819‡ | MR810 prcS452A-HA-Cam | MR810 X P1 (prcS452A-HA-Cam) |
| MR820‡ | MR805 prc-HA-Cam | MR805 X P1(prc-HA-Cam) |
| MR821‡ | MR813 prc-HA-Cam | MR813 X P1(prc-HA-Cam) |
| MR822‡ | MR811 prc-HA-Cam | MR811 X P1(prc-HA-Cam) |
The deletion alleles mepS (spr), mepM (yebA), and nlpI, phoA were sourced from the Keio collection (37). Deletion mutations were tested for authenticity by linkage, PCR, sequence analysis, and phenotype, if known, and introduced into required strain backgrounds by phage P1 transduction (39).
Marker-less (KanS) strains were made by flipping out the Kan cassette using pCP20 that encodes Flp recombinase (40).
The mepS-Flag-Kan, prc-HA-Cam, and prcS452A-HA-Cam constructs are described above in Strain Constructions.
Fig. 2.
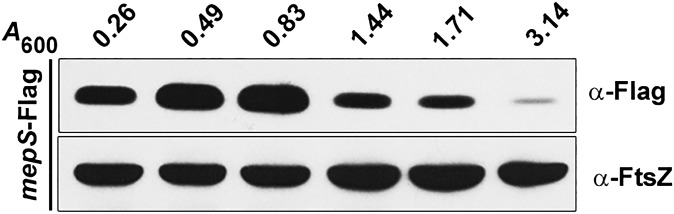
Growth phase specificity of MepS. Strain MR802 (MG1655 mepS-Flag) was grown in LB and fractions were collected at various time intervals (with indicated A600 values) during the growth cycle. Normalized cell extracts (corresponding to 0.25 OD cells) were separated by SDS/PAGE, and MepS was detected by Western analysis using anti-Flag antisera. FtsZ was used as a loading control.
Regulation of MepS Is Dependent on NlpI and Prc.
To understand the basis of the growth-phase specificity of MepS, we identified mutations that altered the activity of alkaline phosphatase (PhoA) fused to the C terminus of mepS (Para::mepS-phoA) (Table S2). Two such mutations that increased the activity of MepS-PhoA were in loci encoding NlpI, an OM lipoprotein of unknown function, and Prc, a periplasmic protease (Fig. S1A). nlpI or prc deletion mutants do not grow on media of low osmotic strength in addition to forming long filaments (15, 16), and absence of MepS was able to suppress the growth and morphological defects of both these mutants (Fig. S1B). Suppression of nlpI or prc mutant phenotypes by mepS deletion was abolished by a plasmid-borne copy of WT mepS, but not by a derivative carrying a mutation, C68A, in the active site of MepS (Fig. S1C). These results suggest that the unfettered d,d-endopeptidase activity of MepS is responsible for the phenotypes of nlpI or prc mutants.
Table S2.
Plasmids used in this study
| Plasmid | Relevant features | Source |
| pBAD18-Amp | ColE1, AmpR, Para | Laboratory collection |
| pBAD33 | p15A, CamR, Para | Laboratory collection |
| pCL1920 | pSC101, SpecR, Plac | Laboratory collection |
| pCP20 | pSC101(Ts), AmpR, CamR, Flp | (40) |
| pET21b | ColE1, AmpR, T7lac | Novagen |
| pPHO7 | ColE1, AmpR, PhoA (AP) | (44) |
| pTRC99a | ColE1, AmpR, Ptrc | Laboratory collection |
| pMN83 | pBAD33-mepS | (9) |
| pMN84 | pET21b-mepS28-188 | (9) |
| pMN88 | pBAD33-mepS-C68A | (9) |
| pMN204 | pTRC99-nlpI | Full-length nlpI |
| pMN208 | pET21b-nlpI19-294 | For NlpI-His purification |
| pMN211 | pCL1920-nlpI-D284 | nlpI lacking 33 bases from the 3′ end |
| pMN212 | pCL1920-nlpI -Q283 | nlpI lacking 36 bases from the 3′ end |
| pMN213 | pCL1920-nlpI -G282 | nlpI lacking 39 bases from the 3′ end |
| pMN214 | pCL1920-prc | Full-length prc |
| pMN216 | pCL1920-nlpI | Full-length nlpI |
| pMN217 | pBAD18-mepS-His | mepS-His cloned in pBAD18 |
| pMN218 | pBAD18-nlpI-His | nlpI-His cloned in pBAD18 |
| pMN219 | pET21b-prc23-682 | For Prc-His purification |
| pMN220 | pET21b-prcK477A | Encoding a mutant Prc (Prc-K477A) |
| pMN221 | pET21b-prcS452A | Encoding a mutant Prc (Prc-S452A) |
| pMN222 | pBAD18-amp-phoA | phoA subcloned from pPHO7 into pBAD18 |
| pMN223 | pMN222-mepS-phoA | mepS-phoA protein fusion |
Fig. S1.

Regulation of mepS by NlpI and Prc. (A) Qualitative patch assay for activity of MepS-PhoA. Cells of MR807 (MG1655 ΔphoA ΔmepS), MR808 (MR807 ΔnlpI::Kan), or MR809 (MR807 Δprc::Kan) carrying plasmid pMN223 (Para::mepS-phoA) were patched onto minimal-121 plates containing 0.01% arabinose, 10−4 M KH2PO4, and 40 μg/mL 5-bromo-4-chloro-3-indolyl phosphate (BCIP) and grown at 37 °C. nlpI and prc mutants showed a marginal but consistent increase of the blue coloration, which is an indicator of MepS-PhoA activity. (B) The absence of mepS suppresses the growth defects of nlpI and prc mutants. Cells of WT (MG1655) and its various mutant derivatives were grown in LB broth, and 5 μL of the indicated dilutions were spotted on LB and LBON (LB without added NaCl) plates and grown at 42 °C. The genetic interactions between mepS, nlpI, and prc were described earlier (16, 17). (C) d,d-endopeptidase activity of MepS is lethal in nlpI and prc mutants. Cells of MR813 (ΔmepS ΔnlpI) or MR814 (ΔmepS Δprc) carrying pBAD33 (Para), pMN83 (Para::mepS), or pMN88 (Para::mepS-C68A) were grown in LB plus Cam broth, and 5 μL of various dilutions were spotted on indicated plates and grown at 42 °C.
Because the experiments above were indicative of regulation of MepS by NlpI and/or Prc, we measured MepS levels in the absence of NlpI or Prc. The data in Fig. 3A show that, in nlpI or prc mutants, the level of MepS is constitutively high throughout the growth cycle, indicating loss of regulation.
Fig. 3.

Regulation of MepS is dependent on NlpI and Prc. (A) MepS-Flag levels in nlpI and prc mutants. Strains MR802 (MG1655 mepS-Flag), MR803 (MR802 ΔnlpI), or MR804 (MR802 Δprc) were grown in LB, and fractions were collected at different time points during growth and analyzed by Western blotting as described in the legend to Fig. 2. (B) Determination of stability of MepS by pulse–chase experiment (in vivo degradation assay). To rapidly growing cultures of the above strains in LB (at an OD600 of ∼0.4), spectinomycin (Spec) was added at a concentration of 300 μg/mL to block translation, and fractions were collected at indicated time points and analyzed as described in the legend to Fig. 2.
To understand the basis of MepS regulation, a pulse–chase experiment was performed after inhibition of protein synthesis by treatment with spectinomycin. Fig. 3B shows that increased MepS in nlpI or prc mutants is a consequence of enhanced posttranslational stability. The half-life of MepS was substantially increased from ∼1–2 min in exponentially growing WT cells to more than 45 min in mutants lacking NlpI or Prc. Consistent with a posttranslational model of regulation, the expression of a chromosomal mepS-lacZ transcriptional fusion was altered neither during the growth cycle nor in nlpI or prc mutants (Table S3).
Table S3.
β-Galactosidase assays of mepS::lacZ fusions
| Strains/genotypes | β-galactosidase values, Miller units* |
| MC4100 ϕ(mepS’::lacZY-Kan) (A600∼0.6)† | 763 ± 53 |
| MC4100 ϕ(mepS’::lacZY-Kan) (A600∼1.2) | 950 ± 64 |
| MC4100 ϕ(mepS’::lacZY-Kan)/pTRC99a-mepS | 820 ± 75 |
| MC4100 ϕ(mepS’::lacZY-Kan)/pTRC99a-mepS (with 100 μM IPTG) | 910 ± 103 |
| MC4100 ϕ(mepS’::lacZY-Kan) ΔnlpI::frt | 760 ± 32 |
| MC4100 ϕ(mepS’::lacZY-Kan) Δprc::frt | 863 ± 27 |
Mean values with SD are shown. Values are taken from three independent experiments. Assays are done as described in Miller (39).
The lacZ fusion strains were made as described (43). Because these strains lack the mepS structural gene, a plasmid-borne copy of mepS (pMN80; pTRC99a-mepS; 9) was used for complementation, and no difference in the activity was observed in the presence or absence of mepS.
To examine whether NlpI and Prc act in concert to regulate MepS, the level of MepS was measured in single and double mutants. As shown in Fig. 4A, MepS levels remain equally high in all of these mutants, suggesting that NlpI and Prc share a common pathway in regulation of MepS. Furthermore, a modest increase in NlpI led to a significant decrease of MepS, which in turn was dependent on the presence of functional Prc, demonstrating the combined requirement of both these factors in MepS regulation (Fig. 4A).
Fig. 4.

Requirement of both NlpI and Prc for regulation of MepS. (A) Levels of MepS in single and double mutants of nlpI and prc. Strains MR802 (MG1655 mepS-Flag), MR803 (MR802 ΔnlpI), MR804 (MR802 Δprc), and MR806 (MR802 ΔnlpI Δprc) were grown in LB to an A600 of 1.0, and MepS was detected by Western analysis as described in the legend to Fig. 2. Plasmid-carrying strains were grown with appropriate antibiotic (Spec) and 1 mM IPTG. FtsZ was used as a loading control. (B) NlpI is not processed by Prc. The indicated strains MG1655 (WT), MR810 (MG1655 ΔmepS), MR812 (MG1655 Δprc), MR814 (MG1655 ΔmepS Δprc) were grown in LB and harvested at an A600 of ∼1.0. Normalized cell extracts were analyzed by Western blotting using anti-NlpI antisera. Plasmid-carrying strains were grown with Spec and 1 mM IPTG. The C-terminal truncations of NlpI (bands in lanes 8 and 9) served as controls to indicate the size of processed NlpI. Processed NlpI is expected to be of 284 amino acids in length (17) whereas full-length is 294 amino acids.
Prc Does Not Process NlpI.
In light of an earlier report that Prc protease processes NlpI into a mature functional derivative by cleaving the latter’s C-terminal 10- to 11-amino acid residues (17), we measured the size of NlpI in WT and prc mutant strains by Western analysis using anti-NlpI antisera. Fig. 4B shows that the size and level of NlpI remain unaltered in the presence or absence of Prc. Importantly, a plasmid-borne copy of nlpI encoding a polypeptide lacking the extreme C-terminal 10 amino acids (NlpI-D284) was able to functionally complement an nlpI mutant but not a prc or prc nlpI mutant, indicating that both NlpI and Prc contribute independently and directly to the stability of MepS (Fig. S2).
Fig. S2.

Complementation using full-length nlpI or nlpI-D284 plasmid derivatives. Cells of MR811 (ΔnlpI), MR812 (Δprc), or MR815 (ΔnlpI Δprc) carrying pCL1920, pMN216 (pCL-nlpI), or pMN211 (pCL-nlpI-D284) were grown in LB broth with Spec, and 5 μL of various dilutions were spotted on indicated plates and grown at 42 °C. IPTG was used at 1 mM.
NlpI Overexpression Phenocopies MepS Deletion.
Because multiple copies of NlpI led to lowered MepS levels, (Fig. 4A), we examined the effect of increased NlpI on the growth and viability of a mutant deleted for the alternative endopeptidase MepM because a strain doubly deficient in MepS and MepM is not viable on rich media (9). A modest increase of NlpI resulted in severe cell lysis in the mepM mutant that was suppressed by introduction of additional copies of MepS (Fig. 5A and Fig. S3). In WT E. coli, as well, increased NlpI conferred the phenotypes of a mepS deletion mutant: i.e., inability to grow on media of low osmolarity and altered cell shape from rods to fat ellipsoids (Fig. 5B). Taken together, these data indicate that increased NlpI decreases MepS levels very effectively in vivo and mimics the MepS− phenotype.
Fig. 5.

NlpI overexpression phenocopies MepS− phenotype. (A) Lethal effect of overexpressed NlpI in ΔmepM mutants. Strains MR816 (BW27783 ΔmepM) carrying either pTRC99a (Ptrc) or pMN204 (Ptrc::nlpI) were grown in LB with Amp, and 5 μL of various dilutions were placed on indicated plates and grown. IPTG was used at 100 μM. (B) Effect of overexpressed NlpI in WT E. coli. MG1655 carrying either pTRC99a (Ptrc) or pMN204 (Ptrc::nlpI) was cultured and grown on nutrient agar (NA) at 42 °C. IPTG was used at 100 μM. ΔmepS strains are known not to grow on NA at 42 °C (24). For microscopy, the cultures were grown, diluted 1:100 either in LBON (for WT and ΔmepS strains) or in LBON plus 100 µM IPTG (for plasmid-carrying strains) and grown at 42 °C until an OD600 of ∼0.4–0.6. Differential interference contrast (DIC) images were taken after concentrating and spotting the culture on agarose pads. Overexpression of NlpI in WT is known to result in loss of rod shape and formation of prolate ellipsoid cells (15). It is not clear why the mutants lacking MepS (or those overproducing NlpI) exhibit fat and wide cell morphology; these cells may possibly be partially deficient in cross-link cleavage, leading to defective growth and elongation of the PG sacculus.
Fig. S3.

Effect of multiple copies of nlpI is suppressible by additional copies of mepS. (A) Strain MR816 /pMN216 carrying either pBAD33 (Para) or pMN83 (Para::mepS) was grown in LB with appropriate antibiotics and subcultured 1:100 in LB broth containing 0.2% arabinose, and, after 1 h growth (∼0.15 OD), 500 μM IPTG was added and grown for a further 1 h, after which cultures were drawn for microscopy. (B) Strains were grown as described above, and live–dead cell staining was done as described in SI Materials and Methods. Green and red represent live and dead cells, respectively. (Scale bars are shown.)
NlpI Enhances the Proteolysis of MepS by Prc.
Our results thus far suggested that NlpI and Prc together modulate the level of MepS in vivo. To test this observation directly in vitro, we purified MepS, NlpI, and Prc, as C-terminal hexahistidine fusion proteins (Fig. 6A, lanes 2, 3, and 4) lacking their signal sequences, and performed degradation assays (Fig. 6A). NlpI and MepS were stable in the presence of one another (Fig. 6A, lane 5) as was Prc in the presence of NlpI (Fig. 6A, lane 6). Coincubation of Prc and MepS led to degradation of the latter to a minimal extent (Fig. 6A, compare lanes 7 and 8), and this process was substantially accelerated in the presence of NlpI (Fig. 6A, compare lanes 9 and 10). A time course experiment also indicated that MepS degradation by Prc is enhanced to a large extent by addition of NlpI (Fig. S4 A and B). Degradation of MepS by Prc was abolished by introduction of mutations at the active site residues of Prc (S452A or K477A) (18), even in the presence of NlpI (Fig. 6B, compare lanes 5, 6, and 7). The proteolytic activity of Prc toward MepS was specific because two other putative periplasmic peptidases, NlpC (a paralog of MepS belonging to the NlpC/P60 family) and YfiH, were not considerably degraded by Prc (Fig. S4C). Overall, these results demonstrate that MepS is a substrate of Prc protease and that NlpI facilitates this degradation.
Fig. 6.

MepS is a substrate of Prc. (A) In vitro degradation assays. The C-terminal hexahistidine-tagged MepS, NlpI, and Prc proteins were mixed in all combinations (as indicated) and incubated for 3 h at 37 °C followed by SDS/PAGE and Coomassie blue staining. Each reaction contained the following (approximately): MepS, 15 μg; NlpI, 2 μg; and Prc, 4 μg. The mixtures in lanes 7 and 9 served as controls (0 h; no incubation). M is a protein marker with the following molecular size range (in kDa): 170, 130, 100, 70, 55, 40, 35, 25, 15, and 10. Purified MepS was always seen on SDS/PAGE as a doublet, and N-terminal sequencing confirmed that both bands belong to MepS itself. (B) Specificity of Prc. Two mutant derivatives of Prc (carrying K477A or S452A alterations at the active site residues) did not show any proteolytic activity (even when higher concentrations were used) against MepS in the presence of NlpI.
Fig. S4.

MepS is a substrate of Prc. (A) Time course degradation assay of MepS. Purified proteins (MepS and Prc) were mixed and incubated at 37 °C as indicated, followed by SDS/PAGE and Coomassie staining. MepS undergoes significant degradation on prolonged incubation with Prc alone (compare lanes 2 and 8), but not when incubated with NlpI (lanes 9 and 10). (B) Time course degradation assay of MepS. Purified proteins (MepS, NlpI, and Prc) were mixed and incubated as indicated at 37 °C, followed by SDS/PAGE and Coomassie staining. MepS degradation is enhanced quite considerably in the presence of NlpI (lanes 2 and 3). (C) Purified YfiH (a putative peptidase) or NlpC (a paralog of MepS) was used as a substrate of Prc. The proteins as indicated were added and incubated for 3 h at 37 °C, followed by SDS/PAGE and Coomassie staining. Lanes 6 and 8 show purified C-terminal His-tagged YfiH and NlpC proteins. YfiH is not degraded at all whereas NlpC seems to be degraded to some extent (lanes 7 and 9).
NlpI Facilitates Interaction of MepS and Prc in Vivo.
To examine the in vivo interactions of MepS with Prc and/or NlpI, pull-down assays were performed in strains carrying a functional chromosomal Prc-HA fusion allele, along with a plasmid-encoded functional MepS bearing a hexahistidine tag at its C terminus (Para::mepS-His). Immunoblot analysis of the MepS-His pull-down fractions showed that NlpI, but not Prc, is copurified with MepS (Fig. 7A and Fig. S5A). The MepS–NlpI interaction occurred even in absence of Prc (Fig. 7A). A mutant derivative of Prc-HA carrying an active site mutation, S452A, that is expected to bind but not cleave MepS (Fig. S5B) also could not be copurified with MepS, suggesting that Prc does not directly interact with MepS in vivo (Fig. 7A).
Fig. 7.

Interactions of MepS, NlpI, and Prc. (A) Western blot showing interaction of MepS-His with NlpI and Prc. Strains MR818 (MG1655 ΔmepS prc-HA-Cam), MR819 (MG1655 ΔmepS prcS452A-HA-Cam), or MR814 (MG1655 ΔmepS Δprc) carrying pMN217 (Para::mepS-His) were grown overnight in LB with Amp and next morning diluted 1:100 into fresh LB and at A600 of ∼0.2–0.3 expression of MepS was induced by addition of 0.05% l-arabinose. Cultures were further grown until an A600 of ∼0.8–1.0, and MepS-His was purified using Ni2+-NTA beads as described in SI Materials and Methods. The purified fractions of MepS-His (pull-down fractions, PL) along with the input samples (IN) were separated on SDS/PAGE, and immunoblot analysis was performed to detect MepS, NlpI, and Prc with anti-His, anti-NlpI, and anti-HA antibodies, respectively. All strains had a deletion of mepS to reduce the interference from the endogenous MepS protein. A strain carrying plasmid-borne, untagged MepS (MG1655 ΔmepS/Para::mepS) was used as a negative control (Fig. S5A). (B) Western blot showing interaction of NlpI-His with MepS and Prc. Strains MR805 (MG1655 ΔnlpI mepS-Flag), MR806 (MG1655 ΔnlpI Δprc mepS-Flag), MR820 (MG1655 ΔnlpI mepS-Flag prc-HA-Cam), MR821 (MG1655 ΔnlpI ΔmepS prc-HA-Cam), and MR822 (MG1655 ΔnlpI prc-HA-Cam) carrying pMN218 (Para::nlpI-His) were grown with 0.05% l-arabinose to an OD of 0.8–1.0. NlpI-His was purified from all these strains using an Ni2+-NTA column, and the purified fractions (PL) along with the input fractions (IN) were separated on SDS/PAGE. Western analysis was done using anti-His, anti-Flag, and anti-HA antibodies to detect NlpI, MepS, and Prc, respectively. In both these experiments, input sample corresponds to ∼0.3 OD cells whereas the pull-down fraction is ∼20-fold concentrated (i.e., from ∼6 OD cells).
Fig. S5.

Interactions of MepS with NlpI and Prc. (A) Western blot showing interaction of MepS with NlpI. Strains MG1655 ΔmepS carrying either Para::mepS or Para::mepS-His were grown with 0.05% arabinose as described in the legend to Fig. 7, and MepS was purified from both these strains using Ni2+-NTA beads. The purified fractions (PL) along with the input samples (IN) were separated by SDS/PAGE, and immunoblot analysis was performed to detect MepS and NlpI using anti-His or anti-NlpI antibodies. The untagged MepS did not show any interaction with NlpI, showing that the bands (NlpI) are specific for the MepS-His tag. (B) Western blot depicting the levels of MepS in WT and in prcS452A mutant. Strain MR802 and its prcS452A-HA-Cam derivative were grown in LB, fractions were collected at various time points, and normalized volumes of cell extracts were separated by SDS/PAGE followed by Western analysis using anti-Flag antibodies. FtsZ was used as loading control.
Pull-down assays were also done using a plasmid-borne NlpI-His (Para::nlpI-His) in strains carrying functional MepS-Flag and Prc-HA fusion alleles at their native chromosomal loci. Here, both MepS and Prc were copurified with NlpI (Fig. 7B), and these interactions were independent of Prc and MepS, respectively (Fig. 7B). Taken together, these results indicate that NlpI is able to form binary complexes each with MepS and Prc in vivo, but MepS is itself unable to form a binary complex with Prc, suggesting that NlpI is required as an adapter protein to bring together the Prc protease with its target protein MepS, an interpretation that is consistent with the genetic and biochemical data.
Discussion
In this study, we demonstrate that a previously unknown proteolytic system comprising NlpI and Prc contributes to growth of the PG sacculus by rapidly degrading the cross-link–specific key murein hydrolytic enzyme MepS. This stringent regulation suggests that the cross-link cleavage activity of MepS is a crucial determinant of PG synthesis and may indeed be the rate-limiting step in PG expansion. To our knowledge, MepS is the first example of an enzyme involved in PG metabolism to be regulated by proteolysis.
The mechanism of degradation of MepS by the NlpI–Prc system seems to be analogous to that described earlier on the proteolysis of the rate-limiting enzyme of the lipopolysaccharide biosynthesis, LpxC, by an essential membrane-anchored protease FtsH and the TPR-containing adapter protein YciM (19).
Role of NlpI–Prc in E. coli.
Contrary to an earlier report that had suggested a proteolytic processing role for Prc to activate NlpI into a mature functional derivative (17), our results show that NlpI is not processed by Prc either in vivo (Fig. 4B) or in vitro (Fig. 6A). Rather, we find that NlpI binds both Prc and MepS, thereby acting like an adapter protein to facilitate degradation of MepS by Prc (Figs. 6 and 7). NlpI is an OM lipoprotein with multiple TPR motifs (which is conserved in γ-proteobacteria of the Enterobacteriaceae, Pasteurellaceae, and Vibrionaceae lineages) (20). It is known that TPR proteins mediate protein–protein interactions to facilitate assembly of multiprotein complexes (21). NlpI binding to Prc has been shown earlier (17). FtsI, the essential division-specific transpeptidase, is also shown to be processed by Prc although the physiological significance of such processing is not clear (22). A previous report had indicated that Prc is a periplasmic protein associated with the cytoplasmic membrane (16); however, as predicted by several algorithms, we find that Prc is a soluble periplasmic protein (Fig. S6). In this context, it is to be noted that Prc is specific to proteins with nonpolar C termini and thus is also called a Tail-specific protease (Tsp) (23). However, in many of our in vitro and in vivo experiments, we have used MepS derivatives with various C-terminal tags; therefore, it is possible that the extent of MepS degradation observed here is an underestimate and that the native cellular MepS in vivo may perhaps be more susceptible to regulation by the NlpI–Prc system.
Fig. S6.

Localization of Prc. The subcellular localization of Prc was examined after partial cell fractionation using the MG1655 prc-Flag-Kan strain. Cells were incubated in Tris-sucrose plus lysozyme plus EDTA buffer, followed by brief sonication for cell lysis. Unbroken cells were removed by a low-speed centrifugation step, and the supernatant (total cell lysate) was centrifuged at 4 °C for 1 h at 1,00,000 × g. Resuspended pellet (membrane fraction) and the supernatant (soluble fraction) were loaded on SDS/PAGE along with the total cell lysate, and Western analysis was done to detect Prc using an anti-Flag antibody. The other marker proteins included Pbp1b and FtsI, which are IM-anchored periplasmic proteins, and MBP, a soluble periplasmic protein. The data indicate that Prc is a soluble protein.
Regulation of MepS seems to be the primary role of NlpI and Prc because most NlpI− or Prc− phenotypes are abrogated by the absence of MepS (this study and refs. 24–27). It is to be noted that the levels of both NlpI and Prc are constitutive and not dependent on growth cycle (Fig. S7). Interestingly, mepS (formerly, spr) was identified initially as a suppressor of prc (24). Our results provide a mechanistic basis for earlier observations from several groups that absence of MepS suppresses various phenotypes of prc and nlpI mutants (24–27).
Fig. S7.

Levels of NlpI and Prc during the bacterial growth cycle. The protein levels of NlpI and Prc were examined using MG1655 carrying a functional prc-Flag-Kan allele. This strain was grown in LB, fractions were taken at the indicated optical density, and normalized cell extracts were analyzed by Western blotting using anti-Flag, anti-NlpI, and anti-FtsZ antisera to detect Prc, NlpI, and FtsZ, respectively. FtsZ was used as a loading control. Strain MR802 (MG1655 mepS-Flag) was also used as a control.
Models for Regulation of MepS.
The data from Fig. 2 show that the levels of MepS significantly decline at the onset of the stationary phase. At the same time, the pulse–chase experiments (Fig. 3B) indicate that MepS has a short half-life of ∼1–2 min, even in exponentially growing cultures, suggesting a rapid and constitutive degradation of MepS throughout the growth cycle. Based on the above findings, the following two scenarios for degradation of MepS can be envisaged. In the first case, the synthesis of MepS could be reduced in the stationary phase, therefore leading to the observed decline in steady-state levels as it is rapidly degraded. However, then the question arises about why cells should indulge in a futile cycle of simultaneous synthesis and degradation unless such a mechanism has evolved to permit instantaneous changes in the levels of MepS in individual cells within a population. The second scenario would be that the observed half-life of MepS represents an average of different cell cycle-specific values for the asynchronously growing culture. In this model, MepS is relatively stable in cells that are in the elongation mode of PG synthesis compared with the cells that are in the division mode of PG synthesis (that is, when the requirement for expansion of the PG sacculus is low). The above two possibilities may not be mutually exclusive and need to be experimentally tested further.
However, additional questions that remain to be addressed relate to the nature of the signal generated during expansion of the PG sacculus and how it is transmitted to NlpI, an OM lipoprotein. The causes of signal generation may include inputs from the PG biosynthetic machinery, alterations in membrane turgor, or the process of cross-link formation itself. Other instances of regulation of PG synthesis by OM lipoproteins are known: The TP activity of PG synthases PBP1a and PBP1b is activated by binding of OM lipoproteins LpoA and LpoB, respectively (6, 7). How these OM lipoproteins sense, bind, and regulate their cognate effectors is an interesting question to be addressed.
Regulation of Other PG Hydrolases.
Most eubacterial genomes encode a multitude of PG hydrolases that function in murein growth, maturation, turnover, recycling, autolysis, and cleavage of septum at cell division (12–14). Even as murein hydrolytic activity is essential for bacterial growth and division, it needs also to be tightly regulated to prevent otherwise lethal degradation of PG. Activation of division-specific amidases in E. coli has been shown to be coupled to formation of a cytokinetic ring at the midcell in which a divisomal component, FtsEX, activates EnvC, a catalytically inactive LytM family peptidase that in turn activates the amidases to cleave septal PG for separation of daughter cells (28).
In Bacillus subtilis, two l,d-endopeptidases, CwlO and LytE, form a minimal essential set of hydrolases for PG enlargement (10, 12, 13), of which CwlO is activated by the FtsEX complex (29) and LytE is regulated by MreBH (30). Likewise, FtsEX activates the hydrolytic activity of the peptidases PcsB and RipC in Streptococcus pneumoniae and Mycobacterium tuberculosis, respectively (31, 32). In contrast, here, we find that MepS is regulated by proteolysis, possibly providing an advantage of a rapid response to changing environmental conditions.
Coupling of Cross-Link Cleavage and Cross-Link Formation.
For an effective enlargement of the PG sacculus, hydrolysis is as important as synthesis although the coupling between these processes is not well understood. In Gram-negative organisms with monolayered PG sacculus, cleavage and resynthesis are believed to be either concomitant or tightly coupled to maintain PG integrity (2, 3, 33–36). On the other hand, in Gram-positive organisms such as B. subtilis, in which the PG sacculus is multilayered, hydrolysis is not coupled to synthesis even though PG hydrolytic activity is essential for growth (10, 12, 13, 29).
It is not yet clear whether cross-link cleavage initiates or follows the cross-link formation in E. coli. However, because some of our preliminary results suggest additional roles for NlpI and/or Prc in PG cross-link formation, we believe that the NlpI–Prc system may facilitate concomitant cleavage and resynthesis of cross-links, resulting in successful expansion of the PG sacculus during growth of bacteria.
Materials and Methods
Detailed strain and plasmid constructions, additional materials, methods, Tables S1–S3, and Figs. S1–S7 are listed in SI Materials and Methods.
Media and Growth Conditions.
Strains were normally grown in LB (1% tryptone, 0.5% yeast extract, 1% NaCl) unless otherwise indicated. LBON has no added NaCl. Nutrient broth has 0.5% peptone and 0.3% beef extract. Solid media had agar to a concentration of 1.5% (wt/vol). Antibiotics were used at the following concentrations (μg⋅mL−1): ampicillin (Amp), 50 μg⋅mL−1; chloramphenicol (Cam), 25 μg⋅mL−1; kanamycin (Kan), 25 μg⋅mL−1; and spectinomycin (Spec), 50 μg⋅mL−1. Concentrations of l-arabinose and isopropyl β-d-thiogalactopyranoside (IPTG) were indicated. Growth temperature was 37 °C unless otherwise specified.
Western Blotting.
Samples were separated using SDS/PAGE and transferred onto a nitrocellulose or PVDF membrane. Membrane was blocked by 5% skimmed milk in 1× TBS-T (Tris-NaCl-Tween 20) for 2 h and then incubated overnight with primary antibodies (1:10,000 for α-NlpI, 1:3,000 for α-His, α-FLAG, and α-HA, and 1:5,000 for α-FtsZ) at 4 °C. Membrane was washed three times with 1× TBS-T and then probed with secondary antibodies (1:10,000) tagged with horseradish peroxidase (HRP) and incubated for 1 h at room temperature. Membrane was overlaid with ECL Prime detection substrate (Amersham) for 5 min, and the blots were developed.
Pull-Down Experiments.
Pull downs were performed as described earlier, but with modifications (6). Strains were grown overnight in LB broth supplemented with Amp and next morning diluted 1:100 into fresh LB (200 mL) and grown at 37 °C until an OD600 of ∼0.2. At this point, 0.05% arabinose was added, and growth was continued until OD600 was 0.8–1.0. Cells were recovered by centrifugation, and the cell pellet was washed, resuspended in 25 mL of the lysis buffer (50 mM Tris⋅Cl, 10 mM MgCl2, 100 mM NaCl, 20% glycerol, 2% Triton X-100, 1 mg/mL lysozyme, 1× mixture protease inhibitor, 20 units of DNase, 20 units of RNase, and 10 mM imidazole; pH 8.0), and incubated on ice for 2 h. This mixture was subjected to brief sonication, and the lysate was gently stirred overnight with glass beads at 4 °C to solubilize the membrane proteins. The next morning, insoluble material of the lysate was removed by centrifugation (14,000 × g, 15 min, 4 °C), at which point 100 μL was set aside for input fractions, and then the supernatant was mixed with 200 μL of Ni2+-NTA agarose and mixed for 2 h at 4 °C. The agarose beads were washed twice with 10 mL of wash buffer (50 mM Tris⋅Cl, 100 mM NaCl, and 20 mM imidazole; pH 8.0) and twice with wash buffer containing 50 mM imidazole, and the bound proteins were eluted with 250 μL of the elution buffer (50 mM Tris⋅Cl, 100 mM NaCl, and 300 mM imidazole; pH 8.0). The eluate was electrophoresed using 12% SDS/PAGE, and the proteins were detected by Western blotting.
SI Materials and Methods
Media, Bacterial Strains, and Plasmids.
Strains were normally grown in LB (1% tryptone, 0.5% yeast extract, 1% NaCl) unless otherwise indicated. LBON has no added NaCl. Nutrient broth has 0.5% peptone and 0.3% beef extract. Solid media had agar to a concentration of 1.5%. Antibiotics were used at the following concentrations ( μg⋅ml−1): ampicillin (Amp), 50 μg⋅ml−1; chloramphenicol (Cam), 25 μg⋅ml−1; kanamycin (Kan), 25 μg⋅ml−1; and spectinomycin (Spec), 50 μg⋅ml−1. Concentrations of l-arabinose and IPTG are indicated. Growth temperature was 37 °C unless otherwise specified.
Bacterial strains and plasmids used in this study are listed in Tables S1 and S2. All strains are derivatives of MG1655 (Coli Genetic Stock Centre), BW25113 (37), or BW27783 (38) unless otherwise indicated. Standard plasmid transformations, P1 phage-mediated transductions, and β-galactosidase measurements were done as described previously (39).
For cloning, MG1655 genomic DNA was used. Pfx polymerase was used routinely for PCR, and cloned fragments were always confirmed by sequencing. Detailed strain and plasmid constructions are described below.
Strain Constructions.
Construction of Δprc::Kan deletion.
A prc insertion–deletion strain was made by recombineering as described (40). Hybrid primers 5′-GCT GTT AAA AAA TCA GGC ACA ATT TCT TGT GCC TGA TTG ATA TTA CTG TAG GCT GGA GCT GCT TC-3′ and 5′-AAC ACC TGG TGT TCT GAA ACG GAG GCC GGG CCA GGC ATG AAC ATG TTC ATA TGA ATA TCC TCC TTA-3′ were used to amplify the KanR gene of pKD4, and the PCR product was electroporated into a strain encoding λ recombination functions (41). Transformants were selected on plates supplemented with 20 μg/mL Kan, and the putative Δprc::Kan mutation was transferred by P1 transduction into MG1655 and confirmed by linkage analysis, phenotype, complementation, and sequencing.
Construction of a strain with mepS-Flag at the chromosomal locus.
The 3XFlag epitope tagging was done at the 3′ end of mepS at its native chromosomal locus as described earlier (42). A pair of hybrid primers [with the forward primer having homology to the C-terminal 40 nucleotides (nt) of mepS gene without a stop codon at its 5′ end and initial 21 nt of Flag epitope sequence of plasmid pSUB11 at the 3′ end (5′-GAAGCGTTACAACGAAGCACGCCGGGTTCTCAGCCGCAGCGACTACAAAGACCATGACGG-3′) and the reverse primer having a 40-nt homology to the immediate downstream sequence of mepS gene at the 5′ end and a 20-nt homology to the plasmid pSUB11 at its 3′ end (5′-ACTCGTCAGGATAGCCAAGGGATTGCATCCAAACGGTTTACATATGAATATCCTCCTTAG-3′)] were used to amplify the 3XFlag sequence along with a KanR marker flanked by FRT (flippase recognition target) sites using pSUB11 as template. The PCR product was electroporated into DY378, and colonies were obtained at 30 °C on LB plates containing Kan (25 µg/mL). The putative mepS-Flag-Kan region was transferred into MG1655 by P1 transduction, and the construct was confirmed by linkage analysis, PCR amplification, and sequencing. This strain did not have any growth defects on any media/temperature and behaved identical to that of a WT strain (MG1655). The KanR resistance cassette was flipped out using pCP20 (40), and the KanS derivative, MR802, was used for all further studies.
Construction of strains with prc-Flag and prc-HA fusions.
The above-described protocol was used to generate prc-Flag and prc-HA epitope tags at the C terminus of the prc gene at the chromosomal locus. Two forward primers, 5′-TGAAAAAGCCAGACCCGCGGAACAACCCGCTCCCGTCAAGGACTACAAAGACCATGACGG-3′ or 5′-TGAAAAAGCCAGACCCGCGGAACAACCCGCTCCCGTCAAGTATCCGTATGATGTTCCTG-3′ having 3XFlag or HA tags, respectively, and a common reverse primer, 5′-TGTTAAAAAATCAGGCACAATTTCTTGTGCCTGATTGATACATATGAATATCCTCCTTAG-3′ were used to amplify either the KanR gene using pSUB11 or CamR gene using pSUB314 as the templates. The PCR products were electroporated and processed as described above. The fusion strains generated were confirmed by sequence analysis. The strains showed no growth defects and grew with similar rates like that of the WT strain.
Generation of prcS452A-HA-Cam strain.
To generate a point mutation at the active site residue of Prc, a mismatch repair-deficient strain was used for recombineering as described earlier (41). A 90-mer oligo having the required mismatch (bold and underlined) to convert a serine codon into an alanine codon at position 452 of Prc (5′-GCA CGA CCG TAA TCC TGC ATT GCC GCG GCA AAG ATT TCT GAA GCC GCA GCA CTG AAG CGG TCA ACC AGC ACC ACC AGC GGG CCT TTA TAG-3′) was made, and the single-stranded oligo was electroporated into HME63 (mutS::amp strain) carrying the prc-HA-Cam allele. The transformants obtained on LB (without any selection) were patched onto nutrient agar (NA) plates at 30 °C. The colonies that were sick on NA, but not on LB, were taken further and analyzed. The presence of a mutation was confirmed by mutant phenotype, sequencing, and complementation with a plasmid carrying the prc gene. The mutation was introduced into required strains by P1 transduction using the Cam marker. As expected, this mutation also stabilized the level of mepS-Flag like that of a Δprc strain (Fig. S5A).
Construction of mepS::lacZY-Kan transcriptional fusion strain.
The ΔmepS::Kan deletion mutation of the Keio collection was converted into lacZY-Kan fusion as described earlier (43). Essentially, the deletion mutation was transferred into strain MC4100 (because it lacks the complete lacIZYA region) by P1 transduction, and then plasmid pCP20 encoding Flp recombinase was introduced into these strains to flip out the KanR determinant at 30 °C. Subsequently, into these KanS derivatives carrying pCP20 plasmid, an R6K-based plasmid, pKGE137, encoding a cassette of promoter-less lacZY reporter along with a KanR marker flanked by FRT sites was introduced. KanR colonies (in which the lacZY-Kan cassette is integrated at the FRT scar sites in the chromosome) were selected on plates supplemented with X-Gal, and the presence of lacZ-Kan cassette at the mepS locus was confirmed by linkage analysis, PCR, and sequencing. The ΔmepS::lacZ-Kan fusion was subsequently transferred into strain MC4100 and its ΔnlpI or Δprc deletion strains.
Plasmid Constructions.
pMN204.
The nlpI gene was amplified using forward and reverse primers 5′-GCTCTAGAGGTCTTCGGGAGTGGGAAATG-3′ and 5′-CCCAAGCTTCTATTGCTGGTCCGATTCTGCCAG-3′, and the resulting fragment was cloned at XbaI-HindIII sites of pTRC99a to generate pMN204, which complements an nlpI deletion mutant very effectively.
pMN208.
The nlpI gene lacking the signal sequence (1–18 amino acids) and stop codon was amplified using forward and reverse primers 5′-GGAATTCCATATGTGCAGTAATACTTCCTGGCGTAAAAGTGAAGTC-3′ and 5′-CCGCTCGAGTTGCTGGTCCGATTCTGCCAGGTCATC-3′, digested, and cloned at the NdeI-XhoI sites of pET21b to generate pMN208.
pMN211, -212, -213, and -216.
For cloning either full-length or 3′-truncated derivatives of nlpI into pCL1920, a common forward primer (5′-CCCAAGCTTATGAAGCCTTTTTTGCGCTGG) and reverse primers (for full-length, 5′-CGCGGATCCCTATTGCTGGTCCGATTCTGCCAGGTC-3′; for D284, 5′-TGCTCTAGACTAGTCCTGGCCCAGGAGCG-3′; for Q283, 5′-TGCTCTAGACTACTGGCCCAGGAGCGATAATTC-3′; and, for G282, 5′- TGCTCTAGACTAGCCCAGGAGCGATAATTC-3′) were used to amplify the nlpI sequences. The PCR products were digested with appropriate enzymes and cloned into pCL1920 at HindIII-BamHI (for full-length) or HindIII-XbaI for the other three derivatives to generate pMN216 (full-length nlpI encoding a polypeptide of 294 amino acids), -211 (nlpI-D284), -212 (nlpI-Q283), and -213 (nlpI-G282). Of these plasmids, pMN216 and -211 complement the nlpI deletion mutant well whereas pMN212 shows a weak complementation and pMN213 is unable to complement.
pMN214.
For cloning the prc gene in pCL1920, the gene was amplified with primers 5′-CTAGTCTAGAATGAACATGTTTTTTAGGCTTACC-3′ and 5′-CTAGTCTAGATTACTTGACGGGAGCGGG-3′ and cloned at the XbaI site of the vector. The orientation was determined by sequencing, and this clone complements a prc deletion mutant (with 1 mM IPTG supplementation).
pMN217 and pMN218.
For cloning a C-terminal hexahistidine-tagged version of mepS or nlpI, these genes were amplified using the forward primers 5′-GAATTCCAAGGGAAAACAAACAACATGGTC-3′and 5′-GGAATTCTGGGAAATGAAGCCTTTTTTGCGC-3′ and the reverse primers 5′-GCTCTAGATTAGTGGTGATGGTGATGATGGCTGCGGCTGAGAACCCG-3′and 5′-GCTCTAGATTAGTGGTGATGGTGATGATGTTGCTGGTCCGATTCTGCC-3′, respectively. Both the reverse primers contained the nucleotide sequences for encoding the hexahistidine tag. The amplified PCR products were digested and cloned at EcoRI-XbaI sites of pBAD18-Amp vector to generate pMN216 (pBAD18-mepS-His) and pMN217 (pBAD18-nlpI-His). Both these vectors were functional and complemented their respective deletion mutants in an arabinose-dependent manner.
pMN219.
The prc gene lacking the signal sequence (1–22 amino acids) and stop codon was amplified using forward and reverse primers 5′-GGAATTCCATATGGAAGATATCACGCGTGCTGATC-3′ and 5′-CCGCTCGAGCTTGACGGGAGCGGGTTGTTCC-3′, digested, and cloned at the NdeI-XhoI sites of pET21b to generate pMN219.
pMN220 and pMN221.
To generate a mutation at the active site residue of Prc (Prc-K477A), two-step PCR was performed using pMN219 as the template. Two oligos that are complementary to each other but have the desired mismatch (to create a codon for alanine instead of lysine) at the center (in bold and underlined) were synthesized. In the first step of PCR, the N-terminal region of prc was amplified using the T7 promoter primer (T7PP) 5′-TAATACGACTCACTATAGGG-3′ and the reverse primer 5′-CGGTATTGCTGAACGGTGCCTGCACCAAACGTCGGTTCACCCA-3′, and the C-terminal region was amplified by forward primer 5′-TGGGTGAACCGACGTTTGGTGCAGGCACCGTTCAGCAATACCG-3′ and the T7 terminator primer (T7TP) 5′-GCTAGTTATTGCTCAGCGG-3′. In the second step, both the amplicons were mixed in a 1:1 molar ratio, and end-filling was carried out in the first 10 cycles of PCR at 35 °C. The PCR was resumed again after addition of T7PP and T7TP for the next 30 cycles. The PCR product was digested and cloned at the NdeI-XhoI sites to generate pET-prc K477A. pMN221 encoding Prc-S452A was also generated similarly using the reverse primer 5′-GCGGCAAAGATTTCTGAAGCCGCAGCACTGAAGCGGTCAACCA-3′ and forward primer 5′-TGGTTGACCGCTTCAGTGCTGCGGCTTCAGAAATCTTTGCCGC-3′.
pMN222.
The pMN222 plasmid was constructed by subcloning the phoA gene from pPHO7 (44) into pBAD18-Amp using the XmaI-SalI sites to generate a signal-less phoA reporter system under the arabinose promoter.
pMN223.
pMN223 was constructed by cloning the full-length mepS gene (amplified from MG1655 using the primers 5′-CTAGCTAGCGGAAAACAAACAACATGGTCAAATCTCAACCGA-3′ and 5′-TCCCCCCGGGAGCTGCGGCTGAGAACCCGGCGT-3′) at the NheI-XmaI sites of pMN222 to generate a mepS-phoA fusion under the arabinose promoter. The pMN223 plasmid was functional and complemented the mepS deletion mutant in an arabinose-dependent manner.
Microscopy and Viability Measurements.
To monitor growth and viability, indicated cells were grown overnight, and the cultures were serially diluted. Then, 5 μL of various dilutions were spotted on indicated plates and grown at 37 °C unless otherwise indicated. Cell viability was measured using the LIVE/DEAD Baclight bacterial viability kit (Molecular Probes, Invitrogen). The immobilized cultures on a thin agarose pad were visualized on a Zeiss apotome microscope by DIC (Nomarski optics) and fluorescence microscopy using GFP and DsRed filters.
Antisera Preparation.
Antibodies were raised against purified NlpI protein (His-tagged at the C terminus) in rabbits. Crude antiserum was collected and initially purified by affinity purification. This purification step was followed by adsorption of the antibodies to a total cell lysate of Escherichia coli deleted for NlpI. The supernatant thus obtained was used as the source of antibodies.
Western Blotting.
For Western blotting, samples were separated by (12%) SDS/PAGE and transferred onto a nitrocellulose/PVDF membrane. Transferred protein bands were visualized by Ponceau S staining and then destained by multiple washes with milli-Q water. Membrane was blocked by 5% skimmed milk in 1× TBS-T for 2 h and then incubated overnight with primary antibodies (1:10,000 for α-NlpI, 1:3,000 for α-His, α-FLAG, and α-HA, and 1:5,000 for α-FtsZ in blocking solution) at 4 °C. Membrane was washed three times with 1× TBST and then probed with appropriate secondary antibodies (1:10,000) tagged with horseradish peroxidase (HRP) and incubated for 1 h at room temperature. Nonspecific binding was removed by washing with 1× TBST thrice. Membrane was overlaid with ECL-Prime detection substrate (Amersham) for 5 min, and the blots were developed.
Protein Purifications.
MepS was purified from strain C41 (λDE3) whereas NlpI, Prc, Prc-S452A, and Prc-K477A were purified from BL21 (λDE3). Just before the step of protein purification, the plasmid clones were transformed into the required strain, and the transformants were inoculated directly into 10 mL of LB broth containing Amp and grown at 37 °C until an OD600 of ∼0.6. These cultures were diluted 1:100 into fresh prewarmed 100–200 mL of LB broth containing Amp and grown at 37 °C until an OD600 of ∼0.6. At this time point, expression of proteins was induced by the addition of 0.2 mM IPTG and grown for an additional 2 h. Cells were recovered by centrifugation at 8,000 × g, 4 °C, 15 min, and cell pellets were stored at −30 °C.
Purification of MepS.
The cell pellet was resuspended in 20 mL of lysis buffer (50 mM NaH2PO4, 1.5 M NaCl, 10 mM imidazole, pH 8.0) and lysed by sonication (10 s on and 10 s off cycle for 15 min). Cell debris was removed by centrifugation at 15,000 × g for 20 min at 4 °C. Supernatant was mixed with 1 mL of Ni2+-NTA agarose (Qiagen) and mixed at 4 °C for 1 h. This mixture was loaded into empty plastic columns (Bio-Rad) and washed with 50 mL of wash buffer-I (50 mM NaH2PO4, 1. 5 M NaCl, 20 mM imidazole, 1% Triton X-100, pH 8.0) followed by 25 mL of wash buffer-II (50 mM NaH2PO4, 300 mM NaCl, 50 mM imidazole, 1% Triton X-100, pH 8.0). Bound proteins were eluted with 10 mL of elution buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole, pH 8.0). Purified protein was concentrated to 2.5 mL using a 3-kDa centrifugal membrane filter (Millipore) followed by buffer exchange using a PD10 column (Amersham). The retained proteins were eluted in a 3.5-mL volume of storage buffer (50 mM NaH2PO4, 100 mM NaCl, and 1 mM DTT). Protein was concentrated to a volume of 500 μL by a 3-kDa centrifugal membrane filter and stored in 50% glycerol at −30 °C.
NlpI and all of the versions of Prc were purified more or less in a similar way, but the buffers used for lysis, washes, and elution had 300 mM NaCl and no Triton X-100. The eluted proteins were processed and stored as described above.
Pull-Down Experiments.
Pull downs were performed as described earlier, but with modifications (6). Strains were grown overnight in LB broth supplemented with Amp and next morning diluted 1:100 into fresh LB (200 mL) and grown at 37 °C until an OD600 of ∼0.2. At this point, 0.05% arabinose was added, and growth was continued until OD600 was 0.8–1.0. Cells were recovered by centrifugation, and the cell pellet was washed, resuspended in 25 mL of the lysis buffer (50 mM Tris⋅Cl, 10 mM MgCl2, 100 mM NaCl, 20% glycerol, 2% Triton X-100, 1 mg/mL lysozyme, 1× mixture protease inhibitor, 20 units of DNase, 20 units of RNase, and 10 mM imidazole; pH 8.0), and incubated on ice for 2 h. This mixture was subjected to brief sonication (four bursts of 10 s on and 10 s off cycle), and the lysate was gently stirred overnight with glass beads at 4 °C to solubilize the membrane proteins. Next morning, insoluble material of the lysate was removed by centrifugation (14,000 × g, 15 min, 4 °C), at which point 100 μL was set aside for input fractions and then the supernatant was mixed with 200 μL of Ni2+-NTA agarose and mixed for 2 h at 4 °C. The agarose beads were washed twice with 10 mL of wash buffer (50 mM Tris⋅Cl, 100 mM NaCl, and 20 mM imidazole; pH 8.0) and once with 50 mM Tris⋅Cl, 100 mM NaCl, and 50 mM imidazole, pH 8.0, and the bound proteins were eluted with 250 μL of the elution buffer (50 mM Tris⋅Cl, 100 mM NaCl, and 300 mM imidazole; pH 8.0). The eluate was electrophoresed using 12% SDS/PAGE, and the proteins were detected by Western blotting.
Acknowledgments
We thank Sujata Kumari and M. R. Sunayana for plasmid constructions; Nilanjan Som for Prc localization studies; Tom Bernhardt for FtsZ antisera; and J. Gowrishankar and N. Madhusudhana Rao for advice. This work is supported by the Department of Biotechnology and the Council of Scientific and Industrial Research, Government of India. S.P. acknowledges financial support from the University Grants Commission of India.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1507760112/-/DCSupplemental.
References
- 1.Weidel W, Pelzer H. Bagshaped macromolecules: A new outlook on bacterial cell walls. Adv Enzymol Relat Areas Mol Biol. 1964;26:193–232. doi: 10.1002/9780470122716.ch5. [DOI] [PubMed] [Google Scholar]
- 2.Park JT. The murein sacculus. In: Neidhardt FC, et al., editors. Escherichia coli and Salmonella: Cellular and Molecular Biology. 2nd Ed. ASM; Washington, DC: 1996. pp. 48–57. [Google Scholar]
- 3.Höltje JV. Growth of the stress-bearing and shape-maintaining murein sacculus of Escherichia coli. Microbiol Mol Biol Rev. 1998;62(1):181–203. doi: 10.1128/mmbr.62.1.181-203.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.den Blaauwen T, de Pedro MA, Nguyen-Distèche M, Ayala JA. Morphogenesis of rod-shaped sacculi. FEMS Microbiol Rev. 2008;32(2):321–344. doi: 10.1111/j.1574-6976.2007.00090.x. [DOI] [PubMed] [Google Scholar]
- 5.Typas A, Banzhaf M, Gross CA, Vollmer W. From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nat Rev Microbiol. 2012;10(2):123–136. doi: 10.1038/nrmicro2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Typas A, et al. Regulation of peptidoglycan synthesis by outer-membrane proteins. Cell. 2010;143(7):1097–1109. doi: 10.1016/j.cell.2010.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paradis-Bleau C, et al. Lipoprotein cofactors located in the outer membrane activate bacterial cell wall polymerases. Cell. 2010;143(7):1110–1120. doi: 10.1016/j.cell.2010.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tomasz A. Building and breaking of bonds in the cell wall of bacteria: The role for autolysins. In: Nombela C, editor. Microbial Cell Wall Synthesis and Autolysis. Elsevier Science; Amsterdam: 1984. pp. 3–12. [Google Scholar]
- 9.Singh SK, SaiSree L, Amrutha RN, Reddy M. Three redundant murein endopeptidases catalyse an essential cleavage step in peptidoglycan synthesis of Escherichia coli K12. Mol Microbiol. 2012;86(5):1036–1051. doi: 10.1111/mmi.12058. [DOI] [PubMed] [Google Scholar]
- 10.Hashimoto M, Ooiwa S, Sekiguchi J. Synthetic lethality of the lytE cwlO genotype in Bacillus subtilis is caused by lack of D,L-endopeptidase activity at the lateral cell wall. J Bacteriol. 2012;194(4):796–803. doi: 10.1128/JB.05569-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dörr T, Cava F, Lam H, Davis BM, Waldor MK. Substrate specificity of an elongation-specific peptidoglycan endopeptidase and its implications for cell wall architecture and growth of Vibrio cholerae. Mol Microbiol. 2013;89(5):949–962. doi: 10.1111/mmi.12323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vollmer W. Bacterial growth does require peptidoglycan hydrolases. Mol Microbiol. 2012;86(5):1031–1035. doi: 10.1111/mmi.12059. [DOI] [PubMed] [Google Scholar]
- 13.Lee TK, Huang KC. The role of hydrolases in bacterial cell-wall growth. Curr Opin Microbiol. 2013;16(6):760–766. doi: 10.1016/j.mib.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Heijenoort J. Peptidoglycan hydrolases of Escherichia coli. Microbiol Mol Biol Rev. 2011;75(4):636–663. doi: 10.1128/MMBR.00022-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ohara M, Wu HC, Sankaran K, Rick PD. Identification and characterization of a new lipoprotein, NlpI, in Escherichia coli K-12. J Bacteriol. 1999;181(14):4318–4325. doi: 10.1128/jb.181.14.4318-4325.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hara H, Yamamoto Y, Higashitani A, Suzuki H, Nishimura Y. Cloning, mapping, and characterization of the Escherichia coli prc gene, which is involved in C-terminal processing of penicillin-binding protein 3. J Bacteriol. 1991;173(15):4799–4813. doi: 10.1128/jb.173.15.4799-4813.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tadokoro A, et al. Interaction of the Escherichia coli lipoprotein NlpI with periplasmic Prc (Tsp) protease. J Biochem. 2004;135(2):185–191. doi: 10.1093/jb/mvh022. [DOI] [PubMed] [Google Scholar]
- 18.Keiler KC, Sauer RT. Identification of active site residues of the Tsp protease. J Biol Chem. 1995;270(48):28864–28868. doi: 10.1074/jbc.270.48.28864. [DOI] [PubMed] [Google Scholar]
- 19.Mahalakshmi S, Sunayana MR, SaiSree L, Reddy M. yciM is an essential gene required for regulation of lipopolysaccharide synthesis in Escherichia coli. Mol Microbiol. 2014;91(1):145–157. doi: 10.1111/mmi.12452. [DOI] [PubMed] [Google Scholar]
- 20.Wilson CG, Kajander T, Regan L. The crystal structure of NlpI: A prokaryotic tetratricopeptide repeat protein with a globular fold. FEBS J. 2005;272(1):166–179. doi: 10.1111/j.1432-1033.2004.04397.x. [DOI] [PubMed] [Google Scholar]
- 21.Blatch GL, Lässle M. The tetratricopeptide repeat: A structural motif mediating protein-protein interactions. BioEssays. 1999;21(11):932–939. doi: 10.1002/(SICI)1521-1878(199911)21:11<932::AID-BIES5>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 22.Hara H, et al. Genetic analyses of processing involving C-terminal cleavage in penicillin-binding protein 3 of Escherichia coli. J Bacteriol. 1989;171(11):5882–5889. doi: 10.1128/jb.171.11.5882-5889.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Silber KR, Keiler KC, Sauer RT. Tsp: A tail-specific protease that selectively degrades proteins with nonpolar C termini. Proc Natl Acad Sci USA. 1992;89(1):295–299. doi: 10.1073/pnas.89.1.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hara H, Abe N, Nakakouji M, Nishimura Y, Horiuchi K. Overproduction of penicillin-binding protein 7 suppresses thermosensitive growth defect at low osmolarity due to an spr mutation of Escherichia coli. Microb Drug Resist. 1996;2(1):63–72. doi: 10.1089/mdr.1996.2.63. [DOI] [PubMed] [Google Scholar]
- 25.Kerr CH, Culham DE, Marom D, Wood JM. Salinity-dependent impacts of ProQ, Prc, and Spr deficiencies on Escherichia coli cell structure. J Bacteriol. 2014;196(6):1286–1296. doi: 10.1128/JB.00827-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schwechheimer C, Rodriguez DL, Kuehn MJ. NlpI-mediated modulation of outer membrane vesicle production through peptidoglycan dynamics in Escherichia coli. MicrobiologyOpen. 2015;4(3):375–389. doi: 10.1002/mbo3.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frandi A, Jacquier N, Théraulaz L, Greub G, Viollier PH. FtsZ-independent septal recruitment and function of cell wall remodelling enzymes in chlamydial pathogens. Nat Commun. 2014;5(5):4200–4210. doi: 10.1038/ncomms5200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang DC, et al. An ATP-binding cassette transporter-like complex governs cell-wall hydrolysis at the bacterial cytokinetic ring. Proc Natl Acad Sci USA. 2011;108(45):E1052–E1060. doi: 10.1073/pnas.1107780108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meisner J, et al. FtsEX is required for CwlO peptidoglycan hydrolase activity during cell wall elongation in Bacillus subtilis. Mol Microbiol. 2013;89(6):1069–1083. doi: 10.1111/mmi.12330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carballido-López R, et al. Actin homolog MreBH governs cell morphogenesis by localization of the cell wall hydrolase LytE. Dev Cell. 2006;11(3):399–409. doi: 10.1016/j.devcel.2006.07.017. [DOI] [PubMed] [Google Scholar]
- 31.Sham LT, Barendt SM, Kopecky KE, Winkler ME. Essential PcsB putative peptidoglycan hydrolase interacts with the essential FtsXSpn cell division protein in Streptococcus pneumoniae D39. Proc Natl Acad Sci USA. 2011;108(45):E1061–E1069. doi: 10.1073/pnas.1108323108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mavrici D, et al. Mycobacterium tuberculosis FtsX extracellular domain activates the peptidoglycan hydrolase, RipC. Proc Natl Acad Sci USA. 2014;111(22):8037–8042. doi: 10.1073/pnas.1321812111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burman LG, Reichler J, Park JT. Evidence for multisite growth of Escherichia coli murein involving concomitant endopeptidase and transpeptidase activities. J Bacteriol. 1983;156(1):386–392. doi: 10.1128/jb.156.1.386-392.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goodell EW, Schwarz U. Cleavage and resynthesis of peptide cross bridges in Escherichia coli murein. J Bacteriol. 1983;156(1):136–140. doi: 10.1128/jb.156.1.136-140.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Holtje JV. “Three for one”: A simple growth mechanism that guarantees a precise copy of the thin, rod-shaped sacculus of Escherichia coli. In: de Pedro MA, Holtje JV, Loffelhardt W, editors. Bacterial Growth and Lysis. Plenum; New York: 1993. pp. 419–426. [Google Scholar]
- 36.Koch AL. The three-for-one model for gram-negative wall growth: A problem and a possible solution. FEMS Microbiol Lett. 1998;162(1):127–134. doi: 10.1111/j.1574-6968.1998.tb12989.x. [DOI] [PubMed] [Google Scholar]
- 37.Baba T, et al. Construction of Escherichia coli K-12 in-frame, single-gene knock-out mutants: The Keio collection. Mol Syst Biol. 2006;2:2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Khlebnikov A, Datsenko KA, Skaug T, Wanner BL, Keasling JD. Homogeneous expression of the P(BAD) promoter in Escherichia coli by constitutive expression of the low-affinity high-capacity AraE transporter. Microbiology. 2001;147(Pt 12):3241–3247. doi: 10.1099/00221287-147-12-3241. [DOI] [PubMed] [Google Scholar]
- 39.Miller JH. A Short Course in Bacterial Genetics: A Laboratory Manual and Handbook for Escherichia coli and Related Bacteria. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1992. [Google Scholar]
- 40.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97(12):6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sharan SK, Thomason LC, Kuznetsov SG, Court DL. Recombineering: A homologous recombination-based method of genetic engineering. Nat Protoc. 2009;4(2):206–223. doi: 10.1038/nprot.2008.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Uzzau S, Figueroa-Bossi N, Rubino S, Bossi L. Epitope tagging of chromosomal genes in Salmonella. Proc Natl Acad Sci USA. 2001;98(26):15264–15269. doi: 10.1073/pnas.261348198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ellermeier CD, Janakiraman A, Slauch JM. Construction of targeted single copy lac fusions using λ Red and FLP-mediated site-specific recombination in bacteria. Gene. 2002;290(1-2):153–161. doi: 10.1016/s0378-1119(02)00551-6. [DOI] [PubMed] [Google Scholar]
- 44.Gutierrez C, Devedjian JC. A plasmid facilitating in vitro construction of phoA gene fusions in Escherichia coli. Nucleic Acids Res. 1989;17(10):3999. doi: 10.1093/nar/17.10.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]