

Significance
By studying premalignant conditions, we can gain a better understanding of the sources of genomic instability and improve cancer prevention and treatment. Because retrotransposition has been observed in many gastrointestinal epithelial cancer types, we focused on L1 mobilization as a source of instability in cancer. Here, we demonstrate that L1 retrotransposition is active in esophageal adenocarcinoma and its precursor, Barrett’s esophagus (BE). We detected clonal populations with precursor cells marked by L1 retrotransposition events either in the normal esophagus or BE. These clones expanded in the BE or esophageal adenocarcinoma (EAC), indicating that somatic L1 insertions are not only potential mutagens in the development of EAC, but also useful markers of tumor clones as well.
Keywords: retrotransposon, LINE-1, Barrett’s esophagus, esophageal adenocarcinoma, retrotransposition
Abstract
Barrett’s esophagus (BE) is a common disease in which the lining of the esophagus transitions from stratified squamous epithelium to metaplastic columnar epithelium that predisposes individuals to developing esophageal adenocarcinoma (EAC). We hypothesized that BE provides a unique environment for increased long-interspersed element 1 (LINE-1 or L1) retrotransposition. To this end, we evaluated 5 patients with benign BE, 5 patients with BE and concomitant EAC, and 10 additional patients with EAC to determine L1 activity in this progressive disease. After L1-seq, we confirmed 118 somatic insertions by PCR in 10 of 20 individuals. We observed clonal amplification of several insertions which appeared to originate in normal esophagus (NE) or BE and were later clonally expanded in BE or in EAC. Additionally, we observed evidence of clonality within the EAC cases; specifically, 22 of 25 EAC-only insertions were present identically in distinct regions available from the same tumor, suggesting that these insertions occurred in the founding tumor cell of these lesions. L1 proteins must be expressed for retrotransposition to occur; therefore, we evaluated the expression of open reading frame 1 protein (ORF1p), a protein encoded by L1, in eight of the EAC cases for which formalin-fixed paraffin embedded tissue was available. With immunohistochemistry, we detected ORF1p in all tumors evaluated. Interestingly, we also observed dim ORF1p immunoreactivity in histologically NE of all patients. In summary, our data show that somatic retrotransposition occurs early in many patients with BE and EAC and indicate that early events occurring even in histologically NE cells may be clonally expanded in esophageal adenocarcinogenesis.
Gastroesophageal reflux disease (GERD) affects a large proportion of Western populations and represents a significant health care burden, partially due to its frequent evolution into Barrett’s esophagus (BE) (1). BE was first described by Norman Barrett in 1950 (2) and is a common disease in which the lining of the esophagus transitions from stratified squamous epithelial cells to a cancer-predisposing metaplastic columnar epithelium (2). The transdifferentiation increases cellular resistance to the low pH from the acid entering the esophagus through the sphincter separating it from the stomach (3). BE occurs in 8–20% of patients with GERD or about 3–8% of the total population (3). Furthermore, recent studies suggest that another 3–8% of the general population may have BE without symptoms (3).
The risk of a patient with BE developing the advanced premalignant lesion, high-grade dysplasia, or frank esophageal adenocarcinoma (EAC) is 0.5%/y; however, the 5-y survival rate from EAC is only 13–16% (3). Moreover, although the risk of malignancy is low, an EAC diagnosis is not usually made until the late stages of the disease when the illness is nearly incurable (3, 4). Early diagnosis of dysplasia and EAC can be accomplished in patients with BE by screening endoscopies with biopsies performed at regular intervals determined by the physician (4). Due to the availability of tissue from these biopsies, detecting how the disease progresses and tracking clonal populations throughout disease progression has provided valuable insights into early cancer development (5, 6).
Various types of mutations can be detected and subsequently monitored by biopsy to determine which clonal population of cells progresses to cancer. One source of mutation in epithelial cancer is retrotransposition (7–11). Retrotransposons compose ∼45% of the human genome (12) and are mobilized via an RNA intermediate to new genomic locations. The long-interspersed element 1 (LINE-1 or L1) is the only autonomous retrotransposon that encodes the proteins necessary for mobilization and reinsertion into the genome. The two proteins encoded by L1 are responsible for the mobilization of other types of retrotransposons, Alu and SVA, as well as processed pseudogenes (13–15). Aside from contributing to genomic variation, retrotransposons can also have functional impact by inserting into transcription factor binding sites, donor and acceptor sites involved in mRNA splicing, enhancer sites, or protein coding regions of genes (12, 16–19). To date, there are more than 100 known retrotransposon insertions that caused single-gene diseases (12, 16–19).
L1 mobilization in epithelial cancer has been observed by many groups at both the protein and DNA level. Interestingly, each individual has a different complement of 80–100 potentially active L1 elements in their genome, which partially explains the large variation of somatic insertions detected in previous studies (7–11, 20). Although it is evident that L1 is active and expressed in many cancer types, this activity has not been robustly evaluated in precancerous lesions such as BE. New somatic insertions of L1 in BE could be used to track clonal progression of disease.
We hypothesized that the alterations in the esophageal lining as it undergoes cellular transdifferentiation present a permissive environment for retrotransposition. To test this hypothesis, we evaluated individuals with BE who progressed to EAC, as well as those with nonprogressive benign BE. We determined the occurrence of retrotransposition in these patients using L1-seq, a high-throughput L1-targeted sequencing method (20), and validated 118 somatic insertions in 10 of the 20 patients evaluated (Fig. 1). Substantial levels of L1 protein expression were also detected in the EAC with immunohistochemistry; moreover, the protein was detected in the normal esophageal (NE) tissue of all patients tested. We conclude that this high prevalence of L1 activity and insertions in BE and EAC, taken together with previous findings in other epithelial cancers, suggests a strong link between cancer and L1 activity. However, it is uncertain to what extent the dysregulation of normal cellular processes is contributing to L1 activation in cancer, as well as whether these somatic insertions are contributing to carcinogenesis in some individuals.
Fig. 1.

(A) Circos diagram mapping the distribution throughout the human genome of 20 validated high-stringency (HS) insertions in BE only (inner red circle), the 765 low-stringency (LS) reference (outer green circle), and 218 LS polymorphic insertions (orange circle) detected with L1-seq. Group contains five individuals. (B) Circos diagram mapping the distribution of the validated 23 HS insertions in BE only (yellow circle), 3 BE and T (orange points), and 23 T only (red circle), as well as the LS reference (752) and LS polymorphic (218) insertions detected with L1-seq (green and orange, respectively). Group contains five individuals. (C) Circos diagram mapping the distribution of 49 validated high stringency insertions in tumor only (red circle), 537 LS reference (green circle), and 282 LS polymorphic (orange circle) insertions. The final group contains 10 individuals. The somatic insertions (e.g., all insertions that are not reference and polymorphic insertions) are all validated; however, the reference and polymorphic insertions have been previously published and are more common in the population and are therefore restricted only by a sequencing read count of 25 or greater and a map score of 0.5 or larger. For the Esophageal cancer group, our map scores were lower overall for the reference insertions; therefore, we restricted these insertions by a read count of 20 and a map score above 0.3.
Results
BE Patients Without Cancer.
To estimate the pervasiveness of retrotransposition in BE, we studied five patients with BE who did not develop high-grade dysplasia or EAC for at least 15 y after their BE specimens were obtained. If L1 is active in patients without cancer, this finding would suggest that the cellular environment in BE per se is permissive for retrotransposition. We obtained matched DNAs from white blood cells (WBCs), NE, and BE and performed L1-seq to enrich DNA libraries for L1 insertions and then subsequently to identify those insertions unique to the metaplasia (20). We classified these “somatic insertions” as those present only in a subset of cells and not inherited from a previous generation, e.g., insertions unique to BE but absent from matched NE and WBC DNA. Alternatively, we reasoned that somatic insertions could occur in a few normal squamous esophageal cells that became clonally amplified in BE. To confirm that an insertion was truly absent, we performed nested PCRs on all samples (Fig. 2).
Fig. 2.

(A) Diagram of the PCR validation scheme for putative insertions: the 3′ end of the LINE-1 insertion is pictured adjacent to a poly-A tail. The nested empty site and filled site primers are flanking the empty and filled site primers. In a nested PCR, the nested primers are used in the first of two reactions. Product (1.5 μL) from the first reaction (with ES and FS primers) are used as template in a second PCR with the nested primers to amplify difficult or rare products. (B) Two examples of validations for insertions present in only tumor and absent from normal DNA. (Left) PCR result depicting both the empty site (ES) and filled site (FS) products for both the normal and tumor DNA samples from patients. Only in the tumor of patient 11 is a filled site band present confirming the insertion is present. (Right) PCR depicting another validation of a somatic insertion present in BE and absent from normal esophageal and white blood cell DNA. There is only a band present in the BE sample for the FS PCR; however, the ES PCR has bands for all three DNA samples as a positive control. (C) An insertion sequence with the unique genomic DNA (blue), target site duplications (purple), LINE-1 sequence (red), and the poly(A) tail sequence (orange).
We confirmed a total of 20 insertions in four of five patients evaluated by PCR and Sanger sequencing. Of the 20 confirmed insertions, 11 were amplified easily with a single PCR (conventional), without the need for a secondary PCR using nested primers (Fig. 2A). We hypothesize that insertions which amplified with a conventional PCR were likely present in a large proportion of cells and were therefore clonal. One insertion in particular was amplified easily with a conventional PCR in BE DNA; notably, this insertion was also observed in normal esophageal DNA only after nested PCR but remained undetectable in WBC DNA (Fig. 3 A and B). We speculated that this somatic insertion could have initially occurred in a single normal squamous cell exposed to high acid content during episodes of GERD, which then transdifferentiated into columnar epithelium, and clonally expanded as BE. This finding suggests that L1 insertions occur in normal squamous esophageal cells at a low frequency and then become more easily detectable after they clonally expand in a disease such as BE or EAC, as previously suggested by Goodier for other tumor types (17).
Fig. 3.

Gels showing clonal expansion of insertions originally present in NE. (A) The first PCR is a conventional PCR done on 1,014 WBC, 1,014 NE, and 1,014 BE DNA and with a water control (no DNA), attempting to amplify the “Filled site” or the insertion in all three samples. A band is present in BE DNA, showing the insertion is likely clonal in BE. (B) Nested PCR showing that the insertion is also present in low concentration in 1,014 NE. (C) A conventional PCR on 1,099 NE, 1,099 BE, and 1,099 T and a water control (no DNA), attempting to amplify the filled site or the insertion in all three samples. A band is present in the tumor DNA only showing the insertion is likely clonal in the tumor. (D) A nested PCR showing that with nested PCR the insertion is also present in NE and in BE DNA. The size difference between the NE, BE, and tumor bands is explained by a difference in the length of the poly(A) tails. The NE and BE insertions have poly(A) tails of greater than 70 nucleotides, whereas the poly(A) tail of the tumor band is only 52 bp (31).
BE Patients with Cancer.
After establishing that L1 is active in patients with benign BE, we evaluated individuals whose disease progressed to EAC. We hypothesized that individuals who develop EAC would have as many or more somatic insertion events due to increased genetic instability in frank cancer (6). We obtained samples from five patients with concomitant BE and EAC. Genomic DNA was isolated from NE, BE, and EAC tissues resected concurrently. After L1-seq, we validated a number of these insertions in two of the five patients. We amplified and successfully Sanger sequenced 11 of 12 tested insertions that occurred in BE tissue alone, 27 of 36 in EAC alone, and 3 insertions that occurred in both BE and matched EAC. Due to the known polyclonal nature of BE, we had not expected all insertions detected in BE to be present in the matched EAC (6). We reasoned that typically, only one clonal population of cells should have evolved into the tumor and thus retained mutations acquired in the precursor lesion; the remaining clonal populations in the BE would not be expected to contain these same mutations (6).
The three insertions that were validated in multiple tissues provided a unique opportunity to look at the contribution of different clonal populations to the precursor lesion and the tumor. We observed three different stages at which a somatic insertion could occur. First, one of the insertions was detected without nested PCR in both BE and matched EAC; therefore, this insertion was likely part of a dominant BE clone that progressed to EAC. The second insertion was readily detected in EAC but required nested PCR to be detected in BE. Finally, a third insertion was amplified with conventional PCR in EAC but was only evident in both NE and BE following nested PCR. This third insertion likely occurred in an NE cell, which evolved into BE, and subsequently clonally expanded in the EAC (Fig. 3 C and D). Altogether, these data are further evidence that insertions occur at a low level in normal or metaplastic tissue and may later expand into a malignant clone. Similar observations, insertions that are easily amplified in the cancer tissue but only amplified in normal tissue following nested PCR reactions, have been observed by others in our laboratory in gastric cancer.
Similar to our previous group of nonprogressive benign BE samples, only two of five individuals had somatic insertions in either BE, EAC, or both tissues. Although fewer patients had insertions in the matched BE-EAC group than in the group with BE or EAC alone, there were on average more somatic insertions validated in the patients with BE and EAC. In individuals with BE alone, we observed an average of 5 insertions per person (20 insertions divided among four patients), whereas in patients with EAC, there were 23.5 insertions on average per person (47 insertions among two patients). Because of the small sample size studied, it was impossible to determine whether this observed difference was statistically significant. Nevertheless, the wide range in the number of insertions per patient and the frequency of patients with insertions is in agreement with other observations (7–11).
EAC Patients.
To further investigate the activity of retrotransposons in EAC, we obtained samples from 10 additional patients with fresh-frozen matched NE and EAC tissue samples. Following L1-seq, we confirmed 49 of 72 randomly selected, high-stringency insertions (Materials and Methods) with PCR and Sanger sequencing. We then selected 20 low-stringency insertions (Materials and Methods) for validation and confirmed six additional somatic insertions. These confirmed insertions occurred in 4 of the 10 individuals’ samples with great variation among individuals regarding the number of somatic insertions. Extrapolating from this large number of low-stringency predicted insertions by L1-seq and our observed 30% validation rate in this group, we speculate that the number of potential L1 insertions per EAC is probably in the hundreds.
Previously, others have shown variability with respect to the number of confirmed somatic insertions per person, as well as the proportion of individuals harboring somatic insertions (7–11). However, many studies have not thoroughly tested the potential clonality of the confirmed insertions. We tested 25 of the confirmed insertions in up to six tissue sections (20 in six of six sections and 2 in two of two sections) (Fig. 4). We observed that 22 of these 25 insertions appeared in all sections tested, whereas the remaining 3 insertions were present in five of six sections tested (Fig. 4). When insertions exist in multiple tissue sections, it suggests that they are likely clonal and may have occurred early during tumorigenesis or even in the precursor lesion (BE). The concept of insertion clonality is important because it supports the conclusion that retrotransposition is active early during tumorigenesis.
Fig. 4.

Representative gels illustrating the presence of specific insertions (numbered 1–5) in multiple sections of tumor tissue. FS refers to the filled site PCR as in Fig. 2. Two additional sections of each tumor were also tested and are positive for the insertions. Altogether, 22 of 25 insertions in EAC tested were present in all sections tested.
Characterization of BE- and EAC-Specific Insertions.
We established that retrotransposition is an active process in some BE and EAC patients by confirming 118 somatic insertions using PCR and Sanger sequencing. The confirmed insertions did not display an obvious bias for chromosomal location (Fig. 1). To identify the precise insertion sites, we confirmed the 5′ ends of 35 of the somatic insertions. For a subset of the insertions, we identified target site duplications (TSDs) and endonuclease cleavage sites, both established hallmarks of retrotransposition (Table S1). However, of 24 endonuclease cleavage sites identified, only 7 were similar (differed by 2 bp or less) to the canonical endonuclease site (12); the remaining 17 sites were more divergent from the canonical sequence. Furthermore, 11 insertions lacked TSDs and clear endonuclease cleavage sites indicating they were likely endonuclease-independent insertions (21). Of the 11 insertions presumed endonuclease independent, 6 insertions had deletions at the site of integration (22). Additional characteristics of these insertions, including mapping statistics of total read count, unique read count, and alignment windows, as well as genes nearby insertion sites, are noted in Table S1. We observed variable lengths among the insertions for which we confirmed 5′ ends, ranging from 111 to 1,579 nucleotides (without the poly-A tail) with 29 of 35 insertions, measuring under 500 nucleotides (Table S1). For the 21 of 35 insertions with TSDs, 12 were longer than 10 nucleotides (Table S1). One insertion contained a 3′ truncated L1 element wherein 100 nucleotides of the 3′ end of the L1 were deleted from the insertion site, suggesting internal reverse transcriptase priming (23). Eleven of 35 insertions contained a 5′ inversion, consistent with previous reports (7–11).
We did not confirm any insertions into exons in either BE or EAC; however, of the 118 confirmed somatic insertions, 48 insertions were into intronic regions of 50 genes. Twenty-three of these 48 insertions were into genes previously associated with cancer (Table S2). Our findings show a statistically significant enrichment of insertions into genes previously associated with cancer (P < 1 × 10−10 by Fisher’s exact test). To accurately test for the observed enrichment, we accounted for the sizes of all of the genes into which insertions occurred, as well as the size of all cancer genes in the genome and the probability that an insertion would hit more of those genes by chance alone. After accounting for gene size, we still observed a significant enrichment of insertions into genes previously associated with cancer (P < 1 × 10−10).
L1 Expression in NE and EAC.
Theoretically, retrotransposition is dependent on L1 protein expression for its activity; therefore, the genetic evidence of somatic insertions strongly suggests L1 proteins are expressed in precancerous lesions and cancer. Expression of the L1 protein has been observed in many cancer types previously but was only rarely detected in histologically normal tissue adjacent to the cancer (24–27). Furthermore, the evidence of somatic insertions in normal esophagus suggests there must be at least transient or a low level of L1 protein expression in the tissue. One of the two proteins encoded by L1, open-reading frame 1p (ORF1p), has been observed in many cancer types and has occasionally been observed in normal tissue adjacent to cancer (24–27). To evaluate ORF1p expression in the patients harboring somatic L1 insertions, we obtained formalin-fixed paraffin-embedded (FFPE) tissues from eight of the aforementioned EAC patients.
We observed ORF1p expression in all eight of these tumor samples by immunohistochemistry (IHC) (Table S3). The level of ORF1p expression varied among individuals, as well as within individual tissue sections where cancerous glands were developing (Fig. 5 A–F). All of the samples with a confirmed somatic insertion showed ORF1p expression. There was no correlation between protein expression and the number of confirmed somatic insertions per individual (Table S3). Interestingly, we detected low-level ORF1p expression in all four of the available matched normal tissues in both the stratified squamous epithelium and the smooth muscle (Fig. 6 A–F). Expression was absent from the progenitor stem cells of the stratified squamous epithelium and seemed to increase with cellular maturation as the cells increased their cytoplasm and radiated away from their progenitors. The expression was absent from the submucosa of the tissue. Expression was observed with two separate monoclonal antibodies that detect different epitopes of the protein (24). Although ORF1p expression in normal tissue has rarely been observed, it supports our finding of somatic insertions in normal esophagus that later expanded in subsequent metaplasia and/or cancer (24–27).
Fig. 5.

(A–F) Representative photomicrographs depicting LINE-1 ORF1p immuno-labeling in esophageal carcinomas. (A) Virtually no background immuno-labeling identified when no primary LINE-1 ORF1p antibody was used in the IHC procedure (i.e., no antibody incubation). Final magnification, ×100. (B) Same case as A, when incubated with LINE-1 ORF1p antibody, indicating the cancer is strongly reactive for ORF1p antigen. Final magnification, ×100. (C) Virtually no background immuno-labeling identified when primary antibody not used. Final magnification, ×100. (D) Same case as C, when incubated with LINE-1 ORF1p antibody, indicating the cancer is reactive for ORF1p antigen. Final magnification, ×100. (E and F) Two additional EAC cases that are reactive for ORF1p antigen. Final magnification, ×160.
Fig. 6.

(A–F) Representative photomicrographs depicting LINE-1 ORF1p expression in normal esophageal tissue. (A and C) Normal esophageal tissue from two distinct individuals stained with H&E. Final magnification, ×100. (B) Same case as A when incubated with LINE-1 ORF1p antibody, indicating the normal esophageal tissue is reactive for ORF1p antigen. Final magnification, ×100. (D) Same case as C when incubated with LINE-1 ORF1p antibody, indicating the normal esophageal tissue is reactive for ORF1p antigen. Final magnification, ×100. (E and F) Normal esophageal tissue from two distinct individuals showing the muscle is reactive for ORF1p antigen.
To investigate whether the expression present in the normal esophagus was limited to patients who had concomitant cancer, we obtained one normal esophagus sample from a biopsy conducted on a patient with gastric ulcers. We also obtained a normal skin biopsy to evaluate the squamous epithelium expression of ORF1p in an epithelial tissue. In both the normal biopsies, dim ORF1p immunoreactivity was evident in the squamous epithelium of the tissue (Fig. S1 A and B). LINE-1 expression in normal epithelial tissues, albeit at low levels, may allow for retrotransposition events. Perhaps a subset of somatic retrotransposition events reported in epithelial cancers actually occur before transformation (7–11). At the same time, the higher levels of LINE-1 expression we see in these cancers may selectively promote somatic insertion events in malignant cells.
Fig. S1.
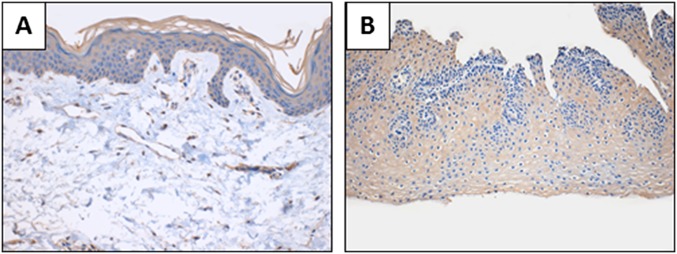
ORF1p expression in normal esophagus and normal skin samples. (A) Normal skin biopsy showing ORF1p expression localized in the stratified squamous epithelium. Final magnification, ×160. (B) Normal esophagus biopsy from patient with stomach ulcers showing ORF1p expression localized in the stratified squamous epithelium. Final magnification, ×100.
Discussion
Improved understanding of carcinogenesis should lead to earlier diagnosis and more effective treatment, but this advance requires the study of precursor lesions. In many ways, BE is ideal for studying clonal expansion in precursor lesions even when disease progression does not occur. BE is accessible and present in a sizable proportion of the Western population, even though it only progresses to EAC in a small subset of patients (3). Cellular processes that are dysregulated during the transdifferentiation from stratified squamous epithelium to metaplastic columnar epithelium may provide a fertile environment for dysregulation of L1.
We found that retrotransposition is active in a subset of individuals with BE and EAC; however, this process does not occur in all patients and is active in patients with long-standing benign disease. Therefore, L1 activity alone is not a reliable predictor of disease progression in BE. BE appears to provide a permissive environment for L1 retrotransposition, which in turn increases the mutational burden and potentially contributes to disease progression. As evidence of this permissive environment, we demonstrated that L1 elements were active in 6 of the 10 BE tissues evaluated by confirming 46 new somatic insertions. Furthermore, we validated 75 insertions in 6 of 15 EAC samples. Where somatic insertions occurred, there was a variable frequency of events, ranging from 1 to 44 insertions, among different individuals. In contrast to our previous colon cancer study (9), we did not observe a linear correlation between the number of insertions and any other characteristic, including age or L1 protein expression.
Many of the insertions validated in BE and EAC had characteristics that differ from typical germ-line somatic insertions (14). First, we observed 8 of 35 (23%) insertions with integration-site deletions, much greater than the 10% seen in the germ line (12). Our failure to detect more than 30% of the 5′ ends may, in a number of the cases, be due to even larger integration site deletions (22). Second, we found 11 insertions which appeared to be endonuclease independent, a much larger number than that observed in germ-line insertions (12, 20). These insertions were presumed to be endonuclease independent due to their lack of both target site duplications and clear endonuclease cleavage sites, both hallmarks of the canonical process of retrotransposition. Third, the majority of the insertions for which we identified the 5′ end were highly truncated, with 29 of 35 below 500 nucleotides in length. Also, we did not detect any 3′ transductions among our confirmed insertions in contrast to the findings of others (8), but L1-seq detects only a small fraction of these events.
Somatic L1 insertions are seldom observed in normal tissues (28) with the most notable exception being those observed in the hippocampus (29, 30). Between our three groups of samples analyzed with L1-seq, we attempted to validate nine high-stringency insertions predicted in normal esophagus only. Even with nested PCR, we were unable to confirm any of these normal-only insertions, a result that we have seen previously for normal-specific insertions (9). Interestingly, we validated two insertions in normal tissue that were also present in BE and EAC. This finding suggests that at least some insertions may occur in normal squamous epithelium cells and are then selected for in the ensuing pathological state. We speculate that indeed many of the BE and EAC insertions occur initially in only one or a small number of normal esophageal cells. Clonal expansion in diseases such as BE and EAC may make it easier to detect the low level of L1 activity in a subset of normal cells. Future studies using single-cell sequencing may allow us to better determine the activity of L1 in normal tissues and whether insertions in BE or in tumor are truly clonal.
Somatic retrotransposition occurs at a detectable rate in squamous cell lung, head and neck, colorectal, endometrial, hepatocellular, breast, prostate, bone, and various other types of cancer (7–11). We now demonstrate that this process occurs in premalignant BE and EAC. Although retrotransposition does not occur in all BE and EAC patients and recurrent insertions were not found, L1 may still participate in carcinogenesis. It appears that, although epithelial cancers are permissive for retrotransposition, there may be other factors mediating this process that allow it to occur in certain individuals more than in others. Identifying the factors underlying the activation of retrotransposition, as well as the contributions it makes to carcinogenesis, will be essential to improve our understanding of genomic instability generated by L1 and the role of retrotransposition in epithelial tumor development.
Materials and Methods
L1-seq.
DNA was isolated from the frozen tissue samples from thinly sliced sections of tissue embedded in OTC freezing media with the DNeasy kit (Qiagen). Our samples were not microdissected to remove all normal tissue largely because half of our samples were either acquired as genomic DNA or previously frozen tissues. Equal amounts of genomic DNA from each individual were pooled by group. Hemispecific PCR amplified the young, active L1 elements from the genome (20). Products between 200 and 500 nucleotides in size are excised from a 1% agarose gel and purified. Following analysis on the Bioanalyzer 2100, the products from each of the eight reactions with the degenerate primers were added in equimolar ratios and sent for next-generation sequencing on the Illumina HiSeq 2500. When results were obtained from the core facility, the reads were aligned by Bowtie2 and sorted based on the presence or absence of L1 sequence. During the sorting of the aligned reads, the previously published polymorphic insertions and reference insertions were identified (20) (Fig. 1). Our bioinformatics analysis was essentially identical to previous analyses (20).
Stringency Analysis.
For an insertion to be considered high stringency in the library containing matched EAC and NE samples, we required a map score of at least 0.5 or greater, 50 total reads, and a window of 100 bp or more spanning the junction of the 3′ end of the L1 and the genomic DNA. Low-stringency insertions had below 50 reads, a map score of 0.5 or greater, and a window of less than 100 bp. These original parameters are similar to those previously used (20); however, because we had more difficulty validating insertions in our other libraries, we reevaluated the thresholds. For both of the remaining libraries, (i) the library containing the matched BE and NE samples and (ii) the library containing matched EAC, BE, and NE samples, we adjusted the thresholds by looking at the few validated insertions from the original high-stringency group. We noted the lowest unique read count, total read count, and window size among the previously validated insertions in each group and used these numbers as our new parameters for high stringency. We also required a higher map score for the redefined high-stringency insertions in both libraries. Consequently, a high-stringency insertion in the library containing matched BE and NE samples required a map score of 0.8 or greater, 3 unique reads, 64 total reads, and a window of at least 107 bp. High stringency insertions in the library containing matched EAC, BE, and NE samples required at least a map score of 0.8, 3 unique reads, 63 total reads, and a window of 140 bp. For each insertion validated, the specific map score, read count, unique read count, and window size (bp) is noted (Table S1).
Random Insertion Selection.
For the group of matched NE and EAC samples, insertions were randomly selected using a random number generator with parameters for both high and low stringency. A list of random numbers between 1 and the total number of predicted insertions (at varying levels of confidence) was then created, and the rows that matched the numbers generated in the .csv file containing the predicted somatic insertions were selected for validation with PCR and sequencing. We made a histogram of the data to be sure the selection was even and random throughout the number range given and finally performed an empirical distribution analysis to evaluate our random selection process.
IHC.
IHC was performed using the EnVision System-HRP (catalog K4006; Dako) according to the manufacturer’s protocol. Primary antibody incubation was performed using the mouse monoclonal ORF1 (1.25 mg/mL) at a 1:3,000 dilution for 40 min at room temperature. Secondary antibody incubation was performed per the manufacturer’s protocol. For the skin biopsy, the sample was stained in an overnight protocol at a 1:1,200 dilution with the monoclonal mouse ORF1 antibody. A second rabbit monoclonal ORF1 antibody was used to confirm initial results. This second antibody was used at a concentration of 1:2,000 dilution with an overnight incubation at 4ο C and secondary antibody incubation as per the manufacturer’s protocol. Orf1 monoclonal mouse antibody recognizes amino acids 35–44, whereas the rabbit monoclonal antibody (JH74) detects the coiled-coil domain including amino acids 137–337 (24).
Supplementary Material
Acknowledgments
We thank Dr. Lindsay Horvath for careful review of the manuscript, Dr. Norman J Barker for assistance with photomicrographs, and Dr. Jeffrey Han for use of the JH74 rabbit monoclonal ORF1p antibody. This work was funded by the following: National Institute of General Medical Science (NIGMS) Grant R01 GM 099875 (to H.H.K.), the Saul-Goldman award (to H.H.K.), the Burroughs Wellcome Fund Career Award for Biomedical Scientists (to K.H.B.), National Institutes of Health (NIH) Grant R01 CA163705 (to K.H.B.), NIH Grant CA190040 (to S.J.M.), NIH Grant CA146799 (to S.J.M.), NIH Grant DK087454 (to S.J.M.), NIH Grant CA173390 (to S.J.M.), an American Cancer Society Clinical Research Professorship (to S.J.M.), and a NIGMS P-50 Grant GM 107632 (to H.H.K. and K.H.B.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The data reported in this paper have been deposited in the dbGaP database, www.ncbi.nlm.nih.gov/gap (accession no. phs000536v2.p2).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1502474112/-/DCSupplemental.
References
- 1.Badillo R, Francis D. Diagnosis and treatment of gastroesophageal reflux disease. World J Gastrointest Pharmacol Ther. 2014;5(3):105–112. doi: 10.4292/wjgpt.v5.i3.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barrett NR. Chronic peptic ulcer of the oesophagus and ‘oesophagitis’. Br J Surg. 1950;38(150):175–182. doi: 10.1002/bjs.18003815005. [DOI] [PubMed] [Google Scholar]
- 3.Sappati Biyyani RS, Chak A. Barrett’s esophagus: Review of diagnosis and treatment. Gastroenterol Rep (Oxf) 2013;1(1):9–18. doi: 10.1093/gastro/got015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Modiano N, Gerson LB. Barrett’s esophagus: Incidence, etiology, pathophysiology, prevention, and treatment. Ther Clin Risk Manag. 2007;3(6):1035–1045. [PMC free article] [PubMed] [Google Scholar]
- 5.Rasking W, et al. Persistent clonal areas and clonal expansion in Barrett’s esophagus. Cancer Res. 1992;52(10):2946–2950. [PubMed] [Google Scholar]
- 6.Maley CC, et al. Selectively advantageous mutations and hitchhikers in neoplasms: p16 lesions are selected in Barrett’s esophagus. Cancer Res. 2013;64(10):3414–3427. doi: 10.1158/0008-5472.CAN-03-3249. [DOI] [PubMed] [Google Scholar]
- 7.Shukla R, et al. Endogenous retrotransposition activates oncogenic pathways in hepatocellular carcinoma. Cell. 2013;153(1):101–111. doi: 10.1016/j.cell.2013.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Helman E, et al. Somatic retrotransposition in human cancer revealed by whole-genome and exome sequencing. Genome Res. 2014;24(7):1053–1063. doi: 10.1101/gr.163659.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Solyom S, et al. Extensive somatic L1 retrotransposition in colorectal tumors. Genome Res. 2012;22(12):2328–2338. doi: 10.1101/gr.145235.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee E, et al. Landscape of somatic retrotransposition in human cancers. Science. 2012;337(6097):967–971. doi: 10.1126/science.1222077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tubio JM, et al. Mobile DNA in cancer. Extensive transduction of nonrepetitive DNA mediated by L1 retrotransposition in cancer genomes. Science. 2014;345(6196):1251343. doi: 10.1126/science.1251343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hancks DC, Kazazian HH., Jr Active human retrotransposons: Variation and disease. Curr Opin Genet Dev. 2012;22(3):191–203. doi: 10.1016/j.gde.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dewannieux M, Esnault C, Heidmann T. LINE-mediated retrotransposition of marked Alu sequences. Nat Genet. 2003;35(1):41–48. doi: 10.1038/ng1223. [DOI] [PubMed] [Google Scholar]
- 14.Ostertag EM, Goodier JL, Zhang Y, Kazazian HH., Jr SVA elements are nonautonomous retrotransposons that cause disease in humans. Am J Hum Genet. 2003;73(6):1444–1451. doi: 10.1086/380207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Esnault C, Maestre J, Heidmann T. Human LINE retrotransposons generate processed pseudogenes. Nat Genet. 2000;24(4):363–367. doi: 10.1038/74184. [DOI] [PubMed] [Google Scholar]
- 16.van der Klift HM, Tops CM, Hes FJ, Devilee P, Wiknen JT. Insertion of an SVA element, a nonautonomous retrotransposon, in PMS2 intron 7 as a novel cause of lynch syndrome. Hum Mutat. 2012;33(7):1051–1055. doi: 10.1002/humu.22092. [DOI] [PubMed] [Google Scholar]
- 17.Goodier JL. Retrotransposition in tumors and brains. Mob. DNA. 2014;5:11. doi: 10.1186/1759-8753-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goodier JL, Kazazian HH., Jr Retrotransposons revisited: The restraint and rehabilitation of parasites. Cell. 2008;135(1):23–25. doi: 10.1016/j.cell.2008.09.022. [DOI] [PubMed] [Google Scholar]
- 19.Beck CR, Garcia-Perez JL, Badge RM, Moran JV. LINE-1 elements in structural variation and disease. Annu Rev Genomics Hum Genet. 2011;12:187–215. doi: 10.1146/annurev-genom-082509-141802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ewing AD, Kazazian HH., Jr High-throughput sequencing reveals extensive variation in human specific L1 content in individual human genomes. Genome Res. 2010;20(9):1262–1270. doi: 10.1101/gr.106419.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morrish TA, et al. DNA repair mediated by endonuclease-independent LINE-1 retrotransposition. Nat Genet. 2002;31(2):159–165. doi: 10.1038/ng898. [DOI] [PubMed] [Google Scholar]
- 22.Gilbert N, Lutz-Prigge D, Moran JV. Genomic deletions created upon LINE-1 retrotransposition. Cell. 2002;110(3):315–325. doi: 10.1016/s0092-8674(02)00828-0. [DOI] [PubMed] [Google Scholar]
- 23.Ostertag EM, Kazazian HH., Jr Twin priming: A proposed mechanism for the creation of inversions in L1 retrotransposition. Genome Res. 2001;11(12):2059–2065. doi: 10.1101/gr.205701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rodić N, et al. Long interspersed element-1 protein expression is a hallmark of many human cancers. Am J Pathol. 2014;184(5):1280–1286. doi: 10.1016/j.ajpath.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Asch HL, et al. Comparative expression of the LINE-1 p40 protein in human breast carcinomas and normal breast tissues. Oncol Res. 1996;8(6):239–247. [PubMed] [Google Scholar]
- 26.Bratthauer GL, Fanning TG. LINE-1 retrotransposon expression in pediatric germ cell tumors. Cancer. 1993;71(7):2383–2386. doi: 10.1002/1097-0142(19930401)71:7<2383::aid-cncr2820710733>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 27.Bratthauer GL, Cardiff RD, Fanning TG. Expression of LINE-1 retrotransposons in human breast cancer. Cancer. 1994;73(9):2333–2336. doi: 10.1002/1097-0142(19940501)73:9<2333::aid-cncr2820730915>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 28.Evrony GD, et al. Single-neuron sequencing analysis of L1 retrotransposition and somatic mutation in the human brain. Cell. 2012;151(3):483–496. doi: 10.1016/j.cell.2012.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baillie JK, et al. Somatic retrotransposition alters the genetic landscape of the human brain. Nature. 2011;479(7374):534–537. doi: 10.1038/nature10531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Upton KR, et al. Ubiquitous L1 mosaicism in hippocampal neurons. Cell. 2015;161(2):228–239. doi: 10.1016/j.cell.2015.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grandi FC, Rosser JM, An W. LINE-1-derived poly (A) microsatellites undergo rapid shortening and create somatic and germline mosaicism in mice. Mobile DNA. 2013;30(3):503–512. doi: 10.1093/molbev/mss251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Quesada V, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet. 2011;44(1):47–52. doi: 10.1038/ng.1032. [DOI] [PubMed] [Google Scholar]
- 33.Wood L, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318(5853):1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 34.Lee W, et al. The mutation spectrum revealed by paired genome sequences from a lung cancer patient. Nature. 2010;465(7297):473–477. doi: 10.1038/nature09004. [DOI] [PubMed] [Google Scholar]
- 35.Futreal PA, et al. A census of human cancer genes. Nat Rev Cancer. 2004;4(3):177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stephens PJ, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486(7403):400–404. doi: 10.1038/nature11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guo G, et al. Frequent mutations of genes encoding ubiquitin-mediated proteolysis pathway components in clear cell renal cell carcinoma. Nat Genet. 2011;44(1):17–19. doi: 10.1038/ng.1014. [DOI] [PubMed] [Google Scholar]
- 38.Dalgliesh GL, et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010;463(7279):360–363. doi: 10.1038/nature08672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Onk CK, et al. Exome sequencing of liver fluke associated cholangiocarcinoma. Nat Genet. 2012;44(6):690–693. doi: 10.1038/ng.2273. [DOI] [PubMed] [Google Scholar]
- 40.Berger MF, et al. The genomic complexity of primary human prostate cancer. Nature. 2011;470(7333):214–220. doi: 10.1038/nature09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kan Z, et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature. 2010;466(7308):869–873. doi: 10.1038/nature09208. [DOI] [PubMed] [Google Scholar]
- 42.Ding L, et al. Genome remodeling in a basal-like breast cancer metastasis and xenograft. Nature. 2010;464(7291):999–1005. doi: 10.1038/nature08989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Puente XS, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475(7354):101–105. doi: 10.1038/nature10113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shah SP, et al. Mutational evolution in a lobular breast tumour profiled at a single nucleotide resolution. Nature. 2009;461(7265):809–813. doi: 10.1038/nature08489. [DOI] [PubMed] [Google Scholar]
- 45.Turajlic S, et al. Whole genome sequencing of matched primary and metastatic acral melanomas. Genome Res. 2012;22(2):196–207. doi: 10.1101/gr.125591.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koboldt DC, et al. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stansky N, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333(6046):1157–1160. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang L, et al. Whole-exome sequencing of human pancreatic cancers and characterization of genomic instability caused by MLH1 haploinsufficiency and complete deficiency. Genome Res. 2012;22(2):208–219. doi: 10.1101/gr.123109.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Clark MJ, et al. U87MG decoded: The genomic sequence of a cytogenetically aberrant human cancer cell line. PLoS Genet. 2010;6(1):e1000832. doi: 10.1371/journal.pgen.1000832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang AG, et al. Identification of intrahepatic cholangiocarcinoma related genes by comparison with normal liver tissues using expressed sequence tags. Biochem Biophys Res Commun. 2006;345(3):1022–1032. doi: 10.1016/j.bbrc.2006.04.175. [DOI] [PubMed] [Google Scholar]
- 51.Akatsuka A, et al. Tumor cells of non-hematopoietic and hematopoietic origins express activation-induced C-type lectin, the ligand for killer cell lectin-like receptor F1. Int Immunol. 2010;22(9):783–790. doi: 10.1093/intimm/dxq430. [DOI] [PubMed] [Google Scholar]
- 52.Ghersi E, Vito P, Lopez P, Abdallah M, D’Adamio L. The intracellular localization of amyloid beta protein precursor (AbetaPP) intracellular domain associated protein-1 (AIDA-1) is regulated by AbetaPP and alternative splicing. J Alzheimers Dis. 2004;6(1):67–68. doi: 10.3233/jad-2004-6108. [DOI] [PubMed] [Google Scholar]
- 53.Casagrande G, te Kronnie G, Basso G. The effects of siRNA-mediated inhibition of E2A-PBX1 on EB-1 and Wnt16b expression in the 697 pre-B leukemia cell line. Haematologica. 2006;91(6):765–771. [PubMed] [Google Scholar]
- 54.San Martin IA, et al. Impaired cell cycle regulation of the osteoblast-related heterodimeris transcription factor Runx2-Cbfbeta in osteosarcoma cells. J Cell Physiol. 2009;221(3):560–571. doi: 10.1002/jcp.21894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Van der Deen M, et al. Genomic promoter occupancy or runt-related transcription factor RUNX2 in Osteosarcoma cells identifies genes involved in cell adhesion and motility. J Biol Chem. 2012;287(7):4503–4517. doi: 10.1074/jbc.M111.287771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Muller T, et al. ASAP1 promotes tumor cell motility and invasiveness, stimulates metastasis formation in vivo, and correlates with poor survival in colorectal cancer patients. Oncogene. 2010;29(16):2393–2403. doi: 10.1038/onc.2010.6. [DOI] [PubMed] [Google Scholar]
- 57.Lai F, et al. WOX1 is essential for UVB irradiation-induced apoptosis and down-regulated via translational blockade in UVB-induced cutaneous squamous cell carcinoma in vivo. Clin Cancer Res. 2005;11(16):5769–5777. doi: 10.1158/1078-0432.CCR-04-2274. [DOI] [PubMed] [Google Scholar]
- 58.Ishii H, et al. Effect of exogenous E2F-1 on the expression of common chromosome fragile site genes, FHIT and WWOX. Biochem Biophys Res Commun. 2004;316(4):1088–1093. doi: 10.1016/j.bbrc.2004.02.159. [DOI] [PubMed] [Google Scholar]
- 59.Potts PR, Yu H. The SMC5/6 complex maintains telomere length in ALT cancer cells through SUMOylation of telomere-binding proteins. Nat Struct Mol Biol. 2007;14(7):581–590. doi: 10.1038/nsmb1259. [DOI] [PubMed] [Google Scholar]
- 60.Vauhkonen H, Vauhkonen M, Sipponen P, Knuutila S. Oligonucleotide array comparative genomic hybridization refines the structure of 8p23.1, 17q12 and 20q13.2 amplifications in gastric carcinomas. Cytogenet Genome Res. 2007;119(1-2):39–45. doi: 10.1159/000109617. [DOI] [PubMed] [Google Scholar]
- 61.Korff S, et al. Frameshift mutations in coding repeats of protein tyrosine phosphatase genes in colorectal tumors with microsatellite instability. BMC Cancer. 2008;8:329. doi: 10.1186/1471-2407-8-329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Blackburn AC, et al. Genetic mapping in mice identifies DMBT1 as a candidate modifier of mammary tumors and breast cancer risk. Am J Pathol. 2007;170(6):2030–2041. doi: 10.2353/ajpath.2007.060512. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.