

Abstract
Purpose:
To assess the association of LTBP2 mutations with primary angle closure glaucoma (PACG).
Methods:
We studied 54 unrelated patients with PACG and one individual with pseudoexfoliation accompanied with angle closure glaucoma; these consisted of 28 female and 27 male subjects aged 27 to 82 (mean, 63) years. The 36 exons and flanking intronic sequences of LTBP2 in all patients were amplified by PCR and sequenced by the Sanger protocol. The sequences were compared to LTBP2 reference sequences. A total of 100 to 400 controls aged at least 60 years old were screened for various variations.
Results:
Out of 24 observed sequence variations, ten were in amino acid coding regions; of these four created synonymous codons while six caused amino acid changes. Based on allele frequencies, biochemical parameters, absence in control individuals, evolutionary conservation of affected amino acids, and bioinformatic predictions on the effects on protein function, it was concluded that only two mutations causing p. Gln1417Arg and p. Gly1660Trp may contribute to PACG. The p. Gly1660Trp mutation was observed in a patient with both PACG and PEX syndrome. P. Gln1417Arg had previously been reported only in a subject with POAG.
Conclusion:
LTBP2 may contribute to PACG. This finding emphasizes that there may be an overlap in the etiology of various forms of glaucoma and the overlaps likely contribute to common features in various forms of glaucoma.
Keywords: Primary Angle Closure Glaucoma, LTBP2, p.Gln1417Arg, p.Gly1660Trp
INTRODUCTION
Glaucoma is a heterogeneous optic neuropathy characterized by a specific pattern of visual field loss and degeneration of the optic nerve which is usually accompanied by increased intraocular optic pressure (IOP).[1] Glaucoma, a major cause of blindness worldwide, is sub-classified into three major forms based on anatomy of the anterior chamber of the eye and age of onset: primary congenital glaucoma (PCG), primary open angle glaucoma (POAG), and primary angle closure glaucoma (PACG).[2] POAG and PACG are by far more common than PCG.[3] In China, approximately 3.5 million individuals are affected with PACG and this form of glaucoma accounts for 90% of glaucoma associated blindness.[4] In a study including approximately 2,000 individuals from a central province in Iran, the prevalence of POAG and PACG, were 3.2% and 0.4% respectively.[5] PACG results from impaired aqueous outflow from the trabecular meshwork due to abnormal anterior apposition of the iris root at the anterior chamber drainage angle.[6] Factors positively correlated with PACG include older age, Far East ancestry, female gender, and anatomical features including shallow anterior chamber, short axial length, and narrow iridocorneal angle.[6,7,8,9,10] The role of genetic background in the etiology of PACG has also been evidenced by familial clustering of the disease and sib-pair studies.[10,11] The genetic component of PACG has most often been queried in association studies, the results of which have implicated a role for several genes such as the matrix metalloproteinase-9 encoding gene (OMIM 120361), HGF (OMIM 142409), COL11A1 (OMIM 120280), PLEKHA7 (OMIM 612686), and HSP70 (OMIM 140550). However, these findings are not generally considered definitive as some were not reproduced in independent studies and others may be population specific. Despite intensive investigations, a PACG causing gene or strongly associated locus in humans has not been identified.[3]
LTBP2 (OMIM 231300) is a large gene with 36 exons which encodes latent transforming growth factor (TGF)-beta binding protein 2. It was identified as a causative gene of PCG in 2009.[2,12] Subsequently, mutations in this gene were found in patients affected with megalocornea, spherophakia, microspherophakia, lens dislocation, and Weill–Marchesani syndrome.[13,14,15,16] All of these disorders are sometimes accompanied by secondary glaucoma.[1] LTBP2 is expressed in various ocular tissues including the trabecular meshwork and ciliary zonules.[2] LTBP2 is an extracellular matrix protein and a member of a protein superfamily composed of fibrillins and LTBP proteins; it interacts with fibrillin containing microfibrils.[2,17] In addition to structural roles, LTBP2 may also affect TGF-β functions. TGF-β has multiple important cellular functions such as modulation of extracellular matrix formation.[16] After identification of a role for LTBP2 in PCG, taking into consideration that a locus that included this gene was previously linked to POAG and that mutations in CYP1B1 (OMIM 601771), another PCG gene, had been identified in POAG patients, Jelodari-Mamaghani and colleagues performed LTBP2 mutation screening in POAG patients.[1,18] They also performed the screening in patients affected with pseudoexfoliation (PEX) because the extracellular matrix is affected in this disorder, LTBP2 protein is a component of PEX material, and many PEX patients also present with glaucoma.[1] These studies identified several sequence variations that appeared to contribute to the disease status of both groups of patients, albeit the variations did not evidence Mendelian inheritance and they revealed incomplete penetrance. Microscopic studies showed that the extracellular matrix of mutation harboring patients was disrupted.[1]
In the current study, the possible contribution of LTBP2 to PACG was considered by mutation screening of the gene in affected patients. Despite known differences in the etiology of various forms of glaucoma, it is expected that their common features reflect partial overlap in the etiologies. In fact, evidence exists for a possible role of LTBP2 in manifestation of PACG. Azmanov et al,[19] while studying a PCG family, observed that an individual affected with angle closure glaucoma and pupillary block harbored a p. Glu229Lys causing mutation in CYP1B1 and also a p. Arg299* causing mutation in LTBP2. Furthermore, Haji-Seyed-Javadi et al[16] studied a family with Marfan syndrome and noted that one member of the family affected with angle closure glaucoma and mitral valve prolapse harbored a p. Arg548* mutation in LTBP2.
It was in light of this background that we set out to perform mutation screening of the large LTBP2 gene in PACG patients. Our findings suggest that LTBP2 may indeed play a role in presentation of the disease.
METHODS
This study adhered to tenets of the Declaration of Helsinki and was approved by the board of Ethics of the University of Tehran and the Ophthalmic Research Center of Shahid Beheshti University of Medical Sciences. Informed consent was obtained from all participants.
The study included 54 unrelated patients with PACG and one individual with PEX accompanied with angle closure glaucoma; these included 28 female and 27 male subjects aged 27 to 82 (mean, 63) years. The patients were consecutively recruited from the Ophthalmology Department of Labbafinejad Medical Center, Shahid Beheshti University of Medical Sciences without regard to familial status of their disease. All patients were diagnosed by a glaucoma specialist (S.Y) and except one subject did not have other ocular diseases and also reported to be free of non-ocular diseases. Patient 407 was diagnosed with both angle closure and PEX.
Slit lamp biomicroscopy, IOP measurement, gonioscopy, and fundus examination were performed on all patients. IOP measurements were obtained using Goldmann applanation tonometry. Criteria for PACG diagnosis included the presence of elevated intraocular pressure (≥22 mmHg), appositional or synechial angle closure >270 degrees, and structural and/or functional damage due to glaucomatous optic neuropathy in at least one eye.
For mutation screening of LTBP2, its exons and flanking intronic sequences were amplified by polymerase chain reaction (PCR) and subsequently sequenced with the Sanger protocol using an ABI 3730XL genetic analyzer instrument (Applied Biosystems, Foster City, CA, USA). Sequences were analyzed with the Sequencher 4.8 software (Gene Codes Corporation, Ann Arbor, MI, USA). LTBP2 reference sequences NC_000014.9, NM_000428.2 and NP_000419.1 were used. The effects of the variant sequences on splicing were predicted by using NNSPLICE 0.9 (http://www.fruitfly.org/seq_tools/splice.html) and Human Splicing Finder V 2.4.1 (http://www.umd.be/HSF). To determine the extent of conservation of amino acids altered due to nucleotide variations, the amino acid sequences of homologous proteins from other species were aligned using the ClustalW2 software (http://www.ebi.ac.uk/Tools/msa/clutalw2). Coding variations deemed to possibly contribute to disease were assessed in control individuals with allele-specific PCR protocols. There were 100 to 400 controls (aged at least 60 years) who were screened for the various variations. The effects of the variations on protein function were predicted with the PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2) and SIFT (http://sift.jcvi.org) bioinformatics tools. The sequences all primers used to amplify and sequence the LTBP2 exons are available upon request.
RESULTS
Sequencing of LTBP2 exons in 55 patients revealed 24 sequence variations of which 14 were intronic or in 3’-UTR regions and 10 were within amino acid coding exons [Table 1]. Five of the variations were novel, and only one of these was within an amino acid coding region. The novel coding variation caused p. Gly1660Trp. Nine out of the 10 non-novel non-coding variations had high allele frequencies (5.5–21.8%) in our cohort [Table 1]. All these nine variations and also the tenth that we observed only in one patient and is reported to have a low allele frequency (0.013) single nucleotide polymorphism (dbSNP) database (http://www.ncbi.nlm.nih.gov/snp/) were predicted not to affect splicing. Among the four novel non-coding variations, only c.4453+4A>G was predicted to possibly affect splicing. Therefore, based on allele frequency and/or predicted effect on splicing, only the c.4453+4A>G variation among the 14 non-coding variations is a potential candidate for being associated with PACG. Though bioinformatics tools predicted c.4453+4A>G may affect splicing, it has been reported that A at the +4 position is found in less than 50% of conventional donor sites, and that the second most common nucleotide at that position is G.[20] With this consideration, the c.4453+4A>G variation was not considered to be cause of PACG.
Table 1.
LTBP2 sequence variations observed among 55 patients with PACG
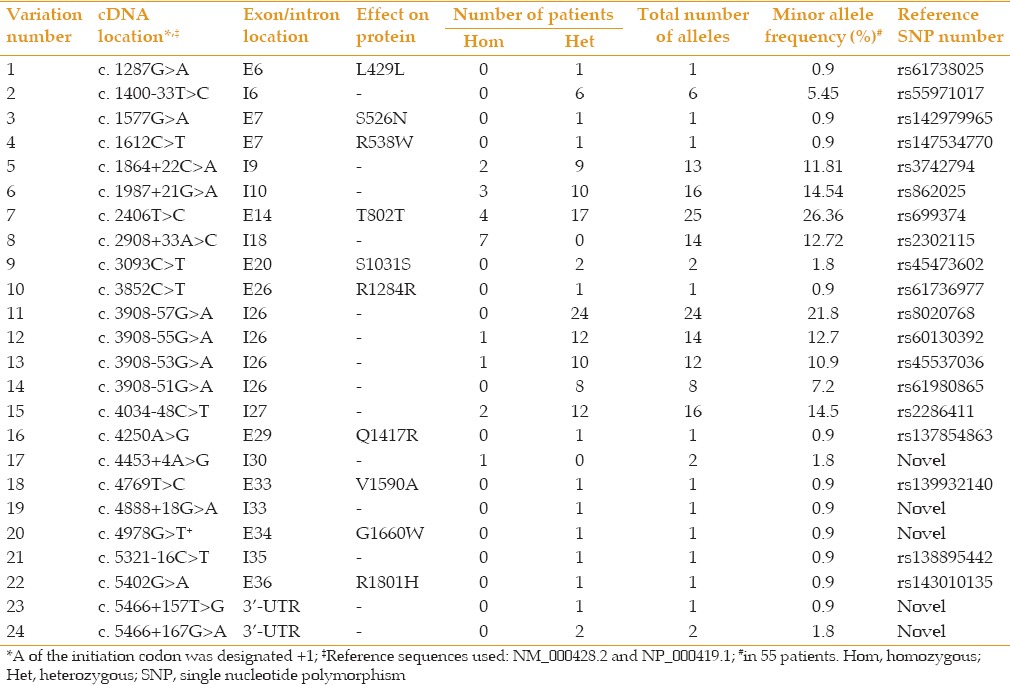
Out of the ten variations that were positioned within codon coding sequences, four affected synonymous codon changes (p.Leu429Leu, p.Thr802Thr, p.Ser1031Ser, and p.Arg1284Arg) of which two variations (p.Ser1031Ser and p.Thr802Thr) were observed in two or more patients. These findings suggest that these four sequence variations are not the cause of disease. The remaining six sequence variations that affected amino acid changes were further considered [Table 2]. They were all observed in the heterozygous state and each one was found in only one patient. In Table 2, motif location of the affected amino acids, consequence of the alteration on charge and size of the affected amino acid, the conservation of the amino acid during evolution, results of screenings in control individuals, minor allele frequencies (MAF) reported in dbSNP database, and bioinformatics predictions on protein function for the six coding variations are detailed. The position of the affected amino acids is also shown in a schematic diagram of the LTBP2 protein in Figure 1. Specifics on evolutionary conservation of the amino acids are evident in Table 3. In a conservative assessment, four of the variations were for various reasons disregarded as disease associated variations, these are described in more detail as follows. The p. Ser526Asn and p. Val1590Ala causing variations were, reported in the dbSNP database as having been observed in 12 and 93 individuals respectively, among approximately 6,000 subjects screened in the National Heart Lung and Blood Institute (NHLBI) (http://evs.gs.washington.edu/EVS) sequencing project. The p. Arg538Trp causing variation had been reported in a patient with PCG, but was reported to also have been present in unaffected members of the patient's family.[21] Finally, the variation which caused p. Arg1801His was found in three out of 400 screened controls. Furthermore, one of the two PACG affected siblings of our proband with this variation did not carry the causative variation.
Table 2.
Missense variations observed in LTBP2
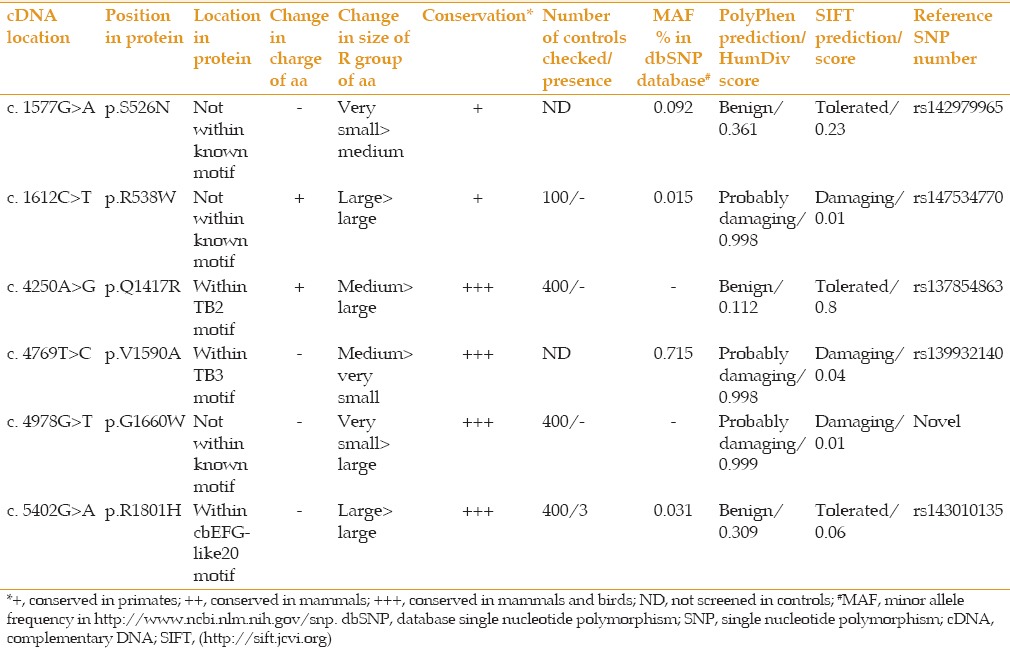
Figure 1.
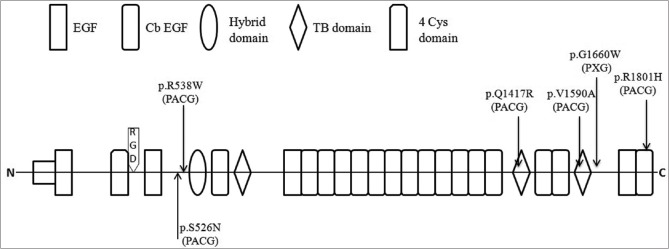
Positions of missense mutations in latent transforming growth factor-beta binding protein 2 (LTBP2) that were observed in PACG patients. Structure symbols used for the various LTBP2 domains are indicated; horizontal lines represent protein regions not known to be specific domains.
Table 3.
Conservation during evolution of amino acids in LTBP2 that were affected by mutations observed in PACG patients

The only remaining variations considered to potential contribute to PACG were p. Gln1417Arg and p. Gly1660Trp [Figure 2]; both of these produce an amino acid differing in size from the wild type amino acid, are evolutionarily conserved among mammals and birds, and were absent in the 800 control chromosomes screened [Tables 2 and 3]. Although P. Gln1417Arg was considered benign by bioinformatics tools, it affects an amino acid positioned in a known functional motif of the LTBP2 protein and results in a change in amino acid charge [Figure 1]. The only previous report of this mutation was in a patient with POAG supporting the proposal for the role of this mutation in a patient with PACG in the present study. This 52-year-old woman showed acute primary angle closure (APAC) with intraocular pressure of 44 mmHg, narrow anterior chamber angle, and optic nerve damage in her right eye [Figure 3]. The second potentially disease causing variation was P. Gly1660Trp which is not positioned within a known motif of the encoded protein, but was predicted to be potentially damaging to protein function by both used bioinformatics tools [Table 2]. This mutation was observed in patient 407 with PACG and PEX. The phenotypic features of the patients harboring p. Gln1417Arg and P. Gly1660Trp are presented in Table 4.
Figure 2.
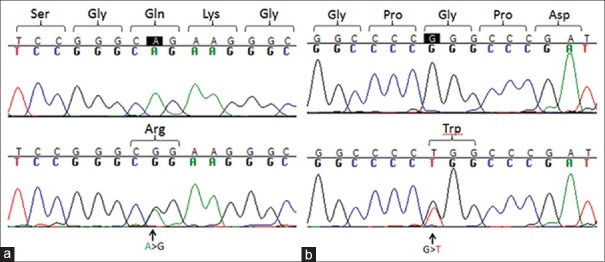
Chromatograms showing LTBP2 mutations that may contribute to PACG. (a) P.Gln1417Arg causing mutation, top: homozygous wild-type c.4250A genotype; bottom: Heterozygous mutated genotype c.4250A>G; (b) P.Gly1660Trp causing mutation, top: homozygous wild-type c.4978G genotype, bottom: Heterozygous mutated genotype c.4978G>T.
Figure 3.
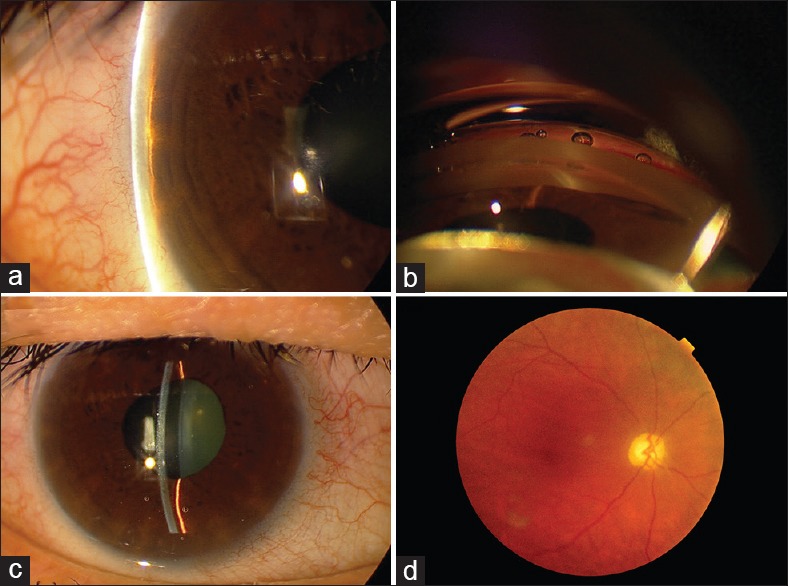
The right eye of a patient with PACG harboring the p.Gln1417Arg-causing mutation. (a) Narrow anterior chamber angle by Van Herick method; (b) Complete angle closure by gonioscopy; (c) A centrally narrow anterior chamber and a distorted and slightly dilated pupil as a sequel of previous APAC; (d) Optic nerve damage, vertical cupping with optic disc pallor are noted in fundus examination.
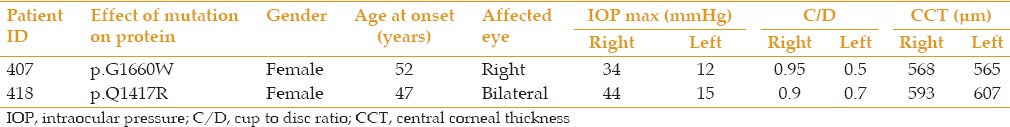
Table 4.
Phenotypic features of two patients with variations in LTBP2 that potentially contribute to PACG status
DISCUSSION
Approximately 16 million people suffer from PACG worldwide and 80% of them are of Asian ancestry, most often from China, Mongolia, Singapore, and India.[22,23] PACG is a complex disorder. The risk of the disease is increased six-fold in first-degree relatives of affected patients, reflecting the genetic component of PACG.[11,24] The heritability is partly accounted for by the heritability of the size of the irido-corneal angle.[25] A number of studies have attempted to identify genes contributing to PACG,[3] and PRSS56 (OMIM 613858) was recently identified as an angle closure glaucoma gene in a murine model and also of posterior microphthalmia in humans.[22] Three PACG loci were also identified in association studies.[24] Nevertheless, the genetic basis of PACG is thought to be largely unknown. As already stated, LTBP2 was considered a candidate gene with a role in PACG owing to earlier reports of LTBP2 sequence variations in PACG patients and also because of the consideration that various forms of glaucoma may have partially overlapping etiologies.[16,19] In the present study, we identify two coding mutations which may have contributed to the PACG disease status of the individuals who carried the mutations. It may well be that these mutations were not the sole reason of PACG manifestations, but rather that they, along with the summed effects of other factors, resulted in disease presentation. The contribution of the mutated LTBP2 proteins may be related to the role of the protein in the extracellular matrix. Our results justify further LTBP2 mutation screenings in larger samples of PACG patients and examination on extracellular matrix in PACG patients who carry putative LTBP2 disease associated mutations.
Financial Support and Sponsorship
Nil.
Conflicts of Interest
There are no conflicts of interest.
Acknowledgements
We thank the patients and their family members for participating in this study. We acknowledge the Iran National Science Foundation and the Ophthalmic Research Center of Shahid Beheshti University of Medical Sciences for funding this research.
REFERENCES
- 1.Jelodari-Mamaghani S, Haji-Seyed-Javadi R, Suri F, Nilforushan N, Yazdani S, Kamyab K, et al. Contribution of the latent transforming growth factor-ß binding protein 2 gene to etiology of primary open angle glaucoma and pseudoexfoliation syndrome. Mol Vis. 2013;19:333–347. [PMC free article] [PubMed] [Google Scholar]
- 2.Narooie-Nejad M, Paylakhi SH, Shojaee S, Fazlali Z, Rezaei Kanavi M, Nilforushan N, et al. Loss of function mutations in the gene encoding latent transforming growth factor beta binding protein 2, LTBP2, cause primary congenital glaucoma. Hum Mol Genet. 2009;18:3969–3977. doi: 10.1093/hmg/ddp338. [DOI] [PubMed] [Google Scholar]
- 3.Shastry BS. Genetic susceptibility to primary angle closure glaucoma (PACG) Discov Med. 2013;15:17–22. [PubMed] [Google Scholar]
- 4.Foster PJ, Johnson GJ. Glaucoma in China: How big is the problem? Br J Ophthalmol. 2001;85:1277–1282. doi: 10.1136/bjo.85.11.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pakravan M, Yazdani S, Javadi M-A, Amini H, Behroozi Z, Ziaei H, et al. A population-based survey of the prevalence and types of glaucoma in central Iran: the Yazd Eye Study. Ophthalmology. 2013;120:1977–1984. doi: 10.1016/j.ophtha.2013.02.029. [DOI] [PubMed] [Google Scholar]
- 6.Salmon JF. Predisposing factors for chronic angle-closure glaucoma. Prog Retin Eye Res. 1999;18:121–132. doi: 10.1016/s1350-9462(98)00007-x. [DOI] [PubMed] [Google Scholar]
- 7.Foster PJ, Alsbirk PH, Baasanhu J, Munkhbayar D, Uranchimeg D, Johnson GJ. Anterior chamber depth in Mongolians: Variation with age, sex, and method of measurement. Am J Ophthalmol. 1997;124:53–60. doi: 10.1016/s0002-9394(14)71644-7. [DOI] [PubMed] [Google Scholar]
- 8.Lim KJ, Hyung SM, Youn DH. Ocular dimensions with aging in normal eyes. Korean J Ophthalmol. 1992;6:19–31. doi: 10.3341/kjo.1992.6.1.19. [DOI] [PubMed] [Google Scholar]
- 9.Congdon NG, Youlin Q, Quigley H, Hung PT, Wang TH, Ho TC, et al. Biometry and primary angle-closure glaucoma among Chinese, white, and black populations. Ophthalmology. 1997;104:1489–1495. doi: 10.1016/s0161-6420(97)30112-2. [DOI] [PubMed] [Google Scholar]
- 10.Yazdani S, Akbarian S, Pakravan M, Afrouzifar M. Prevalence of angle closure in siblings of patients with primary angle-closure glaucoma. J Glaucoma. 2015;24:149–153. doi: 10.1097/IJG.0000000000000091. [DOI] [PubMed] [Google Scholar]
- 11.Wang N, Wu H, Fan Z. Primary angle closure glaucoma in Chinese and Western populations. Chin Med J (Engl) 2002;115:1706–1715. [PubMed] [Google Scholar]
- 12.Ali M, McKibbin M, Booth A, Parry DA, Jain P, Riazuddin SA, et al. Null mutations in LTBP2 cause primary congenital glaucoma. Am J Hum Genet. 2009;84:664–671. doi: 10.1016/j.ajhg.2009.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Désir J, Sznajer Y, Depasse F, Roulez F, Schrooyen M, Meire F, et al. LTBP2 null mutations in an autosomal recessive ocular syndrome with megalocornea, spherophakia, and secondary glaucoma. Eur J Hum Genet. 2010;18:761–767. doi: 10.1038/ejhg.2010.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khan AO, Aldahmesh MA, Alkuraya FS. Congenital megalocornea with zonular weakness and childhood lens-related secondary glaucoma-A distinct phenotype caused by recessive LTBP2 mutations. Mol Vis. 2011;17:2570–2579. [PMC free article] [PubMed] [Google Scholar]
- 15.Kumar A, Duvvari MR, Prabhakaran VC, Shetty JS, Murthy GJ, Blanton SH. A homozygous mutation in LTBP2 causes isolated microspherophakia. Hum Genet. 2010;128:365–371. doi: 10.1007/s00439-010-0858-8. [DOI] [PubMed] [Google Scholar]
- 16.Gibson MA, Hatzinikolas G, Davis EC, Baker E, Sutherland GR, Mecham RP. Bovine latent transforming growth factor beta 1-binding protein 2: Molecular cloning, identification of tissue isoforms, and immunolocalization to elastin-associated microfibrils. Mol Cell Biol. 1995;15:6942. doi: 10.1128/mcb.15.12.6932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haji-Seyed-Javadi R, Jelodari-Mamaghani S, Paylakhi SH, Yazdani S, Nilforushan N, Fan JB, et al. LTBP2 mutations cause Weill-Marchesani and Weill-Marchesani-like syndrome and affect disruptions in the extracellular matrix. Hum Mutat. 2012;33:1182–1187. doi: 10.1002/humu.22105. [DOI] [PubMed] [Google Scholar]
- 18.Suri F, Kalhor R, Zargar SJ, Nilforooshan N, Yazdani S, Nezari H, et al. Screening of common CYP1B1 mutations in Iranian POAG patients using a microarray-based PrASE protocol. Mol Vis. 2008;14:2349–2356. [PMC free article] [PubMed] [Google Scholar]
- 19.Azmanov DN, Dimitrova S, Florez L, Cherninkova S, Draganov D, Morar B, et al. LTBP2 and CYP1B1 mutations and associated ocular phenotypes in the Roma/Gypsy founder population. Eur J Hum Genet. 2011;19:326–333. doi: 10.1038/ejhg.2010.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abril JF, Castelo R, Guigó R. Comparison of splice sites in mammals and chicken. Genome research. 2005;15:111–119. doi: 10.1101/gr.3108805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sharafieh R, Child AH, Khaw PT, Fleck B, Sarfarazi M. LTBP2 gene analysis in the GLC3C-linked family and 94 CYP1B1-negative cases with primary congenital glaucoma. Ophthalmic Genet. 2013;34:14–20. doi: 10.3109/13816810.2012.716486. [DOI] [PubMed] [Google Scholar]
- 22.Nair KS, Hmani-Aifa M, Ali Z, Kearney AL, Ben Salem S, Macalinao DG, et al. Alteration of the serine protease PRSS56 causes angle-closure glaucoma in mice and posterior microphthalmia in humans and mice. Nat Genet. 2011;43:579–584. doi: 10.1038/ng.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nongpiur ME, Khor CC, Jia H, Cornes BK, Chen LJ, Qiao C, et al. ABCC5, a gene that influences the anterior chamber depth, is associated with primary angle closure glaucoma. PLoS Genet. 2014;10:e1004089. doi: 10.1371/journal.pgen.1004089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vithana EN, Khor CC, Qiao C, Nongpiur ME, George R, Chen LJ, et al. Genome-wide association analyses identify three new susceptibility loci for primary angle closure glaucoma. Nat Genet. 2012;44:1142–1146. doi: 10.1038/ng.2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Amerasinghe N, Zhang J, Thalamuthu A, He M, Vithana EN, Viswanathan A, et al. The heritability and sibling risk of angle closure in Asians. Ophthalmology. 2011;118:480–485. doi: 10.1016/j.ophtha.2010.06.043. [DOI] [PubMed] [Google Scholar]