

Abstract
We introduce RLIMS-P version 2.0, an enhanced rule-based information extraction (IE) system for mining kinase, substrate, and phosphorylation site information from scientific literature. Consisting of natural language processing and IE modules, the system has integrated several new features, including the capability of processing full-text articles and generalizability towards different post-translational modifications (PTMs). To evaluate the system, sets of abstracts and full-text articles, containing a variety of textual expressions, were annotated. On the abstract corpus, the system achieved F-scores of 0.91, 0.92, and 0.95 for kinases, substrates, and sites, respectively. The corresponding scores on the full-text corpus were 0.88, 0.91, and 0.92. It was additionally evaluated on the corpus of the 2013 BioNLP-ST GE task, and achieved an F-score of 0.87 for the phosphorylation core task, improving upon the results previously reported on the corpus. Full-scale processing of all abstracts in MEDLINE and all articles in PubMed Central Open Access Subset has demonstrated scalability for mining rich information in literature, enabling its adoption for biocuration and for knowledge discovery. The new system is generalizable and it will be adapted to tackle other major PTM types. RLIMS-P 2.0 online system is available online (http://proteininformationresource.org/rlimsp/) and the developed corpora are available from iProLINK (http://proteininformationresource.org/iprolink/).
Index Terms: Biology and genetics, context analysis and indexing, natural language processing, text mining
1 Introduction
PROTEIN phosphorylation is a type of post-translational modification (PTM), in which a phosphate group is attached to an amino acid residue (site) of a protein (substrate), catalyzed by an enzyme (kinase). In signal transduction networks, it serves as a switch to transmit signals in response to extracellular stimuli and intracellular changes [1]. Protein phosphorylation has been widely studied, and research findings have been curated in several biological knowledgebases, such as Protein Ontology [2], PhosphoSite-Plus [3], Phospho.ELM [4], and UniProt Knowledgebase (UniProtKB) [5]. The manual curation of phosphorylation information reported in literature, however, lags behind because of the ever-increasing publications on this active research topic. Text mining applications to support biocurators have been developed [6], [7], [8], [9], [10]. In this paper, we report on a new version of RLIMS-P, an information extraction (IE) system to extract kinase, substrate, and site of phosphorylation reported in biomedical literature [6], [8]. The system is designed to help biocurators to search and retrieve articles of their interests and to efficiently review and extract phosphorylation information therein.
RLIMS-P consists of a series of natural language processing (NLP) modules to analyze input text, an IE engine to apply lexical, syntactic, and semantic patterns to extract target information, and an additional IE component to extract information beyond patterns. Our goal in developing a new version of RLIMS-P, hereinafter called RLIMS-P 2.0, was to make the system generalizable and easily adaptable to the extraction of other types of PTMs. To this end, we designed a new architecture of the rule-based IE engine and supplemented it with task-independent components to support extraction procedures. The new architecture facilitates the portability of the system to extraction of other PTM types and also improves its maintainability for operational use.
In our previous study [11], we evaluated RLIMS-P 2.0 on the MEDLINE abstracts of the GENIA event extraction (GE) corpus, which was released for the 2011 BioNLP shared task (BioNLP-ST). In this corpus, however, expressions used to report phosphorylation events were limited, and hence insufficient for developing and thoroughly evaluating phosphorylation IE systems. To address this issue and thoroughly evaluate RLIMS-P 2.0, in the current study, diverse MEDLINE abstracts were annotated in-house by experienced biocurators. The performance of RLIMS-P 2.0 was evaluated on these new data sets, and also compared to the results on the existing in-house annotated corpora, including a set of full-text articles sampled from PubMed Central (PMC). Additionally, RLIMS-P 2.0 was evaluated on the test set of the 2013 BioNLP-ST GE task, which has been recently made available. Finally, we report on the full scale application of RLIMS-P 2.0 to abstracts in the MEDLINE database and the full-text articles in the PMC Open Access Subset (PMC OA). Application of the system to PMC OA allowed us to gain insights into the distribution of phosphorylation information reported in different sections of articles.
2 Background
Text mining has been actively studied in the biological domain [12], [13], [14], [15], [16], [17], [18], [19], [20], [21], [22], [23], [24], [25]. Among the various text mining applications are IE systems, which aim to extract facts reported in biology literature, e.g., [26], [27]. Protein phosphorylation information has been tackled as one of the IE targets [6], [8], [9], [10], [28].
2.1 Phosphorylation IE Systems
Besides RLIMS-P, there have been a few other efforts towards the extraction of protein phosphorylation information from biomedical literature [9], [10], [28].
Saric et al. developed a rule-based IE system to extract information on phosphorylation and regulation of gene expression. Their system, STRING-IE, uses syntactic parsing to analyze input text, and dictionary lookup to identify protein names. It then applies hand-coded patterns to extract target relations, e.g., kinase-substrate relations. The system has been applied to MEDLINE abstracts retrieved for four model organisms. The reported precisions range from 0.86 to 0.95 for (de-) phosphorylation and from 0.83 to 0.90 for regulation. The recall performance was estimated to be around 0.3 based on sampled sentences, and an F-score of 0.44.
MinePhos [10] is a rule-based IE system, which extracts kinase, substrate, and site. As noted by the authors, it essentially uses the lexical, syntactic, and semantic patterns of RLIMS-P. The authors reported the addition of five new extraction patterns, but as far as we could tell, these patterns are present in RLIMS-P. The main difference of MinePhos from RLIMS-P is the use of a different named-entity recognition method. It uses a name dictionary compiled from Phospho.ELM along with a machine learning tool. The system was evaluated on two sets of 200 MEDLINE abstracts, and F-scores of 0.863 and 0.864 were reported.
Veuthey et al. [9] reported a tool for supporting curation of modified sites in PTMs for UniProtKB/Swiss-Prot. It focuses on the curation of modified sites, and the tool applies a filter to identify and report potentially relevant sentences and site mentions. This filtering process was evaluated on 100 MEDLINE abstracts, and a precision of 0.71 was reported. A recall estimated using annotations in Uni-ProtKB/Swiss-Prot was 0.93. Using regular expressions, the tool identifies modified sites in the filtered sentences and displays a list of proteins detected through an existing protein name tagger. The authors also applied the tool to 11,000 full-text articles and reported that 90 percent of the detected sites were found in the full-text body, but not in the abstracts.
2.2 BioNLP-ST GE Task
In the GE task of BioNLP-ST 2009 and 2011, sets of MED-LINE abstracts were used to investigate extraction of substrates and sites for phosphorylation events, along with other targets for various events (e.g., gene expression, binding, and regulation). In the 2011 GE task, five full-text articles were added to each of the training, development, and test sets. As for phosphorylation events, the best performance on the abstract set, used both in 2009 and 2011, was reported to be an F-score of 0.8295 for substrates, and 0.8381 for sites [29], [30].
In the recent BioNLP-ST 2013 GE task, new sets of 10, 10, and 14 full-text articles were used as the training, development, and testing sets, respectively [31]. As for phosphorylation events, a new target role, cause, was considered in addition to substrates and phosphorylation sites. However, few phrases are annotated as the cause in the data sets. In the 2013 BioNLP-ST task, ten teams participated in the GE task and F-scores reported for phosphorylation range from 0.5978 to 0.8148. Of them, two teams also tackled extraction of phosphorylation sites, and their F-scores were 0.4628 and 0.5120 [31].
Most top-ranked systems in BioNLP-ST employed machine learning approaches [31]. Kilicoglu and Bergler, on the other hand, demonstrated that a rule-based system could achieve competitive performance in the BioNLP-ST task settings, and their system was reportedly ranked fourth for phosphorylation events in the 2011 GE task [32]. Their system exploits syntactic dependencies of selected triggers and composes semantic interpretation out of them in a bottom-up manner. This approach is similar to RLIMS-P 2.0 in that it first extracts basic relations from a sentence and derives target information. Their system, however, is different from RLIMS-P in several ways, including that it does not support IE beyond sentence, it is not specialized in exploiting distinctive patterns for phosphorylation and PTM, and it extracts basic relations using a syntactic parser, rather than local syntactic-semantic patterns, as described next.
2.3 RLIMS-P version 1.0
The original version of RLIMS-P, hereinafter called RLIMS-P 1.0, is a rule-based IE system designed to extract kinase, substrate, and site reported in abstracts. To our best knowledge, RLIMS-P is the only system, other than MinePhos that is based on RLIMS-P rules, that specifically focuses on these three types of entities, and aims to extract information across sentences beyond anaphoric relations.
The evaluation of the extraction performance of RLIMS-P 1.0 was first reported in [6]. The system was evaluated on MEDLINE abstracts available in the PIR iProLink resource [33], which were originally collected for the curation of phosphorylation information in the PIR-PSD database [34]. The ability to filter relevant abstracts was tested on this data set, and an F-score of 0.94 was reported on 370 abstracts (110 positives and 260 negatives). The system was further tested on the extraction of substrate-site relations, and an F-score of 0.93 was reported on 108 abstracts.
Later RLIMS-P 1.0 was enhanced with heuristic rules to extract substrates and sites across sentences and also rules to integrate information extracted in the whole abstract [8], The system performance was evaluated on 386 abstracts sampled among literature referenced in the phospho.ELM database. An F-score of 0.85 was reported for the extraction of the triple: kinase, substrate and site and an F-score of 0.89 was reported for extraction of the substrate-site relation.
RLIMS-P 1.0 achieves a high precision owing to its detailed IE patterns. A high recall of RLIMS-P is facilitated by a large number of such patterns, realized by complex and often redundant regular expressions in conditional control-flows. These patterns cover numerous variations in textual expression that may be encountered in phosphorylation literature. Despite the good performance attained in this manner, this configuration made it difficult to maintain the system and daunting to generalize it for other IE targets, such as different PTMs. In designing RLIMS-P version 2.0, the challenge we encountered was to keep the pattern collection simple and concise, and make it readily portable for other IE tasks, while still maintaining good pattern coverage as well as accuracy.
3 RILMS-P version 2.0 Algorithms and Approaches
The architecture of the RLIMS-P 2.0 system is shown in Fig. 1. The system consists of two major pipelines, an NLP pipeline and an IE pipeline.
Fig.1.
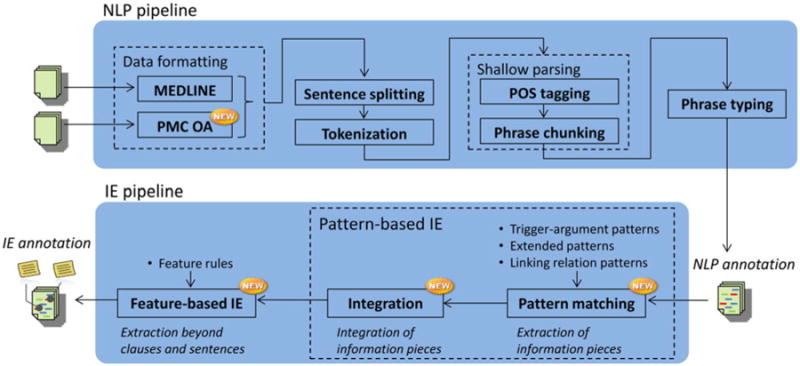
The overview of RLIMS-P 2.0. The system consists of an NLP pipeline extending RLIMS-P 1.0 and a newly designed IE pipeline.
3.1 NLP Pipeline
The NLP pipeline, extending from that of RLIMS-P 1.0, consists of multiple steps: (i) data formatting, (ii) sentence splitting and tokenization, (iii) shallow parsing, and (iv) phrase typing (Fig. 1). The data formatting step is to transform the input data of different types for common subsequent processing, where the data can be either MEDLINE abstracts or PMC OA full-text articles. A new set of programs was prepared to preprocess full-text articles from PMC OA. Besides low-level data processing for full-text articles, such as parsing of PMC XML data, conversion of special symbols, and removal of citation in text, the main functionality of these programs is to extract article sections as defined in the PMC XML format. For each section extracted from an article, its title and text are regarded in the same manner as those in the abstracts for the subsequent processes (see Section 3.7). Figure captions are also extracted and processed in the same manner.
The other components in the NLP pipeline include a shallow parser built on part-of-speech (POS) tagging and phrase chunking and a type assignment module. These steps are essentially based on an existing named-entity recognition system [35]. To improve its performance in RLIMS-P 2.0, dictionary-based recognition of protein names has been integrated and rules facilitating phrase chunking were revised.
The shallow parser of RLIMS-P breaks input text sentences into phrase chunks. Shallow parsing, rather than deep parsing, is employed in this system because it can provide sufficient abstraction of phrases for IE goals in the biomedical domain, and because the processing time was found to be less than that of deep parsing. Certain syntactic constructions, including phrase coordination, appositives, and parenthetical expressions, are detected in this stage in order to help improve the later IE process. Neighboring chunks found in these constructions are further grouped together so that the sentence is syntactically simplified for the subsequent procedures. The chunk types detected by the parser include noun phrases (NP) and verb groups (VG). VG is further annotated with the voice information. The following example illustrates the utility of these analyses beyond simple pattern matching:
“RIG-INP has been shownVP to be ableVP to phosphorylateVP STAT1NP at both Tyr701 and Ser727NP …” [PMC3497619]
In the consecutive VGs above, the last one, “to phosphorylate”, is detected to be in the active voice. Then, NP at the subject position, “RIG-I”, can be recognized as an agent of this action and, as described later, it will be eventually extracted as a kinase phrase. Note that the coordinated phrases referring to two phosphorylation sites, “Tyr701” and “Ser727”, are grouped together in one NP, which helps simplify the design of IE patterns.
The phrase type assignment module identifies semantic types of noun phrases and also terms embedded in the other phrases. These types include Protein (gene/protein), Protein Part (such as amino acid residue, domain, region, and motif), Chemical, Cell, Species, and Others (other types of biological entities). Semantic types play an important role in specifying IE patterns in RLIMS-P. For instance, see the two examples below
“SrcphosphorylatesTyr284 in TGF-beta type II receptor” [PMID17440088].
“CaMKII δ phosphorylatestitin in mouse LV skinned fibers” [PMID 23220127].
In these examples, the sentences fit the same lexico-syntactic pattern of “X phosphorylates Y in Z.” But the phrases at the positions, Y and Z, belong to different semantic types. For example, at the position Y, we find “Tyr284” (a residue) vs. “titin” (a protein). Consequently they fill the roles of site and substrate respectively during the extraction from the two sentences. The semantic type information is important for accurate IE. The type assignment module of RLIMS-P was extended with the addition of a dictionary-lookup component and some additional filtering rules.
Note that the NLP pipeline (Fig. 1) is a common pre-processing step for many biomedical IE tasks, not specific to phosphorylation. With the appropriate set of semantic types as well as the right level of abstraction of input text, this pipeline is suitable as preprocessing for extracting PTM information in general.
3.2 IE Based on Syntactic Arguments
In RLIMS-P, the IE mechanism is invoked by the presence of trigger words, which are selected keywords commonly found with phosphorylation information. The major trigger words in RLIMS-P include the word ‘phosphorylation’, its verbal forms (phosphorylate, phosphorylates, phosphorylated, and phosphorylating), and its adjectival forms (phosphorylated and phospho-).
In the basic IE mechanism of RLIMS-P 2.0 (the pattern matching component in Fig. 1), target entities are sought among the arguments of a trigger word. This mechanism is implemented as matching of patterns using lexical, syntactic, and semantic constraints. A detailed pattern specifying a phosphorylation mention can be precise in extracting target entities, but a large collection of near-redundant patterns is required to achieve good pattern coverage. A major cause of the redundancy is due to the combinations of pattern variations. Consider the two patterns below with text snippets that they match:
-
NPtype=protein,role=kinase VGhead=“phosphorylate”,voice=active
NPtype=protein_part,role=site
“Src phosphorylates Tyr284 …” [PMID17440088]
-
NPtype=protein, role=kinase VGhead=“phosphorylate”,voice=active
NPtype=protein,role=substrate
“CaMKII δ phosphorylates titin …” [PMID 23220127].
Here, NPtype=protein,role=kinase, for example, represents a noun phrase of type protein, whose role in the phosphorylation event is a kinase. In this notation, a pattern to be matched (e.g., NPtype=protein) and a role to be identified (e.g., role=kinase) are conflated to save space. Now, imagine a case where the subject is a relative pronoun, e.g., “which phosphorylates ….” As this variation is possible for both patterns above, each of the two patterns may be duplicated and modified. Now we have four patterns with a fair overlap. If we multiply patterns in this manner, the number of patterns can quickly explode.
As the first step to alleviate this situation, each IE pattern is designed to extract one target (i.e., trigger-kinase, trigger-substrate, or trigger-site), instead of aiming to extract all the target entities present. This design is possible as the extraction of any target is generally independent from each other. This mechanism significantly eases the creation and maintenance of IE patterns. Under this new design, in the previous example, we would have one pattern for the kinase (trigger-kinase), two patterns for the substrate (trigger-substrate). Combinations of these patterns effectively cover different situations. Given the previous example of a relative pronoun (“which phosphorylates …”), a new trigger-kinase pattern can be added to the collection without modifying the existing patterns, and the resulting collection covers more situations through pattern combinations
The second step in simplifying IE patterns was to focus on the two arguments, agent and theme, where a substrate or a site (part of substrate) can serve as the theme. To illustrate our approach centered around this notion, let us consider an example outside the biology domain–a case of a glass that is part of a window. The glass and the window can appear in similar ways with a verb like “break”, e.g., “I broke the glass” and “I broke the window.” When it can be inferred from context that “the glass” is part of “the window”, then the former can be said to imply the latter. Anyway, the main point is that the two words can be used in similar ways with respect to the verb “break.” This example underlies our approach of treating either a site or a substrate as the theme of the predicate “phosphorylate”. In simplified patterns, we do not differentiate the site and the substrate, and call them theme arguments of phosphorylation. Based on the argument type, we can decide later whether an entity extracted as a theme is a site or a substrate. In the case of phosphorylation, the agent argument is generally taken to be a kinase.
With the above two changes, IE patterns are now simplified for two kinds of relations, <trigger, agent> and <trigger, theme>. Each new pattern is anchored by one trigger, and then one argument is sought. The key idea of the new design is that each pattern can be created and maintained separately, while their combinations can effectively cover the triplet <trigger, agent, theme>. Examples of simplified patterns are shown below
Theme patterns:
VGhead=“phosphorylate”,voice=active NPtype={protein,_ part},role=theme
NPtype={protein,protein_part},role=theme VGhead=“phosphorylated”,voice=passive
Agent patterns:
NPtype=protein,role=agent VGhead=“phosphorylate”,voice=active
VGhead=“phosphorylated”,voice=passive by NPtype=protein,role=agent
As stated earlier, a theme is a substrate if it is a protein, and a site if it is a protein part. An agent is a kinase if it is a protein.
Readers might notice that for a sentence like “SrcphosphorylatesTyr284 in TGF-beta type II receptor”, only the kinase (agent), “Src”, and the site (theme), “Tyr284”, are extracted with these patterns. The extraction of the substrate in this case will be handled separately by linking relations, as discussed later in Section 3.4. The set of lexico-syntactic patterns discussed in this section can be found on the RLIMS-P website at http://proteininformationresource.org/rlimsp/.
Lastly we note that the extraction mechanism based on trigger-argument as well as the techniques to simplify it are applicable to extraction of PTM information in general because the underlying ideas are not specific to the case of phosphorylation, e.g.,
“PCAFacetylatescdk2 at lysine 33” [PMID 19773423]
“Parkinis ubiquitinated by Nrdp1” [PMID18541373]
“Glycosylation of Ser-16 is negatively affected” [PMID10187769].
3.3 IE with Extended Patterns
So far, we have discussed the situations where the agent or theme appears in a particular syntactic argument position with respect to the trigger. However, consider the sentence
“LMP1activatedNF-kappa Bvia phosphorylation” [PMID11780335].
As for the trigger word, “phosphorylation”, in this expression, we argue that its arguments are omitted here because they are shared with the preceding predicate “activated.” It is awkward for the authors to write “LMP1activatedNF-kappa Bvia phosphorylation of NF-kappa B by LMP1.” In other words, these trigger words following via, and similarly by, upon, after, through, etc., appear to have elided arguments. Note that the same argument can hold for some variants of this expression, e.g., “activation of NF-kappa B by LMP1 via phosphorylation.” There is a similar, but slightly different case for the trigger in the gerund form, e.g., “… by phosphorylating ….” In this case, the theme element is usually mandatory, but the agent (kinase) is not present. In all these cases, the arguments involved in the phosphorylation event are inferable in the local context, and they can be extracted with patterns.
We have examined trigger occurrences falling in this class, and based on our observations, we generalized them as a new class of patterns. The following examples show a few of the patterns in this new class.
NPhead={“activation”,”inhibition”,…} of NPtype=protein,role=theme {by, via, upon, after, through} NPhead=“phosphorylation” “… inhibition ofGSK3β by phosphorylation” [PMID21837363]
NPtype=protein,role=theme VGvoice=passive,head={activate,inhibit,…} {by, via, upon, after, through} NPhead=“phosphorylation” “… the GEFis inhibited upon phosphorylation” [PMID23378025]
NPtype=protein,role=agent VGvoice = active,head={activate,inhibit,…} NPtype≠protein {by, via, upon, after, through} VGhead=“phosphorylating” NPtype={protein,protein part},role=theme “p38 MAPK negatively regulates the proteasome activity by phosphorylatingThr-273 …” [PMID10074427].
These phenomena observed for the phosphorylation are common to other predicates pertaining to PTMs and the patterns mined for phosphorylation can be generalized, e.g.,
“… regulation ofGATA-2by acetylation.” [PMID15001660]
“JosD1, a membrane-targeted deubiquitinating enzyme, is activated by ubiquitination and …” [PMID23625928]
“… the histone methyltransferase Dot1mediates global genomic repair by methylatinghistone H3” [PMID21460225].
3.4 IE Enhanced by Linking Relations
Even when an argument of a trigger word is found, it may not be the name phrase expected in the IE task, and its identity may be stated elsewhere in the text. One obvious case is an anaphoric expression, such as “this protein is phosphorylated.” The reference needs to be resolved in order to identify an appropriate entity mention in this case. Once the anaphoric expression and the antecedent phrase are linked, then this linking relation can be used to associate the trigger with the appropriate target entity. In RLIMS-P 2.0, we generalize this idea to include three other linking relations: member-collection, identity, and part-whole relations. In all these cases, we expect that the phrases extracted by the trigger-argument patterns do not provide appropriate entity names. Instead, phrases linked from them through these relations do. So the trigger-argument relations together with the linking relations can finally associate the trigger with the target mentions. In RLIMS-P 2.0, the modules for the extraction of the different linking relations of the basic phosphorylation IE relations operate independently of each other. In the following sections, we first discuss different kinds of phrase linking relations considered in the system and then describe the process to integrate them for information extraction.
3.4.1 Member-Collection
Given a trigger in text, a phrase at the argument position may refer to a class or group of proteins, while a specific protein belonging to that class/group may be mentioned in the nearby context. Unlike the anaphoric relation, specific instances (members) typically follow the mention of a class or group (collection). Currently, we find such relations by the use of keywords like “include” and “such as.” Below is a typical pattern for extracting member-collection relations:
NPtype=x,role=collection such as NPtype=x,part,role=member “phosphorylation of stress-activated signaling proteins, such asc-Jun N-terminal kinase (JNK) and/or p38 mitogen-activated protein kinase (MAPK).” [PMID21310627].
In this example, “JNK” and “MAPK” (members) are linked from the phrase “stress-activated signaling proteins” (collection), which is also the argument of the trigger word “phosphorylation.” Putting together these pieces of information, “JNK” and “MAPK” can be extracted as substrates. Note that the trigger-argument relation (“phosphorylation” of “stress-activated signaling proteins”) and the member-collection relation (“stress-activated signaling proteins” include “c-Jun N-terminal kinase and/or p38 mitogen-activated protein kinase”) are extracted independently and the extraction patterns are developed and managed independently.
3.4.2 Identity
In some cases, the entity is ambiguously stated in one place, but may be clearly named in another place. Below is a pattern to identify such an instance and an example snippet
NPtype=x,role=entity1 VGhead=“identified”,voice=passive as NPtype=x,part,role=entity2 “p130 Crk-associated substrate (Cas)entity1, a putative c-Src substrate, was originally identified asa highly phosphorylated proteinentity2” [PMID10480886].
In this example, the entity referred to as “a highly phosphorylated protein” is “p130 Crk-associated substrate.” To extract this relation using the pattern-based approach in RLIMS-P, an appositive phrase, “a putative c-Src substrate”, can be overlooked after sentence simplification [36], i.e., a sentence without the appositive is first generated. Then, the relation between the two phrases can be identified by a rather simple pattern, triggered by the keyword “identified.” With this relational information, “Cas” can be linked with the immediate theme phrase, and eventually extracted as a substrate in the reported phosphorylation event. We note that extraction of this relation, which we call “identity” relation, is independent of extraction of trigger-argument relations or any other linking relations.
Apart from the explicit keyword “identity” used in the example above, we also extract abbreviations and their corresponding expanded phrases from parenthetical expressions and other selected constructions, such as those involving “is”, as shown below:
“The site1, identified asSer378, is also the site2 of phosphorylation” [PMID8491187].
Here, the immediate argument of the phosphorylation trigger, “the site2”, is first linked with “The site1.” Note that this relation can be readily identified, triggered by the keyword “is”, because the clause, “identified as Ser378”, can be overlooked during pattern matching, similar to the aforementioned appositive case. Meanwhile, given this clause similar to the appositive configuration, the phrase “The site11” is known to refer to “Ser378.” Putting the information together, “Ser378” is extracted as the site of phosphorylation.
3.4.3 Part-Whole
In the new way of specifying extraction patterns in RLIMS-P 2.0, there is a need for an additional mechanism to link a detected site (part) with a corresponding substrate (whole) or vice versa. In fact, detection of the part-whole relation is desirable as a generic mechanism in order to extract remotely stated substrate and site. As for part-whole relation, we focus on relation between protein parts (e.g., a residue and a region), and that of a protein part and a protein (e.g., a residue/region and a protein). These relations may be found inside a noun phrase or between noun phrases
Ntype=protein,role=whole Ntype=protein part,role=part “ICAM-1wholeTyr518part” [PMID21474822]
NPtype=protein,role=whole VGhead={“contains”,”have”,…},voice=active NPtype=protein_part,role=part
“AMPKwhole contains a glycogen-binding domainpart” [PMID21067629]
NPtype=protein_part,role=part {at, in, on, of} NPtype=protein,role=whole “… phosphorylatesTyr284part in TGF-beta type II receptorwhole …” [PMID17440088].
The new approach requires the processing of all the site-protein mentions in the document, regardless of the trigger presence. In fact, this is a better way of associating a detected phosphorylation site with a remotely mentioned substrate because it reduces the reliance on ad hoc heuristic rules.
Once a relation between a residue position and a protein is known (e.g., in the latter example “Tyr 284” belongs to “TGF-beta type II receptor”), whether from a phosphorylation event mention or from any other instances (e.g., “the mutation of Tyr284 in TGF-beta type II receptor”), such associations can be remembered throughout the same document and any of the position mentions can be associated with the corresponding protein name when it is not stated locally.
Since these linking relations are general, the technique can be used for extracting different types of PTM other than phosphorylation. The example sentence below shows that the relations would be useful also in extracting other PTM types, e.g.,
“Ubc9can sumoylatetargetssuch asRanGAP” [PMC3025465]
“Themethylatedproteinwas identified asPP2Ac” [PMC2278024]
“acetylation at Lysine-14inthe N-terminal tailsofthe nucleosomal protein histone H3” [PMID 16197509].
3.5 Integration of Relations
After patterns for all relations are applied, pieces of the independently extracted information are integrated for each trigger (see Fig. 1). The integration procedure involves assembly of immediate argument phrases for a trigger (agents and themes), traversal of the linked phrases starting at the immediate arguments, and extraction of actual target entities (kinase, substrate, and site). To illustrate the integration procedure, we will use the following example again:
“SrcphosphorylatesTyr284 in TGF-beta type II receptor …“ [PMID17440088].
The immediate arguments extracted for the highlighted triggers are “Src” (agent) and “Tyr284” (theme). The agent is of the Protein type and is recorded as the kinase. As for the theme, the phrase is of the Protein Part type, and the amino acid type and the residue position are extracted, Tyrosine at position 284. Meanwhile, as noted before, a part-whole relation has been established between “Tyr284” and “TGF-beta type II receptor.” The integration process connects “Tyr284” with the phrase known to be its whole, “TGF-beta type II receptor”, and identifies that protein as the substrate. This integration process is facilitated by traversing the part-whole link. Similarly, if a phrase extracted as an argument is an anaphor (see the introductory paragraph of Section 3.4) or if it is in an identity relation with another phrase (see Section 3.4.2), such links traversed to extract phrases referring to the same entity. Linking relations between two phrases are extracted independently of each other and independently of trigger-argument relations. They are used to connect phrases with multiple links away (e.g., see Section 3.4.2). The integration process involves appropriate traversal of links (e.g., part → whole, but not whole → part) in order to identify all the candidate phrases. From the candidate phrases, site information (amino acids and/or their positions) or protein names are extracted.
The extraction of part-whole relations has been studied in the field [37], [38], including a case in the context of IE [39]. Compared to the existing studies, the mechanism we propose in this paper extracts and uses various relations in a knowledge-intensive manner, where we review and analyze each individual relation type and use it for a specific IE purpose, e.g., within the same document, a site-protein relation (residue position-protein) extracted in one place is remembered and referenced later to infer the protein name from the same residue position.
3.6 IE Using Context-Based Features
One of the unique characteristics in RLIMS-P was IE beyond local contexts [8]. For example, see the following example, in particular the second trigger “phosphorylation2.”
“We also show that stimulation of HeLa cells with the phorbol ester TPA enhances phosphorylation1 of PTP1B. […] The site, identified as Ser378, is also the site of phosphorylation2 by protein kinase C (PKC) in vitro.” [PMID8491187]
For the second trigger, besides the kinase “protein kinase C”, the site phrase “Ser378” can be extracted using the phrase linkage in RLIMS-P 2.0, but not the substrate. Now that the phosphorylation site is detected, there must be a substrate protein in the context, and it should be sought in the document. There is, however, no local context pattern that could associate the current trigger with the substrate, which is “PTP1B.” RLIMS-P 1.0 implemented heuristics rules to tackle the situation where the substrate appears in a sentence different from the one with the trigger and the site. RLIMS-P 2.0, instead, uses simple features (the notion of features as in machine learning) where these features try to capture the same underlying ideas behind the first version's heuristic rules. Our hypothesis is that if a substrate is not stated as an argument of the trigger and, hence, not extracted by a pattern, the substrate protein must be one currently in focus. This is because the phosphorylation site alone would not be reported by the authors unless the substrate protein is already known to the readers and this must be clear to the readers that the reported site belongs to that protein. We selected features that help identify the substrate protein in focus.
The features currently considered in the system include
Is the candidate already extracted as a substrate for a preceding trigger? For example, in the previous example, the candidate “PTP1B” in the first sentence would have been extracted as a substrate when we consider the trigger in the second sentence.
Does the candidate appear in the title or the first sentence of the abstract? For example, “PTP1B” appears in both the title and the first sentence in the abstract.
Is the candidate repeatedly mentioned in the abstract and, if so, is it frequently mentioned in the document? For example, in this abstract “PTP1B” is repeated 11 times, and it is the most repeated protein name.
Does the candidate appear in the same sentence? For example, for the second trigger in the example, no candidate appears in the same sentence, and this feature does not apply (no candidate for the second trigger can have this feature being “yes”).
Is the candidate the subject of the clause in the matrix sentence of the form: “We {found, discovered, show, …}”? For example, one mention of “PTP1B” in the abstract is found as “We show that PTP1B is […]” in the abstract.
As shown above, these features are essentially the properties indicative of the discourse focus in the given document. In the above example, the entity “PTP1B” is extracted as the substrate for the clear emphasis on this entity throughout the abstract.
This extraction of features was developed with a machine learning approach in mind. However, compared to the number of instances addressed by the rule-based method, the number of instances targeted by the feature-based approach was small. Observing that the current features could be predictive of the target individually and independently, we decided to weight them equally in our feature-based method, instead of preparing costly training data for machine learning. Specifically, given candidate phrases, one that satisfies more conditions that are described above as features is selected in the final output.
We also use the feature-based approach when the substrate is extracted by a pattern, but the site cannot be. We do not look for the site across sentences, and consider only when there is a site(s) in the same sentence as the trigger. The features in this case included determining whether the candidate site is known to be a part of any protein (based on the part-whole relations extracted as discussed in Section 3.4.3); whether the site is mentioned in the subject position within the sentence; whether the sentence mentions that the site has been mutated (detected with simple patterns).
3.7 IE from Full-Text Articles
Since abstracts of articles need to be succinct, authors may choose not to report all the phosphorylation events or they may only report substrates without stating sites and/or kinases. Our goal in mining full-text articles is to extract rich information beyond what is reported in abstracts.
In order to extract phosphorylation information from full-text articles, the basic, extended patterns and patterns for linking relations do not need to be changed. However, the feature-based extraction across sentences will require attention. This is because the methods are essentially based on the hypothesis that, when an expected entity (substrate or site) does not appear in the local context of a trigger, it must be an entity currently in focus that can be readily identifiable by readers.
The concept of focus and the identification of the focused entity relies on the notion of the discourse and the scope of the focus [40], [41], as the focus changes at the discourse boundaries. In case of MEDLINE abstracts, it is reasonable to assume that each abstract is a self-contained unit in which the focus does not change. Clearly, a full-text article has many such units. Through our close examination of full-text articles, we came to find that even a single section, like Results, is too large as a discourse unit for applying RLIMS-P as each section covers multiple topics and entities and, therefore, the focus changes within the section. We found sections of selected sections, in particular those of Results, Discussion, and Conclusion, form appropriate discourse units, since each of these sections typically focuses on one aspect of the authors' finding.
Using sections as discourse units, we have adapted RLIMS-P to full-text articles in a straightforward manner. We treat each section as an “abstract” and reformat them accordingly for application of the system. A section title is treated as an “abstract” title. Now the context-based features and associated rules can be applicable to full-text articles, which are provided as a collection of “abstracts” (the re-formatting module in Fig. 1).
4 Results and Discussion
4.1 BioNLP-ST GE Corpus
Extraction of phosphorylation information has been considered in the BioNLP-ST GE shared task. To evaluate RLIMS-P 2.0, we use the online evaluation system that has been made available by the BioNLP organizer. Compared to 2009 and 2011 BioNLP-ST corpora, the corpus prepared for the 2013 task was annotated with a new role, the cause of phosphorylation. The cause could be any entity involved in an upstream event or even such an event. Extraction of causes was included in the core task of the GE task, along with extraction of substrates and triggers. Although RLIMS-P was not developed to extract upstream entities or events, it was evaluated in the core task for the arguments it extracted, and also in the optional task for phosphorylation sites. There are a few differences observed in the annotations of the BioNLP corpus and the design of RLIMS-P, focusing on the curation of phosphorylation information. For instance, RLIMS-P is designed not only to extract individual proteins as kinases or substrates, but also to report a protein family name, when appropriate. Additionally, it has a negation filter that avoids detection of phosphorylation event in the scope of a negation. To conform to the BioNLP task settings, some of these functionalities in RLIMS-P 2.0 were disabled during the evaluation.
The results of RLIMS-P 2.0 on the 2013 BioNLP-ST GE test corpus are shown in Table 1. The first row gives the performance for the core task. The F-score of 0.8679 improves upon the previous top F-score of 0.8148 in the 2013 GE task. In fact, both precision and recall of RLIMS-P 2.0 exceed the corresponding scores of any of the top five systems (ranked by F-scores) in the 2013 BioNLP-ST GE task. The second and third rows show the results specifically for the trigger-substrate and -site, respectively. Again these results exceed the performance of the systems from 2013 GE task, and particularly notable is the F-score of 0.8588 on trigger-site extraction, improving upon the previously reported F-score of 0.5120.
Table 1. Evaluation Results on 2013 BioNLP-ST GE Test Corpus.
| RLIMS-P 2.0 | 2013 BioNLP-ST | |||
|---|---|---|---|---|
|
|
||||
| Precision | Recall | F-score | Highest F-score | |
| Core task (trigger, substrate, and cause) | .8625 | .8734 | .8679 | .8148 |
| Theme –Phosphorylation (trigger and substrate) | .8875 | .8987 | .8931 | .8395 |
| Site-Phosphorylation (trigger and site) | .8690 | .8488 | .8588 | .5120 |
Precision = True Positives/(True Positives + False Positives), Recall = True Positives/(True Positives + False Negatives), F-score = 2 × Precision × Recall/ (Precision + Recall).
4.2 In-house MEDLINE Corpus
4.2.1 Motivation of the New Phosphorylation Corpus
To thoroughly evaluate a phosphorylation IE system, a corpus covering a wide variety of expressions in terms of phosphorylation events is expected. The data sets used in the BioNLP-ST GE task include abstracts and also paragraphs from full-text articles. Expressions used in the corpus, however, could be limited due to the specific focus of the corpus (e.g., the abstracts in the 2011 corpus were retrieved using the search terms “human”, “blood cell”, and “transcription factor”). As seen in the study by Landeghem et al. [27], the selection of the corpus could have a significant impact on the performance of biological event extraction systems, including performance on phosphorylation events. In our prior work [11], we evaluated RLIMS-P 2.0 on the 2011 GE corpus. We further examined the evaluation results and analyzed the patterns pertaining to phosphorylation events in the 2011 GE corpus as detailed in Section 4.2.4. We believe the limitation observed in the 2011 GE corpus is applicable to the 2013 corpus as well, since the 2013 was also compiled with a specific focus (articles selected for the 2013 GE corpus were retrieved from PubMed Central using the keywords “NFkB”, “pathway”, and “regulation”). The number of articles annotated in the 2013 corpus is limited (e.g., 14 articles were annotated as the test set). Notably, while the new role, cause, is annotated for phosphorylation events in the 2013 corpus, there are few instances annotated in the corpus.
The goal in our project and the requirement for RLIMS-P is to support the database curation of phosphorylation information. For instance, curation of kinases as well as substrates and sites is important in our practical goal. The GE corpus with a specific biological focus and different annotation criteria is limited for our goal of developing and evaluating a phosphorylation IE system to support biocuration. We felt it would be desirable to have a corpus with a wider coverage of focus and patterns. For these reasons, we decided to create an annotated corpus, consisting of diverse MEDLINE abstracts, specifically for phosphorylation information extraction. In the next section, we describe the development of this in-house corpus.
4.2.2 Preparation of the Corpus
In order to obtain different types of textual forms of phosphorylation mentions, we collected and annotated three different kinds of MEDLINE abstract sets, with a focus on kinase, substrate, and site, respectively. The set focusing on kinases was compiled by sampling abstracts pertaining to kinases. To that end, PAK1, PAK2, and PAK3 proteins were selected, and MEDLINE abstracts referring to these proteins and containing phosphorylation triggers were collected. Similarly, the set focusing on substrates was compiled by sampling abstracts pertaining to AXIN1, CTNB1, and EFI4EBP1. In order to collect abstracts rich in phosphorylation site mentions, PMIDs referenced in the records of Phospho.ELM database were sampled, and the respective MEDLINE abstracts were retrieved.
For the kinase, substrate, and site collections, 45, 45, and 60 abstracts, respectively, were randomly selected. These abstracts were annotated by two expert curators. Annotation criteria have been discussed and developed over an additional collection of abstracts prior to the annotation of these evaluation sets. Annotation discrepancies found on the evaluation corpora were discussed by the curators and they were resolved jointly.
4.2.3 Evaluation Results
The evaluation results can be found in Table 2. The overall performance on the in-house MEDLINE corpus was comparable with that observed on the BioNLP-ST GE corpus for both substrates and sites.
Table 2. Evaluation Results on the In-House Corpora.
| Kinase | Substrate | Site | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
||||||||
| Prec. | Rec. | F-score | Prec. | Rec. | F-score | Prec. | Rec. | F-score | ||
| MEDLINE (abstract) corpus | Kinase-based | .96 | .90 | .93 | .97 | .93 | .95 | .94 | .95 | .95 |
| Substrate-based | .68 | .88 | .76 | .95 | .90 | .93 | 1.0 | .98 | .99 | |
| Site-based | .92 | .92 | .92 | .94 | .88 | .90 | .97 | .93 | .95 | |
| Total | .91 | .91 | .91 | .95 | .89 | .92 | .96 | .94 | .95 | |
| Full-text corpus | .88 | .88 | .88 | .93 | .89 | .91 | .94 | .91 | .92 | |
Prec: Precision = True Positives/(True Positives + False Positives), Rec: Recall = True Positives/(True Positives + False Negatives), F-score = 2 × Precision × Recall/(Precision + Recall)
The system performance appears to differ within the three sets of the in-house corpus (kinase-based, substrate-based, and site-based). In particular, extraction of kinases in the substrate-based set lagged behind (F-score of 0.76). This could be attributed to the fact that there were only a small number of kinases annotated in the substrate-based set and the resulting measure could have a high variance, i.e., 21 kinases annotated in the substrate-based set, as opposed to 132 and 157 kinases annotated in the kinase-based and site-based corpus. The high F-score for site extraction on this set (0.99) may be explained, at least in part, for the same reason, i.e., 42 sites annotated in the substrate-based set, as opposed to 80 and 225 sites annotated in the kinase-based and the site-based corpus. The skewed distribution across the three sets shows the significance of sampling different kinds of documents when evaluating phosphorylation IE systems, and developing systems as well.
4.2.4 Comparison of the Corpora
We analyzed our in-house abstract corpus and compared it with abstracts in the training corpus of the BioNLP GE task (therefore, abstracts used in the 2009 and 2011 GE task). The following differences were observed:
In the GE corpus, 31 percent of triggers annotated with substrates are also annotated with sites. Few instances among them are annotated with positions, in addition to amino acid types. In the training corpus, nine triggers were annotated with positions (21 percent of the triggers annotated with the sites). In contrast, in the in-house corpus, 46 percent of triggers are annotated with sites, and 78 percent of them involve site positions (289 triggers).
In the GE corpus, there are only six triggers (4 percent), for which the substrate is not in the same sentence as the trigger. In five of these cases, however, anaphoric expressions referring to the substrates (in the previous sentences) do appear in the same sentence as the trigger. This motivated some participants in the GE task to focus only on relations reported within a sentence [42]. In the in-house corpus, cross-sentence relations (not including anaphoric expressions) are more frequently annotated. For example, they constitute 15 percent of trigger-substrate relations in the site-focused set, e.g., “These results indicated that the phosphorylation of serine 202 was necessary” [PMID 15133036] where the phosphorylation site “serine 202” is known to belong to GFAT2 from a preceding sentence (“The protein sequence around the serine 202 of GFAT2 was …”).
- In the GE corpus, majority of trigger-argument relations for phosphorylation events can be captured by using just a handful of patterns. For instance, assuming phrase coordination and anaphoric relations are properly handled, five major patterns could cover 90 percent of trigger-substrate relations in the training corpus. These five patterns are:
- - phosphorylation of <substrate>
- - <substrate> phosphorylation
- - <substrate> (be) phosphorylated
- - phosphorylate <substrate>
- - phosphorylated (form of) <substrate>.
In the in-house corpus, these five patterns only cover about 55 percent of the relations, and the rest of the relations require more patterns or different extraction techniques discussed in this paper, including extraction of implicit relations (“… by phosphorylation”), linking relations, and feature-based extractions (focused entities in the discourse).
These observations support our belief that the in-house corpus might better serve the purpose of training and evaluation of phosphorylation IE systems than the BioNLP-ST GE corpus.
4.3 In-House Full-Text Corpus
One of the goals in redesigning the RLIMS-P system was to adapt the system to full-text article mining. Apart from the low-level text processing implemented, we facilitate mining of phosphorylation information by treating each article section as a separate document, just as the MEDLINE abstract. Writing styles in the full-text articles, however, can be different from those in abstracts [43] and that might affect the performance of RLIMS-P. To evaluate the system in processing full-text articles, we prepared an annotated full-text corpus for our earlier work [11]. We report the development of this corpus, and include the RLIMS-P 2.0 evaluation results on this corpus (Table 2).
4.3.1 Preparation of the Corpus
A collection of sections derived from 100 full-text articles has been prepared. These articles were sampled among the document set originally compiled for the BioCreative III Interactive Text Mining (IAT) Task [44]. Abstract, Results, Discussion and Conclusion sections containing (potential) trigger words were extracted for annotation. The annotation is restricted to these sections because we are interested in capturing facts from experimental results for the database curation purpose. The resulting collection consists of 264 sections. The corpus was annotated by two expert curators, and any discrepancies in the annotation were discussed and resolved. We believe this corpus would be a useful resource in the field as it consists of a large number of diverse documents and target entities, compared to the 2013 BioNLP-ST GE corpus. Again, like with the in-house abstract corpus, it is annotated for substrate, site and kinase.
4.3.2 Evaluation Results
The evaluation results are shown in Table 2. The performance on substrate was comparable with those obtained on MEDLINE sets, although the performance for kinase and site dropped a little.
The slight decrease in performance for site is likely due to the less constrained writing in full-text articles, including uncommon and/or more complex expressions, e.g., “Tyr402 and ERK1/2 phosphorylation” where a site belonging to a particular protein is syntactically coordinated with another protein or “the phosphorylation-deficient mutant at S369-S373-S377 […] mutations at the other four phosphorylation sites” where the coreference resolution necessary for the site mention is difficult (finding five sites and then excluding a specific site at “S369-S373-S377” to report the remaining four). The decreased performance for kinase can also be attributed to the similar diversity in expressions.
4.4 Large-Scale Text Mining
We ran RLIMS-P 2.0 on all the abstracts from the MEDLINE database as well as all the full-length articles from the PMC OA database. These results are stored in our local database for efficient retrieval and are being made publicly available through the RLIMS-P website. In this section, we discuss several aspects of the information extracted from these two data collections.
4.4.1 IE from MEDLINE Abstracts
We have applied RLIMS-P 2.0 to all the abstracts (titles and abstracts) available in the entire MEDLINE database (2013 MEDLINE Baseline Database released by National Library of Medicine). As majority of the abstracts in MEDLINE do not concern with protein phosphorylation, we first filtered the abstracts based on the presence of selected trigger words. The abstracts with the trigger words (about 1 percent of the MEDLINE records) were processed with RLIMS-P 2.0, and phosphorylation information (substrates) was detected in over 150,000 abstracts with substrate proteins. In 16 percent of these abstracts, phosphorylation site (with positions) were detected, and in 23 percent of them, kinase information was found.
4.4.2 IE from PMC OA Full-Text Articles
Our evaluation study on the in-house full-text corpus suggests that there is rich information in the full-text article body. In particular, we expected to find more detailed phosphorylation information, namely site and kinase information in full-text articles. In order to verify this in a larger sample of articles, we analyzed the entire PMC OA subset containing 682,000 articles, downloaded in fall of 2013. After filtering the input records based on the presence of trigger words, we obtained a set of 78,000 articles. RLIMS-P 2.0 detected phosphorylation information in 45,000 articles. Of over 300,000 tuples (tuples unique in each section) that were found in those articles, 55 percent were detected in the results/discussion sections. The remaining instances were distributed in introduction/background (14 percent), figure caption (12 percent), materials/methods (10 percent), abstract (8 percent), and conclusion (<1 percent).
In those articles, RLIMS-P 2.0 detected 42,000 kinases, each of which is unique in the article. As for phosphorylation sites, it reported 37,000 amino acids with positions, each of which is unique within an article. Of them, 91 percent were found in the bodies of the full-text articles, but absent in the corresponding abstracts. In this regard, we reviewed the full-text corpus annotated in-house and found that there were 98 site mentions, including 61 unique positions. Among them, 75 percent are mentioned only in the body of the full-text articles (i.e., Results, Discussion, and Conclusion sections). This, as well as the observation on the PMC OA subset, confirms the significance and the impact of mining full-text articles for phosphorylation site information. The results further suggest that full-text processing would benefit mining of other types of information involving phosphorylation, such as the impact of phosphorylation on protein-protein interactions [45].
On the PMC OA subset, we further investigated the significance of mining information additionally from figure captions. Notably, 20 percent of the detected sites were found in the figure captions, where 24 percent of those were extracted only from the figure captions.
5 Conclusion
In this work we designed an enhanced, generalizable architecture of a rule-based IE engine, and implemented it for phosphorylation IE in RLIMS-P 2.0. State-of-the-art performance was observed on the 2013 BioNLP-ST GE test corpus for phosphorylation information. A second contribution of this work was the creation of several annotated corpora that can be used for training and evaluating phosphorylation IE systems. Our analysis revealed the need for such corpora that cover a diverse range of patterns of phosphorylation mentions. Three sets of MED-LINE abstracts focusing on different aspects were compiled and annotated by expert curators. Additionally, a large set of text collection from full-text articles was annotated. RLIMS-P 2.0 show uniformly good performance across these corpora, indicating that the system is robust and it adapts well to phosphorylation IE from various types of documents, including full-text articles.
Finally, RLIMS-P 2.0 was applied to the entire collection of MEDLINE abstracts and PMC OA full-text articles. The results confirm that rich phosphorylation information is available in full-text articles and RLIMS-P 2.0 can be used to help curators retrieve and annotate the information. A web interface to RLIMS-P 2.0 has been developed [46] and made publicly available at the PIR website [47].1 All literature corpora are available at the PIR iProLINK website as well.2
In summary, the current study demonstrates both the good performance and scalability of RLIMS-P 2.0 for full-scale mining of protein phosphorylation information in abstracts and in full-text articles, enabling its adoption for biocuration and for knowledge discovery [48], [49]. Indeed, RLIMS-P 2.0 has been evaluated by curators from PhosphoGrid [49], Phospho.ELM [4] and Protein Ontology (PRO) [2] in BioCreative Interactive Text Mining task [46], [51], and it has been integrated into their curation workflows. The current work focuses on protein phosphorylation information, but the IE pipeline employing the enhanced, generalizable architecture can be readily ported to the extraction of PTM types other than phosphorylation. We are in the process of porting RLIMS-P for several other PTM types, including acetylation, ubiquitination, methylation, and glycosylation.
Acknowledgments
Research reported in this article was supported by the National Library of Medicine of the National Institutes of Health under award number G08LM010720. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This material is also based upon work supported by the National Science Foundation under Grant No. ABI-1062520. The authors thank the BioNLP workshop organizer for making the annotated corpora publicly available and the NLM for the MEDLINE and PMC datasets. The authors acknowledge the work of Drs. Narayanaswamy and Ravikumar, who played an integral role in the development of the original RLIMS-P system. They also thank Mr. Yifan Peng, Dr. Oana Tudor, Ms. Amy Siu and other members of the text mining group at the University of Delaware for discussion and contribution to the system development.
Biographies

Manabu Torii received the PhD degree in computer science from the University of Delaware in 2006. He was a postdoctoral fellow in the Department of Biostatistics, Bioinformatics, and Biomathematics in Georgetown University Medical Center (GUMC) and subsequently joined the Imaging Science and Information Systems (ISIS) Center at GUMC as a research assistant professor. At the time of this work, he was a research assistant professor in the Department of Computer and Information Sciences and the Center for Bioinformatics and Computational Biology (CBCB) at the University of Delaware. He is currently a scientist in the Medical Informatics group at Kaiser Permanente Southern California. His research interests include natural language processing, machine learning, and their application in the biomedical and clinical domain.

Cecilia N. Arighi received the PhD degree in biochemistry from the University of Buenos Aires in 2001. She was awarded the Latin American PEW fellowship to conduct a postdoctoral research at the National Institute of Child Health and Human Development, NIH, and subsequently became a research assistant professor at the Protein Information Resource, first at GUMC and then at the University of Delaware. She is currently a research associate professor in the Department of Computer and Information Sciences and CBCB at the University of Delaware. She has been actively engaged in various biocuration-related project including UniProt, the Protein Ontology, and the BioCreative challenges. Her research interest is in the accurate representation of protein information (e.g., sequence, evolution, function, post-translational modifications, and pathways), that can be reasoned both by humans and computers to provide the basis for hypothesis generation.

Gang Li received the bachelor's degree from the Beijing Institute of Technology in 2011. He is cur rently working toward the PhD degree from the Department of Computer and Information Sciences at the University of Delaware. His research interests include biomedical text mining, natural language processing, and machine learning.

Qinghua Wang received the PhD degree in biochemistry, molecular biology and biophysics from the University of Minnesota in 2005 with a focus on structural biology. She was a postdoctoral fel low at the University of Massachusetts Amherst and studied the complex in-cell protein interactions from 2006 to 2009. Since 2010, she has been a member of CBCB, PIR, and the Department of Computer and Information Sciences at the University of Delaware. She is currently an associate scientist, and has contributed to various biocuration-related projects such as curation of Swiss-Prot entries and PIR Site Rules for UniProtKB database, and the development and evaluation of text mining tools. In addition, she studies cellular and molecular mechanisms with omics-data analysis and other bioinformatic methods. Her research interests include bioinformatics, structural biology, systems biology, and text mining.

Cathy H. Wu received the PhD degree from Purdue University in 1984. She is the Edward G. Jefferson chair and director of CBCB at the University of Delaware. She is also the director of PIR. She has conducted bioinformatics research for 25 years and is the PI/Co-PI on several consortium projects, including the UniProt and the Protein Ontology. She serves on several advisory boards, including the ACM SIGBio, and has served on more than 50 international conference organizing committees. Her research encompasses protein functional annotation, biomedical text mining and ontology, systems biology, and translational bioinformatics. She has published more than 200 peer-reviewed papers and 12 books, conference proceedings and journal special issues, as well as given more than 150 invited talks.

K. Vijay-Shanker received the PhD degree in computer science from the University of Pennsylvania in 1987. He joined the Department of Computer Science at the University of Delaware and currently a professor in the department. He served as an editorial board member of the journals Grammars and Computational Linguistics. He has been a committee member for a number of academic conferences, including his role of cochair for the 38th Annual Meeting of the Association of Computational Linguistics and the fourth Workshop on Tree-Adjoining Grammars and Related Formalisms, TAG+, and the role of area chair (Grammars and Parsing) for the 37th Annual Meeting of the Association of Computational Linguistics.
Footnotes
Contributor Information
Manabu Torii, Email: manabu.torii@kp.org, Medical Informatics Group, Kaiser Permanente Southern California, 11975 El Camino Real, San Diego, CA 92130.
Cecilia N. Arighi, Email: arighi@dbi.udel.edu, Center for Bioinformatics & Computational Biology, University of Delaware, 15 Innovation Way, Newark, DE 19711.
Gang Li, Email: ligang@udel.edu, Center for Bioinformatics & Computational Biology, University of Delaware, 15 Innovation Way, Newark, DE 1971.
Qinghua Wang, Email: wangq@dbi.udel.edu, Center for Bioinformatics & Computational Biology, University of Delaware, 15 Innovation Way, Newark, DE 19711.
Cathy H. Wu, Email: wuc@dbi.udel.edu, Center for Bioinformatics & Computational Biology, University of Delaware, 15 Innovation Way, Newark, DE 19711.
K. Vijay-Shanker, Email: vijay@udel.edu, Department of Computer and Information Sciences, University of Delaware, 101 Smith Hall, Newark, DE 19716.
References
- 1.Hunter T. Why nature chose phosphate to modify proteins. Philos Trans Royal Soc London B Biol Sci. 2012 Sep;367(1602):2513–2516. doi: 10.1098/rstb.2012.0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Natale DA, Arighi CN, Blake JA, Bult CJ, Christie KR, Cowart J, D'Eustachio P, Diehl AD, Drabkin HJ, Helfer O, Huang H, Masci AM, Ren J, Roberts NV, Ross K, Ruttenberg A, Shamovsky V, Smith B, Yerramalla MS, Zhang J, Aljanahi A, Celen I, Gan C, Lv M, Schuster-Lezell E, Wu CH. Protein Ontology: A controlled structured network of protein entities. Nucleic Acids Res. 2013 Nov;42:415–421. doi: 10.1093/nar/gkt1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hornbeck PV, Kornhauser JM, Tkachev S, Zhang B, Skrzypek E, Murray B, Latham V, Sullivan M. PhosphoSitePlus: A comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012 Jan;40(Database issue):D261–270. doi: 10.1093/nar/gkr1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dinkel H, Chica C, Via A, Gould CM, Jensen LJ, Gibson TJ, Diella F. Phospho.ELM: A database of phosphorylation sites–update 2011. Nucleic Acids Res. 2011 Jan;39(Database issue):D261–D267. doi: 10.1093/nar/gkq1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.The UniProt Consortium. Reorganizing the protein space at the Universal Protein Resource (UniProt) Nucleic Acids Res. 2012 Jan;40(Database issue):D71–D75. doi: 10.1093/nar/gkr981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hu ZZ, Narayanaswamy M, Ravikumar KE, Vijay-Shanker K, Wu CH. Literature mining and database annotation of protein phosphorylation using a rule-based system. Bioinformatics. 2005 Jun;21(11):2759–2765. doi: 10.1093/bioinformatics/bti390. [DOI] [PubMed] [Google Scholar]
- 7.Kim JD, Ohta T, Pyysalo S, Kano Y, Tsujii J. Overview of BioNLP'09 shared task on event extraction. Proc Workshop BioNLP: Shared Task. 2009:1–9. [Google Scholar]
- 8.Narayanaswamy M, Ravikumar KE, Vijay-Shanker K. Beyond the clause: Extraction of phosphorylation information from medline abstracts. Bioinformatics. 2005 Jun;21(Suppl 1):i319–327. doi: 10.1093/bioinformatics/bti1011. [DOI] [PubMed] [Google Scholar]
- 9.Veuthey AL, Bridge A, Gobeill J, Ruch P, McEntyre JR, Bougueleret L, Xenarios I. Application of text-mining for updating protein post-translational modification annotation in UniProtKB. BMC Bioinformatics. 2013 Mar;14(1):104. doi: 10.1186/1471-2105-14-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu Y, Teng D, Lei Y. MinePhos: A literature mining system for protein phosphorylation information extraction. IEEEACM Trans Comput Biol Bioinformat. 2011 Apr;9(1):311–315. doi: 10.1109/TCBB.2011.85. [DOI] [PubMed] [Google Scholar]
- 11.Torii M, Arighi CN, Wang Q, Wu CH, Vijay-Shanker K. Text mining of protein phosphorylation information using a generalizable rule-based approach. Proc ACM Conf Bioinformat, Comput Biol Biomed Inf. 2013:201. doi: 10.1109/TCBB.2014.2372765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Altman RB, Bergman CM, Blake J, Blaschke C, Cohen A, Gannon F, Grivell L, Hahn U, Hersh W, Hirschman L, Jensen LJ, Krallinger M, Mons B, O'Donoghue SI, Peitsch MC, Rebholz-Schuhmann D, Shatkay H, Valencia A. Text mining for biology–The way forward: Opinions from leading scientists. Genome Biol. 2008;9(Suppl 2):S7. doi: 10.1186/gb-2008-9-s2-s7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ananiadou S, McNaught J. Text Mining for Biology and Bio-medicine. Boston, MA, USA: Artech House; 2006. [Google Scholar]
- 14.Blaschke C, Valencia A. The functional genomics network in the evolution of biological text mining over the past decade. New Biotechnol. 2013 Mar;30(3):278–285. doi: 10.1016/j.nbt.2012.11.020. [DOI] [PubMed] [Google Scholar]
- 15.de Bruijn B, Martin J. Getting to the (c) ore of knowledge: Mining biomedical literature. Int J Med Inf. 2002 Dec;67(1–3):7–18. doi: 10.1016/s1386-5056(02)00050-3. [DOI] [PubMed] [Google Scholar]
- 16.Cohen KB, Hunter L. Getting started in text mining. PLoS Comput Biol. 2008 Jan;4(1):e20. doi: 10.1371/journal.pcbi.0040020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hersh WR. Information Retrieval: A Health and Biomedical Perspective. New York, NY, USA: Springer; 2010. [Google Scholar]
- 18.Hirschman L, Park JC, Tsujii J, Wong L, Wu CH. Accomplishments and challenges in literature data mining for biology. Bioinformatica. 2002 Dec;18(12):1553–1561. doi: 10.1093/bioinformatics/18.12.1553. [DOI] [PubMed] [Google Scholar]
- 19.Krallinger M, Valencia A, Hirschman L. Linking genes to literature: Text mining, information extraction, and retrieval applications for biology. Genome Biol. 2008;9(Suppl 2):S8. doi: 10.1186/gb-2008-9-s2-s8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rodriguez-Esteban R. Biomedical text mining and its applications. PLoS Comput Biol. 2009 Dec;5(12):e1000597. doi: 10.1371/journal.pcbi.1000597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rzhetsky A, Seringhaus M, Gerstein M. Seeking a new biology through text mining. Cell. 2008 Jul;134(1):9–13. doi: 10.1016/j.cell.2008.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rzhetsky A, Seringhaus M, Gerstein MB. Getting started in text mining: Part two. PLoS Comput Biol. 2009 Jul;5(7):e1000411. doi: 10.1371/journal.pcbi.1000411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shatkay H, Craven M. Mining the Biomedical Literature. Cambridge, MA, USA: MIT Press; 2012. [Google Scholar]
- 24.Simpson MS, Demner-Fushman D. Biomedical text mining: A survey of recent progress. In: Aggarwal CC, Zhai C, editors. Mining Text Data. Boston, MA, USA: Springer; 2012. pp. 465–517. [Google Scholar]
- 25.Zweigenbaum P, Demner-Fushman D, Yu H, Cohen KB. Frontiers of biomedical text mining: current progress. Brief Bio-informat. 2007 Sep;8(5):358–375. doi: 10.1093/bib/bbm045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hunter L, Lu Z, Firby J, Baumgartner WA, Jr, Johnson HL, Ogren PV, Cohen KB. OpenDMAP: An open source, ontology-driven concept analysis engine, with applications to capturing knowledge regarding protein transport, protein interactions and cell-type-specific gene expression. BMC Bioinformatics. 2008;9:78. doi: 10.1186/1471-2105-9-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van Landeghem S, De Bodt S, Drebert ZJ, Inze D, Van de Peer Y. The potential of text mining in data integration and network biology for plant research: A case study on Arabidopsis. Plant Cell. 2013 Mar;25(3):794–807. doi: 10.1105/tpc.112.108753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saric J, Jensen LJ, Ouzounova R, Rojas I, Bork P. Extraction of regulatory gene/protein networks from Medline. Bioinformatica. 2006 Mar;22(6):645–650. doi: 10.1093/bioinformatics/bti597. [DOI] [PubMed] [Google Scholar]
- 29.Kim JD, Nguyen N, Wang Y, Tsujii J, Takagi T, Yonezawa A. The genia event and protein coreference tasks of the BioNLP shared task 2011. BMC Bioinformat. 2012;13(Suppl 11):S1. doi: 10.1186/1471-2105-13-S11-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim JD, Pyysalo S, Ohta T, Bossy R, Nguyen N, Tsujii J. Overview of BioNLP Shared Task 2011. Proc BioNLP Shared Task 2011 Workshop. 2011:1–6. [Google Scholar]
- 31.Kim JD, Wang Y, Yamamoto Y. The genia event extraction shared task. Proc BioNLP Shared Task 2013 Workshop. 2013:8–15. [Google Scholar]
- 32.Kilicoglu H, Bergler S. Biological event composition. BMC Bioinformatics. 2012;13(Suppl 11):S7. doi: 10.1186/1471-2105-13-S11-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu ZZ, Mani I, Hermoso V, Liu H, Wu CH. iProLINK An integrated protein resource for literature mining. Comput Biol Chem. 2004 Dec;28(5/6):409–416. doi: 10.1016/j.compbiolchem.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 34.Barker WC, Garavelli JS, McGarvey PB, Marzec CR, Orcutt BC, Srinivasarao GY, Yeh LS, Ledley RS, Mewes HW, Pfeiffer F, Tsugita A, Wu C. The PIR-international protein sequence database. Nucleic Acids Res. 1999 Jan;27(1):39–43. doi: 10.1093/nar/27.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Narayanaswamy M, Ravikumar KE, Vijay-Shanker K. A biological named entity recognizer. Proc Pac Symp Biocom-put. 2003:427–438. doi: 10.1142/9789812776303_0040. [DOI] [PubMed] [Google Scholar]
- 36.Peng Y, Tudor CO, Torii M, Wu CH, Vijay-Shanker K. iSimp: A sentence simplification system for biomedicail text. Proc IEEE Int Conf Bioinformat Biomed. 2012:1–6. [Google Scholar]
- 37.Pyysalo S, Ohta T, Rak R, Sullivan D, Mao C, Wang C, Sobral B, Tsujii J, Ananiadou S. Overview of the ID, EPI and REL tasks of BioNLP Shared Task 2011. BMC Bioinformatics. 2012;13(Suppl 11):S2. doi: 10.1186/1471-2105-13-S11-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ravikumar K, Liu H, Cohn JD, Wall ME, Verspoor K. Literature mining of protein-residue associations with graph rules learned through distant supervision. J Biomed Semantics. 2012 Oct;3(Suppl 3):S2. doi: 10.1186/2041-1480-3-S3-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Van Landeghem S, Bjorne J, Abeel T, De Baets B, Salakoski T, Van de Peer Y. Semantically linking molecular entities in literature through entity relationships. BMC Bioinformatics. 2012;13(Suppl 11):S6. doi: 10.1186/1471-2105-13-S11-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mizuta Y, Korhonen A, Mullen T, Collier N. Zone analysis in biology articles as a basis for information extraction. Int J Med Inf. 2006 Jun;75(6):468–487. doi: 10.1016/j.ijmedinf.2005.06.013. [DOI] [PubMed] [Google Scholar]
- 41.Teufel S, Moens M. Summarizing scientific articles: Experiments with relevance and rhetorical status. Comput Linguistic. 2002 Dec;28(4):409–445. [Google Scholar]
- 42.Bjorne J, Heimonen J, Ginter F, Airola A, Pahikkala T, Salakoski T. Extracting complex biological events with rich graph-based features sets. Proc Workshop BioNLP: Shared Task. 2009:10–18. [Google Scholar]
- 43.Cohen KB, Johnson HL, Verspoor K, Roeder C, Hunter LE. The structural and content aspects of abstracts versus bodies of full text journal articles are different. BMC Bioinformatics. 2010;11:492. doi: 10.1186/1471-2105-11-492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arighi CN, Roberts PM, Agarwal S, Bhattacharya S, Cesareni G, Chatr-Aryamontri A, Clematide S, Gaudet P, Giglio MG, Harrow I, Huala E, Krallinger M, Leser U, Li D, Liu F, Lu Z, Maltais LJ, Okazaki N, Perfetto L, Rinaldi F, Sætre R, Salgado D, Srinivasan P, Thomas PE, Toldo L, Hirschman L, Wu CH. BioCreative III interactive task: An overview. BMC Bioinformatics. 2011;12(Suppl 8):S4. doi: 10.1186/1471-2105-12-S8-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tudor CO, Arighi CN, Wang Q, Wu CH, Vijay-Shanker K. The eFIP system for text mining of protein interaction networks of phosphorylated proteins. Database J Biol Databases Curation. 2012;2012:bas044. doi: 10.1093/database/bas044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Torii M, Li G, Li Z, Oughtred R, Diella F, Celen I, Arighi CN, Huang H, Vijay-Shanker K, Wu CH. RLIMS-P: An online text-mining tool for literature-based extraction of protein phosphorylation information. Database J Biol Databases Curation. 2014;2014:bau081. doi: 10.1093/database/bau081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu C, Nebert DW. Update on genome completion and annotations: Protein information resource. Human Genomics. 2004 Mar;1(3):229–233. doi: 10.1186/1479-7364-1-3-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ross KE, Arighi CN, Ren J, Natale DA, Huang H, Wu CH. Use of the protein ontology for multi-faceted analysis of biological processes: A case study of the spindle checkpoint. Front Genetics. 2013;4:62. doi: 10.3389/fgene.2013.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ross KE, Arighi CN, Ren J, Huang H, Wu CH. Construction of protein phosphorylation networks by data mining, text mining, and ontology integration: analysis of the spindle checkpoint. Database J Biol Databases Curation. 2013:bat038. doi: 10.1093/database/bat038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stark C, Su TC, Breitkreutz A, Lourenco P, Dahabieh M, Breitkreutz BJ, Tyers M, Sadowski I. PhosphoGRID: A database of experimentally verified in vivo protein phosphorylation sites from the budding yeast Saccharomyces cerevisiae. Database J Biol Databases Curation. 2010;2010:bap026. doi: 10.1093/database/bap026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Matis-Mitchell S, Roberts P, Tudor CO, Arighi CN. BioCreative IV interactive task. Proc 4th BioCreative Challenge Evaluation Workshop. 2013:190–203. [Google Scholar]