

Abstract
NRAS-mutant melanomas are extremely aggressive and highly resistant to currently available therapeutic modalities. Hence, new targets and therapeutic strategies for NRAS-driven melanomas are needed. As blocking NRAS directly has not been possible thus far, targeting downstream NRAS effectors, such as MEK, is being evaluated as an alternative therapeutic approach. However, blocking this pathway alone has limited efficacy. In this issue, Posch et al. report on a combination approach co-targeting PLK1 and MEK in NRAS-mutant melanomas. This combination triggers a dual blockade of the cell cycle machinery, leading to apoptosis, and it may provide a new strategy to treat NRAS-mutant melanoma.
Mutant NRAS in melanoma
RAS is mutated in approximately 30% of human cancers. For example, neoplasms of the skin, pancreas, and urinary tract carry activating mutations in the RAS isoforms NRAS, KRAS, and HRAS, respectively (Prior et al., 2012). In melanoma, approximately 25% of tumors harbor NRAS mutations. Most NRAS mutations affect codon 61, locking the small G protein in the active GTP-bound form and leading to persistent RAS signaling (Ascierto et al., 2013; Burd et al., 2014). Oncogenic NRAS activates several signaling pathways, including the mitogen activating protein kinase (MAPK), PI3K/mTOR, and Ral GDP dissociation stimulator (RalGDS), resulting in aberrant cell proliferation and increased tumor cell survival. Attempts to inhibit oncogenic NRAS directly have not been successful to date, prompting a search for alternative strategies to blunt NRAS signaling. Suppression of the NRAS effector pathway MAPK (RAF/MEK/ERK) with MEK inhibitors (MEKi) has been evaluated in clinical trials of NRAS mutant melanoma; however, response rates were barely 20% and short-lived (Ascierto et al., 2013). Immune checkpoint inhibitors are only approved for BRAF-mutant patients thus far, and their role in treating NRAS-mutant melanoma remains to be established. Additionally, it has been reported recently that NRAS mutant melanomas are associated with lower levels of lymphocyte infiltration, suggesting a more immunosuppressive microenvironment and possibly poor responses to immunotherapy (Thomas et al., 2015). Consequently, identifying novel targets and/or cotargeting other NRAS-specific vulnerabilities are essential for designing effective treatments for these types of tumors. In this issue of the Journal of Investigative Dermatology, Posch et al. (2015) report on suppressing a mitotic master Ser/Thr kinase, polo-like kinase 1 (PLK1) in combination with MEK inhibition as a therapeutic strategy for NRAS-mutant melanoma (Posch et al., 2015).
Polo-like kinase: a new therapeutic target for NRAS-mutant melanoma
PLK1 has emerged as a therapeutic target in cancer as it regulates cell cycle progression and genome integrity, and it is highly expressed in many types of tumors. Expression of PLK1 is inversely correlated with patients’ survival in non-small cell lung, head and neck, and esophageal cancer (Yim and Erikson, 2014). PLK1 plays a key role in M phase progression; it regulates centrosomal maturation, mitotic spindle assembly, chromosomal segregation, and cytokinesis. During the G2/M transition, PLK1 phosphorylates the mitotic regulator cyclinB-Cdk1 and the phosphatase Cdc25, which activates Cdk1, facilitating mitotic entry (Yim and Erikson, 2014). In addition, PLK1 plays a key role in maintaining DNA integrity and DNA damage responses (Yim and Erikson, 2014).
Preclinical studies indicate that PLK1 is, indeed, a promising therapeutic target, especially for RAS-mutant tumors (Luo et al., 2009; Yim and Erikson, 2014). Luo, Elledge, and colleagues identified essential mitotic genes, including PLK1, in a genome-wide short hairpin RNA (shRNA) screen in human KRAS-mutant cancer cells (Luo et al., 2009). Inhibition of mitotic regulators such as PLK1 led to profound G2/M arrest and caused synthetic lethality in KRAS-mutant tumor cells. Likewise, genetic or pharmacological inhibition of PLK1 caused apoptosis selectively in KRAS-mutated tumors (Yim and Erikson, 2014), consistent with the notion that RAS oncogenes create mitotic stress (Luo et al., 2009). This suggests that these types of tumors could be hypersensitive to mitotic disruption. PLK1 inhibitors (PLK1i) have been evaluated in clinical trials as treatment for advanced malignancies, including acute myeloid leukemia (AML) and solid tumors such as non-small cell lung, pancreatic, prostate, ovarian, and urothelial cancer (Gjertsen and Schoffski, 2015). Compounds currently under clinical evaluation include NMS-P937, GSK461364, BI2536, and BI6727 (volasertib) (Strebhardt, 2010). Notably, the potent and selective PLK1 inhibitor volasertib has been granted breakthrough therapy designation by the Food and Drug Administration (FDA).
Previous studies in melanoma also support the promise of PLK1 as a therapeutic target. PLK1 is overexpressed in cultured melanoma cell lines and in patient samples (Schmit et al., 2009). Inhibition of PLK1 by shRNA or small molecule inhibitors induces cell cycle arrest, mitotic catastrophe, and apoptosis in melanoma cells (Schmit et al., 2009). Posch and colleagues targeted PLK1 and MEK simultaneously, as a potential therapeutic strategy for NRAS-driven melanoma (Posch et al., 2015). The investigators demonstrated increased PLK1 expression in NRAS-mutant melanomas; PLK1 mRNA levels were higher in NRAS mutant melanoma cell lines compared to levels expressed in cells lines harboring wild-type (WT) NRAS. Furthermore, ectopic expression of mutant NRAS in melanocytes upregulated PLK1 mRNA and protein levels. The increased expression of PLK1 was notably associated with the most frequent NRAS mutation (Q61) in melanoma (Ascierto et al., 2013; Burd et al., 2014).
These findings prompted the investigators to evaluate the value of cotargeting PLK1 and the MAPK pathway, which is often activated in melanoma and seems to play a critical role in NRAS-mutant melanoma. Inhibition of PLK1 or MEK alone impacted the expression of cell cycle regulatory genes, triggering proliferation arrest. Specifically, the MEK inhibitor trametinib led to a G0/G1 arrest, whereas the PLK1i volasertib (BI6727) arrested the cells in G2/M. Notably, volasertib synergized with trametinib triggering a dual cell cycle arrest and increased apoptosis in melanoma cell lines and xenograft models. The investigators further demonstrated that p53 depletion or CHK inhibition abrogated the effectiveness of this combination, suggesting that the effect of PLK1 inhibitors might be partially dependent on the CHK/p53 axis.
Mechanistic explanations
One of the key effectors of NRAS is the MEK/MAPK pathway. Yet, inhibition of MEK alone often elicits inadequate and/or incomplete responses in tumors with NRAS mutations. Combination therapies including MEK inhibitors have been evaluated in NRAS-mutated melanoma. In particular, combinations of MEK inhibitors with CDK4/6 or PI3K/mTOR inhibitors are being tested in preclinical studies and in clinical trials (Kwong et al., 2012; Posch et al., 2013). Unfortunately, the efficacy of these combinations is frequently limited by toxicity. Furthermore, responses to these combinations are often dependent on the status of other concurrent genetic alterations; for example, loss of p16/CDKN2A can modulate the response to a MEK/CDK1 combination. The current study by Posch et al. demonstrated that concurrent blockade of the central mitotic kinase PLK1 and the NRAS downstream effector MEK induces apoptosis synergistically in NRAS-mutant melanoma cells. PLK1 inhibitors likely synergize with MEK inhibitors by two mechanisms: (1) independent dual cell cycle arrest: while MEK inhibition predominantly causes G1 arrest, PLK inhibitors lead to a G2/M arrest; and (2) increased induction of apoptosis. By combining PLK1i with MEKi, cells that might escape from arrest in one phase of the cell cycle can be trapped in the other. Hence, this dual cell cycle blockade would be more effictive than strategies that arrest cells in a single phase. Because PLK1 plays key roles in DNA damage repair and cell cycle progression, it is possible that PLK1 inhibition might induce apoptosis by triggering mitotic catastrophe. Of note, missense mutations in PLK1 are found in approximately 2.5 % of melanomas (cBioPortal). However, it appears that the effects of PLK1 blockade are independent of PLK1 mutation status, although the studies that support this effect included a limited number of melanomas with PLK1 mutations.
Several studies have revealed a link between PLK1 and the tumor suppressor p53, whereby the two proteins regulate each other in a negative fashion: while phosphorylation of p53 by PLK1 inhibits its activity, p53 transcriptionally represses PLK1 expression (Yim and Erikson, 2014). Posch and colleagues propose that the efficacy of PLK1i is somewhat dependent on p53, as silencing of p53 diminished the effect of the PLK1i and MEK/PLK1i combination. It is important to mention that although mutations in p53 are infrequent in melanoma, the tumor suppressor is often inactivated through different mechanisms, such as overexpression of its negative regulator MDM2/4. In contrast to the findings in Posch et al., previous studies have suggested that loss of p53 is associated with sensitivity to PLK1i (Yim and Erikson, 2014); the underlying reason for this tumor or drug-specific difference is not yet well defined, suggesting a need for additional investigation. To extend this paradigm to other NRAS-driven cancers, the authors also explored this combination in neuroblastoma and lung cancer and showed encouraging results. Overall, this study demonstrates a new paradigm for NRAS-driven tumors, one that warrants further scrutiny.
Perspective and future directions
Targeting the cell cycle seems to be a promising approach in treating NRAS-mutant melanoma. For example, a phase 1b/2 study combining LEE011, an inhibitor of the G1 phase cyclin dependent kinases CDK4/6, with the MEK inhibitor MEK162 (NCT01719380) showed favorable antitumor activity in patients with NRAS mutant melanoma (Sosman et al., 2014). However, because this combination causes mainly a G1 phase cell cycle arrest, it is plausible that a subset of tumor cells will escape drug-induced G1 blockade, leading to transient responses and eventually to tumor recurrence. Hence, the strategy proposed by Posch et al., hitting the cell cycle machinery at two different phases, may offer a more effective approach to induce robust and persistent cell cycle arrest.
Because trametinib and PLK1i are undergoing clinical investigation, this combination could be translated into treatment strategies for patients with melanoma. However, additional rigorous preclinical studies that take into account the complexity, plasticity, and heterogeneity of melanoma will be needed to support such trials. Besides identifying a promising combination therapy, this study also raises questions that merit further investigation. For example, it would be interesting to determine whether PLK1 is a mediator of NRAS oncogenic activity or if PLK1 mitigates stress created by oncogenic NRAS. Moreover, a number of studies indicate that PLK1 has non-mitotic functions. For instance, it has been suggested that PLK1 can regulate PI3K and mTORC1/2 (Gjertsen and Schoffski, 2015). Are any of the effects observed in this study mediated by the RAS downstream effectors PI3K or mTORC1/2? Because PLK1 has been associated with melanoma metastasis (Kneisel et al., 2002), would PLK1 inhibition affect metastasis? Furthermore, when using ATP-competitive PLK1 inhibitors such as BI2536 and BI6727, the functions of other PLK family members should be considered, as some of these drugs can also inhibit PLK2 and PLK3 (Strebhardt, 2010). This is important because PLK2 and PLK3 are considered tumor suppressors. Additionally, it has been reported that PLK2/3 can mediate DNA and oxidative stress responses in cancer (Strebhardt, 2010); hence, suppressing these PLK isoforms could counteract the efficacy of PLK1 inhibition. If this were the case, more selective PLK1 inhibitors may be needed. It is also worth noting that the mechanism by which NRAS regulates PLK1 remains to be determined. In addition, it is not yet known if other driver genes upregulate PLK1 or if other genetic alterations (such as loss of PTEN, which positively regulates PLK1 (Yim and Erikson, 2014), affect response to PLK inhibitors.
Other issues need further evaluation before these findings can be translated from bench to bedside. For example, while PLK1 inhibitors seem to have a manageable safety profile (Gjertsen and Schoffski, 2015), the tolerability of MEK/PLK1i combinations remains to be determined. In addition, identification of predictive biomarkers of responses to MEK/PLK1i will be valuable. Certainly, assessing the efficacy of this combination in the context of a functional immune system would be absolutely necessary. Answering these questions would provide valuable information to advance this promising combination and to provide rational and effective treatment options for patients with NRAS-mutant melanoma.
Figure 1. PLK1 inhibitors synergize with MEK inhibitors in NRAS(Q61) mutant melanoma.
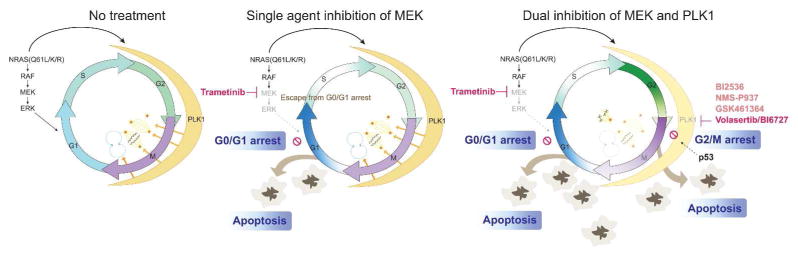
NRAS (Q61) mutants activate the MAPK pathway and increase PLK1 expression (thick black arrows). PLK1 promotes centrosomal maturation, spindle assembly, chromosomal segregation and cytokinesis (orange arrows). Suppression of MEK (magenta) leads to cell cycle arrest in G0/G1 and apoptosis. Inhibition of PLK1 synergizes with MEKi. PLK1i (volasertib; magenta) arrests cells in G2/M and traps cells that escape from G0/G1 by MEKi. PLK1i can also cause monopolar spindles and mitotic catastrophe, leading to apoptosis. This activity may be partly mediated by p53. Faded colors indicate suppression of MAPK pathway activity and M phase progression. Selected PLK1 inhibitors currently in clinical trials are included (pink). Arrowheads indicate stimulation. Blunt lines indicate inhibition.
CLINICAL IMPLICATIONS.
New therapies have emerged for BRAF-mutant melanoma, but NRAS-mutant melanoma continues to have poor prognosis and limited therapeutic options.
PLK1 has emerged as a promising therapeutic target in cancer. Oncogenic NRAS induces high expression of PLK1, providing the rationale to target this kinase in melanoma.
An array of small molecule PLK1 inhibitors are undergoing clinical investigation, making the translation of this approach feasible.
Co-inhibition of MEK- and PLK1-triggered dual cell cycle blockade may be superior to drug combinations that arrest tumor cells in a single cell cycle phase.
Acknowledgments
Work in our laboratory is supported by NIH/NCI grants K01 CA175269, PO1 CA114046, P30 CA010815, the American Cancer Society, the V Foundation for Cancer Research, and the Melanoma Research Alliance.
Footnotes
Conflict of Interest
The authors state no conflict of interest.
References
- Ascierto PA, Schadendorf D, Berking C, et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. The Lancet Oncology. 2013;14:249–56. doi: 10.1016/S1470-2045(13)70024-X. [DOI] [PubMed] [Google Scholar]
- Burd CE, Liu W, Huynh MV, et al. Mutation-specific RAS oncogenicity explains NRAS codon 61 selection in melanoma. Cancer discovery. 2014;4:1418–29. doi: 10.1158/2159-8290.CD-14-0729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gjertsen BT, Schoffski P. Discovery and development of the Polo-like kinase inhibitor volasertib in cancer therapy. Leukemia. 2015;29:11–9. doi: 10.1038/leu.2014.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneisel L, Strebhardt K, Bernd A, et al. Expression of polo-like kinase (PLK1) in thin melanomas: a novel marker of metastatic disease. Journal of cutaneous pathology. 2002;29:354–8. doi: 10.1034/j.1600-0560.2002.290605.x. [DOI] [PubMed] [Google Scholar]
- Kwong LN, Costello JC, Liu H, et al. Oncogenic NRAS signaling differentially regulates survival and proliferation in melanoma. Nature medicine. 2012;18:1503–10. doi: 10.1038/nm.2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Emanuele MJ, Li D, et al. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137:835–48. doi: 10.1016/j.cell.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posch C, Cholewa BD, Vujic I, et al. Combined Inhibition of MEK and Plk1 has Synergistic Anti-Tumor Activity in NRAS Mutant Melanoma. The Journal of investigative dermatology. 2015 doi: 10.1038/jid.2015.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posch C, Moslehi H, Feeney L, et al. Combined targeting of MEK and PI3K/mTOR effector pathways is necessary to effectively inhibit NRAS mutant melanoma in vitro and in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:4015–20. doi: 10.1073/pnas.1216013110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer research. 2012;72:2457–67. doi: 10.1158/0008-5472.CAN-11-2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmit TL, Zhong W, Setaluri V, et al. Targeted depletion of Polo-like kinase (Plk) 1 through lentiviral shRNA or a small-molecule inhibitor causes mitotic catastrophe and induction of apoptosis in human melanoma cells. The Journal of investigative dermatology. 2009;129:2843–53. doi: 10.1038/jid.2009.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosman JA, Kittaneh M, Lolkema MP. A phase 1b/2 study of LEE011 in combination with binimetinib (MEK162) in patients with NRAS-mutant melanoma: Early encouraging clinical activity. J Clin Oncol. 2014;32(suppl):5s. abstr 9009. [Google Scholar]
- Strebhardt K. Multifaceted polo-like kinases: drug targets and antitargets for cancer therapy. Nature reviews Drug discovery. 2010;9:643–60. doi: 10.1038/nrd3184. [DOI] [PubMed] [Google Scholar]
- Thomas NEES, Alexander A, et al. Association between NRAS and BRAF mutational status and melanoma-specific survival among patients with higher-risk primary melanoma. JAMA Oncol. 2015;1:359–68. doi: 10.1001/jamaoncol.2015.0493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim H, Erikson RL. Plk1-targeted therapies in TP53- or RAS-mutated cancer. Mutation research Reviews in mutation research. 2014;761:36–9. doi: 10.1016/j.mrrev.2014.02.005. [DOI] [PubMed] [Google Scholar]