

Significance
Viral infection activates IRF3 and NF-κB and induces the production of type I interferons and proinflammatory cytokines. In this study, we found that ubiquitin-specific protease 25 (USP25) was required for viral infection-triggered activation of IRF3 and NF-κB and subsequent production of type I IFNs and proinflammatory cytokines. Importantly, the degradation of TRAF3 and TRAF6 was accelerated in Usp25−/− cells compared with wild-type counterparts after viral infection, which could be inhibited by the proteasome inhibitor MG132 and the autophagy inhibitor 3MA, respectively. Reconstitution of TRAF3 and TRAF6 into Usp25−/− MEFs restored virus-triggered production of type I IFNs and proinflammatory cytokines. This study provides insights into an elegant “check and balance” process during innate antiviral responses.
Keywords: USP25, innate antiviral immunity, TRAF3, TRAF6
Abstract
Host pathogen-recognition receptors detect nucleic acid from invading viruses and initiate a series of signaling pathways that lead to the production of type I interferons (IFNs) and proinflammatory cytokines. Here, we found that a viral infection-induced deubiquitinase (DUB), ubiquitin-specific protease 25 (USP25) was required for host defense against RNA and DNA viruses. The activation of transcription factors IRF3 and NF-κB was impaired and the production of type I IFNs and proinflammatory cytokines was inhibited in Usp25−/− cells compared with the wild-type counterparts after RNA or DNA viruses infection. Consistently, USP25 deficient mice were more susceptible to H5N1 or HSV-1 infection compared with the wild-type mice. USP25 was associated with TRAF3 and TRAF6 after infection by RNA or DNA viruses and protected virus-induced proteasome-dependent or independent degradation of TRAF3 and TRAF6, respectively. Moreover, reconstitution of TRAF3 and TRAF6 into Usp25−/− MEFs restored virus-triggered production of type I IFNs and proinflammatory cytokines. Our findings thus reveal a previously uncovered positive feedback regulation of innate immune responses against RNA and DNA viruses by USP25.
Nucleic acid generated by the invading viruses acts as a critical class of pathogen-associated molecular patterns (PAMPs) to induce the production of type I interferons (IFNs) and proinflammatory cytokines (1, 2). Recognition of viral nucleic acid by the innate immune system depends on germ-line-encoded pattern recognition receptors (PRRs) (3). To date, at least three types of PRRs including Toll-like receptors (TLRs), RIG-I-like receptors (RLRs) and cytosolic dsDNA sensors have been characterized to recognize viral nucleic acid in different types of cells or in distinct subcellular organelles. For example, TLR3 recognizes double-stranded RNA (dsRNA) in the endosomes in most types of cells other than plasmacytoid dendritic cells (pDCs), whereas TLR7/8 and TLR9 recognize single-stranded RNA (ssRNA) and unmethylated CpG DNA in the endo-lysosomes in pDCs, respectively (4–6). In cytosol, RLRs and cGMP-AMP (cGAMP) synthase (cGAS) are responsible for the recognition of viral RNA and DNA, and thus are called “universal” sensors for RNA and DNA viruses, respectively (7, 8). In addition, several other PRRs such as RNA polymerase III, DAI, DDX41, IFI16, and Lsm14A have been identified as cell-type or ligand specific DNA sensors (9–13). Following recognition of viral nucleic acid, the PRRs undergo conformational changes to recruit downstream adaptor proteins including TRIF, MyD88, VISA (also called MAVS, Cardif, and IPS-1), and MITA (also called STING) that mediate distinct antiviral signaling pathways converging at the kinase level of IKKα/β/γ and TBK1 (14–20). The IKK complex and TBK1 activate the transcription factors of NF-κB and IRF3, respectively, leading to the expression of type I IFNs and proinflammatory cytokines.
Tumor necrosis factor receptor-associated factor (TRAF) family proteins have been suggested to link the downstream kinases to the adaptor proteins. Among all of the identified TRAFs, TRAF3 and TRAF6 act as scaffolding proteins by recruiting downstream kinases TBK1 or IKKα/β/γ complex to the adaptor proteins and thus are essential for virus-triggered signaling (21–23). It has been demonstrated that the adaptor protein MAVS directly interacts with TRAF3 and TRAF6 through its multiple TRAF-interacting motifs (TIMs) (16, 24). TRAF3 recruits TBK1 to promote the phosphorylation and activation of IRF3, whereas TRAF6 recruits the IKKα/β/γ complex to activate NF-κB. Thus, TRAF3 and TRAF6 function as an essential signaling “hub” that integrates upstream signals from PRRs and transmits the signals to downstream kinases and transcription factors.
It has been recognized that the activity of TRAF3 and TRAF6 is regulated by various deubiquitinating enzymes (DUBs) (25–28). We have previously showed that USP25 removes K48-linked ubiquitination of TRAF3 and K63-linked ubiquitination of TRAF6 in TLR4- and IL-17–mediated signaling pathways, respectively (29, 30). In this study, we found that RNA and DNA virus infection up-regulated Usp25 expression and induced USP25-TRAF3/6 association. USP25-deficient mice were more susceptible to influenza virus H5N1 and herpes simplex virus 1 (HSV-1) infection than the wild-type mice. In addition, USP25 protected TRAF3 and TRAF6 from degradation and thus positively regulated virus-induced production of type I IFNs and proinflammatory cytokines. Our findings thus demonstrate a positive feedback regulation of innate antiviral immune responses by USP25.
Results
USP25 Positively Regulates RNA Virus-Induced Signaling.
In our previous study, we observed that treatment of the TLR4 ligand lipopolysaccharide (LPS) induces the expression of Usp25 in various types of cells (29). In a parallel experiment, we found that the expression of Usp25 and the abundance of USP25 protein were increased in MEFs after Sendai virus (SeV) or vesicular stomatitis virus (VSV) infection but not by TNF-α treatment (Fig. S1 A and B and Table S1). We thus examined the effects of USP25 deficiency on RNA virus-triggered signaling and found that the expression of Ifnb, Ifna4, Tnf, and/or Il6 was inhibited in Usp25−/− MEFs or MLFs compared with the wild-type counterparts after SeV or VSV infection or transfection of the RNA analog poly(I:C) (Fig. 1A and Fig. S2A). Consistent with the results from gene induction analysis, Usp25−/− MEFs or MLFs produced decreased amount of IFN-α, IL-6, and TNF-α proteins after SeV or VSV infection than did the wild-type cells (Fig. 1B and Fig. S2B). In addition, SeV- or VSV-induced phosphorylation of IRF3 and IκBα was substantially impaired in Usp25−/− MEFs or MLFs (Fig. 1C and Fig. S2C). These data suggest that USP25 is required for RNA virus-triggered activation of IRF3 and NF-κB and the induction of type I IFNs and proinflammatory cytokines in primary mouse fibroblasts.
Fig. S1.

Viral infection induces the expression of Usp25. (A) The mRNA levels of Usp25 were increased after infection with RNA viruses in mouse embryonic fibroblasts (MEFs). MEFs were transfected with poly(I:C) or stimulated with Sendai virus (SeV), vesicular stomatitis virus (VSV) or TNF-α for the indicated time points before qPCR was performed. (B) The abundance of USP25 protein was increased after SeV or VSV infection in MEFs. Immunoblot analysis of USP25 protein in the whole lysate of MEFs infected with SeV or VSV. (C) The mRNA levels of Usp25 were increased in different types of cells after SeV or HSV-1 infection. Mouse lung fibroblasts (MLFs), plasmacytoid dendritic cells (pDCs) or bone marrow-derived dendritic cells (BMDCs) were stimulated with SeV or HSV-1 for 6 h before qPCR was performed. (D) A schematic structure of mouse Usp25 gene promoter. ISRE, IFN-stimulatory response element; NF-κB, nuclear factor kappaB binding site. Data are representative of at least three independent experiments (mean and SD of three replicates in A and C).
Table S1.
Quantitative real-time PCR (qPCR) primers
| Gene name | Primer sequence |
| Il6 | TATGAAGTTCCTCTCTGCAAGAGA |
| TAGGGAAGGCCGTGGTT | |
| Tnf | AATGGCCTCCCTCTCATCAGT |
| GCTACAGGCTTGTCACTCGAATT | |
| Ifnb | TACAACAGATACGCCTGGAT |
| AGTCCGCCTCTGATGCTTAA | |
| Ifna4 | AGGATCACTGTGTACCTGAGA |
| TCTCCACACTTTGTCTCAGGA | |
| Traf3 | GCCGTTCAAGCAGAAAGTGA |
| TCTTGAAGCTGCTGCTGTTG | |
| Traf6 | GAGTTTGACCCACCTCTGGA |
| TTTCATTGTCAACTGGGCACT | |
| HSV-1 UL30 gene | CATCACCGACCCGGAGAGGGAC |
| GGGCCAGGCGCTTGTTGGTGTA | |
| β-Actin | TGGAATCCTGTGGCATCCATGAAAC |
| TAAAACGCAGCTCAGTAACAGTCCG | |
| Usp25 | TCCGGCACCAAGGCACATCAC |
| ACGGCATGGAGGCGGTAAGG | |
| Ifnb promoter | ATTCCTCTGAGGCAGAAAGGACC |
| GCAAGATGAGGCAAAGGCTGTCA |
Fig. 1.

USP25 positively regulates RNA virus-induced signaling in primary mouse embryonic fibroblasts (MEFs). (A) The expression of Ifnb, Ifna4, Tnf, and/or Il6 was inhibited in Usp25−/− MEFs than in wild-type counterparts after SeV or VSV infection or transfection of the dsRNA analog poly(I:C). Wild-type or Usp25−/− MEFs were infected with SeV or VSV or transfected with poly(I:C) (1 μg) for the indicated time points before qPCR analysis was performed. (B) Usp25−/− MEFs produced decreased amount of IFN-α, IL-6, and TNF-α proteins after SeV or VSV infection than did the wild-type cells. Wild-type or Usp25−/− MEFs were infected with SeV or VSV for the indicated time points and the supernatants were collected for ELISA analysis. (C) SeV- or VSV-induced phosphorylation of IRF3 and IκBα was substantially impaired in Usp25−/− MEFs. Wild-type or Usp25−/− MEFs were infected with SeV or VSV for the indicated time points. Cells were lysed and the cell lysates were subject to immunoblot analysis with the indicated antibodies. (D and E) The replication of GFP-tagged Newcastle disease virus (NDV) was potentiated in Usp25−/− MEFs compared with the wild-type MEFs. Wild-type or Usp25−/− MEFs were infected with GFP-NDV (MOI = 0.01, D; MOI = 0.1, E) for 18 or 24 h. Cells were harvested for flow cytometry or microscopy imaging and immunoblot analysis. Data are representative of three independent experiments (mean and SD of three replicates in A and B).
Fig. S2.

USP25 positively regulates RNA virus-induced signaling in primary mouse lung fibroblasts (MLFs). (A) The expression of Ifnb, Ifna4, Tnf, and/or Il6 was inhibited in Usp25−/− MLFs than in wild-type counterparts after SeV or VSV infection or transfection of the RNA analog poly(I:C). (B) Usp25−/− MLFs produced decreased amount of IFN-α, IL-6 and TNF-α proteins after SeV or VSV infection than did the wild-type cells. (C) SeV- or VSV-induced phosphorylation of IRF3 and IκBα was substantially impaired in Usp25−/− MLFs. Data are representative of two independent experiments (means and SD of three replicates in A and B).
Because USP25 deficiency dampened RNA virus-triggered signaling, we examined the role of USP25 in cellular antiviral response. We found that the replication of GFP-tagged Newcastle disease virus (NDV) was potentiated in Usp25−/− MEFs compared with the wild-type MEFs as monitored by the expression and intensity of GFP protein (Fig. 1 D and E). These results indicate that USP25 restricts viral replication and positively regulates cellular antiviral response.
To determine whether USP25 is involved in RNA virus-triggered signaling in other types of cells, we generated wild-type and Usp25−/− bone marrow-derived macrophages (BMDMs) and dendritic cells (BMDCs) and FLT3L-induced plasmacytoid dendritic cells (pDCs) and infected these cells with SeV or VSV. Upon SeV or VSV infection, Usp25−/− cells expressed lower levels of Ifna4, Ifnb, Tnf, and/or Il6 mRNA and produced reduced IFN-α and IL-6 proteins than did the wild-type counterparts (Fig. S3 A and B). In addition, SeV-induced phosphorylation of IRF3 and IκBα was impaired in Usp25−/− BMDCs compared with wild-type BMDCs (Fig. S3C). These data together suggest that USP25 positively regulates RNA virus-induced signaling in various hematopoietic innate immune cells.
Fig. S3.

USP25 is required for RNA virus-triggered signaling in mouse macrophages and dendritic cells. (A) Usp25−/− cells expressed lower levels of Ifna4, Ifnb, Tnf, and/or Il6 mRNA than did the wild-type counterparts after SeV or VSV infection. Wild-type or Usp25−/− BMDMs, BMDCs, FLT3L-pDCs were infected with SeV or VSV for the indicated time points before qPCR analysis was performed. (B) Usp25−/− cells produced reduced IFN-α and IL-6 proteins than did the wild-type counterparts after SeV or VSV infection. Wild-type or Usp25−/− BMDMs, BMDCs, FLT3L-pDCs were infected with SeV or VSV for 12 h and the supernatants were collected for ELISA analysis. (C) SeV-induced phosphorylation of IRF3 and IκBα was impaired in Usp25−/− BMDCs compared with wild-type BMDCs. Wild-type or Usp25−/− BMDCs were infected with VSV for 6–12 h. Cells were lysed and the lysates were subject to immunoblot analysis with the indicated antibodies. Data are representative of three independent experiments (mean and SD of three replicates in A and B).
USP25 Is Required for DNA Virus-Induced Signaling.
We found that the DNA virus HSV-1 infection also resulted in increased expression of Usp25 in MLFs, pDCs and BMDCs (Fig. S1C), and thus examined the effects of USP25 deficiency on HSV-1–triggered signaling by real-time qPCR analysis with wild-type and Usp25−/− cells. HSV-1–induced expression of Ifnb and Il6 was inhibited in Usp25−/− MLFs, BMDCs, or FLT3L-pDCs (Fig. 2A). Consistent with these data, Usp25−/− MLFs, BMDCs, or FLT3L-pDCs produced reduced levels of IFN-α and IL-6 cytokines after HSV-1 infection or CpG treatment compared with the wild-type counterparts (Fig. 2B). In addition, HSV-1 infection-induced phosphorylation of IRF3 and IκBα was impaired in Usp25−/− BMDCs compared with the wild-type BMDCs (Fig. 2C). It has been demonstrated that the STING pathway senses dsDNA generated by invading DNA viruses in various types of cells. We thus examined whether USP25 was involved in dsDNA-triggered STING-dependent pathway. The results suggested that USP25 deficiency significantly impaired various cytosolic dsDNA-triggered induction of type I IFNs and proinflammatory cytokines in MLFs or BMDCs (Fig. S4). Taken together, these data suggest that USP25 positively regulates HSV-1– and cytosolic DNA-triggered production of type I IFNs and proinflammatory cytokines in various types of cells.
Fig. 2.

USP25 is required for HSV-1–induced production of type I IFNs and proinflammatory cytokines. (A) HSV-1–induced expression of Ifnb and Il6 was inhibited in Usp25−/− cells. Wild-type or Usp25−/− MLFs, BMDCs, and FLT3L-pDCs were infected with HSV-1 or treated with CpG for the indicated time points before qPCR analysis was performed. (B) Usp25−/− cells produced reduced levels of IFN-α and IL-6 cytokines after HSV-1 infection compared with the wild-type counterparts. Wild-type or Usp25−/− BMDMs, BMDCs, FLT3L-pDCs were infected with HSV-1 or treated with CpG for 12 h and the supernatants were collected for ELISA analysis. (C) HSV-1 infection-induced phosphorylation of IRF3 and IκBα was impaired in Usp25−/− BMDCs compared with the wild-type BMDCs. Wild-type or Usp25−/− BMDCs were infected with HSV-1 for 6–12 h. Cells were lysed and cell lysates were subject to immunoblot with the indicated antibodies. Data are representative of three independent experiments (mean and SD of three replicates in A and B).
Fig. S4.

Role of USP25 in cytosolic double-stranded DNA-triggered induction of type I IFNs and proinflammatory cytokines. (A and B) USP25 deficiency impaired dsDNA-triggered induction of type I IFNs and proinflammatory cytokines. Wild-type and Usp25−/− BMDCs or MLFs were mock transfected or transfected with polydA:dT, ISD, or HSV120 (3 μg). Eight hours later, cells were collected for qPCR analysis (A). Sixteen hours later, the supernatants were harvested for ELISA analysis (B). Data are representative of three independent experiments (mean and SD of three replicates). *P < 0.05, **P < 0.01, ***P < 0.001.
USP25-Deficient Mice Are More Susceptible to H5N1 and HSV-1 Infection.
We next examined the role of USP25 in host defense against RNA and DNA viruses in vivo. After infection with VSV, Usp25−/− mice produced significantly decreased amounts of IFN-α and IL-6 cytokines than did wild-type mice (Fig. S5A). The infiltration of Ly6c+ monocytes in the lungs of Usp25−/− mice was impaired compared with that of wild-type mice (Fig. S5B). In addition, Usp25−/− mice exhibited more severe weight loss after infection with the H5N1 influenza A virus compared with wild-type littermates. Importantly, all of the Usp25−/− mice (10 out of 10) died within 1 wk, whereas more than one-third (4 out of 11) wild-type mice survived for 2 wk after H5N1 challenge (Fig. 3A). Consistent with these observations, the expression of Ifnb, Ifna4, and Tnf was significantly inhibited in the lungs and livers of Usp25−/− mice compared with the wild-type mice after another stain of influenza A virus H1N1(PR8) challenge (Fig. 3B). We next infected wild-type and Usp25−/− mice with HSV-1 and monitored their survival. Three out of five Usp25−/− mice died at the 5th day after HSV-1 infection and all of the Usp25−/− mice died 7 d after HSV-1 infection. In contrast, only one wild-type mouse died at the sixth day after HSV-1 infection and four of the five wild-type mice survived even 10 d after infection (Fig. 3C). In addition, we found that Usp25−/− mice produced significantly decreased amount of IFN-α and IL-6 cytokines than did the wild-type mice after HSV-1 challenge (Fig. 3D). Hematoxylin-eosin staining and immunohistochemistry analysis of lung tissues showed that less Ly6C+ monocytes existed in the lungs of Usp25−/− mice than in the lungs of wild-type mice after HSV-1 infection (Fig. 3E). These data collectively suggest that USP25 protects mice from RNA and DNA virus infection by promoting the induction of type I IFNs and proinflammatory cytokines.
Fig. S5.
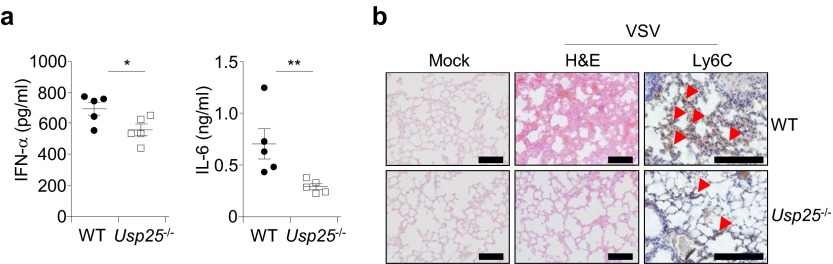
Usp25−/− mice produced decreased amounts of IFN-α and IL-6 cytokines and exhibited less infiltration of monocytes after VSV infection than did wild-type mice. (A) Usp25−/− mice produced significantly decreased amounts of IFN-α and IL-6 cytokines than did wild-type mice. Wild-type and Usp25−/− mice were infected with VSV (2 × 107) through tail vein. Twelve hours after infection, the sera were collected and the amount of IFN-α or IL-6 in the sera was determined by ELISA analysis. (B) The infiltration of monocytes in the lungs of Usp25−/− mice was impaired compared with that of wild-type mice. Lung tissues from mice treated in A were isolated and subject to histological analysis with hematoxylin and eosin staining (H&E) 24 h after infection. [Original magnification, ×20 (H&E) or ×40 (Ly6C).] (Scale bars, 100 μm.) Data are representative of two independent experiments. *P < 0.05, **P < 0.01.
Fig. 3.

USP25-deficient mice are more susceptible to the infection with H5N1 and HSV-1. (A) Usp25−/− mice exhibited earlier mortality and more severe weight loss after H5N1 infection than did wild-type mice. Wild-type (n = 10) and Usp25−/− mice (n = 11) were infected with the H5N1 influenza virus (1 × 103 EID50 per mouse) and the survival and weight loss were monitored every day. (B) The expression of Ifnb, Ifna4, and Tnf was inhibited in the lungs and livers of Usp25−/− mice after H1N1(PR8) virus challenge compared with the wild-type mice. Mice (n = 4) were infected with H1N1(PR8) and lungs and livers were collected for qPCR analysis 24 h after infection. (C and D) Usp25−/− mice exhibited higher mortality rate and produced decreased amount of IFN-α and IL-6 cytokines after HSV-1 challenge than did the wild-type mice. Wild-type and Usp25−/− mice (n = 5) were infected with HSV-1 (1 × 106 pfu). The survival of mice was monitored every day (C). The sera were collected at 12 h after infection followed by ELISA analysis (D). (E) Usp25−/− mice showed less Ly6C+ monocytes infiltration in the lungs than did wild-type mice. Wild-type and Usp25−/− mice (n = 3) were infected with HSV-1 (2 × 106 pfu). Twenty-four hours after infection, lungs from mice were isolated and subject to histological analysis. [Original magnification, × 20 (H&E) or ×40 (Ly6C).] (Scale bars, 100 μm.) Arrow heads, infiltrated Ly6C+ monocytes. Data are representative of two independent experiments. *P < 0.05, **P < 0.01.
The DUB Activity of USP25 Is Required for Virus-Triggered Signaling.
We next determined whether the deubiquitination (DUB) activity was required for promoting virus-triggered signaling. We found that reconstitution of Usp25−/− MEFs with wild-type USP25 but not USP25(C178S) restored SeV-, VSV-, or HSV-1–triggered expression of Ifnb, Ifna4, and Il6 (Fig. 4A). In addition, SeV- or HSV-1–induced production of IFN-α and IL-6 cytokines was rescued by reconstitution with wild-type USP25 but not USP25(C178S) (Fig. 4B). Consistent with these observations, SeV-triggered phosphorylation of IRF3 and IκBα was restored by the reconstitution of Usp25−/− MEFs with USP25 but not by reconstitution with USP25(C178S) (Fig. 4C), indicating that the DUB activity of USP25 is required for mediating virus-triggered production of type I IFNs and proinflammatory cytokines. Furthermore, the expression of SeV P protein was restricted in Usp25−/− MEFs reconstituted with wild-type USP25 but not USP25(C178S) (Fig. 4D). Similarly, the RNA level of HSV-1 UL30 gene in Usp25−/− MEFs reconstituted with wild-type USP25 was substantially lower than that in Usp25−/− MEFs reconstituted with USP25(C178S) (Fig. 4E). These data together suggest that the DUB activity of USP25 is required for restricting virus replication.
Fig. 4.

The DUB activity of USP25 is required for mediating virus-triggered signaling. (A) Reconstitution of Usp25−/− MEFs with wild-type USP25 but not USP25(C178S) restored SeV-, VSV- or HSV-1–triggered expression of Ifnb, Ifna4, and Il6. Usp25−/− MEFs were reconstituted with empty vector, USP25 (WT) or USP25(C178S) (CS) followed by SeV, VSV, or HSV-1 infection. Cells were harvested for qPCR analysis at 8 h after infection. (B) SeV- or HSV-1–induced production of IFN-α and IL-6 cytokines was rescued by reconstitution with USP25 but not USP25(C178S). Cells were treated as in A and the supernatants were collected for ELISA analysis at 12 h after infection. (C) SeV-triggered phosphorylation of IRF3 and IκBα was restored by the reconstitution of Usp25−/− MEFs with USP25 but not by reconstitution with USP25(C178S). Usp25−/− MEFs were reconstituted with empty vector, USP25 (WT), or USP25(C178S) (CS) and infected by SeV followed by immunoblot analysis with the indicated antibodies. (D and E) The replication of SeV or HSV-1 was inhibited in Usp25−/− MEFs reconstituted with USP25 but not USP25(C178S). Usp25−/− MEFs were reconstituted with empty vector, USP25 (WT) or USP25(C178S) (CS) and infected with SeV or HSV-1 (MOI = 1). One hour later, the viruses were removed and the cells were cultured with full medium for 24 h before immunoblot (D) or qPCR analysis (E) were performed. Data are representative of two independent experiments (mean and SD of three replicates in A, B, and E).
USP25 Interacts with and Stabilizes TRAF3 and TRAF6.
When examining the effects of USP25 deficiency on virus-triggered signaling, we repeatedly observed that the protein levels of TRAF3 and TRAF6 was substantially decreased in Usp25−/− cells than in the wild-type counterparts after virus infection (Fig. S6A). However, the mRNA levels of Traf3 or Traf6 were comparable between wild-type and Usp25−/− cells with or without viral infection (Fig. S6 B and C). In addition, virus-triggered degradation of TRAF3 and TRAF6 in Usp25−/− MEFs was inhibited by reconstitution of USP25 but not USP25(C178S) (Fig. 4C), indicating that USP25 stabilizes TRAF3 and TRAF6 protein through deubiquitination. Interestingly, we found that USP25 interacted with TRAF3 or TRAF6 after SeV or HSV-1 infection in primary MEFs (Fig. 5A). Sequence analysis identified two conserved TRAF-interacting motifs (TIMs) [Px(Q/E)E or PxExx(aromatic/acid residue)] in USP25, 62-PPQEE-66 and 797-PPETDY-802 (Fig. S7A). Simultaneously, mutation of these two TIMs (62-FPNEE-66 and 797-FPNTDY-802) [designated as USP25(ΔT1+2)] impaired its ability to associate with TRAF3 and TRAF6 (Fig. S7B), indicating that the TIMs of USP25 are involved in mediating USP25-TRAF interaction. Furthermore, reconstitution of USP25(ΔT1+2) into Usp25−/− MEFs failed to fully rescue SeV-triggered induction of Ifnb and Tnf (Fig. S7C), suggesting that USP25-TRAF association is a prerequisite for mediating virus-triggered induction of type I IFNs and proinflammatory cytokines.
Fig. S6.

USP25 regulates the stability of TRAF3 and TRAF6 proteins. (A) Wild-type or Usp25−/− MEFs were left untreated or treated with CHX (100 μg/mL) for one hour followed by SeV or HSV-1 infection. Cells were lyzed and the immunoblot analysis was performed at 8 h after infection. (B and C) Wild-type and Usp25−/− BMDCs or MLFs were infected with SeV or HSV-1 for the indicated time points before qPCR analysis was performed. Data are representative of two (A) or at least three (B and C) independent experiments (graphs show mean and SD of three replicates).
Fig. 5.

USP25 interacts with TRAF6 and TRAF3 after virus infection. (A) USP25 interacted with TRAF3 or TRAF6 after SeV or HSV-1 infection in primary MEFs. MEFs were infected with SeV or HSV-1 for the indicated time points. Cells were lysed and the cell lysates were immunoprecipitated with anti-TRAF3 or anti-TRAF6. The immunoprecipitates were analyzed by immunoblot with anti-USP25, anti-TRAF3, or anti-TRAF6 (top four panels). The expression levels of the proteins in the lysates were analyzed by immunoblot with the indicated antibodies (bottom four panels). (B) K48-linked ubiquitination of TRAF3 was enhanced in Usp25−/− BMDCs after SeV or HSV-1 infection compared with the wild-type BMDCs. Wild-type and Usp25−/− BMDCs were infected with SeV or HSV-1for the indicated time points followed by denature immunoprecipitation (Denature-IP) and immunoblot assays with the indicated antibodies. (C) SeV-induced K48-linked ubiquitination of TRAF3 was inhibited in Usp25−/− MEFs reconstituted with USP25 but not those with USP25(C178S). Usp25−/− MEFs were reconstituted with empty vector, USP25 or USP25(C178S) and infected with SeV followed by Denature-IP and immunoblot assays with the indicated antibodies. Data are representative of three independent experiments.
Fig. S7.

USP25 interacts with TRAF3 and TRAF6 through the TRAF-interacting motifs (TIMs). (A) An alignment of the conserved TIMs of USP25. (B) Mutation of the TIMs of USP25 impaired TRAF-USP25 interaction. HEK293 cells were transfected with HA-TRAF3 or HA-TRAF6 (3 μg) and Flag-tagged USP25, USP25(ΔT1), USP25(ΔT2) or USP25(ΔT1+2) (5 μg). Twenty hours later, coimmunoprecipitation and immunoblot analysis was performed with the indicated antibodies. (C) Reconstitution of USP25(ΔT1+2) into Usp25−/− MEFs failed to fully restore the expression of Ifnb and Tnf after SeV or HSV-1 infection. Usp25−/− MEFs were reconstituted with an RVKM-IRES-GFP empty vector (Vec), RVKM-USP25, or RVKM-USP25(ΔT1+2) through retrovirus-mediated gene transfer. Forty hours later, the expression of transduced proteins was examined by immunoblot analysis (Left) and cells were infected with SeV or HSV-1 for 12 h followed by qPCR analysis (Right). Data are representative of two independent experiments (mean and SD of three replicates in C). *P < 0.05, **P < 0.01, ***P < 0.001.
We further found that K48-linked ubiquitination of TRAF3 was enhanced in Usp25−/− BMDCs after SeV or HSV-1 infection compared with the wild-type cells (Fig. 5B). Reconstitution of USP25 but not USP25(C178S) into Usp25−/− MEFs inhibited SeV-induced K48-linked ubiquitination of TRAF3 (Fig. 5C). It has been shown that RNA virus infection induces degradation of TRAF3 to activate noncanonical NF-κB p100/p52, which up-regulates H3K9 trimethylation (H3K9me3) of Ifnb promoter by attenuating the recruitment of JMJD2A (31). However, the activation of noncanonical NF-κB and the H3K9me3 modification of Ifnb promoter were comparable between wild-type and Usp25−/− cells after SeV or HSV-1 infection (Fig. S8), indicating that USP25-mediated stabilization of TRAF3 is independently of the noncanonical NF-κB signaling (discussed below). In contrast, the H3K4me3 modification of Ifnb promoter was inhibited in Usp25−/− BMDCs, which might be a result of impaired activation of IRF3 in these cells (Fig. S8C). Together, these data suggest that USP25 removes K48-linked polyubiquitin chains from TRAF3 and thereby stabilizes TRAF3 after viral infection.
Fig. S8.

Role of USP25 in virus-triggered noncanonical NF-κB pathways. (A and B) USP25 deficiency had no effect on SeV- or HSV-1–triggered accumulation of NIK or p52 in BMDCs. Wild-type and Usp25−/− BMDCs were infected with SeV or HSV-1 followed by immunoblot with the indicated antibodies. (C) H3K4me3 and H3K9me3 of the Ifnb promoter in wild-type and Usp25−/− BMDCs. Wild-type and Usp25−/− BMDCs were left uninfected or infected with SeV or HSV-1 for 8 h followed by ChIP analysis with the indicated antibodies. Data are representative of three (A and B) or two (C) independent experiments. Graphs show means and SD of four replicates. **P < 0.01, ***P < 0.001.
We next investigated whether USP25 stabilized TRAF6 through a similar mechanism. Interestingly, however, SeV- or HSV-1–triggered K48-linked ubiquitination of TRAF6 was comparable between wild-type and Usp25−/− MEFs (Fig. S9A), which is consistent with our previous observations that USP25 does not remove K48-linked polyubiquitin chains from TRAF6 (29). Moreover, SeV-induced degradation of TRAF6 in Usp25−/− MEFs was blocked by treatment with the autophagy inhibitor 3MA, whereas SeV-induced degradation of TRAF3 in Usp25−/− MEFs was inhibited by proteasome inhibitor MG132 (Fig. S9B). These data collectively suggest that USP25 inhibits virus-triggered degradation of TRAF3 and TRAF6 and thereby promotes the induction of type I IFNs and proinflammatory cytokines after viral infection.
Fig. S9.

USP25 stabilizes TRAF6 through proteasome-independent pathways after virus infection. (A) USP25 deficiency resulted in accelerated degradation of TRAF6 without altering the K48-linked ubiquitination of TRAF6 after viral infection. Wild-type and Usp25−/− BMDCs were infected with SeV or HSV-1 for the indicated time points followed by denature-IP and immunoblot assays. (B) SeV-induced degradation of TRAF6 in Usp25−/− MEFs was inhibited by the autophagy inhibitor 3MA. Wild-type and Usp25−/− MEFs were pretreated with MG132 (10 nM) or 3MA (0.5 μg/mL) for 2 h and infected with SeV for 8 h before immunoblot analysis was performed with the indicated antibodies. Data are representative of two independent experiments.
Complement of TRAF3/6 into USP25-Deficient MEFs restores Virus-Triggered Signaling.
Because TRAF3 and TRAF6 are required for virus-triggered activation of IRF3 and NF-κB, respectively, we reasoned that reconstitution of TRAF3 or TRAF6 into Usp25−/− cells might rescue virus-triggered activation of IRF3 and NF-κB. Intriguingly, reconstitution with TRAF3 or TRAF6 into Usp25−/− MEFs restored SeV-triggered phosphorylation of IRF3 and IκBα, respectively (Fig. 6A). Consistently, SeV- or HSV-1–triggered expression of Ifnb and Ifna4 or Il6 was fully restored in Usp25−/− MEFs reconstituted with TRAF3 or TRAF6, respectively (Fig. 6 B and C). In addition, simultaneous reconstitution of both TRAF3 and TRAF6 into Usp25−/− MEFs fully restored SeV- or HSV-1–induced expression of Ifna4, Ifnb, and Il6 (Fig. 6D). In contrast, reconstitution of MAVS (an adaptor protein upstream of TRAF3 and TRAF6) into Usp25−/− MEFs failed to fully rescue SeV-triggered expression of type I IFNs and proinflammatory cytokines (Fig. S10). These data suggest that USP25 positively regulates virus-triggered activation of IRF3 and NF-κB and subsequent induction of type I IFNs and proinflammatory cytokines through stabilizing TRAF3 and TRAF6, respectively.
Fig. 6.

Reconstitution of TRAF3 and TRAF6 into Usp25−/− MEFs rescues virus-triggered induction of type I INFs and proinflammatory cytokines. (A–C) Reintroduction of Traf3 and Traf6 into Usp25−/− MEFs restored SeV-induced activation of IRF3 and NF-κB, respectively. Wild-type and Usp25−/− MEFs were transfected with a retrovirus empty vector (RVKM) or RVKM-Traf3, a lentivirus empty vector (phage-puro) or phage-puro-Traf6 through retrovirus- or lentivirus-mediated gene transfer. (A) Cells were infected with SeV for the indicated time points followed by immunoblot analysis with the indicated antibodies. (B and C) Cells were infected with SeV or HSV-1 for 8 h before qPCR analysis was performed. (D) Wild-type or Usp25−/− MEFs were cotransfected with RVKM and phage-puro (Vec) or RVKM-Traf3 and phage-puro-Traf6 (T3+T6). The expression of Traf3 and Traf6 was examined by immunoblot analysis (Left). Cells were infected with SeV or HSV-1 for 8 h followed by qPCR analysis (graphs). Data are representative of two (A) or three (B–D) independent experiments (mean and SD of three replicates in B–D).
Fig. S10.
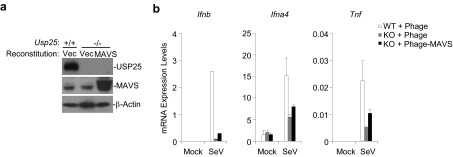
Reconstitution of MAVS into Usp25−/− MEFs partially restored SeV-triggered expression of type I IFNs and proinflammatory cytokines. (A) Wild-type or Usp25−/− MEFs were transfected with Phage-puro (Vec) or Phage-puro-Flag-MAVS followed by selection with puromycin (1 μg/mL) for 2 d. The puromycin-resistant cells were subject to immunoblot with the indicated antibodies. (B) The expression of type I IFNs and proinflammatory cytokines in cells obtained from A. Cells were infected with SeV or left uninfected for 12 h before qPCR analysis was performed. Data are representative of two independent experiments.
Discussion
TRAF3 and TRAF6 are critically involved in virus-triggered signaling by linking upstream adaptor proteins to downstream kinases and transcription factors. In this study, we demonstrated that USP25 positively regulated RNA and DNA virus-triggered innate immune responses by stabilizing TRAF3 and TRAF6. Viral infection induced the expression of USP25 and the interaction between USP25 and TRAF3 or TRAF6. Deficiency of USP25 resulted in destabilized TRAF3 and TRAF6, impaired activation of IRF3 and NF-κB, and reduced production of type I IFNs and proinflammatory cytokines after viral infection. In addition, Usp25−/− mice produced decreased levels of antiviral cytokines after viral infection and exhibited increased susceptibility to H5N1 and HSV-1 infection compared with the wild-type littermates. These data collectively suggest that USP25 is indispensable for host defense against RNA and DNA viruses.
Sequence analysis identified multiple ISRE and κB binding sites in the promoter of the USP25 gene (Fig. S1D). Interestingly, the ISRE sites are preferentially recognized by IRF7 but not IRF3 or ISGF3 (Genomatix Software). It has been demonstrated that little IRF7 protein exists in unstimulated cells and type I IFN stimulation induces robust expression of IRF7. Thus, it is likely that viral infection results in induction of type I IFNs and activation of NF-κB, which collaborate to induce the transcription of USP25 gene to form a positive feedback regulation loop of innate antiviral response. Obviously, the hypothesis awaits further investigations.
We have observed that USP25 deficiency resulted in the degradation of TRAF6 and reduced activation of NF-κB after viral infection, whereas reconstitution TRAF6 into Usp25−/− MEFs rescued virus-triggered activation of NF-κB and subsequent induction of proinflammatory cytokines. In addition, reconstitution of wild-type USP25 but not USP25(C178S) inhibited virus-triggered degradation of TRAF6 and activation of NF-κB, indicating that USP25 regulates the stability of TRAF6 protein dependently on its DUB activity. Interestingly, however, virus-triggered K48-linked ubiquitination of TRAF6 was comparable between wild-type and Usp25−/− MEFs and virus-triggered degradation of TRAF6 was inhibited by the autophagy inhibitor 3MA but not the proteasome inhibitor MG132, which is consistent with our previous report that USP25 does not regulate K48-linked ubiquitination of TRAF6 (29). In contrast to a previous study that shows USP25 removes K63-linked polyubiquitin chains from TRAF6 and thus inhibits SeV-triggered signaling (32), we observed very minor difference in regard of the K63-linked ubiquitination of TRAF6 in wild-type and Usp25−/− BMDCs following SeV or HSV-1 infection. The reason behind the discrepancies in our data and theirs is currently unknown. Considering overexpression of USP25 may lead to an overall decrease of cellular ubiqutination signals (32), we have provided genetic evidence showing that USP25 is a positive regulator for RNA and DNA virus-triggered induction of proinflammatory cytokines. Several studies have suggested that ubiquitin-modified species or aggregates are recognized and degraded by the selective autophagy pathway, which is mediated by the action of specific adaptors to connect the ubiquitin modification with the autophagy pathway (33, 34). It is possible that USP25 catalyzes deubiquitination of TRAF6 (which is neither K63- nor K48-linkage) and thereby inhibits the autophagy-dependent degradation of TRAF6. Alternatively, USP25 may indirectly regulate the stability of TRAF6 protein by catalyzing deubiquitination of other uncharacterized proteins. Nonetheless, our data have clearly demonstrated that USP25 protects TRAF6 from proteasome-independent degradation after viral infection.
We demonstrated that SeV- or HSV-1–triggered K48-linked ubiquitination of TRAF3 was potentiated and the degradation of TRAF3 was accelerated in Usp25−/− cells compared with wild-type cells. Consistently, SeV-induced degradation of TRAF3 in Usp25−/− MEFs was inhibited by the proteasome inhibitor MG132, suggesting that USP25 protects TRAF3 from degradation by catalyzing deubiquitination of TRAF3 and thereby positively regulates RNA and DNA virus-triggered type I IFN induction. A recent study has shown that RNA virus infection causes degradation of TRAF3 and the processing of p100 into p52 (31). The noncanonical NF-κB p52 binds to the Ifnb promoter and suppresses its activation by attenuating the recruitment of JMJD2A to the Ifnb promoter. Although the degradation of TRAF3 was accelerated in Usp25−/− cells, it is unlikely that USP25 regulates virus-triggered signaling through TRAF3-NIK-noncanonical NF-κB pathways. First, we observed that the accumulation of p52 and the H3K9me3 modification of the Ifnb promoter were comparable between wild-type and Usp25−/− cells after SeV or HSV-1 infection. Second, USP25 deficiency impaired SeV- or HSV-1–induced activation of IRF3 and IκBα, which is not affected by the NIK-p52 pathways. Third, USP25 deficiency in pDCs also resulted in decreased production of type I IFNs and proinflammatory cytokines, which is supposed not to be regulated by the noncanonical NF-κB pathways. It is possible that there are two pools of TRAF3 in the cytosol, one of which mediates TBK1-IRF3 pathway to activate type I IFN expression and the other mediates NIK-p52 pathway to inhibit virus-triggered type I IFN induction. These two pools of TRAF3 may be differentially regulated by distinct DUBs. In this context, a previous report and our present study have clearly shown that OTUD7B and USP25 regulate TRAF3-p52 and TRAF3-IRF3 pathways, respectively (35). Collectively, our characterization of USP25-mediated stabilization of TRAF3 and TRAF6 proteins provides insight into the mechanisms on host defense against RNA and DNA viruses and highlights a new target for treatment or vaccination of virus-induced diseases.
Materials and Methods
Mice.
Usp25−/− was generated as described (30). Mice were maintained in the specific pathogen-free facility of Wuhan University. Age and sex matched Usp25+/+ and Usp25−/− littermates were used for all experiments. All animal experiments were in accordance with protocols approved by the Institutional Animal Care and Use Committee of Wuhan University.
Other Materials and Methods.
Other materials and methods used in this study are described in SI Materials and Methods.
SI Materials and Methods
Viral Infection.
Cells were seeded into 24-well plates (2 × 105 cells per well) or six-well plates (106 to 107 cells per well). Twenty-four hours later, cells were infected with NDV-GFP, SeV, VSV, or HSV-1 for the indicated time points. The cells were collected for qPCR or immunoblot assays, and the supernatants were saved for ELISA analysis. For mice infection, age- and sex-matched Usp25+/+ and Usp25−/− littermates were injected i.v. with VSV (2 × 107 pfu per mouse) or HSV-1 (1 × 106 pfu per mouse) or injected intranasally with H5N1 strain (A/Chicken/Hubei/489) (1 × 103 EID50 per mouse) or H1N1(PR8) and the survival of animals was monitored every day. The sera or lungs from mice infected with VSV or HSV-1 were collected for qPCR or histological analysis at 12 or 24 h after infection, respectively. For virus replication assays, viruses were removed at 1 h after infection. Cells were washed with PBS for twice followed by culture with full medium for the indicated time points.
Antibodies and Reagents.
Mouse control IgG (Santa Cruz Biotechnology, sc-2025) and rabbit control IgG (Millipore, 12–370), HRP-conjugated goat-anti mouse or rabbit IgG (Thermo Scientific, PA1-86717 and SA1-9510), mouse anti-Myc (Sungene Biotech, KM8003), mouse anti-Flag (KM8002), mouse anti-β-Actin (KM9001), mouse anti-HA (COVANCE, MMS-101R), anti-pIκBα (9246L), anti-Ubiquitin (sc-8017), anti-TRAF6 (sc-7221), anti-TRAF3 (sc-1828), anti-IRF3 (sc-9082), Rabbit anti-Ubiquitin, lys48-specific (MILLIPORE, 05–1307),anti-Ubiquitin, lys63-specific (MILLIPORE, 05–1308), anti-IκBα (sc-371), anti-NF-κΒ2(p100/52) (CST, 4882P), anti-NIK (4994P), and anti-p-IRF3(4947S) were purchased from the indicated manufactures. Rabbit anti-SeV P protein was raised against recombinant P protein of SeV. Rabbit anti-USP25 was described (36) and kindly provided by Gemma Marfany (University de Barcelona, Barcelona). PolydA;dT, IFN-stimulating DNA and HSV120 were described (37). MG132, 3MA, CpG DNA and poly(I:C) were purchased from Sigma. Recombinant mouse TNFα and FLT3L were purchased from Peprotech.
Cell Culture.
Primary MEFs were prepared from E12.5–14.5 embryos and cultured as described (29, 30). Bone marrow cells were isolated from the femurs of the indicated mice. The cells were cultured in DMEM containing 20% (vol/vol) FBS, 1% streptomycin-penicillin and 10 μM β-mercaptoethanol with 20% (vol/vol) L929 cell supernatant for BMDM differentiation or with 20 ng/mL GM-CSF for BMDC differentiation. The medium was changed every two days. At the 7–9 d, BMDMs or BMDCs were harvested and stained with CD11b (BioLegend, 101206) or CD11c (BD Biosciences, 557401) and subjected to flow cytometry to confirm the purity, respectively. BMDMs and BMDCs (>90% purity) were used for subsequent analysis. For generation of pDCs, bone marrow cells were cultured in RPMI medium containing containing 10% FBS, 1% streptomycin–penicillin and recombinant mouse FLT3L (20 ng/mL) for a week and purified by flow cytometry based on the specific surface markers (CD11c+CD11b−B220+). Primary mouse lung fibroblasts were isolated from ∼8- to 10-wk-old mice. Lungs were minced and digested in calcium- and magnesium-free HBSS buffer supplemented with 10 mg/mL type II collagenase (Worthington) and 20 μg/mL DNase I (Sigma-Aldrich) for 3 h at 37 °C with shaking. Cell suspensions were filtered through progressively smaller cell strainers (100 and 40 μm). Filtered cells were plated in the culture media [1:1 (vol/vol) DMEM/Ham’s F-12 containing 10% FBS, 15 mM Hepes, 2 mM l-glutamine, 50 U/mL penicillin, and 50 μg/mL streptomycin]. One hour later, adherent fibroblasts were rinsed with HBSS and cultured for experiments.
Real-Time Quantitative PCR and ELISA.
Cells treated with various stimuli were harvested in TriZol (Invitrogen, 15596–018), and the first-strand cDNA was synthesized with a reverse transcription kit (Invitrogen, 4368814). Gene expression was examined with a Bio-Rad SFX connect system with an iQ SYBR Green Real-Time PCR kit (Bio-Rad Laboratories). Data were normalized to the expression of β-Actin. Gene-specific primers are listed in Table S1. The ELISA kits for IFN-β, TNF-α, and IL-6 (BD Biosciences) and IFN-α (R&D Systems) were used to detect the indicated cytokines in the supernatants or sera.
Immunohistochemistry Analysis.
Lungs from mice were fixed in formalin and embedded into paraffin blocks. The paraffin blocks were sectioned (5 μm) for H&E staining. The immunohistochemistry analysis was performed on the 5-μm sections. The sections were placed on polylysinecoated slides, deparaffinized in xylene, rehydrated through graded ethanol, quenched for endogenous peroxidase activity in 3% (vol/vol) hydrogen peroxide, and processed for antigen retrieval by microwave heating for 7 min in 10 mM citrate buffer (pH 6.0). The anti-Ly6C antibody (M100L4; Tianjin Sungene Biotech) was diluted 1:100 in phosphate buffer saline (PBS) containing 1% BSA and incubated at room temperature for over 6 h. Immunostaining was performed using the Maixin_Bio Detection kit peroxidase/diaminobenzidine (DAB) rabbit/mouse (Kit-9710, DAB-0031; Maixin_Bio), which resulted in a brown-colored precipitate at the antigen site. Subsequently, sections were counterstained with hematoxylin (Zymed Laboratories) for 5 min and coverslipped. Pictures were acquired using a HistoFAXS system.
Constructs, Coimmunoprecipitation, and Immunoblot.
Mammalian expression plasmids for Flag- or Myc-tagged USP25 and its truncated mutants, TRAF6, TRAF3 and Flag-USP25(C178S) are described (29, 30). Coimmunoprecipitation and immunoblot and ubiquitination assays were performed as previous described with modifications (29, 30). Cells were lysed and the protein concentration was quantified with a BCA quantification kit (Thermo Scientific). To detect the protein levels of USP25, β-Actin, IRF3, and IκBα, 10 μg of proteins were loaded for the immunoblot analysis. For detection of pIRF3, pIκBα, TRAF3, and TRAF6, 45 μg proteins were loaded for the immunoblot analysis. The antibodies were diluted in 3–5% (wt/vol) fat-free milk (BD Biosciences) or 1% BSA (Sigma) in TBS-T (1:500–1:2,000), and the membranes were incubated at 4 °C on an orbital shaker for overnight. Finally, Clarity Western ECL Substrate (Bio-Rad) and SuperSignal West Femto Stable Peroxide (Thermo Scientific) were used to detect the HRP signals.
Chromatin Immunoprecipitation Assays.
Chromatin immunoprecipitation assays were performed as described (38). Briefly, 5 × 106 cells were fixed with 1% formaldehyde and quenched by glycine. The cells were washed three times with PBS and then harvested in ChIP lysis buffer (50 mM Tris⋅HCl pH 8.0, 1% SDS, 5 mM EDTA) followed by sonication until the sizes of DNA were 400–600 bp. The lysate was centrifuged at 4 °C for 15 min and ChIP dilution buffer (20 mM Tris⋅HCl, pH 8.0, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100) was added to the supernatant (4:1 volume). The resulted lysate was then incubated with antibodies at 4 °C overnight. The protein G beads were added into the lysate on the next morning and incubated at 4 °C for 3 h. DNA was eluted using the ChIP elution buffer (0.1 M NaHCO3, 1% SDS, 30 μg/mL proteinase K) through incubation at 65 °C overnight, and DNA was purified with a DNA purification kit (Sangon). The purified DNA was assayed by quantitative PCR with SFX connect system with an iQ SYBR Green Real-Time PCR kit (Bio-Rad Laboratories).
Site-Directed Mutagenesis.
Site-directed mutagenesis was performed by using a mutagenesis kit according to the instructions from the manufacture (A14604; Life Technologies).
Virus-Mediated Gene Transfer.
Retrovirus-mediated gene transfer was performed as described (29, 30). For lentivirus-mediated gene transfer, phage-6tag-MAVS, phage-6tag-puro-TRAF6 or the empty vector was cotransfected with the packaging vectors pSPAX2 and pMD2G into HEK293T cells. Eight hours after transfection, the medium was changed with fresh full medium (10% FBS, 1% streptomycin-penicillin and 10 μM β-mercaptoethanol). Forty hours later, the supernatants were harvested to infect MEFs.
Statistical Analysis.
Differences between experimental and control groups were determined by Student’s t test (where two groups of data were compared) or by two-way ANOVA analysis (where more than two groups of data were compared). P values less than 0.05 were considered statistically significant. For animal survival analysis, the Kaplan–Meier method was adopted to generate graphs, and the survival curves were analyzed with log-rank analysis.
Acknowledgments
We thank Drs. Yan Zhou, Zan Huang, Xiao-Dong Zhang (Wuhan University), members of B.Z. laboratory, and members of the core facilities of College of Life Sciences for technical help. This work was supported by grants from the Ministry of Science and Technology of China (2014CB542601 and 2012CB910201), the National Natural Science Foundation of China (31371427), the Ministry of Education of China (201427), and Large-scale Instrument and Equipment Sharing Foundation of Wuhan University.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. A.T. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1509968112/-/DCSupplemental.
References
- 1.Nakhaei P, Genin P, Civas A, Hiscott J. RIG-I-like receptors: Sensing and responding to RNA virus infection. Semin Immunol. 2009;21(4):215–222. doi: 10.1016/j.smim.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 2.Wu J, Chen ZJ. Innate immune sensing and signaling of cytosolic nucleic acids. Annu Rev Immunol. 2014;32:461–488. doi: 10.1146/annurev-immunol-032713-120156. [DOI] [PubMed] [Google Scholar]
- 3.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140(6):805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 4.Heil F, et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303(5663):1526–1529. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- 5.Hemmi H, et al. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408(6813):740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 6.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413(6857):732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 7.Yoneyama M, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5(7):730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 8.Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339(6121):786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chiu YH, Macmillan JB, Chen ZJJ. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell. 2009;138(3):576–591. doi: 10.1016/j.cell.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takaoka A, et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. 2007;448(7152):501–505. doi: 10.1038/nature06013. [DOI] [PubMed] [Google Scholar]
- 11.Zhang Z, et al. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat Immunol. 2011;12(10):959–965. doi: 10.1038/ni.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Unterholzner L, et al. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol. 2010;11(11):997–1004. doi: 10.1038/ni.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Y, et al. LSm14A is a processing body-associated sensor of viral nucleic acids that initiates cellular antiviral response in the early phase of viral infection. Proc Natl Acad Sci USA. 2012;109(29):11770–11775. doi: 10.1073/pnas.1203405109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamamoto M, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301(5633):640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 15.Wesche H, Henzel WJ, Shillinglaw W, Li S, Cao Z. MyD88: An adapter that recruits IRAK to the IL-1 receptor complex. Immunity. 1997;7(6):837–847. doi: 10.1016/s1074-7613(00)80402-1. [DOI] [PubMed] [Google Scholar]
- 16.Xu LG, et al. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell. 2005;19(6):727–740. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 17.Seth RB, Sun L, Ea CK, Chen ZJJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122(5):669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 18.Kawai T, et al. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol. 2005;6(10):981–988. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- 19.Zhong B, et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity. 2008;29(4):538–550. doi: 10.1016/j.immuni.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 20.Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455(7213):674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oganesyan G, et al. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature. 2006;439(7073):208–211. doi: 10.1038/nature04374. [DOI] [PubMed] [Google Scholar]
- 22.Häcker H, et al. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature. 2006;439(7073):204–207. doi: 10.1038/nature04369. [DOI] [PubMed] [Google Scholar]
- 23.Saha SK, et al. Regulation of antiviral responses by a direct and specific interaction between TRAF3 and Cardif. EMBO J. 2006;25(14):3257–3263. doi: 10.1038/sj.emboj.7601220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paz S, et al. A functional C-terminal TRAF3-binding site in MAVS participates in positive and negative regulation of the IFN antiviral response. Cell Res. 2011;21(6):895–910. doi: 10.1038/cr.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li S, et al. Regulation of virus-triggered signaling by OTUB1- and OTUB2-mediated deubiquitination of TRAF3 and TRAF6. J Biol Chem. 2010;285(7):4291–4297. doi: 10.1074/jbc.M109.074971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kayagaki N, et al. DUBA: A deubiquitinase that regulates type I interferon production. Science. 2007;318(5856):1628–1632. doi: 10.1126/science.1145918. [DOI] [PubMed] [Google Scholar]
- 27.Nakhaei P, et al. The E3 ubiquitin ligase Triad3A negatively regulates the RIG-I/MAVS signaling pathway by targeting TRAF3 for degradation. PLoS Pathog. 2009;5(11):e1000650. doi: 10.1371/journal.ppat.1000650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boone DL, et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol. 2004;5(10):1052–1060. doi: 10.1038/ni1110. [DOI] [PubMed] [Google Scholar]
- 29.Zhong B, et al. Ubiquitin-specific protease 25 regulates TLR4-dependent innate immune responses through deubiquitination of the adaptor protein TRAF3. Sci Signal. 2013;6(275):ra35. doi: 10.1126/scisignal.2003708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhong B, et al. Negative regulation of IL-17-mediated signaling and inflammation by the ubiquitin-specific protease USP25. Nat Immunol. 2012;13(11):1110–1117. doi: 10.1038/ni.2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jin J, et al. Noncanonical NF-κB pathway controls the production of type I interferons in antiviral innate immunity. Immunity. 2014;40(3):342–354. doi: 10.1016/j.immuni.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhong H, et al. Ubiquitin-specific proteases 25 negatively regulates virus-induced type I interferon signaling. PLoS One. 2013;8(11):e80976. doi: 10.1371/journal.pone.0080976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stolz A, Ernst A, Dikic I. Cargo recognition and trafficking in selective autophagy. Nat Cell Biol. 2014;16(6):495–501. doi: 10.1038/ncb2979. [DOI] [PubMed] [Google Scholar]
- 34.Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–132. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- 35.Hu H, et al. OTUD7B controls non-canonical NF-κB activation through deubiquitination of TRAF3. Nature. 2013;494(7437):371–374. doi: 10.1038/nature11831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bosch-Comas A, Lindsten K, Gonzàlez-Duarte R, Masucci MG, Marfany G. The ubiquitin-specific protease USP25 interacts with three sarcomeric proteins. Cell Mol Life Sci. 2006;63(6):723–734. doi: 10.1007/s00018-005-5533-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou Q, et al. The ER-associated protein ZDHHC1 is a positive regulator of DNA virus-triggered, MITA/STING-dependent innate immune signaling. Cell Host Microbe. 2014;16(4):450–461. doi: 10.1016/j.chom.2014.09.006. [DOI] [PubMed] [Google Scholar]
- 38.Wang X, et al. MLL1, a H3K4 methyltransferase, regulates the TNFα-stimulated activation of genes downstream of NF-κB. J Cell Sci. 2012;125(Pt 17):4058–4066. doi: 10.1242/jcs.103531. [DOI] [PubMed] [Google Scholar]