

Significance
A persistent memory T-cell population is the basis for successful T-cell–based vaccine against pathogens. Numerous extracellular and intracellular molecules have been demonstrated to play critical roles in the differentiation of memory T cells; however, the mechanisms that control the long-term maintenance of memory T cells remain incompletely understood. Here we show that continuous transforming growth factor-β signaling is required to maintain the identity of memory T cells.
Keywords: TGF-β, memory T cell, acute infection, CD8+
Abstract
The long-term maintenance of memory T cells is essential for successful vaccines. Both the quantity and the quality of the memory T-cell population must be maintained. The signals that control the maintenance of memory T cells remain incompletely identified. Here we used two genetic models to show that continuous transforming growth factor-β signaling to antigen-specific T cells is required for the differentiation and maintenance of memory CD8+ T cells. In addition, both infection-induced and microbiota-induced inflammation impact the phenotypic and functional identity of memory CD8+ T cells.
Infectious diseases pose a significant public health burden, accounting for nearly one-fifth of annual deaths worldwide. Vaccines remain the most effective way to prevent infectious diseases. Functionally sustained memory T cells are the ideal cell population to be generated by T-cell–based vaccines. Considerable efforts have been made to elucidate the mechanisms that mediate the establishment of long-lived immunologic memory (1–6); however, the signals that control the differentiation and maintenance of memory T cells remain incompletely identified.
During the early stages of an immune response, proinflammatory cytokines IL-12 and type I IFN promote the expansion of effector CD8+ T cells by sustaining the expression of the high-affinity IL-2 receptor CD25 (7, 8). In addition to its role in T-cell proliferation, IL-2 also functions as a differentiation factor for effector CD8+ T cells by promoting the differentiation of short-lived effector cells [SLECs; IL-7Rα−KLRG1+ (killer cell lectin-like receptor subfamily G, member 1)] and inhibiting the differentiation of memory precursor effector cells (MPECs; IL-7Rα+KLRG1−) (9–12). Furthermore, IL-10 and IL-21 signals promote MPEC differentiation through a STAT3-dependent mechanism (13, 14). During the late stages of an immune response, IL-15 and IL-7 are required to maintain the population of memory CD8+ T cells (15, 16); however, after the clearance of an infection, whether memory CD8+ T cells require any additional signals to maintain their phenotypic and functional identity remains unknown.
Recent findings have revealed that effector and memory CD8+ T cells display nearly endless diversity based on the expression of surface and intracellular molecules that serve as the markers of antigen-experienced T cells (17). Thus, it is conceivable that memory CD8+ T cells might not be a fixed cell lineage, but instead represent an active differentiation state. Even in the absence of cognate antigens, memory CD8+ T cells may constantly receive diverse environmental signals; however, how memory T cells maintain their relatively stable characters under such circumstances remains unexplored.
Here we show that TGF-β signaling to CD8+ T cells controls the differentiation of memory T cells at both early and late stages. By deleting TGF-β receptor in antigen-specific T cells at different time points following an acute infection, we demonstrate that during the effector phase of an immune response, TGF-β restrains the inflammatory signals associated with the infection. At the memory phase, both the TGF-β signal and the basal inflammation induced by microbiota cooperate to shape the memory T-cell population. Taken together, our findings show that continuous TGF-β signaling is required to maintain the identity of memory CD8+ T cells following acute infections.
Results
Aberrant Effector and Memory T-Cell Differentiation in the Absence of TGF-β Signaling.
Previous findings have suggested that TGF-β inhibits SLEC differentiation and promotes MPEC differentiation during CD8+ effector and memory T-cell development (18–20). Paradoxically, a recent study has demonstrated that TGF-β inhibits the differentiation of central memory T cells (21), which are derived from MPECs.
To reconcile this discrepancy and to study the T-cell intrinsic function of TGF-β signaling in antigen-specific T cells, we generated Tgfbr2f/fdLck-cre (hereinafter referred to as Tgfbr2−/−) OT-1 TCR transgenic mice (22), which carry CD8+ T cells specific for the Kb-Ova257–264 epitope. Using these T cells and the experimental setup described in Fig. S1A, we observed that after acute bacterial infection, OT-1 T-cell expansion was slightly (less than twofold) but consistently decreased in the absence of TGF-β signaling. The number of memory T cells was comparable in control and Tgfbr2−/− OT-1 populations (Fig. S1B). Consistent with previous findings, Tgfbr2−/− cells contained a significantly higher percentage of KLRG1+ cells and a lower percentage of IL-7Rα+KLRG1− cells (Fig. S1C). Thus, in the absence of TGF-β signaling, although a comparable number of memory T cells was generated, the composition of memory T cells was significantly altered.
Fig. S1.

Aberrant effector and memory T-cell differentiation in the absence of TGF-β signaling. (A) Schematic of the experiments. Naïve OT-1 T cells were purified from congenically marked control and Tgfbr2−/− mice and mixed at a 1:1 ratio. Then 104 mixed OT-1 cells were transferred into each B6 recipient mouse, followed by infection with 2,000 cfu of LM-ova administered i.v. on day 2. OT-1 T-cell response in the peripheral blood and spleen was measured by flow cytometry at different time points. (B) Percentages of control and Tgfbr2−/− OT-1 T cells in total CD8+ T cells. (C) Percentages of KLRG1+ (Left) and IL-7Rα+KLRG1− (Right) populations within OT-1 T cells. Combined results from five or six independent experiments are shown in B and C. **P < 0.01, Student t test.
TGF-β Signaling at the Early Stages of Effector T-Cell Differentiation.
To elucidate the mechanisms underlying TGF-β–dependent effector and memory T-cell differentiation, we aimed to determine when T cells received TGF-β signaling during an immune response. To this end, we first examined the role of TGF-β during the early stages of effector T-cell differentiation. At 2-1/2 d postinfection, Tgfbr2−/− OT-1 cells exhibited comparable levels of T-cell activation as control cells (Fig. S2A); however, at 3-1/2 d postinfection, when effector T cells began to up-regulate the effector T-cell marker KLRG1, a significantly higher percentage of Tgfbr2−/− OT-1 T cells expressed KLRG1 compared with cotransferred control OT-1 T cells (Fig. S2 B and C).
Fig. S2.

TGF-β controls the early differentiation of effector T cells. The experimental setup was similar to that shown in Fig. S1, except that 4 × 105 mixed OT-1 T cells were transferred into each B6 recipient mouse. (A and B) At 2.5 d (A) and 3.5 d (B and C) after infection, OT-1 T cells in the spleen were examined by flow cytometry. Representative FACS profiles pregated on donor OT-1 T cells and host-derived endogenous CD8+ T cells are shown in A (n = 5). Representative FACS profiles pregated on donor OT-1 T are shown in B (n = 5). (C) Percentage of KLRG1+ cells in OT-1 T cells. Each symbol represents data from an individual recipient mouse. **P < 0.01, Student t test. Representative data from two independent experiments are shown.
TGF-β is generally considered an anti-inflammatory cytokine for CD8+ T cells. If the major role of TGF-β signaling during acute infections is to restrain infection-induced inflammatory signals, then limiting infection would largely rescue the defects in Tgfbr2−/− T cells. To test this hypothesis, at 1 d after bacterial inoculation, we administered ampicillin to limit bacterial infection (Fig. S3A). We found that ampicillin treatment indeed significantly enhanced MPEC differentiation and suppressed SLEC differentiation at 7 d after infection (Fig. S3 B and C, Upper). Surprisingly, however, the effects of ampicillin treatment were not long-lasting; shortly after the contraction phase, we observed no significant difference between ampicillin-treated and untreated Tgfbr2−/− OT-1 T cells (Fig. S3 B and C, Lower), suggesting that TGF-β might antagonize non–infection-associated signals at late stages of memory T-cell differentiation and maintenance.
Fig. S3.

Limiting infection partially rescues the defects in Tgfbr2−/− T cells at early stages. (A) Schematic of the experiments. The experimental setup was similar to that shown in Fig. S1. At 1 d after LM-ova infection, 2 mg of ampicillin or saline control was injected i.p. into each recipient mouse. OT-1 T cells in the peripheral blood were examined at the indicated times. (B and C) Percentages of MPECs (B) and SLECs (C) in Tgfbr2−/− OT-1 T cells. Each symbol in B and C represents data from an individual recipient mouse. P values were calculated using the Student t test. Combined results from two independent experiments are shown.
Taken together, our findings indicate that TGF-β inhibits infection-associated inflammatory signals at early stages of effector T-cell differentiation, and continues to suppress other non–infection-associated signals after the infection is cleared.
TGF-β Signaling at the Late Stages of Memory T-Cell Differentiation.
To specifically determine the role of TGF-β during late stages of memory T-cell differentiation, we used a mouse line carrying a Cre-Estrogen Receptor T2 (ER-cre) allele targeted to the ubiquitously expressed ROSA26 locus (23) to temporally control the expression of TGF-β receptor II (TGF-βRII). As shown in Fig. 1A, congenically marked naïve OT-1 T cells were purified from Tgfbr2f/fER-cre and control mice, and adoptively transferred into B6 mice, followed by infection with Listeria monocytogenes-expressing chicken ovalbumin (LM-ova). Tamoxifen (TAM) or solvent alone was administered every other day for five treatments starting at day 30 postinfection. At 3–4 wk after the initial TAM injection, TGF-βRII expression was significantly decreased on Tgfbr2f/fER-cre OT-1 T cells compared with that of cotransferred control cells (Fig. 1B). Indeed, the deletion efficiency mediated by ER-cre was comparable to that seen in dLck-cre mice (Fig. 1B). Inducible deletion of TGF-βRII did not lead to a significant alteration of memory T-cell number (Fig. 2). Solvent alone did not induce any detectable deletion of TGF-βRII or phenotypic changes in Tgfbr2f/fER-cre cells (Fig. 3A).
Fig. 1.

Delayed deletion of the TGF-β receptor alters memory T-cell population. (A) Schematic of the experiments. Naïve OT-1 T cells were purified from congenically marked control and Tgfbr2f/fER-cre mice and mixed at a 1:1 ratio. Then 104 mixed OT-1 cells were transferred into each B6 recipient mouse, followed by infection with 2,000 cfu of LM-ova administered i.v. on the second day. Starting at day 30 postinfection, 1 mg TAM or solvent alone was injected i.p. every other day for a total of five treatments. OT-1 T cells in the spleen were examined at the indicated times. (B) The mean fluorescence intensity (MFI) of TGF-βRII on OT-1 T cells was determined by flow cytometry. (C) Percentages of MPEC-like (Left), SLEC-like (Middle), and IL-2–producing cells (Right) within OT-1 T cells. Combined results from two independent experiments are shown. Each symbol in B and C represents data from an individual recipient mouse. **P < 0.01; *P < 0.05; NS, not significant, Student t test.
Fig. 2.
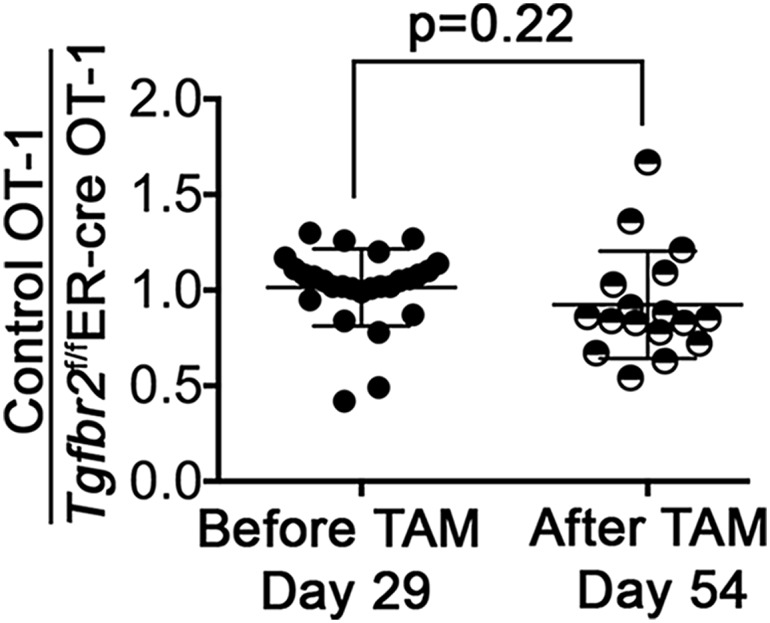
Delayed abrogation of TGF-βRII does not affect the number of memory T cells. The experimental setup was similar to that shown in Fig. 1. The ratios of control OT-1 cells to Tgfbr2f/fER-cre OT-1 cells before (day 29 postinfection, peripheral blood) and after (day 54 postinfection, spleen) TAM treatment are shown. Each symbol represents data from an individual recipient mouse. P values were calculated by the Student t test.
Fig. 3.

Delayed deletion of the TGF-β receptor has a broad impact on memory T cells. The experimental setup was similar to that shown in Fig. 1. OT-1 T cells in the spleen were examined at day 58 postinfection. (A) Representative FACS profiles pregated on OT-1 T cells (n = 5–10). (B) MFIs of CD122, Granzyme B, Eomes, T-bet, Bcl-6, and Foxo1. Each symbol in B represents data from an individual recipient mouse. **P < 0.01; NS, not significant, Student t test. Representative data from two independent experiments are shown.
Importantly, delayed deletion of TGF-βRII after the formation of memory T cells led to dramatic changes in the phenotype and function of splenic CD8+ memory T cells. The percentages of “MPEC-like” cells and IL-2–producing cells were significantly decreased, whereas the percentage of “SLEC-like” cells was significantly increased, in Tgfbr2f/fER-cre OT-1 T cells after TAM treatment (Fig. 1C). Consistent with our findings that TGF-β controls the early differentiation of effector T cells (Figs. S2 and S3), the phenotype of TAM-treated Tgfbr2f/fER-cre cells was significantly different from that of Tgfbr2f/fdLck-cre cells (Fig. 1C), confirming that TGF-β controls the differentiation of memory CD8+ T cells at both early and late stages of an immune response.
Because TGF-β directly inhibits the expression of KLRG1 in CD8+ T cells (24), there are two possible explanations for the observation shown in Fig. 1C. First, TGF-β may only control the expression of KLRG1 and some associated molecules. Second, delayed deletion of the TGF-β receptor has a broad impact on memory T cells. To distinguish these two possibilities and to further characterize the effects of delayed deletion of TGF-β receptor on memory T cells, we measured the expression of a panel of memory T-cell markers. The expression of CD62L, CXCR3, CD27, and CD122 was substantially decreased in Tgfbr2f/fER-cre cells after TAM treatment (Fig. 3 A and B), whereas the expression of effector molecule granzyme B was significantly enhanced after delayed TGF-β receptor ablation (Fig. 3B). Furthermore, the expression of memory T-cell–associated transcription factors Eomes (25), Bcl-6 (13), and Foxo1 (26, 27) was significantly reduced, and the expression of effector T-cell–associated transcription factor T-bet (28, 29) was enhanced in Tgfbr2f/fER-cre memory T cells after TAM treatment (Fig. 3B).
Taken together, our findings indicate that at the memory phase of an immune response, continuous TGF-β signaling to T cells is required to maintain the transcription program of memory T cells. Even after pathogen clearance and in the absence of any detectable infections, TGF-β signaling is constantly shaping the population of memory T cells.
Microbiota-Induced Basal Inflammation Impacts the Composition of Memory T Cells.
One potential signal constantly delivered to memory T cells is the basal inflammatory signal induced by microbiota. To examine this possibility, we administered a mixture of antibiotics during TAM treatment at the memory phase following acute infection (Fig. S4A) in an attempt to limit basal inflammation (Fig. S4B). Indeed, antibiotic treatment significantly altered the phenotype of both control and Tgfbr2f/fER-cre memory T cells. Significantly more MPEC-like cells and IL-2–producing cells and fewer SLEC-like cells were observed for both control and Tgfbr2f/fER-cre cells (Fig. S4C).
Fig. S4.

Basal inflammation induced by microbiota impacts the composition of memory T cells. (A) Schematic of the experiments. The experimental setup was similar to that shown in Fig. 1. During TAM treatment, a mixture of antibiotics (1 mg/mL ampicillin + 2 mg/mL neomycin + 100 U/mL polymyxin B) was administered in the drinking water, and 2.5 mg of vancomycin was given via i.p. injection on a daily basis. (B) Percentage of inflammatory monocytes (defined as CD11b+Ly6C+CD11cintSSClo) in the spleen. OT-1 T cells in the spleen were examined on day 54 postinfection. (C) Percentages of MPEC-like (Left), SLEC-like (Middle), and IL-2–producing (Right) cells. Each symbol in B and C represents data from an individual recipient mouse. **P < 0.01; *P < 0.05; NS, not significant, Student t test.
Interestingly, antibiotic treatment specifically enhanced the expression of Bcl-2 in KLRG1− memory T cells (Fig. S5). However, even after antibiotic treatment, there were significant differences between control and TGF-β–unresponsive memory T cells (Figs. S4C and S5). Thus, microbiota-induced basal inflammation affects the memory T-cell population, but might not be the only signal restrained by a continuous TGF-β signal.
Fig. S5.

Defective expression of Bcl-2 in the absence of TGF-β signaling. The experimental setup was similar to that shown in Fig. S4. After TAM treatment, in the presence or absence of antibiotics, the expression of Bcl-2 in KLRG1+ (K+) and KLRG1− (K−) OT-1 T cells was measured by intracellular flow cytometry staining. Each pair of symbols represents data from an individual recipient mouse. **P < 0.01, paired Student t test; *P < 0.05, unpaired Student t test.
TGF-β Inhibits the Proliferation of KLRG1+ Memory T Cells.
Previous studies have focused on the Bim-mediated apoptosis of KLRG1+ effector T cells in response to TGF-β signaling (19, 20). However, we found that significantly reduced Bcl-2 expression in TGF-β–unresponsive memory T cells, especially for KLRG1+ cells (Fig. S5). In addition, significantly increased Tgfbr2−/− KLRG1+ T cells expressed cell cycle-associated protein Ki67 at the memory stage of the immune response (Fig. 4 A and B). A similar effect of enhanced Ki67 expression was observed in KLRG1+ Tgfbr2f/fER-cre cells after TAM treatment (Fig. 4C). Although less dramatic, significantly more KLRG1+ Tgfbr2−/− T cells incorporated BrdU at the memory stage (Fig. 4 D and E). Collectively, our findings confirm that continuous TGF-β signaling controls the homeostasis of KLRG1+ T cells by regulating both proliferation and survival.
Fig. 4.

TGF-β inhibits KLRG1+ T-cell proliferation. The experimental setup was similar to that shown in Fig. S1 (for A, B, D, and E) or in Fig. 1 (for C). OT-1 T cells in the spleen were examined at day 27 (A and B), day 54, (C), and day 41 (D and E) postinfection. Representative FACS profiles pregated on OT-1 T cells are shown in A and D. The percentages of Ki67+ cells within KLRG1− and KLRG1+ OT-1 T-cell populations are shown in B and C. The percentages of BrdU+ cells within KLRG1− and KLRG1+ OT-1 T-cell populations are shown in E. Each pair of symbols in B, C, and E represents data from an individual recipient mouse. **P < 0.01; *P < 0.05; NS, not significant, paired Student t test.
Defective Recall Response in the Absence of TGF-β Signaling.
Even though Tgfbr2−/− memory T cells exhibited significant phenotypic changes, the number of memory T cells was not significantly altered (Fig. S1 and Fig. 5B). To test the functional consequence of TGF-β–dependent differentiation and shaping of memory T cells, we heterogeneously challenged host mice harboring both control and Tgfbr2−/− memory T cells with a different pathogen carrying the same antigen, vesicular stomatitis virus-expressing chicken ovalbumin (VSV-ova), for OT-1 T cells (Fig. 5A), then measured the recall response 4 d later. In the absence of TGF-β signaling, the reexpansion of memory T cells was almost completely abolished (Fig. 5B). Furthermore, in a less competitive setting, after adoptive transfer of memory T cells, the recall response of Tgfbr2−/− memory T cells was substantially reduced (Fig. S6). Thus, TGF-β signaling to T cells is required to maintain the phenotypic and functional identity of memory T cells.
Fig. 5.

Defective recall response in Tgfbr2−/− memory T cells. (A) Schematic of the experiments. The experimental setup was similar to that shown in Fig. S1. At 143 d after LM-ova infection, recipient mice were rechallenged with 2 × 106 pfu of VSV-ova via i.v. injection. OT-1 T cells were examined in the peripheral blood on day 142 and in the spleen on day 147. (B) Percentage of OT-1 T cells in total CD8+ T cells. Each symbol in B represents data from an individual recipient mouse. **P < 0.01; NS, not significant, Student t test. Combined results from two independent experiments are shown.
Fig. S6.

Defective recall response in the absence of TGF-β signaling. (A) Schematic of the experiments. Similar to the experiment shown in Fig. S1, at 237 d after LM-ova infection, control and Tgfbr2−/− memory OT-1 T cells were FACS-sorted and mixed at a 1:1 ratio. (B) Purity of sorted memory OT-1 T cells. Here 5 × 103 mixed memory OT-1 T cells were adoptively transferred into each naïve B6 recipient, followed by LM-ova infection. At 6 d later, lymphocytes were isolated from the spleen and the liver, and the recall response was determined by flow cytometry. (C) Representative FACS profiles. (D) The ratio of control to Tgfbr2−/− OT-1 T cells. Each symbol in D represents data from an individual recipient.
Discussion
In this study, using two genetic models including both constitutive and inducible deletion of the TGF-β receptor on antigen-specific CD8+ T cells, we have shown that T-cell intrinsic TGF-β signaling is required at both early and late stages of the immune response to shape effector and memory T-cell populations.
During acute infections, on encountering a specific antigen, naïve T cells undergo massive expansion followed by contraction. The cells that survive contraction are generally considered memory T cells. Various proinflammatory and antiinflammatory cytokines have been demonstrated to regulate effector and memory T-cell differentiation through the control of effector T-cell expansion or MPEC/SLEC differentiation. Among these cytokines, TGF-β has been found to limit the differentiation of SLECs (18, 19). It is generally assumed that TGF-β controls the differentiation of MPECs and SLECs by restraining infection-induced inflammatory signals. We have shown that TGF-β–unresponsive CD8+ T cells exhibit significant defects as early as 3.5 d after Listeria infection in vivo (Fig. S2). The only cytokines found to function during the maintenance phase of memory T cells are homeostatic cytokines IL-15 and IL-7. After clearance of the infection, under homeostatic conditions, whether memory T cells require other signals to maintain their identity remains unknown.
Here we have shown that even after the formation of memory T cells and weeks after pathogen clearance, deletion of the TGF-β receptor leads to dramatic phenotypic and functional alterations of memory CD8+ T cells (Fig. 1). A constant TGF-β signal is required to maintain the expression levels of several essential transcription factors that mediate the differentiation of memory T cells (Fig. 3). In addition, micriobiota-induced basal inflammation affects the composition of memory T cells (Fig. S4). Our results also suggest that memory CD8+ T cells may represent an active differentiation state that constantly incorporates various environmental cues.
During early stages of LM-ova infection, ampicillin treatment efficiently limits infection-associated inflammation. The same treatment also reduces the duration of pathogen specific antigen presentation (30). The potential interaction between TGF-β signaling and the duration of antigen display awaits future investigation.
We consistently observed slightly (less than twofold) but significantly decreased accumulation of Tgfbr2−/− OT-1 T cells at the peak of the immune response (Fig. S1). This defect was not OT-1 or Listeria infection-specific, given that we also observed a similar defect in Tgfbr2−/− P14 T cells and a lymphocytic choriomeningitis virus infection model (31). In contrast, a recent study using Tgfbr2f/fCD4-cre OT-1 T cells and LM-ova infection found comparable expansion of OT-1 T cells in the presence and absence of TGF-β signaling (18). These discrepant findings may be related to differences in experimental design.
In our experiments, to mimic the low frequency of antigen-specific T cells in a naïve animal, we adoptively transferred 104 OT-1 T cells (∼1,000–2,000 cells/mouse considering a 10–20% intake rate) into each recipient mouse. In the previous study using Tgfbr2f/fCD4-cre OT-1 T cells, 105 OT-1 T cells were transferred into each recipient (18). When we increased the number of OT-1 T cells transferred into each mouse, the slight expansion defect in Tgfbr2−/− OT-1 T cells disappeared. The mechanisms underlying this defect await further investigation.
In contrast to our observation that Tgfbr2−/− memory T cells exhibit defective recall response in a highly competitive setting (Fig. 5), a recent study found apparently normal recall responses for memory CD8+ T cells in the absence of TGF-β signaling (18). These discrepant findings may be related to differences in experimental design and animal models. Indeed, when a small number of Tgfbr2−/− memory T cells were FACS-sorted and transferred into a naïve animal followed by reinfection, a less dramatic defect in recall response was observed (Fig. S6). However, compared with a naïve host, an immunized host with competitive environment is physiologically more relevant for vaccination in humans.
TGF-β has been suggested to directly induce apoptosis in SLECs during the effector phase of an immune response (19, 20). In contrast, we have shown that TGF-β may promote the survival of memory T cells by up-regulating Bcl-2 expression (Fig. S5). The discrepancy may be related to differences in experimental systems. Previous work has focused on the effector phase, whereas we focused on the memory phase of an immune response. In addition, different animal models were used to suppress TGF-β signaling in T cells.
We have shown that TGF-β may directly inhibit the proliferation of KLRG1+ T cells (Fig. 4). The molecular mechanisms that mediate TGF-β–controlled proliferation of KLRG1+ T cells are currently under investigation. A recent study demonstrated that CD27−KLRG1+ effector-like CD8+ T cells in the memory population persist for a long time and mediate immediate potent protection against subsequent bacterial reinfection (32). The phenotype of these effector-like CD8+ T cells resembles that of the memory T cells that are unresponsive to TGF-β. The relationships among TGF-β signaling, environmental signals, and these CD27−KLRG1+ T cells is currently under investigation.
Taken together, our findings in the present study demonstrate that memory CD8+ T cells constantly incorporate environmental signals to maintain their phenotypic and functional character. TGF-β is an essential guardian of memory T-cell identity. From the early stages of differentiation to the late stages of maintenance, continuous TGF-β signaling to CD8+ T cells is critical for the formation and maintenance of memory T cells.
Materials and Methods
Mice, Bacteria, and Viruses.
Tgfbr2f/f dLck-cre OT-1 mice have been described previously (22). C57BL/6 (stock no. 000664) and ER-cre (stock no. 008463) mice were obtained from The Jackson Laboratory. All recipient mice were used at 6–12 wk of age. All experiments were conducted in accordance with University of Texas Health Science Center at San Antonio Institutional Animal Care and Use Committee guidelines.
Mice were infected i.v. with 2 × 103 cfu of LM-ova or with 2 × 106 pfu of VSV-ova. Bacteria and viruses were grown and quantified as described previously (33).
Adoptive Transfer.
Naive OT-1 T cells were isolated from spleen and lymph nodes using a CD8 isolation kit (Miltenyi Biotec) following the manufacturer’s instructions, but with the addition of biotin-αCD44 antibody during the biotin antibody mixture incubation step. The indicated number of naïve OT-1 T cells were transferred i.v. into sex-matched recipient mice.
Antibiotic Treatment.
For the experiments shown in Fig. S3, 2 mg of ampicillin was administered i.p. at 1 d after LM-ova infection. For the experiments shown in Fig. S4, mice were given drinking water containing 1 mg/mL ampicillin + 2 mg/mL neomycin + 100 U/mL polymyxin B starting at day 30 postinfection, along with 2.5 mg of vancomycin i.p. daily.
TAM Treatment.
TAM (Sigma-Aldrich) was dissolved in corn oil at 10 mg/mL. Then 1 mg of TAM was administered i.p. every other day for five treatments starting at day 30 postinfection.
BrdU Labeling.
At day 38 postinfection, mice were provided with drinking water containing 0.8 mg/mL BrdU (Sigma-Aldrich) ad libitum. In addition, 1 mg of BrdU was administered i.p. on day 38 postinfection. Three days later, BrdU incorporation in splenic OT-1 T cells was determined by flow cytometry using a BrdU staining kit (BD Biosciences).
Antibodies and Flow Cytometry.
Single-cell suspensions were prepared from the spleen and peripheral blood at the indicated time points postinfection. Cells were typically stained with antibodies specific for CD8, CD127, CD27, CD122, KLRG1, CD69, CD25, CD45.1, CD45.2, CXCR3, CD62L, CD44, IFN-γ, IL-2, Eomes and T-bet (eBioscience, Biolegend, and BD Biosciences), Bcl-6 (Santa Cruz Biotechnology), Granzyme B (Invitrogen), Foxo1 (Cell Signaling Technology), and TGF-βRII (R&D Systems). For intracellular cytokine staining, cells were stimulated ex vivo with 10 nM SIINFEKL peptide for 4 h at 37 °C in the presence of brefeldin A, followed by surface staining and treatment with a BD Biosciences Cytofix/Cytoperm Kit. For intracellular transcription factor staining, after surface staining, cells were treated with a Foxp3/TF buffer set (eBioscience), followed by intracellular transcription factor staining. Cells were analyzed using FACSCanto and LSRII flow cytometry systems (BD Biosciences) and FlowJo software (Tree Star).
Acknowledgments
We thank Dr. Michael J. Bevan for his generous support throughout the project.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1510119112/-/DCSupplemental.
References
- 1.Chang JT, Wherry EJ, Goldrath AW. Molecular regulation of effector and memory T cell differentiation. Nat Immunol. 2014;15(12):1104–1115. doi: 10.1038/ni.3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol. 2012;12(11):749–761. doi: 10.1038/nri3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jameson SC, Masopust D. Diversity in T cell memory: An embarrassment of riches. Immunity. 2009;31(6):859–871. doi: 10.1016/j.immuni.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaech SM, Wherry EJ. Heterogeneity and cell-fate decisions in effector and memory CD8+ T cell differentiation during viral infection. Immunity. 2007;27(3):393–405. doi: 10.1016/j.immuni.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Masopust D, Vezys V, Wherry EJ, Ahmed R. A brief history of CD8 T cells. Eur J Immunol. 2007;37(Suppl 1):S103–S110. doi: 10.1002/eji.200737584. [DOI] [PubMed] [Google Scholar]
- 6.Williams MA, Bevan MJ. Effector and memory CTL differentiation. Annu Rev Immunol. 2007;25:171–192. doi: 10.1146/annurev.immunol.25.022106.141548. [DOI] [PubMed] [Google Scholar]
- 7.Starbeck-Miller GR, Xue HH, Harty JT. IL-12 and type I interferon prolong the division of activated CD8 T cells by maintaining high-affinity IL-2 signaling in vivo. J Exp Med. 2014;211(1):105–120. doi: 10.1084/jem.20130901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim MT, Harty JT. Impact of inflammatory cytokines on effector and memory CD8+ T cells. Front Immunol. 2014;5:568. doi: 10.3389/fimmu.2014.00295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kalia V, et al. Prolonged interleukin-2Ralpha expression on virus-specific CD8+ T cells favors terminal-effector differentiation in vivo. Immunity. 2010;32(1):91–103. doi: 10.1016/j.immuni.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 10.Pipkin ME, et al. Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity. 2010;32(1):79–90. doi: 10.1016/j.immuni.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arsenio J, et al. Early specification of CD8+ T lymphocyte fates during adaptive immunity revealed by single-cell gene-expression analyses. Nat Immunol. 2014;15(4):365–372. doi: 10.1038/ni.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mitchell DM, Ravkov EV, Williams MA. Distinct roles for IL-2 and IL-15 in the differentiation and survival of CD8+ effector and memory T cells. J Immunol. 2010;184(12):6719–6730. doi: 10.4049/jimmunol.0904089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cui W, Liu Y, Weinstein JS, Craft J, Kaech SM. An interleukin 21-interleukin 10-STAT3 pathway is critical for functional maturation of memory CD8+ T cells. Immunity. 2011;35(5):792–805. doi: 10.1016/j.immuni.2011.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Siegel AM, et al. A critical role for STAT3 transcription factor signaling in the development and maintenance of human T cell memory. Immunity. 2011;35(5):806–818. doi: 10.1016/j.immuni.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boyman O, Krieg C, Homann D, Sprent J. Homeostatic maintenance of T cells and natural killer cells. Cell Mol Life Sci. 2012;69(10):1597–1608. doi: 10.1007/s00018-012-0968-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aiello FB, Graciotti L, Procopio AD, Keller JR, Durum SK. Stemness of T cells and the hematopoietic stem cells: Fate, memory, niche, cytokines. Cytokine Growth Factor Rev. 2013;24(6):485–501. doi: 10.1016/j.cytogfr.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Newell EW, Sigal N, Bendall SC, Nolan GP, Davis MM. Cytometry by time-of-flight shows combinatorial cytokine expression and virus-specific cell niches within a continuum of CD8+ T cell phenotypes. Immunity. 2012;36(1):142–152. doi: 10.1016/j.immuni.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ishigame H, Mosaheb MM, Sanjabi S, Flavell RA. Truncated form of TGF-βRII, but not its absence, induces memory CD8+ T cell expansion and lymphoproliferative disorder in mice. J Immunol. 2013;190(12):6340–6350. doi: 10.4049/jimmunol.1300397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanjabi S, Mosaheb MM, Flavell RA. Opposing effects of TGF-beta and IL-15 cytokines control the number of short-lived effector CD8+ T cells. Immunity. 2009;31(1):131–144. doi: 10.1016/j.immuni.2009.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tinoco R, Alcalde V, Yang Y, Sauer K, Zuniga EI. Cell-intrinsic transforming growth factor-beta signaling mediates virus-specific CD8+ T cell deletion and viral persistence in vivo. Immunity. 2009;31(1):145–157. doi: 10.1016/j.immuni.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takai S, Schlom J, Tucker J, Tsang KY, Greiner JW. Inhibition of TGF-β1 signaling promotes central memory T cell differentiation. J Immunol. 2013;191(5):2299–2307. doi: 10.4049/jimmunol.1300472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang N, Bevan MJ. TGF-β signaling to T cells inhibits autoimmunity during lymphopenia-driven proliferation. Nat Immunol. 2012;13(7):667–673. doi: 10.1038/ni.2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ventura A, et al. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445(7128):661–665. doi: 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- 24.Schwartzkopff S, et al. TGF-beta down-regulates KLRG1 expression in mouse and human CD8 T cells. Eur J Immunol. 2015 doi: 10.1002/eji.201545634. [DOI] [PubMed] [Google Scholar]
- 25.Banerjee A, et al. Cutting edge: The transcription factor eomesodermin enables CD8+ T cells to compete for the memory cell niche. J Immunol. 2010;185(9):4988–4992. doi: 10.4049/jimmunol.1002042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hess Michelini R, Doedens AL, Goldrath AW, Hedrick SM. Differentiation of CD8 memory T cells depends on Foxo1. J Exp Med. 2013;210(6):1189–1200. doi: 10.1084/jem.20130392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim MV, Ouyang W, Liao W, Zhang MQ, Li MO. The transcription factor Foxo1 controls central-memory CD8+ T cell responses to infection. Immunity. 2013;39(2):286–297. doi: 10.1016/j.immuni.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Intlekofer AM, et al. Requirement for T-bet in the aberrant differentiation of unhelped memory CD8+ T cells. J Exp Med. 2007;204(9):2015–2021. doi: 10.1084/jem.20070841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Joshi NS, et al. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity. 2007;27(2):281–295. doi: 10.1016/j.immuni.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Williams MA, Bevan MJ. Shortening the infectious period does not alter expansion of CD8 T cells but diminishes their capacity to differentiate into memory cells. J Immunol. 2004;173(11):6694–6702. doi: 10.4049/jimmunol.173.11.6694. [DOI] [PubMed] [Google Scholar]
- 31.Zhang N, Bevan MJ. Transforming growth factor-β signaling controls the formation and maintenance of gut-resident memory T cells by regulating migration and retention. Immunity. 2013;39(4):687–696. doi: 10.1016/j.immuni.2013.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Olson JA, McDonald-Hyman C, Jameson SC, Hamilton SE. Effector-like CD8+ T cells in the memory population mediate potent protective immunity. Immunity. 2013;38(6):1250–1260. doi: 10.1016/j.immuni.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wakim LM, Woodward-Davis A, Bevan MJ. Memory T cells persisting within the brain after local infection show functional adaptations to their tissue of residence. Proc Natl Acad Sci USA. 2010;107(42):17872–17879. doi: 10.1073/pnas.1010201107. [DOI] [PMC free article] [PubMed] [Google Scholar]