

Significance
Small molecule probes have proved indispensable in dissecting bacterial systems. Their combinations have further expanded their utility as tools by enabling the study of interacting pathways. As such, screens for synergy between compounds have been widely used to reveal functional connections among cellular components. The utility of antagonism, however, has largely been overlooked. This study highlights the value of antagonistic interactions in elucidating genetic networks and mechanisms of drug action. Herein, we report on the discovery of clomiphene, an inhibitor of bacterial cell wall synthesis, uncovered through a systematic screen for antagonism. The discovery of clomiphene shed light on the pathways of cell wall biogenesis and, importantly, represents a new promising lead for the fight against infection.
Keywords: wall teichoic acids, antagonism, antibacterial, undecaprenyl phosphate, UppS
Abstract
Drug combinations are valuable tools for studying biological systems. Although much attention has been given to synergistic interactions in revealing connections between cellular processes, antagonistic interactions can also have tremendous value in elucidating genetic networks and mechanisms of drug action. Here, we exploit the power of antagonism in a high-throughput screen for molecules that suppress the activity of targocil, an inhibitor of the wall teichoic acid (WTA) flippase in Staphylococcus aureus. Well-characterized antagonism within the WTA biosynthetic pathway indicated that early steps would be sensitive to this screen; however, broader interactions with cell wall biogenesis components suggested that it might capture additional targets. A chemical screening effort using this approach identified clomiphene, a widely used fertility drug, as one such compound. Mechanistic characterization revealed the target was the undecaprenyl diphosphate synthase, an enzyme that catalyzes the synthesis of a polyisoprenoid essential for both peptidoglycan and WTA synthesis. The work sheds light on mechanisms contributing to the observed suppressive interactions of clomiphene and in turn reveals aspects of the biology that underlie cell wall synthesis in S. aureus. Further, this effort highlights the utility of antagonistic interactions both in high-throughput screening and in compound mode of action studies. Importantly, clomiphene represents a lead for antibacterial drug discovery.
Small molecules have long proven their utility as an alternative to genetic mutation in perturbing and understanding biological systems (1). Interactions between small molecules are comparable to those observed among genetic mutations. In combination, one molecule may enhance (synergism) or suppress (antagonism) the effect of another molecule. In recent years, drug combinations have been extensively used to explore connections between cellular processes. Specifically, synergies have provided unique means to expose pathway interactions (2, 3) and, in turn, understand the mode of action of small molecules (4, 5). Underpinning these interactions are complex genetic networks that define the outcome of drug combinations (6, 7). Indeed, synergistic pairs often exhibit their effects by targeting related processes and impacting genetic interactions that bridge those pathways. Further, synergistic drug combinations are now essential therapeutic strategies in the clinic in areas such as cancer, HIV, and infectious diseases (8). Where antagonistic interactions are generally undesirable from a therapeutic perspective, the utility of this class of interactions to reveal biological function and underlying network connectivity has been largely overlooked. Like synergism, antagonism typically has a genetic basis that reflects relationships between cellular targets and mechanisms of drug action (7). Recent reports of antagonistic drug interactions have shed light on novel connections that govern important bacterial physiology. For example, the suppressive effects of antibiotics targeting protein and DNA synthesis in Escherichia coli revealed antagonistic connections between the regulation of ribosomal genes and the DNA stress response (9). A more recent study surveyed suppressive interactions among antifungals and described the mechanism of the suppressive activities of bromopyruvate and staurosporine (10). Interestingly, but perhaps counterintuitively, other studies have suggested that antagonistic drug pairs can even slow the evolution of drug resistance (11, 12). Nevertheless, the utility of antagonism among small molecules has yet to be fully explored as a tool to study biological function. Certainly, there have been no systematic searches for antagonistic interactions to exploit suppressive network connections and, in turn, uncover novel inhibitors of the targeted pathways.
Bacterial cell wall synthesis is an antibacterial target that is celebrated for its druggability and, increasingly, for its genetic complexity. Indeed, the dispensability of wall teichoic acid (WTA) genes in gram-positive bacteria has emerged in recent years as a prototypical example of genetic antagonism. WTAs are phosphate-rich polymers that make up a large proportion of the cell wall of gram-positive bacteria and, in the pathogen Staphylococcus aureus, have a key role in cell division and virulence (13, 14). The synthesis of WTA in S. aureus is initiated by the action of two nonessential gene products: TarO and TarA. TarO (undecaprenyl-phosphate N-acetylglucosaminyl 1-phosphate transferase) transfers an N-acetyl-glucosamine-1-phosphate moiety to an undecaprenyl phosphate carrier lipid, followed by the transfer of N-acetylmannosamine by TarA (N-acetylglucosaminyldiphospho-undecaprenol N-acetyl-β-d-mannosaminyltransferase). The resulting glycolipid is a substrate for so-called late-step gene products that append a polymer with ribitol-phosphate repeats before export to the external surface and attachment to peptidoglycan (PG). Paradoxically, the genes encoding the late-steps in WTA synthesis have an essential phenotype, but become dispensable in strains with a deletion in either of the early-step genes, tarO or tarA (encoding for an N-acetylglucosamine-1-phosphate transferase and an N-acetylmannosamine transferase, respectively) (15). Interestingly, blocking the early steps in WTA biosynthesis leads to β-lactam sensitivity in methicillin-resistant S. aureus (MRSA) (2, 16). These observations highlight the complexity of cell wall synthesis in gram-positive bacteria and provide a rationale for combination therapy. Further, the idiosyncratic genetic antagonism of the WTA biosynthetic pathway and interactions with additional components of cell wall synthesis provide a unique opportunity to screen for new chemical matter with utility as probes to better understand this genetic complexity.
To this end, we conducted a search for compounds that antagonize the lethal activity of targocil (17) (Scheme 1), a probe of TarG, the essential gene product that makes up the transmembrane transporter that exports WTAs to the cell surface. Screening a library of previously approved drugs we discovered that clomiphene (Scheme 1) a widely used fertility drug, was a potent antagonist of targocil. Mechanistic characterization revealed that its target was the undecaprenyl diphosphate synthase (UppS), responsible for the synthesis of the lipid carrier, undecaprenyl phosphate (Und-P), and we solved a cocrystal structure of clomiphene with UppS from E. coli. We report on the ability of clomiphene to potentiate the activity of β-lactam antibiotics against MRSA, revealing UppS as a key component of the network that supports β-lactam resistance in MRSA. As such, clomiphene is new cell-permeable probe of the synthesis of Und-P and represents a potential lead for antibiotic drug discovery.
Scheme 1.
Chemical structures of clomiphene, targocil, and ticlopidine.
Results
A Screen for Inhibitors That Antagonize Targocil’s Activity.
Our work began with a high-throughput screen to identify molecules that antagonized the activity of a lethal concentration of targocil, a probe of TarG, against S. aureus (strain Newman).
We screened a library of 1,600 off-patent US Food and Drug Administration (FDA)-approved molecules (Pharmakon; Microsource) (SI Appendix, Fig. S1). Of the 1,600 drug pairs tested, we found 68 potentially antagonistic interactions (Fig. 1A). Confirmation of antagonism with targocil in dose resulted in 62 confirmed actives (Fig. 1A). Given the gene dispensability patterns that govern WTA synthesis, we reasoned that our screening approach would inherently identify potential probes of the dispensable early steps of WTA synthesis. Indeed, inhibitors of TarO and TarA would protect S. aureus from the activity of targocil. We previously reported on an inhibitor of the first step of WTA synthesis (2) and, in this study, were most interested in uncovering probes that target alternate and essential pathways linked to late-step WTA synthesis. We therefore focused on molecules that were intrinsically growth inhibitory to S. aureus, and were thus unlikely to be inhibitors of the early steps of WTA synthesis. These 12 priority actives were followed up using a literature search for favorable pharmacology and toxicology (Fig. 1A). Among them, one molecule, clomiphene, was of particular interest because it is a widely used fertility drug with well-established pharmacology and toxicology. Clomiphene is a nonsteroidal selective estrogen receptor modulator that can block estrogen receptors, stimulating ovulation in anovulatory women (18). Importantly, clomiphene may be a good candidate for repurposing, because it appears to have few reported adverse side effects (19). In combination with targocil, clomiphene led to a fractional inhibitory concentration index (FICI) of 8.0, indicative of profound antagonism (Fig. 1B). On its own, clomiphene had a minimum inhibitory concentration (MIC) of 8 μg/mL against both S. aureus and the model organism Bacillus subtilis.
Fig. 1.

Screen to uncover molecules that antagonize the activity of targocil identifies clomiphene. (A) Schematic diagram of the workflow of the screen conducted from 1,600 previously approved drugs to the lead molecule, clomiphene. Compounds were eliminated at each stage according to the criteria indicated. A final literature search for previously-reported pharmacology and toxicology data put clomiphene in a favorable light. (B) Microdilution checkerboard analysis showing the antagonistic combined effect of clomiphene and targocil against CA-MRSA USA300, where the extent of inhibition is shown as a heat plot, such that the darkest blue color represents full bacterial growth. (C) Checkerboard analysis showing the antagonistic combined effect of ticlopidine on clomiphene against CA-MRSA USA300. Full growth is represented as the darkest shade of blue and complete growth inhibition, as white.
To confirm the observed antagonism, we combined clomiphene with ticlopidine (Scheme 1) another inhibitor of WTA biosynthesis but one that targets an early-step, TarO, in S. aureus. The interaction again exhibited clear antagonism, yielding an FICI ≥8.3 (Fig. 1C). Further, working in the model gram-positive B. subtilis, where the genetic tools are particularly strong, we showed that clomiphene suppressed the lethality observed on depleting the WTA biosynthetic enzyme TagB (SI Appendix, Fig. S2). Taken together, these results confirm that the interaction between clomiphene and targocil is functional in nature and likely reflects pathway connections among their target genes. The question arises, then, as to the identity of the target(s) of clomiphene.
Sensitization of MRSA to β-Lactam Antibiotics Point to a Clomiphene Target.
In recent years, WTA synthesis has been implicated in methicillin resistance in strains of MRSA. Indeed, we and others have shown that inhibition of WTA synthesis can render MRSA susceptible once again to β-lactam antibiotics (2, 16). We reasoned that a small molecule that perturbs a pathway connected to late-step WTA synthesis, as identified through our antagonism screen, might similarly resensitize MRSA to the action of β-lactams. We thus combined clomiphene with a variety of antibiotics against the highly problematic CA-MRSA USA 300 strain to determine FICI values (Table 1). Remarkably, clomiphene potentiated the activity of all β-lactams (and cephalosporins) surveyed, with FICIs ranging from 0.3 to 0.5 (Table 1). On the other hand, combining clomiphene with non–β-lactam antibiotics having diverse mechanisms of action was generally additive (FICI = 1), the exception being bacitracin, an antibiotic known to inhibit the dephosphorylation of undecaprenyl diphosphate; bacitracin and clomiphene exhibited potent synergy (FICI = 0.38; Table 1). We also investigated the same inhibitor combinations in B. subtilis to assess whether the observed interactions with clomiphene were specific to the MRSA phenotype (SI Appendix, Table S1). Here, we also surveyed fosmidomycin, which is known to target the 1-deoxy-d-xylulose-5-phosphate (DOXP) pathway for polyisoprenoid synthesis. This pathway is absent in S. aureus, which instead uses the mevalonate pathway. As with S. aureus, clomiphene had synergistic activity with all β-lactams tested, as well as with bacitracin against B. subtilis. In addition, clomiphene was highly synergistic with fosmidomycin, resulting in an FICI of 0.38. Overall, these results show that clomiphene synergizes with β-lactam antibiotics, not only in MRSA but also in B. subtilis.
Table 1.
Combinations of clomiphene and antibiotics against CA-MRSA USA 300
| Antibiotic | MIC antibiotic (µg/mL) | FIC antibiotic | MIC clomiphene (µg/mL) | FIC clomiphene | FIC index |
| Ampicillin | 8 | 0.125 | 16 | 0.25 | 0.375 |
| Cloxacillin | 128 | 0.125 | 16 | 0.125 | 0.250 |
| Nafcillin | 32 | 0.063 | 16 | 0.25 | 0.313 |
| Piperacillin | 256 | 0.125 | 16 | 0.25 | 0.375 |
| Cefmandole | 8 | 0.125 | 16 | 0.25 | 0.375 |
| Cefaclor | 64 | 0.125 | 16 | 0.25 | 0.375 |
| Cephalexin | 128 | 0.125 | 16 | 0.25 | 0.375 |
| Cefadroxil | 128 | 0.125 | 16 | 0.25 | 0.375 |
| Cefuroxime | 256 | 0.063 | 16 | 0.25 | 0.313 |
| Vancomycin | 1 | 0.25 | 16 | 0.5 | 0.750 |
| Bacitracin | 256 | 0.125 | 16 | 0.25 | 0.375 |
| Chloramphenicol | 8 | 1 | 16 | 1 | 2 |
| Cycloserine | 128 | 0.25 | 16 | 0.5 | 0.75 |
| Erythromycin | 0.25 | 1 | 16 | 1 | 2 |
| Fosfomycin | 32 | 0.125 | 16 | 0.5 | 0.625 |
| Kanamycin | 2 | 1 | 16 | 1 | 2 |
| Norfloxacin | 1 | 1 | 16 | 1 | 2 |
| Novobiocin | 0.25 | 1 | 16 | 1 | 2 |
| Spectinomycin | 128 | 1 | 16 | 1 | 2 |
| Tetracycline | 2 | 0.5 | 16 | 0.5 | 1 |
FIC, fractional inhibitory concentration = [X]/MICX, where [X] is the lowest inhibitory concentration of drug in the presence of the codrug; FIC index = FICclomiphene + FICantibiotic.
The observation that clomiphene can reverse methicillin resistance in MRSA aided in narrowing its list of candidate cellular targets. In recent years, numerous studies have defined auxiliary factors that play key roles in the expression of methicillin resistance in MRSA (20–22). Among them, a recent chemical-genetic study made use of antisense interference to identify genes that, when partially depleted, restored the susceptibility of MRSA to β-lactams (22). This list provided a starting point for our search for the cellular target of clomiphene.
The identified genes were largely related to cell wall synthesis, the majority participating in PG synthesis, as well as other aspects of cell wall biogenesis (22). Further, based on the observed synergies for clomiphene with fosmidomycin (in B. subtilis) and bacitracin, we reasoned that the staphylococcal enzymes required for mevalonate and Und-P synthesis, encoded by mva, fni, and uppS, were also candidate targets. Using similar antisense technology as described by Tan et al. (22), we assessed whether depletion of the various genes by antisense induction would result in enhanced susceptibility to clomiphene. All genes tested are listed in SI Appendix, Table S2, together with the relative sensitivity observed on their depletion, in the presence of clomiphene (compared with solvent). Depletion of most of the genes involved in late-step PG synthesis led to sensitivity to clomiphene (SI Appendix, Table S2) and was consistent with our observed chemical-chemical interaction data with β-lactams that target PG synthesis. As expected, there was no sensitivity detected on depletion of TarL, a late-step WTA synthesis gene product, for which we would predict an antagonistic interaction with clomiphene. Depletion of genes involved in cell division (and various other genes originally identified as auxiliary factors for methicillin resistance) also remained unaffected on addition of clomiphene (SI Appendix, Table S2). Interestingly, depletion of uppS considerably enhanced the activity of clomiphene (SI Appendix, Table S2), and an analysis of the antisense inducer, xylose, with clomiphene also showed a clear potentiating relationship (SI Appendix, Fig. S3). These data led to the hypothesis that clomiphene targets Und-P synthesis, adversely affecting pathways such as WTA and PG that are dependent on the carrier lipid Und-P. Further, this hypothesis suggested that UppS might be an auxiliary factor involved in methicillin resistance of MRSA.
Clomiphene Inhibits the Synthesis of Undecaprenyl Phosphate.
Und-P biosynthesis involves the initial sequential condensation of dimethylallyl diphosphate (DMAPP) with 2 molecules of isopentenyl diphosphate (IPP) to form farnesyl diphosphate (FPP) which then condenses with 8 molecules of IPP (as catalyzed by a UppS), generating undecaprenyl diphosphate (Und-PP). UppP, a nonessential undecaprenyl diphosphate phosphatase—thought to be the target of the antibiotic bacitracin—then dephosphorylates Und-PP to generate Und-P, which acts as the carrier lipid in the synthesis of PG and WTA polymers. To assess clomiphene’s effect on Und-P synthesis, we first used a phenotype-based assay reliant on chemical supplementation of Und-P. Indeed, metabolite supplementation has been shown to be an effective approach to chart cellular targets (23). To validate the assay, we first tested the outcome of chemically supplementing intermediates of isoprenoids and Und-P pathways in B. subtilis, which enabled the use of the control compound, fosmidomycin. Addition of exogenous IPP, FPP, or Und-P to cells suppressed the activity of fosmidomycin, which inhibits an enzyme upstream of the synthesis of the surveyed metabolites, validating the assay (SI Appendix, Fig. S4A). In S. aureus (Fig. 2A) and B. subtilis (SI Appendix, Fig. S5), addition of exogenous Und-P completely suppressed the activity of clomiphene. Furthermore, the addition of IPP and FPP, whose synthesis is upstream of the hypothesized target of clomiphene, had no effect (SI Appendix, Fig. S6).
Fig. 2.
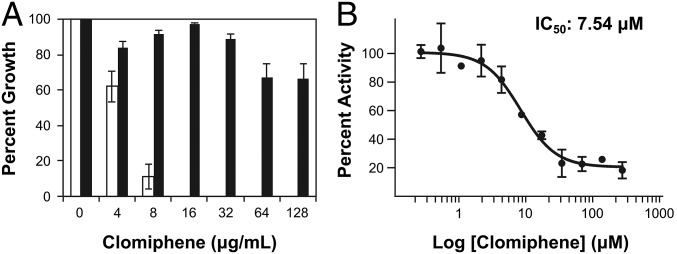
Clomiphene inhibits the synthesis of undecaprenyl phosphate. (A) Addition of exogenous Und-P to clomiphene-treated S. aureus can suppress the inhibitory activity of clomiphene. Shown in white bars is the effect of increasing concentrations of clomiphene on S. aureus in the absence of additional Und-P. Addition of exogenous Und-P in black bars can suppress the growth inhibition almost completely. (B) Dose–response curve of clomiphene against recombinant UppS. The dose–response curve was fitted to the four-parameter logistic nonlinear regression curve yielding an IC50 value of 7.5 μM.
Given its inhibitory effect against S. aureus, we reasoned clomiphene likely targeted the sole essential step in Und-P synthesis, mediated by UppS. We tested this hypothesis using cell-based and in vitro assays. Overexpression of UppS in a hyperpermeable strain of E. coli (MC1061), which was otherwise susceptible to clomiphene, led to a twofold suppression of activity of clomiphene, consistent with UppS being the cellular target (SI Appendix, Fig. S7). In addition, we observed that clomiphene-treated cells had increased levels of staphyloxanthin, the golden pigment produced by S. aureus (SI Appendix, Fig. S8). The starting substrate for the synthesis of staphyloxanthin is FPP, which is transformed by enzymes encoded by crt genes to form this carotenoid (24). We reasoned that inhibition of UppS by clomiphene should lead to an augmented pool of FPP, which could result in increased staphyloxanthin synthesis. Finally, we assayed the inhibitory activity of clomiphene against recombinant S. aureus UppS through a coupled spectrophotometric assay that monitors the production of diphosphate from FPP and IPP. The dose–response curve is displayed in Fig. 2B and shows that clomiphene is an inhibitor of UppS with an IC50 of 7.5 μM (Fig. 2B). With EcUppS, the IC50 was 15 μM using the assay reported earlier (25) (SI Appendix, Fig. S9). Together, these results strongly support the hypothesis that clomiphene acts as an inhibitor of Und-P synthesis, specifically through inhibition of UppS.
UppS-Clomiphene Crystal Structure.
To investigate the mechanism of UppS inhibition by clomiphene, we solved the structure of E. coli UppS (EcUppS) to 2.15-Å resolution in the presence of clomiphene. We turned to the E. coli UppS homolog, as it has been shown to have strong sequence and structural similarities with the S. aureus UppS and higher crystallization success rates than the S. aureus enzyme (26). Although our cocrystal structure is not fully resolved (refined to ∼80% occupancy with the diethylamino group omitted due to disorder) and only clearly bound to one monomer in the asymmetric unit (in keeping with previous UppS/inhibitor complexes; Materials and Methods), density consistent with clomiphene is observed at the hydrophobic center of the protein in the channel that links binding sites 1, 2, and 3 with binding site 4 (Fig. 3A and SI Appendix, Fig. S10 for 2mFo-DFc, simulated annealing mFo-DFc omit map and Phenix feature enhanced map (27). Sites 1–3 are likely involved in diphosphate binding, whereas the channel and site 4 normally accommodate the C55 side-chain of the UPP product (25). The chloride and ring moieties of the bound clomiphene are observed to form strictly hydrophobic interactions with the surrounding apolar residues of the channel. Chlorine substituents increase compound lipophilicity and are commonly observed to form multiple hydrophobic interactions (28). A single hydrogen bond is observed from the phenoxy oxygen of clomiphene to the carboxylate side chain oxygen of Glu96. As mentioned, the terminal diethylamino tail is disordered in the structure, perhaps not surprising given the close proximity of the electropositive cluster of Lys44, Lys48, and Arg51 (Lys44, Lys48, and the general basicity of this region is conserved in S. aureus and B. subtilis) at the perimeter of the binding site, suggesting a possible point of design for optimized clomiphene derivatives.
Fig. 3.

Crystal structure of clomiphene bound to E. coli UppS (PDB ID 5CQJ). (A) Clomiphene (magenta) binds in the hydrophobic channel linking binding sites 1 (above image) and 4 (below image). The gray colored region in the ligand indicates atoms unresolved in the electron density and omitted from refinement. (B) Comparison of clomiphene binding position with substrate and inhibitor. Clomiphene (magenta) binds in the hydrophobic channel linking binding sites 1 and 4. The binding position overlaps with the binding of FPP in sites 1 and 4 (substrate, white, PDB ID code 1V7U) and high affinity bisphosphonate inhibitor in site 4 (yellow, PDB ID code 2E98).
To further validate the clomiphene binding, we used visual inspection and various refinement tests to exclude the presence of density corresponding to other possible components of the crystallization condition and also solved the EcUppS structure in the absence of clomiphene, revealing an empty hydrophobic channel [as observed in a previous apo EcUppS structure, Protein Data Bank (PDB) ID code 3QAS, under slightly different crystallization conditions]. Comparison with the binding of the substrate FPP shows that clomiphene overlaps with the side-chains of substrates bound in sites 1 and 2 (Fig. 3B) and also overlaps with the side-chains of many of the inhibitors reported previously, notably binding to site 4, where occupancy was found to correlate with increased UppS inhibition (Fig. 3).
Effect of Clomiphene on Morphology.
To further characterize the action of clomiphene, we investigated its effect on cell morphology. Following treatment with clomiphene at sublethal concentrations, we observed that B. subtilis changed from normal to greatly enlarged rods, whereas the solvent-treated B. subtilis was unaltered (Fig. 4). Quantitative imaging confirmed this, with clomiphene having a significant effect (P < 0.0001) on cell width and overall swelling of the bacilli (Fig. 4). Interestingly, clomiphene did not lead to a phenotype characteristic of PG inhibition, but rather to the phenotype observed with a tagO null, lacking in WTA polymers, which has been shown to lose its rod shape and increase its cell volume (29). Increased concentrations of clomiphene eventually lead to a lytic phenotype, suggesting a further drop in Und-P levels, which can no longer support the synthesis of PG.
Fig. 4.

Quantitative imaging of clomiphene-treated B. subtilis. Imaging reveals that clomiphene treatment leads to signs of swelling, with clomiphene having an extremely significant effect (P < 0.0001) on cell width and overall swelling of the bacilli compared with solvent-treated swelling.
Shedding Light on the Genetic Complexity Surrounding WTA Synthesis.
The dispensability of the early steps in WTA synthesis clearly indicates that the polymer itself is dispensable for growth, at least in vitro. We have previously speculated that the essential phenotype of the late steps in WTA synthesis comes from a toxic buildup of undecaprenol-linked WTA intermediates or from the sequestration of the Und-P carrier lipid in these intermediates and that the latter would ultimately adversely affect PG assembly (30). Having chemically rescued the cells from the activity of clomiphene, we were prompted to further investigate the “toxic intermediate versus sequestration” question using Und-P as a tool. We first tested the ability of exogenous Und-P to suppress the inhibitory activity of targocil. We reasoned that if supplying Und-P were to reverse targocil’s activity, this would suggest that the cause of the lethality is likely sequestration from PG synthesis, rather than an accumulation of toxic intermediates, because the Und-P pool would be replenished for PG synthesis. Addition of exogenous Und-P rendered targocil inactive and allowed cells to grow similarly to untreated cells (Fig. 5). To corroborate this observation, we ran an analogous experiment where the late-step enzyme TagF could be depleted by controlling its expression from a xylose-inducible promoter. Here, we also observed suppression by Und-P of the lethal phenotype associated with depletion of TagF in the absence of the inducer xylose (SI Appendix, Fig. S11). Overall, these results suggest that sequestration of Und-P from PG synthesis, rather than an accumulation of toxic intermediates, is the likely reason for only blocks in late-acting steps being deleterious to bacterial growth.
Fig. 5.

Addition of exogenous Und-P renders targocil inactive against S. aureus. White bars represent percent growth of S. aureus treated with increasing concentrations of targocil. Addition of Und-P (150 μM), shown in black bars, suppresses the inhibitory activity of targocil.
Discussion
Systematic screens for synergy between double gene KOs (31–33) and small molecules (2, 3) have been successfully used to establish functional connections among genetic modules in microbial systems. To date, the utility of antagonistic interactions as tools to study biological function and uncover modes of action of novel drugs has been largely overlooked. In this study, we used a known probe of late-step WTA synthesis to carry out a screen to identify molecules that antagonize its activity and, ultimately, to chart connections in cell wall biogenesis. We focused on one molecule, clomiphene, a widely used fertility drug with safe toxicology and pharmacological profiles. Follow-up work on clomiphene identified its cellular target as UppS, the synthase responsible for synthesizing undecaprenyl diphosphate, the direct precursor of the lipid carrier, Und-P. Given that Und-P is the key carrier lipid in PG biosynthesis, it was not surprising that treatment with clomiphene had significant effects on cell wall biogenesis.
We hypothesized that the connectivity between WTA and PG syntheses through the synthesis of the common lipid carrier Und-P underlies the observed antagonism between clomiphene and WTA inhibitors and synergy with PG inhibitors. Although the pathways for the biosynthesis of PG and WTA polymers have been thoroughly investigated, the connection between them remains speculative. Early work on the relationship between the biosynthesis of these two wall polymers has suggested that the lipid monophosphate carrier is shared between the two pathways (34) and is generally assumed to be available in small amounts (35). More recently, our group reported on the effect of depleting the late-step gene tagF on PG synthesis in B. subtilis, observing a reduction in PG synthesis, thus suggesting the lipid intermediate is shared (36). The suppressive interaction of clomiphene and targocil and that of ticlopidine toward clomiphene is consistent with these functional connections in cell wall biogenesis. Put simply: inhibition of UppS by clomiphene lowers the pool of Und-P available for WTA synthesis such that fewer WTA polymers are synthesized, thereby rendering targocil less effective. Conversely, addition of ticlopidine may abolish the flux into WTA synthesis such that the pool of Und-P is more readily available to participate in the essential synthesis of PG, thus rendering clomiphene less effective against S. aureus. Indeed, the activity of clomiphene, which ultimately leads to a decreased pool of Und-P through its inhibition of UppS can be reversed in the presence of ticlopidine, because the latter abolishes WTA synthesis and renders the pool of Und-P available primarily for PG synthesis. Moreover, the ability of clomiphene to potentiate β-lactam antibiotics may reflect a serial inhibition of targets, such that inhibition of UppS lowers the availability of Und-P and thus lipid II, which is used as a substrate in the transglycosylation and transpeptidation reaction of PBP’s. Thus, the latter enzymes may become more susceptible to the inactivating action of β-lactam antibiotics. These interactions suggest, therefore, that clomiphene may be a promising lead for treating staphylococcal infections, both in combination therapy by restoring drug sensitivity in drug-resistant strains or as a monotherapy, through the inhibition of Und-P biosynthesis.
The biosynthesis of Und-P has been of considerable interest as an avenue for antimicrobial development. Indeed, UppS represents an attractive target due to its essential role in the biosynthesis of the different cell wall elements. To date, there have been no validated inhibitors of UppS but there are some interesting leads (25, 37). For instance, various bisphosphonate compounds that mimic diphosphate substrates have been found to inhibit UppS in vitro via competition with substrate for enzyme binding (25, 38). Bisphosphonates have, however, been shown to be susceptible to rapid removal from the circulatory system by binding to bone mineral (39). Other common inhibitors of UppS, such as tetramic acids, are metal chelators and have cytotoxic properties (40). Recently, diamidines have been found to have potent activity both in vitro and in vivo (25). Efforts are ongoing to identify novel nontoxic inhibitors of UppS (25, 41). In this respect, clomiphene represents a potentially important lead given it is largely devoid of side effects when used in humans (19).
Our screening approach revealed how antagonism can be a powerful tool to understand biological function where we explored functional connections among UppS and early- and late-step gene products of WTA synthesis, as well as PBPs, the cellular targets of β-lactams. Our study also reinforced the power of drug interactions to uncover the mode of action of uncharacterized drugs, based on their patterns of interactions with common antibiotics (4). Our pathway-specific screening platform allowed for hypothesis-driven discovery, enabling mechanistic follow-ups and eliminating nuisance compounds. Overall, exploiting pathway-specific synthetic viability through an antagonism approach, as reported herein, provides a unique conceptual framework for charting novel cellular connections while identifying novel inhibitors of bacterial physiology.
Materials and Methods
Details for the antagonism screen with targocil can be found in SI Appendix, SI Materials and Methods. All phenotypic assays to investigate clomiphene’s mode of action are detailed in SI Appendix, SI Materials and Methods. The in vitro UppS assay was carried out as previously described (42), with more details in SI Appendix, SI Materials and Methods. Detailed methods for structure determination and refinement can be found in SI Appendix, SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank T. Roemer (Merck) for providing the antisense S. aureus strains and M. Nosella for assistance with UppS isolation. This work was supported by the Canadian Institutes of Health Research (MOP-81330 and Canada-UK Partnership on Antibiotic Resistance Award 114045), by Canada Research Chair awards (to E.D.B. and N.C.J.S.), and in part by the US Public Health Service (National Institutes of Health Grant CA 158191 to E.O.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 5CQB and 5CQJ).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1511751112/-/DCSupplemental.
References
- 1.Falconer SB, Czarny TL, Brown ED. Antibiotics as probes of biological complexity. Nat Chem Biol. 2011;7(7):415–423. doi: 10.1038/nchembio.590. [DOI] [PubMed] [Google Scholar]
- 2.Farha MA, et al. Inhibition of WTA synthesis blocks the cooperative action of PBPs and sensitizes MRSA to β-lactams. ACS Chem Biol. 2013;8(1):226–233. doi: 10.1021/cb300413m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lehár J, et al. Chemical combination effects predict connectivity in biological systems. Mol Syst Biol. 2007;3:80. doi: 10.1038/msb4100116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Farha MA, Brown ED. Chemical probes of Escherichia coli uncovered through chemical-chemical interaction profiling with compounds of known biological activity. Chem Biol. 2010;17(8):852–862. doi: 10.1016/j.chembiol.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 5.Yeh P, Tschumi AI, Kishony R. Functional classification of drugs by properties of their pairwise interactions. Nat Genet. 2006;38(4):489–494. doi: 10.1038/ng1755. [DOI] [PubMed] [Google Scholar]
- 6.Cokol M, et al. Systematic exploration of synergistic drug pairs. Mol Syst Biol. 2011;7:544. doi: 10.1038/msb.2011.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yeh PJ, Hegreness MJ, Aiden AP, Kishony R. Drug interactions and the evolution of antibiotic resistance. Nat Rev Microbiol. 2009;7(6):460–466. doi: 10.1038/nrmicro2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keith CT, Borisy AA, Stockwell BR. Multicomponent therapeutics for networked systems. Nat Rev Drug Discov. 2005;4(1):71–78. doi: 10.1038/nrd1609. [DOI] [PubMed] [Google Scholar]
- 9.Bollenbach T, Quan S, Chait R, Kishony R. Nonoptimal microbial response to antibiotics underlies suppressive drug interactions. Cell. 2009;139(4):707–718. doi: 10.1016/j.cell.2009.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cokol M, et al. Large-scale identification and analysis of suppressive drug interactions. Chem Biol. 2014;21(4):541–551. doi: 10.1016/j.chembiol.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chait R, Craney A, Kishony R. Antibiotic interactions that select against resistance. Nature. 2007;446(7136):668–671. doi: 10.1038/nature05685. [DOI] [PubMed] [Google Scholar]
- 12.Michel JB, Yeh PJ, Chait R, Moellering RC, Jr, Kishony R. Drug interactions modulate the potential for evolution of resistance. Proc Natl Acad Sci USA. 2008;105(39):14918–14923. doi: 10.1073/pnas.0800944105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pasquina LW, Santa Maria JP, Walker S. Teichoic acid biosynthesis as an antibiotic target. Curr Opin Microbiol. 2013;16(5):531–537. doi: 10.1016/j.mib.2013.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sewell EW, Brown ED. Taking aim at wall teichoic acid synthesis: New biology and new leads for antibiotics. J Antibiot (Tokyo) 2014;67(1):43–51. doi: 10.1038/ja.2013.100. [DOI] [PubMed] [Google Scholar]
- 15.D’Elia MA, et al. Lesions in teichoic acid biosynthesis in Staphylococcus aureus lead to a lethal gain of function in the otherwise dispensable pathway. J Bacteriol. 2006;188(12):4183–4189. doi: 10.1128/JB.00197-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Campbell J, et al. Synthetic lethal compound combinations reveal a fundamental connection between wall teichoic acid and peptidoglycan biosyntheses in Staphylococcus aureus. ACS Chem Biol. 2011;6(1):106–116. doi: 10.1021/cb100269f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Swoboda JG, et al. Discovery of a small molecule that blocks wall teichoic acid biosynthesis in Staphylococcus aureus. ACS Chem Biol. 2009;4(10):875–883. doi: 10.1021/cb900151k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anonymous; Practice Committee of the American Society for Reproductive Medicine Use of clomiphene citrate in infertile women: A committee opinion. Fertil Steril. 2013;100(2):341–348. doi: 10.1016/j.fertnstert.2013.05.033. [DOI] [PubMed] [Google Scholar]
- 19.Homburg R. Clomiphene citrate: End of an era? A mini-review. Hum Reprod. 2005;20(8):2043–2051. doi: 10.1093/humrep/dei042. [DOI] [PubMed] [Google Scholar]
- 20.Berger-Bächi B, Rohrer S. Factors influencing methicillin resistance in staphylococci. Arch Microbiol. 2002;178(3):165–171. doi: 10.1007/s00203-002-0436-0. [DOI] [PubMed] [Google Scholar]
- 21.de Lencastre H, Tomasz A. Reassessment of the number of auxiliary genes essential for expression of high-level methicillin resistance in Staphylococcus aureus. Antimicrob Agents Chemother. 1994;38(11):2590–2598. doi: 10.1128/aac.38.11.2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tan CM, et al. Restoring methicillin-resistant Staphylococcus aureus susceptibility to β-lactam antibiotics. Sci Transl Med. 2012;4(126):126ra35. doi: 10.1126/scitranslmed.3003592. [DOI] [PubMed] [Google Scholar]
- 23.Zlitni S, Ferruccio LF, Brown ED. Metabolic suppression identifies new antibacterial inhibitors under nutrient limitation. Nat Chem Biol. 2013;9(12):796–804. doi: 10.1038/nchembio.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pelz A, et al. Structure and biosynthesis of staphyloxanthin from Staphylococcus aureus. J Biol Chem. 2005;280(37):32493–32498. doi: 10.1074/jbc.M505070200. [DOI] [PubMed] [Google Scholar]
- 25.Zhu W, et al. Antibacterial drug leads targeting isoprenoid biosynthesis. Proc Natl Acad Sci USA. 2013;110(1):123–128. doi: 10.1073/pnas.1219899110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu W, et al. Antibacterial drug leads: DNA and enzyme multitargeting. J Med Chem. 2015;58(3):1215–1227. doi: 10.1021/jm501449u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adams PD, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bissantz C, Kuhn B, Stahl M. A medicinal chemist’s guide to molecular interactions. J Med Chem. 2010;53(14):5061–5084. doi: 10.1021/jm100112j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.D’Elia MA, Millar KE, Beveridge TJ, Brown ED. Wall teichoic acid polymers are dispensable for cell viability in Bacillus subtilis. J Bacteriol. 2006;188(23):8313–8316. doi: 10.1128/JB.01336-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Watkinson RJ, Hussey H, Baddiley J. Shared lipid phosphate carrier in the biosynthesis of teichoic acid and peptidoglycan. Nat New Biol. 1971;229(2):57–59. doi: 10.1038/newbio229057a0. [DOI] [PubMed] [Google Scholar]
- 31.Babu M, et al. Genetic interaction maps in Escherichia coli reveal functional crosstalk among cell envelope biogenesis pathways. PLoS Genet. 2011;7(11):e1002377. doi: 10.1371/journal.pgen.1002377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lehner B, Crombie C, Tischler J, Fortunato A, Fraser AG. Systematic mapping of genetic interactions in Caenorhabditis elegans identifies common modifiers of diverse signaling pathways. Nat Genet. 2006;38(8):896–903. doi: 10.1038/ng1844. [DOI] [PubMed] [Google Scholar]
- 33.Tong AH, et al. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science. 2001;294(5550):2364–2368. doi: 10.1126/science.1065810. [DOI] [PubMed] [Google Scholar]
- 34.Anderson RG, Hussey H, Baddiley J. The mechanism of wall synthesis in bacteria. The organization of enzymes and isoprenoid phosphates in the membrane. Biochem J. 1972;127(1):11–25. doi: 10.1042/bj1270011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mengin-Lecreulx D, Texier L, Rousseau M, van Heijenoort J. The murG gene of Escherichia coli codes for the UDP-N-acetylglucosamine: N-acetylmuramyl-(pentapeptide) pyrophosphoryl-undecaprenol N-acetylglucosamine transferase involved in the membrane steps of peptidoglycan synthesis. J Bacteriol. 1991;173(15):4625–4636. doi: 10.1128/jb.173.15.4625-4636.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.D’Elia MA, et al. Probing teichoic acid genetics with bioactive molecules reveals new interactions among diverse processes in bacterial cell wall biogenesis. Chem Biol. 2009;16(5):548–556. doi: 10.1016/j.chembiol.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 37.Silver LL. Viable screening targets related to the bacterial cell wall. Ann N Y Acad Sci. 2013;1277:29–53. doi: 10.1111/nyas.12006. [DOI] [PubMed] [Google Scholar]
- 38.Guo RT, et al. Bisphosphonates target multiple sites in both cis- and trans-prenyltransferases. Proc Natl Acad Sci USA. 2007;104(24):10022–10027. doi: 10.1073/pnas.0702254104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jahnke W, et al. Allosteric non-bisphosphonate FPPS inhibitors identified by fragment-based discovery. Nat Chem Biol. 2010;6(9):660–666. doi: 10.1038/nchembio.421. [DOI] [PubMed] [Google Scholar]
- 40.Athanasellis G, Igglessi-Markopoulou O, Markopoulos J. Tetramic and tetronic acids as scaffolds in bioinorganic and bioorganic chemistry [published online ahead of print May 25, 2010] Bioinorg Chem Appl. 2010 doi: 10.1155/2010/315056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Durrant JD, et al. Non-bisphosphonate inhibitors of isoprenoid biosynthesis identified via computer-aided drug design. Chem Biol Drug Des. 2011;78(3):323–332. doi: 10.1111/j.1747-0285.2011.01164.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Webb MR. A continuous spectrophotometric assay for inorganic phosphate and for measuring phosphate release kinetics in biological systems. Proc Natl Acad Sci USA. 1992;89(11):4884–4887. doi: 10.1073/pnas.89.11.4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.