

Significance
DNA replication occurs in the nucleus of eukaryotic cells, which is also the site of replication of many DNA viruses. In contrast, poxviruses replicate entirely within the cytoplasm, using enzymes encoded within their genomes. Whether poxviruses replicate their linear, covalently closed, double-stranded DNA exclusively by nicking and strand displacement or by leading and lagging strand synthesis at replication forks similar to eukaryotes remained unclear. Furthermore, poxvirus genome replication origins had not been mapped, and even their existence has been questioned. Here, using directional deep sequencing, we provide evidence for the leading/lagging strand replication mode with origins located near the end of the genome and at the concatemer junction of replicative intermediates.
Keywords: vaccinia virus, DNA replication, DNA replication fork, DNA replication origin, Okazaki fragments
Abstract
Poxviruses reproduce in the host cytoplasm and encode most or all of the enzymes and factors needed for expression and synthesis of their double-stranded DNA genomes. Nevertheless, the mode of poxvirus DNA replication and the nature and location of the replication origins remain unknown. A current but unsubstantiated model posits only leading strand synthesis starting at a nick near one covalently closed end of the genome and continuing around the other end to generate a concatemer that is subsequently resolved into unit genomes. The existence of specific origins has been questioned because any plasmid can replicate in cells infected by vaccinia virus (VACV), the prototype poxvirus. We applied directional deep sequencing of short single-stranded DNA fragments enriched for RNA-primed nascent strands isolated from the cytoplasm of VACV-infected cells to pinpoint replication origins. The origins were identified as the switching points of the fragment directions, which correspond to the transition from continuous to discontinuous DNA synthesis. Origins containing a prominent initiation point mapped to a sequence within the hairpin loop at one end of the VACV genome and to the same sequence within the concatemeric junction of replication intermediates. These findings support a model for poxvirus genome replication that involves leading and lagging strand synthesis and is consistent with the requirements for primase and ligase activities as well as earlier electron microscopic and biochemical studies implicating a replication origin at the end of the VACV genome.
Poxviruses comprise a large family of complex enveloped DNA viruses that infect vertebrates and insects and includes the agent responsible for human smallpox (1). In contrast to the nuclear location exploited for genome replication by many other DNA viruses, the 130- to 230-kbp linear double-stranded DNA genomes of poxviruses are synthesized within discrete, specialized regions of the cytoplasm known as virus factories or virosomes. Furthermore, most, if not all, proteins required for DNA replication are virus-encoded (2). Poxvirus genomes, as shown for the prototype species vaccinia virus (VACV), have covalently closed hairpin termini, so that the DNA forms a continuous polynucleotide chain (3). The hairpin is a 104-nucleotide (nt) A+T-rich incompletely base-paired structure that exists in two inverted and complementary forms. As expected for a genome with such covalently closed ends, VACV replicative intermediates are head-to-head or tail-to-tail concatemers (4, 5). The concatemers exist only transiently because they are cleaved by a virus-encoded Holliday junction resolvase into unit length genomes with hairpin ends before incorporation into virus particles (6, 7). Because VACV genomes and concatemers resemble the replicative intermediates of the much smaller parvoviruses, the rolling hairpin model of replication originally proposed for the latter family was extended to poxviruses and has become the current scheme for poxvirus genome replication (2). In the rolling hairpin model, DNA synthesis occurs by strand displacement without discontinuous synthesis of the lagging strand and accordingly implies no role for Okazaki fragments. However, in contrast to parvoviruses, poxviruses have not been shown to encode an endonuclease that introduces a nick to provide a free 3′-OH for priming DNA synthesis. On the other hand, all poxviruses do encode a primase fused to a helicase, which is essential for VACV DNA replication (8). The presence of the essential primase–helicase and the requirement for the viral ligase or the host DNA ligase 1 (9) imply that poxvirus DNA replication is RNA-primed and could involve discontinuous synthesis of the lagging strand.
Although not followed up on for nearly 4 decades, early studies on poxvirus DNA replication described putative Okazaki fragments of about 1,000 nt in length and RNA primers on the 5′-ends of newly made chains of VACV DNA (10, 11). Additionally, the specific activity of [3H]thymidine incorporated during a short pulse under conditions of synchronous VACV DNA synthesis was highest in fragments from the ends of the genome, suggesting that replication originated close by (12). In agreement with the apparent terminal localization of origins, variable size double-stranded DNA loops were described at one end of replicating VACV DNA (13). Collectively, these data are compatible with a discontinuous or semidiscontinuous mode of genome replication, with origins located near the termini. Subsequently, however, the existence of specific VACV origins came into question by the demonstration that any circular DNA replicates in VACV or Shope fibroma virus-infected cell cytoplasm, and that replication is not enhanced by inclusion of any VACV DNA sequence (14, 15). Moreover, all VACV proteins known to be required for genome replication are also required for origin-independent plasmid replication (16). In contrast to a circular plasmid, however, efficient replication of a linear minichromosome with covalently closed ends requires a specific 150-bp telomere segment derived from the poxvirus genome, whereas a smaller 65-bp segment is insufficient (17). Those findings are compatible with the location of a replication origin within the terminal 150 bp. However, the latter region contains the consensus resolution sequence that is required for the conversion of the concatemer junction into the covalently closed ends of mature genomes (18, 19). Therefore, the possibility remained that the concatemer resolution sequence is required for the continuation of minichromosome replication, whereas initiation potentially could occur at any position. Thus, the existence of distinct origins of replication near the ends of the VACV genome remained an open question.
Genome replication in most cellular life forms is bidirectional and semidiscontinuous, resulting in a switch in the direction of the Okazaki fragments at the origins of replication and transition from continuous to discontinuous synthesis. This asymmetry in the distribution of RNA-primed nascent strands has been used to map initiation points at nucleotide accuracy and to generate high-resolution maps of replication origins (20–22). We used the same basic strategy but applied directional deep sequencing to map the 5′-ends of short nascent fragments to plus or minus strands of the genome instead of hybridization or primer extension and sequencing gels used in the original classical studies. A major advantage of deep sequencing for determination of origins is that no prior information on their location is required, as documented by recent studies with yeast showing that the transition points in the direction of Okazaki fragments identified by directional deep sequencing were in good agreement with the position of replication origins identified by other methods (23–25). In addition, the ability to computationally discriminate viral and host DNA provided by deep sequencing is important for the analysis of virus DNA replication. This next generation sequencing approach provided evidence for VACV replication origins at the nucleotide level.
Results
Directional Libraries of Single-Stranded Fragments of VACV DNA.
Our strategy for investigating replication origins was to perform directional (strand-specific) sequencing of VACV RNA-primed nascent DNA. The basic procedures for generating the Experimental Library, as well as Control Libraries, which will be discussed later, are outlined in Fig. S1. We isolated VACV DNA from the cytoplasmic fraction obtained at 4 h after infection, which is a time of active viral DNA synthesis. The DNA was heat-denatured, and short single strands up to 2 kb in length, which should include Okazaki fragments and nascent leading strands, were collected from the top of a neutral sucrose gradient. The next step was enrichment of RNA-primed nascent strands by lambda exonuclease (Lexo) treatment, which eliminates single-stranded DNA with phosphorylated 5′-ends but preserves hybrid molecules containing 5′-terminal RNA moieties. This procedure is widely used in the study of replication initiation and is considered to be the most stringent method to isolate nascent DNA molecules that contain 5′-terminal RNA primers (26). Two cycles of T4 polynucleotide kinase (PNK) and Lexo treatment of the short single-stranded DNA fragments isolated from the sucrose gradient resulted in a 10- to 20-fold reduction in the amount of DNA, as estimated by quantitative real-time PCR (qPCR) with selected VACV primers. Next, all samples were treated with NaOH to remove RNA primers, generating 5′-OH at the RNA/DNA junctions.
Fig. S1.

Outline of protocol used to prepare DNA fragment libraries. Cytoplasmic DNA was isolated from HeLa cells at 4 h after infection and size-selected by neutral sucrose gradient sedimentation. The Experimental Library was subjected to two rounds of treatment with bacteriophage T4 PNK and Lexo. The RNA primers were removed by treatment with NaOH, and single-stranded DNA adaptors were added to the 5′- and 3′-ends of the fragments. The fragments were gel-purified and sequenced on an Illumina platform. For the Control Library on the Left, Lexo treatments were omitted. For the Control Library on the Right, RNA primers were removed by NaOH treatment preceding the Lexo steps.
To ensure unambiguous separation of the reads mapping to each of the genome DNA strands, we ligated different single-stranded adaptors to the 5′- and 3′-ends of the single-stranded VACV DNA fragments. The libraries were amplified by PCR, and fragments of estimated size of 150–650 bp were extracted from agarose gels. Fig. S2A shows the size distribution of a typical library before loading onto the Illumina sequencer platform. The majority of the fragments were between 200 and 500 bp in length, including the 122 bp from the two adaptors. The inherent bias toward isolation of smaller fragments during purification, library amplification, and sequencing resulted in a substantial shift of the size distribution between the starting material and the sequenced fragments. The length distribution of the sequenced fragments had a mode of ∼50 nt and a median of ∼65 nt (Fig. S2B). Reads shorter than 35 nt were automatically discarded by the software. The size distribution was highly consistent between different libraries and did not appreciably differ between libraries produced before or after Lexo treatment. After sequencing, the paired-end reads were aligned to the VACV genome using Bowtie 2 (27) and visualized with the integrative genome viewer (28) and Mochiview (29) software.
Fig. S2.

Size distribution of a typical library of VACV DNA fragments. (A) The fragment sizes were determined before sequencing using the High Sensitivity DNA Analysis kit for the Agilent 2100 Bioanalyzer. The fragment sizes included two adaptors with the total added length of 122 nt. (B) The sizes of fragments, excluding those that mapped to the areas of multiple direct repeats, were determined by sequence analysis.
Deep Sequencing and Mapping of RNA-Primed DNA Fragments.
Illumina paired-end reads were first aligned to the plus and minus strands of the mature monomer VACV genome (Fig. 1A). As expected, fragments were displayed along the entire genome. However, a distinct asymmetry was noted near the two termini, which is more apparent in the expanded view of the right terminus, in which far fewer reads mapped to the lower DNA strand (Fig. 1B). Such asymmetry suggests the presence of an origin of replication near the terminus; the closely spaced rightward reads (putative Okazaki fragments) represent the lagging strand, and the near absence of the start sites of reads following the high peak of leftward reads (Fig. 1B, the green peak at the very right end) on the opposite strand could be interpreted as the continuous synthesis of the leading strand.
Fig. 1.

The 5′-end density plot of reads mapped to the VACV genome. (A) DNA was isolated at 4 h after infection, enriched for small RNA-primed fragments using Lexo, as outlined in Fig. S1. The number of 5′-ends of reads started at each nucleotide is displayed along the entire VACV genome. Reads in blue are directed to the right; reads in green are directed to the left. ITR, inverted terminal repetition. Double-headed arrows depict the terminal ∼200-bp region. (B) The Right end (1 kb) of the plot is shown at high resolution.
The covalently closed ends of the VACV genome consist of 104 nt incompletely base-paired A+T-rich sequences, which exist in two forms that are inverted and complementary (5). The adjacent ∼3,500 bp largely consists of sets of direct repeat sequences interrupted by a unique 325-bp sequence. The hairpin loops of mature genomes are derived by cleavage and resolution of the completely base-paired concatemer junction of replicative intermediates, which represent the most abundant DNA structures at 2.5–4 h after infection (4, 5, 30). Therefore, we also aligned the reads to the head-to-head dimer such that the concatemer junction was positioned in the center of the plot. Fig. 2A shows the 5′-end density reads aligned to the 12-kb region that encompasses the junction of two monomers in a head-to-head orientation. The position of the apex of the junction and the adjacent sets of eighteen and thirteen 70-bp repeats and a set of eight 54-bp repeats are indicated above the figure. Reads that map to the tandem direct repeats are recognizable by the periodic pattern of the peaks. The highest 5′-end density peaks were within the unique 378-bp region surrounding the apex of the concatemer junction. A clear asymmetry was apparent in the distribution of reads around the center of the junction. The 5′-end density peaks of the rightward reads (top strand to the left of the center) close to the apex were consistently higher than those of the leftward reads (bottom strand) at the corresponding position. Furthermore, the 5′-density peaks of adjacent rightward reads (to the right of the center) in the area of direct repeats were considerably lower than the closely spaced leftward reads in the corresponding position. We interpreted the regions of depleted peaks as representing continuous synthesis of the leading strand from the origin near the apex and the regions of closely spaced peaks as Okazaki fragments directed to the center of the replicative fork. A bidirectional origin of replication was suggested by transitions on both strands from continuous to discontinuous synthesis at the center of the concatemer junction.
Fig. 2.

The central 12 kb of the 5′-end density plot of reads mapped to the VACV head-to-head dimer. (A) The library was prepared from Lexo-treated samples, the same as in Fig.1. Boxes 1, 2, and 3 represent the first set of 70-bp repeats, the second set of 70-bp repeats, and the set of 54-bp repeats on each side of the apex of the concatemer junction (arrow), respectively. The double-headed arrow depicts the ∼400-bp region around the apex of the junction. (B) The fragment library was prepared by omitting Lexo treatment, as depicted in Fig. S1. The fragments were sequenced and the reads were aligned as in A. (C) The library was prepared from samples treated with NaOH before Lexo treatment, as in Fig. S1. The fragments were sequenced and the reads were aligned as in A.
The specific distribution of reads defining the origin was apparent in the plots of Lexo-treated samples (Fig. 2A, Experimental Library of Fig. S1) but not in the untreated samples (Fig. 2B, Left, Control Library of Fig. S1) or the samples treated with NaOH to remove potential RNA primers followed by Lexo (Fig. 2C, Right, Control Library of Fig. S1). The latter control shows that the observed pattern was specific for RNA–primed short nascent strands and could not be explained by the bias of Lexo.
A model depicting replication initiating at an origin within the hairpin of the VACV genome monomer and bidirectionally within the concatemer junction of replication intermediates is shown in Fig. 3.
Fig. 3.

Model depicting replication origins at the terminal hairpin and concatemer junction. The VACV genome monomer with hairpin ends that is packaged in virus particles is shown at the Top. DNA synthesis initiating within one hairpin is shown below. Replication results in the formation of a dimer by conversion of the incompletely base-paired hairpins into completely base-paired concatemer junctions. In this model, the concatemer junction serves as a bidirectional origin. Finally, the concatemers are resolved into unit genomes by the Holliday junction resolvase. Leading strands are shown as blue arrows, and lagging strand fragments are shown as short green arrows. The apex of the concatemer junction is indicated by red dashes.
Potential Initiation Points Within the Area Around the Concatemer Junction and Terminal Hairpin.
To finely map the potential start sites of leading strands, we analyzed the 378-bp region around the center of the concatemer junction at higher resolution (Fig. 4A). This unique area, which is located between the multiple tandem direct repeats, contained the most pronounced 5′-end density peaks. In the Lexo-treated samples, a prominent peak of 5′-ends was located 35 nt to the left of the concatemer junction (Fig. 4B), corresponding to the apex of the monomer hairpin (Fig. 4C), and consists of reads directed toward the apex. Given that the minimum length of the sequenced fragments was 35 nt, all of the fragments that start at this point cross the apex of the concatemer or extend around the hairpin of the monomer. There is substantial depletion of rightward reads beyond this point, as one would expect if the DNA chains starting in this position represented bona fide leading strands. In addition, several other minor putative start sites were detected that could correspond to additional (alternative) initiation sites for leading strands or initiation sites of Okazaki fragments. The most pronounced of these sites are two quasi-symmetrical points at 143 and 135 nt from the apex (to the left and to the right of the apex, respectively; the apex is not an exact axis of symmetry because the 104-nt hairpin is not perfect but rather contains several unpaired bases) that could represent the alternative leading strand start sites in both directions. The other initiation points located further from the apex are more difficult to interpret because they fall within the tandem repeat region and accordingly are repeated multiple times at regular intervals.
Fig. 4.

High-resolution 5′-end density plot of the concatemer junction region. (A) The region shown corresponds to the 378-nt area that encompasses the concatemer junction, which is indicated by the double-headed arrow in Fig. 2A. The distances from the apex are shown in nucleotides for the major peaks. (B) A schematic representation of the 5′-end density peaks, with arrows indicating putative initiation points. At the Bottom, an enlargement of the central area is shown at nucleotide resolution. The concatemer resolution signals located inside the strong late VACV promoter are shown in green and underlined. (C) Terminal hairpin sequences. Note that the sequences of the two hairpins are inverted and complementary. The arrow shows the hairpin sequence that corresponds to the major initiation point depicted in the concatemer in B.
To ensure that the observed 5′-end density distributions were not affected by a bias that could have been introduced by the ligation of the 5′ adaptor, libraries were prepared using two different ligation methods (details in Materials and Methods), and two independent preparations of VACV DNA were sequenced. Fig. S3 shows that the patterns of 5′-end density peaks of reads were closely similar in all three libraries.
Fig. S3.

Consistency of read maps in concatemer junction area. DNA fragment libraries were enriched for RNA-primed DNA fragments by Lexo treatment and prepared for sequencing by two different procedures described in Materials and Methods. The 5′-end density plots of reads mapped to the area around the head-to-head concatemer junction (samples after Lexo treatment) are shown. The libraries were prepared by two different methods from independently acquired materials. Sample 1 was used for library preparation by methods 1 (A) and 2 (B), respectively. (C) Sample 2 was used for library preparation by method 1. The apex of the concatemer junction and the region containing the concatemer resolution signals are indicated.
Consensus Nucleotides at the Start and End of VACV DNA Fragments.
Thus, far, we concentrated on evidence for the location of an origin of genome replication near the concatemer junction. However, putative Okazaki fragments mapped throughout the genome. Because paired-end sequencing was used, we could map the starts and stops of fragments at nucleotide resolution. Analysis of the frequency of different nucleotides at the 5′-ends of the sequenced fragments, which corresponds to the frequency of the first DNA nucleotide of the RNA/DNA junction, showed certain preferences in this position. The 5′-termini of the sequenced fragments mapped to almost all positions on the VACV genome, but a clear consensus emerged when the ends corresponding to the 5% highest peaks were aligned. The 5′ nucleotide (0 in Fig. 5A) was most frequently a purine, and the preceding and following nucleotide were most often T. In contrast, no consensus was observed when the 5′-ends of all putative Okazaki fragments were aligned. An analysis of the highest peaks also revealed that the fragments most often end at Ts before purines (Fig. 5B). Again, when the ends of all fragments were analyzed, no consensus emerged. Given that only one method was used to ligate the 3′ adaptor, we cannot rule out that the preferential detection of Ts on the ends of sequenced fragments reflects the ssDNA ligase bias. The comparison of the consensus patterns of the preferred starts and ends of the fragments implies that many fragments end exactly before the starts of preceding fragments. Fig. 6 shows that this was actually the case for a considerable fraction of the fragments that start at the preferred positions (peaks). However, it is important to point out that these data represent the alignment of fragments derived from millions of infected cells, and therefore we cannot conclude that fragments from an individual replicating DNA molecule are so closely juxtaposed. Nevertheless, these results suggested that the flap endonuclease or ligation step may be rate limiting in DNA synthesis.
Fig. 5.
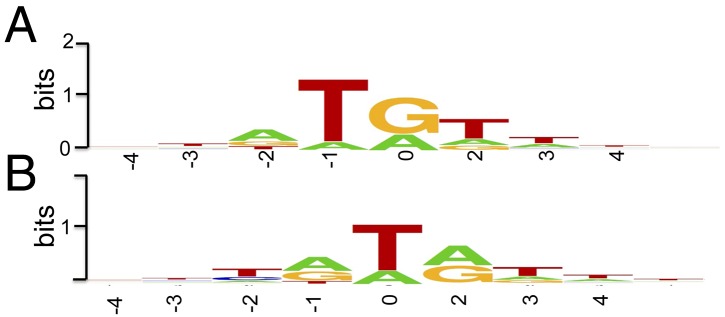
Consensus of 5′-end and 3′-ends of the most abundant putative Okazaki fragments. (A) The 5′- ends. Position 0 is the first deoxynucleotide at the RNA/DNA junction. (B) The 3′-ends. Position 0 is the 3′ nucleotide of the fragment. The consensus was rendered using the SeqLogo software.
Fig. 6.
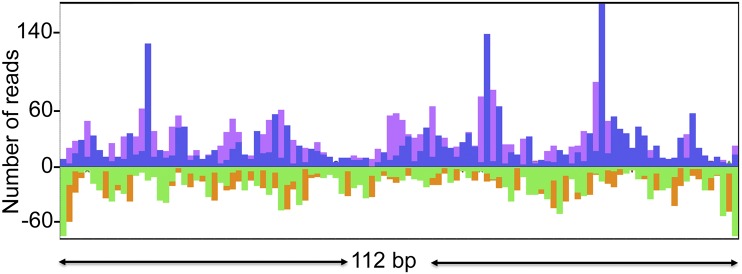
Alignment of the 5′- and 3′-ends of sequenced fragments to a representative segment of DNA. The plot shows the 5′-end (blue and green) and 3′-end (purple and orange) density peaks for the two strands of VACV DNA in an arbitrarily selected 112-bp area of the VACV genome.
Discussion
DNA replication origins are found at a single location or at a few unique locations in bacterial and archaeal chromosomes, whereas hundreds to many thousands are present in the much larger eukaryotic genomes (31). In Saccharomyces cerevisiae and some viruses, origins were identified as autonomous replication sequences that confer replication ability to transfected episomes (31). However, the autonomous replication strategy to define origins was unsuccessful when applied to poxviruses: plasmids regardless of sequence efficiently replicate in poxvirus-infected cells, raising questions of whether specific origins are needed for the viral replication machinery (14–16). To investigate the mechanism of VACV replication, we exploited approaches recently proven effective for mapping eukaryotic replication origins by deep sequencing (26, 32). However, in contrast to the conventional strategy, we sequenced predominantly Okazaki fragments instead of leading strands and used strand-specific deep sequencing. The procedure, which consists of sequencing purified RNA-primed nascent strands, avoids treatment with inhibitors or cell synchronization, and thus the results are likely to reflect the authentic replication mechanism. Additional advantages of deep sequencing included the ability to computationally discriminate viral and host DNA and the absence of a requirement for prior information on the distribution of replication origins. Lexo treatment was used to enrich DNA fragments with RNA primers, as previously described (26). The clear difference in the profiles of reads when Lexo treatment was omitted or if NaOH was used to remove RNA primers before Lexo indicated that VACV nascent chains start from RNA primers. By using directional sequencing, we obtained high resolution and preserved the polarity of the reads, allowing the origins to be defined as the sites of transition between continuous and discontinuous DNA synthesis. In principle, this procedure is equivalent to the primer extension step of the classical replication initiation point (RIP) mapping procedure that had been used for the fine mapping of many origins long before the advent of deep sequencing (21). However, a crucial advantage of the deep sequencing strategy is that, unlike the traditional RIP mapping approach, it does not require preliminary information on the approximate location of the origin(s). Furthermore, we were unable to use RIP mapping to verify the location of the main replication start site of VACV inferred from the sequencing results because the snapback of the ends of the viral genome precluded in vitro primer extension.
Our analysis is consistent with VACV DNA synthesis starting as a replication fork at an origin located within the terminal hairpin of a mature monomeric genome following infection and subsequently near the center of the concatemer junction of replication intermediates (Fig. 3). However, because the hairpin and concatemer junction sequences are identical, we could not discriminate between the two. Indeed, when inserted into a plasmid, the DNA comprising the concatemer junction is extruded as cruciform structures resembling the two isoforms of the terminal hairpin (33, 34). Whether the junction exists as a cruciform within the VACV replicative intermediates in infected cells is not known. The peak of 5′-end density reads, corresponding to the rightward leading strand, was detected at 35 nt from the apex and may represent a main start site of VACV DNA replication. This precise sequence is present at only one of the isomeric hairpins at the end of the mature genome but is present in every concatemer junction. We also detected several minor potential start sites in both directions, which could represent additional or alternative initiation points of the leading strand and/or initiation sites of Okazaki fragments.
The arrangement of the identified initiation points within the 378-nt area around the apex of the VACV concatemer junction strikingly resembles the patterns of initiation points around origins of replication that have been mapped by other methods in diverse genomes. This similarity extends even to the asymmetry between the rightward and leftward directions of the leading strand synthesis (20, 22, 35). Interestingly, the 35-nt start site is located close to a well-characterized, strong late promoter (36). The transcript synthesized from this promoter does not contain an extended ORF, and its role (if any) in VACV reproduction remains unknown. A possibility is that the interaction of the virion RNA polymerase with this late promoter helps unwind the DNA duplex during initiation of replication. The identification of a single most prominent start site of leading strand synthesis implies that the initiation of VACV replication is asymmetric, i.e., starts mostly on one end. This site potentially could be used on monomeric genomes during the first round of replication, whereas later in infection the concatemer junction area could be used for replication initiation. The localization of the origin of replication in the hairpin loop is not unexpected in the light of early experiments that suggested initiation at the genome ends and also because this loop is the most AT-rich region in the VACV genome.
We could not accurately determine the size of the Okazaki fragments because of the bias toward short fragments that is present at each step of library preparation and sequencing, and in particular because fragments carrying RNA primers were selected. Nevertheless, the observed length distributions suggest that the fragments are considerably shorter than the ∼1-kb long fragments that have been reported previously (10); however, there was no data on the presence of RNA primers in those long fragments. Previous in vitro studies had shown a preference for the VACV primase to start at a purine followed by a pyrimidine (37). In the present study, some sequence specificity was found at the junction between the RNA primer and the DNA moiety in nascent VACV fragments. In the most abundant fragments, the DNA started at a purine preceded by a T. Alignment of the nascent fragments on the VACV genome indicated preferred DNA start and stop sites that appeared to be juxtaposed. However, as we mentioned in Results, this is a consensus alignment and cannot be interpreted as indicating juxtaposition of fragments in the same DNA molecule. If the latter were the case, one would expect there to be DNA sequence gaps between the stop and start sites of RNA-primed fragments unless the RNA formed a flap that was displaced by the VACV DNA polymerase. Displacement of RNA by the VACV DNA polymerase is a possibility because it has been shown in vitro that yeast and archaeal DNA polymerases displace RNA more readily than DNA, thus facilitating the degradation of RNA primers (38, 39).
In addition to poxviruses, covalently closed hairpins are present at the ends of several classes of replicons. In particular, such terminal structures are characteristic of the group of nucleocytoplasmic large DNA viruses of eukaryotes that possess linear genomes, namely, asfarviruses, phycodnaviruses, and mimiviruses. All of these viruses appear to have evolved from a common ancestor, as indicated by the conservation of many core genes, including the signature fusion protein consisting of a primase and a helicase domain like the VACV D5 protein (40). The conservation of the primase–helicase, along with the terminal hairpins, implies that all these viruses use similar mechanisms of genome replication including the location of the origin within the hairpin loop, as shown here. By contrast, prokaryotic hairpin-containing replicons appear to replicate via different mechanisms. The archaeal rudiviruses encode an initiation endonuclease and are thought to use a rolling hairpin-like replication route (41). The bacteria in the genus Borrelia have been shown to contain the origin in the middle of their linear chromosomes (42). Thus, replicons with similar, covalently closed genomic ends appear to have evolved widely different mechanisms for replication initiation.
In summary, we used deep sequencing to analyze poxvirus DNA replication. The data obtained fit a model in which DNA replication starts at one end of the genome and involves RNA-primed Okazaki fragments. This model (Fig. 3) departs from the current rolling hairpin model, yet is strongly supported by an earlier electron microscopic study, which showed a variable length double-stranded DNA loop at one end of replicating VACV DNA molecules (13) and by biochemical studies (10–12). Moreover, the sequences determined to be optimal for replication of a linear DNA template with hairpin ends (17) contained the origin sequences described here. Technical challenges have undoubtedly contributed to the paucity of poxvirus DNA replication studies in the past. However, deep sequencing and other methodological advances should accelerate future work in this field.
Materials and Methods
Preparation of Newly Made Chains of VACV DNA.
Rabbit kidney (RK13) cell monolayers were infected with the WR strain of VACV; 4 h after infection, cells were collected and washed with PBS. All subsequent operations were at 4 °C. Cells were resuspended in 20 mM Tris pH 8.0, 10 mM NaCl, 5 mM EDTA and Dounce-homogenized under conditions to disrupt cellular membrane while keeping the nuclear membrane intact. After centrifugation at 800 × g for 3 min, the nuclei were discarded, and the virus factories were pelleted at 20,000 × g for 20 min. For further steps, the protocol described by Cayrou et al. (26) was followed with some modifications. DNA was extracted from the pellets with DNAzol (Life Technologies) in the presence of proteinase K (200 µg/mL) and precipitated with ethanol. The DNA pellet was dissolved in 20 mM Tris pH 8.0, 10 mM NaCl, 5 mM EDTA, 0.5% SDS, and incubated with proteinase K at 37° for 2 h. Proteinase K was removed by phenol/chloroform extraction, and the DNA was ethanol-precipitated. DNA was denatured for 10 min at 100 °C and subjected to neutral sucrose gradient centrifugation. Aliquots of the gradient fractions were analyzed in alkaline agarose gels, and fragments less than 2 kb were collected and ethanol-precipitated. Approximately 45% of the DNA was heated for 2 min at 95 °C and then phosphorylated with T4 PNK. After phenol/chloroform extraction and ethanol precipitation, the DNA was treated with Lexo (10 µg/mL overnight, Fermentas). This procedure of denaturation, T4 PNK, and Lexo treatment was repeated twice. The total amount of DNA after two Lexo treatments was diminished about 10- to 20-fold, as estimated by qPCR with specific VACV primers. Therefore, 10% of the starting material before Lexo treatment was kept as a control. At the next step, both Lexo and untreated samples were incubated in 150 mM NaOH, 1 mM EDTA overnight at 37 °C to digest RNA primers. Approximately 45% of the DNA was used as another control and treated with 150 mM NaOH and 1 mM EDTA overnight at 37 °C before the Lexo treatment and subsequent steps.
Preparation of Sequencing Libraries.
Single-stranded DNA adaptors matching those from the ScriptSeq mRNA-seq library preparation kit (Epicentre) were synthesized by Integrated DNA Technologies (IDT). To control for possible bias, two methods were used for the ligation of adaptors. In the first method the single-stranded 3′-end adaptor, with the 3′-end blocked by the 3SPC3 spacer, was directly ligated to ssDNA fragments with 5.0 units/µl of CircLigase II (Epicentre) at 60° for 3 h in 33 mM Tris-acetate (pH7.5), 66 mM potassium acetate, 5 mM DTT, 2.5 mM MnCl2, 20% (vol/vol) polyethylene glycol, 100 nM of adaptor; the 5′-end single-stranded adaptor was also directly ligated to 5′-ends of the fragments exactly the same way as 3′-adaptor (after the 5′-ends of the fragments were phosphorylated with T4 PNK). In the second method (adapted from ref. 43), only the last 10 nt of the 3′-end adaptor covalently linked to biotin was ligated to the 3′-ends of the fragments, and the fragments were purified on Dynabeads MyOne Streptavidin C1 (Invitrogen). The primer complementary to the full-length 3′-end adaptor was annealed to the purified fragments while still on magnetic beads and was extended by Bst DNA polymerase, large fragment (New England BioLabs) to produce double-stranded DNA. The ends of double-stranded DNA were polished by T4 DNA polymerase in the presence of four dNTPs and ligated to double-stranded 5′-end adaptor (made by annealing the plus strand of the adaptor with its minus strand phosphorylated at the 5′-end) with T4 DNA ligase. The complementary strands of the fragments ligated to the complementary strands of the 5′-end adaptor were eluted from the beads with 50 mM NaOH, 2 mM EDTA and PCR-amplified (15 cycles) in the FailSafe PCR PreMix E by FailSafe PCR enzyme from ScriptSeq mRNA-seq library preparation kit, using ScriptSeq Index RCR Primers (Epicentre). The 150- to 650-nt amplified fragments were extracted from 2% (wt/vol) agarose gels, sequenced on the Illumina Genome Analyzer Iix, and paired-end reads were analyzed.
Analysis of the Sequenced Reads.
The adaptor sequences were computationally trimmed from the ends of the reads, and those shorter than 35 nt and without a pair were discarded. Paired-end reads were mapped to the VACV WR genome (GenBank GI:29692106) or a template created by linking two VACV monomers in head-to-head configuration using Bowtie 2 (27) allowing two mismatches, and the alignment file was split into two (plus and minus). BAM files were visualized with the Integrated Genome Viewer (IGV, Broad Institute). The density plots of the 5′-ends of the first and the second (the 3′-ends of the fragments) reads of the pairs were generated as WIG files and visualized by MochiView software. The information about the size of the fragments was extracted from SAM files.
Acknowledgments
We thank Wolfgang Resch of the Scientific Computing Branch, Center for Information Technology, for useful suggestions on sequence analysis and Eugene Koonin of the National Center for Biotechnology Information of the National Library of Medicine for helpful discussions and critical reading of the manuscript. The research was supported by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Footnotes
The authors declare no conflict of interest.
Data deposition: Sequence reads were deposited in the National Center for Biotechnology Information Sequence Read Archive database (accession no, SRP061373).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1514809112/-/DCSupplemental.
References
- 1.Moss B. 2013. Poxviridae. Fields Virology, eds Knipe DM, Howley PM (Wolters Kluwer/Lippincott Williams & Wilkins, Philadelphia), 6th Ed, Vol 2, pp 2129–2159.
- 2.Moss B. Poxvirus DNA replication. Cold Spring Harb Perspect Biol. 2013;5(9):a010199. doi: 10.1101/cshperspect.a010199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baroudy BM, Venkatesan S, Moss B. Incompletely base-paired flip-flop terminal loops link the two DNA strands of the vaccinia virus genome into one uninterrupted polynucleotide chain. Cell. 1982;28(2):315–324. doi: 10.1016/0092-8674(82)90349-x. [DOI] [PubMed] [Google Scholar]
- 4.Moyer RW, Graves RL. The mechanism of cytoplasmic orthopoxvirus DNA replication. Cell. 1981;27(2 Pt 1):391–401. doi: 10.1016/0092-8674(81)90422-0. [DOI] [PubMed] [Google Scholar]
- 5.Baroudy BM, Venkatesan S, Moss B. Structure and replication of vaccinia virus telomeres. Cold Spring Harb Symp Quant Biol. 1983;47(Pt 2):723–729. doi: 10.1101/sqb.1983.047.01.083. [DOI] [PubMed] [Google Scholar]
- 6.Garcia AD, Moss B. Repression of vaccinia virus Holliday junction resolvase inhibits processing of viral DNA into unit-length genomes. J Virol. 2001;75(14):6460–6471. doi: 10.1128/JVI.75.14.6460-6471.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garcia AD, Aravind L, Koonin EV, Moss B. Bacterial-type DNA Holliday junction resolvases in eukaryotic viruses. Proc Natl Acad Sci USA. 2000;97(16):8926–8931. doi: 10.1073/pnas.150238697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Silva FS, Lewis W, Berglund P, Koonin EV, Moss B. Poxvirus DNA primase. Proc Natl Acad Sci USA. 2007;104(47):18724–18729. doi: 10.1073/pnas.0709276104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paran N, De Silva FS, Senkevich TG, Moss B. Cellular DNA ligase I is recruited to cytoplasmic vaccinia virus factories and masks the role of the vaccinia ligase in viral DNA replication. Cell Host Microbe. 2009;6(6):563–569. doi: 10.1016/j.chom.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Esteban M, Holowczak JA. Replication of vaccinia DNA in mouse L cells. I. In vivo DNA synthesis. Virology. 1977;78(1):57–75. doi: 10.1016/0042-6822(77)90078-2. [DOI] [PubMed] [Google Scholar]
- 11.Pogo BGT, O’Shea MT. The mode of replication of vaccinia virus DNA. Virology. 1978;84(1):1–8. doi: 10.1016/0042-6822(78)90213-1. [DOI] [PubMed] [Google Scholar]
- 12.Pogo BG, Berkowitz EM, Dales S. Investigation of vaccinia virus DNA replication employing a conditional lethal mutant defective in DNA. Virology. 1984;132(2):436–444. doi: 10.1016/0042-6822(84)90048-5. [DOI] [PubMed] [Google Scholar]
- 13.Esteban M, Flores L, Holowczak JA. Model for vaccinia virus DNA replication. Virology. 1977;83(2):467–473. doi: 10.1016/0042-6822(77)90197-0. [DOI] [PubMed] [Google Scholar]
- 14.DeLange AM, McFadden G. Sequence-nonspecific replication of transfected plasmid DNA in poxvirus-infected cells. Proc Natl Acad Sci USA. 1986;83(3):614–618. doi: 10.1073/pnas.83.3.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Merchlinsky M, Moss B. Sequence-independent replication and sequence-specific resolution of plasmids containing the vaccinia virus concatemer junction: Requirements for early and late trans-acting factors. In: Kelly T, Stillman B, editors. Cancer Cells 6/Eukaryotic DNA Replication. Cold Spring Harbor Lab; Cold Spring Harbor, NY: 1988. pp. 87–93. [Google Scholar]
- 16.De Silva FS, Moss B. Origin-independent plasmid replication occurs in vaccinia virus cytoplasmic factories and requires all five known poxvirus replication factors. Virol J. 2005;2(1):23. doi: 10.1186/1743-422X-2-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Du S, Traktman P. Vaccinia virus DNA replication: Two hundred base pairs of telomeric sequence confer optimal replication efficiency on minichromosome templates. Proc Natl Acad Sci USA. 1996;93(18):9693–9698. doi: 10.1073/pnas.93.18.9693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Merchlinsky M. Mutational analysis of the resolution sequence of vaccinia virus DNA: essential sequence consists of two separate AT-rich regions highly conserved among poxviruses. J Virol. 1990;64(10):5029–5035. doi: 10.1128/jvi.64.10.5029-5035.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.DeLange AM, McFadden G. Efficient resolution of replicated poxvirus telomeres to native hairpin structures requires two inverted symmetrical copies of a core target DNA sequence. J Virol. 1987;61(6):1957–1963. doi: 10.1128/jvi.61.6.1957-1963.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hay RT, DePamphilis ML. Initiation of SV40 DNA replication in vivo: Location and structure of 5′ ends of DNA synthesized in the ori region. Cell. 1982;28(4):767–779. doi: 10.1016/0092-8674(82)90056-3. [DOI] [PubMed] [Google Scholar]
- 21.Gerbi SA, Bielinsky AK. Replication initiation point mapping. Methods. 1997;13(3):271–280. doi: 10.1006/meth.1997.0526. [DOI] [PubMed] [Google Scholar]
- 22.Kohara Y, Tohdoh N, Jiang XW, Okazaki T. The distribution and properties of RNA primed initiation sites of DNA synthesis at the replication origin of Escherichia coli chromosome. Nucleic Acids Res. 1985;13(19):6847–6866. doi: 10.1093/nar/13.19.6847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith DJ, Whitehouse I. Intrinsic coupling of lagging-strand synthesis to chromatin assembly. Nature. 2012;483(7390):434–438. doi: 10.1038/nature10895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McGuffee SR, Smith DJ, Whitehouse I. Quantitative, genome-wide analysis of eukaryotic replication initiation and termination. Mol Cell. 2013;50(1):123–135. doi: 10.1016/j.molcel.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yanga W, Lib X. Next-generation sequencing of Okazaki fragments extracted from Saccharomyces cerevisiae. FEBS Lett. 2013;587(15):2441–2447. doi: 10.1016/j.febslet.2013.06.014. [DOI] [PubMed] [Google Scholar]
- 26.Cayrou C, Grégoire D, Coulombe P, Danis E, Méchali M. Genome-scale identification of active DNA replication origins. Methods. 2012;57(2):158–164. doi: 10.1016/j.ymeth.2012.06.015. [DOI] [PubMed] [Google Scholar]
- 27.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robinson JT, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29(1):24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Homann OR, Johnson AD. MochiView: Versatile software for genome browsing and DNA motif analysis. BMC Biol. 2010;8:49. doi: 10.1186/1741-7007-8-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baroudy BM, Moss B. Sequence homologies of diverse length tandem repetitions near ends of vaccinia virus genome suggest unequal crossing over. Nucleic Acids Res. 1982;10(18):5673–5679. doi: 10.1093/nar/10.18.5673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leonard AC, Méchali M. DNA replication origins. Cold Spring Harb Perspect Biol. 2013;5(10):a010116. doi: 10.1101/cshperspect.a010116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Besnard E, et al. Unraveling cell type-specific and reprogrammable human replication origin signatures associated with G-quadruplex consensus motifs. Nat Struct Mol Biol. 2012;19(8):837–844. doi: 10.1038/nsmb.2339. [DOI] [PubMed] [Google Scholar]
- 33.Merchlinsky M, Garon CF, Moss B. Molecular cloning and sequence of the concatemer junction from vaccinia virus replicative DNA. Viral nuclease cleavage sites in cruciform structures. J Mol Biol. 1988;199(3):399–413. doi: 10.1016/0022-2836(88)90613-4. [DOI] [PubMed] [Google Scholar]
- 34.Dickie P, Morgan AR, McFadden G. Cruciform extrusion in plasmids bearing the replicative intermediate configuration of a poxvirus telomere. J Mol Biol. 1987;196(3):541–558. doi: 10.1016/0022-2836(87)90031-3. [DOI] [PubMed] [Google Scholar]
- 35.Bielinsky AK, Gerbi SA. Chromosomal ARS1 has a single leading strand start site. Mol Cell. 1999;3(4):477–486. doi: 10.1016/s1097-2765(00)80475-x. [DOI] [PubMed] [Google Scholar]
- 36.Hu F-Q, Pickup DJ. Transcription of the terminal loop region of vaccinia virus DNA is initiated from the telomere sequences directing DNA resolution. Virology. 1991;181(2):716–720. doi: 10.1016/0042-6822(91)90905-q. [DOI] [PubMed] [Google Scholar]
- 37.De Silva FS, Paran N, Moss B. Products and substrate/template usage of vaccinia virus DNA primase. Virology. 2009;383(1):136–141. doi: 10.1016/j.virol.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stith CM, Sterling J, Resnick MA, Gordenin DA, Burgers PM. Flexibility of eukaryotic Okazaki fragment maturation through regulated strand displacement synthesis. J Biol Chem. 2008;283(49):34129–34140. doi: 10.1074/jbc.M806668200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beattie TR, Bell SD. Coordination of multiple enzyme activities by a single PCNA in archaeal Okazaki fragment maturation. EMBO J. 2012;31(6):1556–1567. doi: 10.1038/emboj.2012.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iyer LM, Koonin EV, Leipe DD, Aravind L. Origin and evolution of the archaeo-eukaryotic primase superfamily and related palm-domain proteins: Structural insights and new members. Nucleic Acids Res. 2005;33(12):3875–3896. doi: 10.1093/nar/gki702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oke M, et al. A dimeric Rep protein initiates replication of a linear archaeal virus genome: implications for the Rep mechanism and viral replication. J Virol. 2011;85(2):925–931. doi: 10.1128/JVI.01467-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Picardeau M, Lobry JR, Hinnebusch BJ. Physical mapping of an origin of bidirectional replication at the centre of the Borrelia burgdorferi linear chromosome. Mol Microbiol. 1999;32(2):437–445. doi: 10.1046/j.1365-2958.1999.01368.x. [DOI] [PubMed] [Google Scholar]
- 43.Gansauge MT, Meyer M. Single-stranded DNA library preparation for the sequencing of ancient or damaged DNA. Nat Protoc. 2013;8(4):737–748. doi: 10.1038/nprot.2013.038. [DOI] [PubMed] [Google Scholar]