

Significance
Neuropathic pain is a complex disorder, characterized by affective and sensory symptoms. Efficient treatment of this condition should target both pain-modulating pathways and mood/affect networks. We show that tricyclic antidepressants (TCAs), which modulate spinal pain processing, also act in the brain reward center to alleviate allodynia and depression-like behaviors. We reveal how one key protein of nucleus accumbens (NAc)-specific signaling affects several molecules/pathways with emerging roles in antinociceptive and antidepressant mechanisms. Our study provides information about the cellular adaptations induced by TCAs in the NAc and novel targets for pain treatment.
Keywords: desipramine, duloxetine, HDAC5, spared nerve injury, gene expression
Abstract
The striatal protein Regulator of G-protein signaling 9-2 (RGS9-2) plays a key modulatory role in opioid, monoamine, and other G-protein–coupled receptor responses. Here, we use the murine spared-nerve injury model of neuropathic pain to investigate the mechanism by which RGS9-2 in the nucleus accumbens (NAc), a brain region involved in mood, reward, and motivation, modulates the actions of tricyclic antidepressants (TCAs). Prevention of RGS9-2 action in the NAc increases the efficacy of the TCA desipramine and dramatically accelerates its onset of action. By controlling the activation of effector molecules by G protein α and βγ subunits, RGS9-2 affects several protein interactions, phosphoprotein levels, and the function of the epigenetic modifier histone deacetylase 5, which are important for TCA responsiveness. Furthermore, information from RNA-sequencing analysis reveals that RGS9-2 in the NAc affects the expression of many genes known to be involved in nociception, analgesia, and antidepressant drug actions. Our findings provide novel information on NAc-specific cellular mechanisms that mediate the actions of TCAs in neuropathic pain states.
Regulator of G-protein signaling 9-2 (RGS9-2) is an intracellular modulator of G-protein–coupled receptor (GPCR) function, which is expressed in medium spiny neurons and cholinergic interneurons of the striatum (1, 2). RGS9-2 influences the magnitude and time course of GPCR signaling by promoting GTPase activity of the Gα subunit and by preventing activation of Gα effectors (3). This modulation can also influence the duration of interactions between the Gβγ subunits and their effector molecules. In addition to Gα subunits, RGS9-2 interacts with several scaffolds and signal transduction proteins that affect its function, expression, and cellular localization. Interactions with the Gβ5 subunit and the adaptor protein R7BP determine the stability and cellular localization of RGS9-2, respectively (3, 4). Several recent studies have provided information on the regulation and function of RGS9-2 complexes in the striatum and how these complexes affect pharmacologic responses (2, 5, 6). In particular, RGS9-2 has been shown to modulate the actions of various psychotropic, antiparkinsonian, neuroleptic, and opiate analgesic drugs (1, 7). The nucleus accumbens (NAc) is a striatal brain region that is a major site of antidepressant drug action (8). Recent studies provided information on signal transduction events triggered by tricyclic antidepressants (TCAs) in NAc neurons and identified several second messengers, transcription factors, and epigenetic molecules involved in their therapeutic actions (9–11). TCAs, such as desipramine (DMI) and nortriptyline (NTL), and selective serotonin/norepinephrine reuptake inhibitors (SNRIs) have also been used to treat neuropathic pain, a complex chronic disorder that is highly comorbid with anxiety and depression (12, 13) and is characterized by thermal hyperalgesia, mechanical allodynia, and dysesthesia (14). TCAs act mainly as serotonin/norepinephrine transporter inhibitors, but additional actions on other GPCRs or ion channels may also contribute to their therapeutic effect (15, 16). In neuropathic pain models, TCAs modulate monoaminergic responses at the spinal cord level, but less is known about their actions in supraspinal sites.
Here, we use genetically modified mice, viral-mediated gene transfer, and several biochemical and molecular biology assays to probe the mechanism via which RGS9-2 in the NAc modulates TCA responses under neuropathic pain states (17, 18). We use the murine spared-nerve injury (SNI) model of neuropathic pain, which produces several symptoms of neuropathic pain, including mechanical allodynia, thermal hyperalgesia, anxiety, and depression (17–19). The results indicate that RGS9-2 plays a prominent role in the onset and efficacy of TCAs with regard to the antiallodynic and antidepressant effects. RGS9-2 negatively regulates the activity of several protein kinase A (PKA) targets, as well as complexes between Gβγ subunits and the GPCR kinase 2 (GRK2) and histone deacetylase 5 (HDAC5). Furthermore, our findings indicate that ablation of Rgs9 prevents the nuclear shuttling and gene-silencing effects of HDAC5 observed during the initial stages of DMI treatment that contribute to the delayed onset of action. RNA-sequencing (RNA-seq) analyses revealed that RGS9-2 affects the expression of a variety of genes involved in TCA responses and/or nociceptive behaviors. Together, these findings provide information on the cellular actions of TCAs in the NAc under neuropathic pain states and identify several factors that affect TCA onset of action and therapeutic efficacy.
Results
Our earlier work demonstrated that RGS9-2 modulates depression-like behaviors developed at 2 mo after nerve injury, but does not affect the intensity of mechanical allodynia or thermal hyperalgesia (19). Here, we use the murine SNI model to test for a role of RGS9-2 in the antiallodynic and antidepressant actions of TCAs. As shown in Fig. 1A, Rgs9–wild-type (Rgs9WT) and Rgs9-knockout (Rgs9KO) mice underwent SNI surgery and began treatment with DMI 2 wk later (a time when maximal allodynia has been established in both genotypes). Mechanical allodynia was evaluated by using the Von Frey test at several time points before and during DMI treatment. In agreement with our earlier findings (19), baseline mechanical allodynia was indistinguishable between Rgs9KO and Rgs9WT mice. Nevertheless, 10 d into a chronic low-dose DMI treatment (5 mg/kg, i.p., twice a day), Von Frey responses were significantly increased in Rgs9KO mice (Fig. 1B). SNI or DMI treatment did not affect the locomotor activity of Rgs9WT or Rgs9KO mice (Fig. S1). Administration of the TCA NTL (3 mg/kg, i.p., twice a day; Fig. 1C) produced an antiallodynic response in Rgs9KO mice only at 2 wk of treatment. The SNRI duloxetine (DLX; 5 mg/kg, i.p., twice a day; Fig. 1D) produced a similar phenotype: Rgs9KO–SNI mice showed an earlier antiallodynic response in the Von Frey test compared to their wild-type controls. An additional cohort of mice was used to determine whether Rgs9KO mice show earlier antiallodynic responses when a high dose of DMI is administered (15 mg/kg, i.p., twice a day). As shown in Fig. 1E, both genotypes showed attenuation of allodynia; however, Rgs9KO mice responded much earlier than Rgs9WT mice. Together, these data suggest that prevention of RGS9-2 action shifts the DMI dose–response curve to the left and also accelerates the onset of the antiallodynic effect. To evaluate the role of RGS9-2 in the antidepressant efficacy of DMI, SNI mice were further treated with DMI (10 mg/kg, i.p., 24 h; 10 mg/kg, i.p., 5 h; and 20 mg/kg, s.c., 1 h before testing) after the 2-wk DMI treatment and monitoring of Von Frey responses and then evaluated in the forced swim test (FST). As shown in Fig. 1F, Rgs9KO–SNI mice that showed increased sensitivity to the antiallodynic actions of DMI also showed enhanced sensitivity to DMI in the FST. Baseline FST immobility time is not different between genotypes, because Rgs9KO mice develop depression-like behavior at 8 wk after SNI (19) and FST assays here are performed at 5 wk after SNI.
Fig. 1.

A role for RGS9-2 in the NAc in the antiallodynic and antidepressant actions of DMI. (A) Timeline and treatment protocol for the DMI studies with Rgs9WT and Rgs9KO. (B) Using the Von Frey assay, we monitored mechanical allodynia responses in SNI groups of Rgs9WT and Rgs9KO mice, at several time points after treatment with a low DMI dose (5 mg/kg, i.p., twice a day). Rgs9KO mice show an antiallodynic response to this dose of DMI within 10 d. *P < 0.05 (two-way ANOVA followed by the Bonferroni post hoc test; n = 9–11 per group). (C and D) A similar phenotype was observed using a low dose of the TCA NTL (3 mg/kg, i.p., twice a day; n = 7 or 8 per group) (C) and the SNRI DLX (5 mg/kg, i.p., twice a day) (D). ***P < 0.001 between Rgs9KO and all treatment groups (two-way ANOVA followed by the Bonferroni post hoc test; n = 7 per group). (E) When a higher DMI dose is used (15 mg/kg, i.p., twice a day), Rgs9KO mice start responding after 10 d of treatment and show a significant antiallodynic response by week 2, whereas Rgs9WT mice respond at 3 wk of treatment. **P < 0.01 (two-way ANOVA followed by Bonferroni post hoc test; n = 8 per group). (F) At 1 wk after the end of the Von Frey assay, Rgs9WT and Rgs9KO mice were tested in the FST. (DMI was administered at 10 mg/kg, i.p., 24 h; 10 mg/kg, i.p., 5 h; and 20 mg/kg, s.c., 1 h before testing). Although DMI did not have an antidepressant effect on Rgs9WT mice, it decreased by 50% the immobility time in Rgs9KO mice. *P < 0.05 (two-way ANOVA followed by Bonferroni post hoc test; n = 5–7 per group). BL, baseline. Data are represented as mean ± SEM.
Fig. S1.

Assessment of locomotor activity in SNI groups of mice. No genotype, drug, or SNI effect was observed on locomotor activity of Rgs9WT and Rgs9KO mice. Mice were treated chronically with saline or DMI (5 mg/kg, twice a day for 3 wk), and locomotor activity was tested 3 h after the last (morning) DMI injection (n = 8–14 per group). Data are presented as mean ± SEM.
To investigate the contribution of RGS9-2 in the NAc to the antiallodynic effects of DMI, we used a viral-mediated gene-transfer approach. Rgs9WT and Rgs9KO mice were treated with a low dose of DMI (5 mg/kg, i.p., twice a day) starting at two weeks after SNI, and on day 23 they were stereotaxically injected with herpes simplex viruses (HSVs) expressing Rgs9-2 or control β-galactosidase (LacZ) (2). DMI administration resumed 1 d after surgery. The HSV viruses were selected because of their rapid onset of action (peak expression at 3 d after stereotaxic infection). Animals were divided into three groups: Rgs9KO–HSV–Rgs9-2, Rgs9KO–HSV–LacZ, and Rgs9WT–HSV–LacZ. As shown in Fig. 2A, restoration of RGS9-2 expression in the NAc of Rgs9KO mice rescues the DMI phenotype observed in the Von Frey assay. A separate series of gene transfer studies were performed for NAc-specific Rgs9-2 overexpression, by using a longer-lasting adeno-associated virus (AAV) gene-transfer method. This time, mice were infected with AAV–Rgs9-2 or AAV–GFP (control) constructs, and SNI surgery was performed 2 wk after infection, when the viral expression is maximal (5). Although Rgs9-2 overexpression in the NAc does not affect baseline mechanical allodynia responses, it prevents the antiallodynic actions of a standard DMI regimen (10 mg/kg, i.p., twice a day for 2 wk; Fig. 2B).
Fig. 2.

RGS9-2 actions in the NAc modulate the antiallodynic effects of DMI. (A) Overexpression of Rgs9-2 in the NAc via stereotaxic infection with an HSV–Rgs9-2 vector rescues the increased sensitivity to DMI phenotype observed in the Von Frey assay. At 3 d after HSV–Rgs9-2 infection, Rgs9KO animals show maximal allodynia, whereas the Rgs9KO mice infected with HSV–LacZ maintain the antiallodynic response. *P < 0.05 for Rgs9KO–HSV–LacZ vs. Rgs9KO–HSV–Rgs9-2 and for Rgs9KO–HSV–LacZ vs. Rgs9WT–HSV–LacZ (two-way ANOVA followed by the Bonferroni post hoc test; n = 8–11 per group). (B) Increased RGS9-2 activity in the NAc (via stereotaxic infection with an AAV–Rgs9-2 vector) blocks the antiallodynic effects of DMI (10 mg/kg, i.p., twice a day) in C57BL/6 mice. *P < 0.05 (two way ANOVA followed by the Bonferroni post hoc test; n = 7–10 per group). Data are represented as mean ± SEM.
Coimmunoprecipitation (co-IP) assays were performed next, to determine whether DMI treatment (10 mg/kg, i.p., twice a day) affects the association of RGS9-2 with its interacting partner Gβ5, an effect known to promote RGS9-2 stability. After 2 wk of DMI treatment (a time point at which C57BL/6 mice have not developed an antiallodynic response), there was an increase in complexes between RGS9-2 and the Gβ5 subunit (Fig. 3B). We hypothesized that in the NAc, RGS9-2 complexes modulate the function of monoamine and other GPCRs that constitute direct or indirect DMI targets and affect signal-transduction events regulated by either Gα or Gβγ subunits (Fig. 3A). Therefore, preventing RGS9-2 activity in the NAc would lead to an earlier onset of DMI action and increase drug efficacy via enhanced Gα and Gβγ signaling. To this end, we treated Rgs9WT and Rgs9KO mice with DMI and collected NAc tissue 2 wk after DMI treatment (15 mg/kg, i.p., twice a day), a time point when Rgs9KO, but not wild-type, mice show an antiallodynic response. Our first set of Western blot analyses monitored the effect of DMI on PKA target proteins. As shown in Fig. 3 C and D, DMI treatment leads to a decrease in glutamate receptor subunit 1 (GluR1) (S845) and cAMP response element-binding protein (CREB) (S133) phosphorylation sites without affecting total protein levels.
Fig. 3.

RGS9-2 modulates signal transduction events triggered by DMI. (A) Cartoon summarizing the role of RGS9-2 in modulation of G protein effector activation after DMI administration. (B) Co-IP assays from ventral striatal tissue reveal that DMI administration (15 mg/kg for 14 d) promotes complexes between RGS9-2 and its interacting partner Gβ5. *P < 0.05 (t test; n = 3 per group). (C) Western blot analysis from NAc tissue of Rgs9WT and Rgs9KO mice at 2 wk after DMI (15 mg/kg, twice a day) treatment (a time point at which only Rgs9KO mice show an antiallodynic response) reveals that DMI leads to a reduction in phosphorylation of the GluR1 subunit at S845. *P < 0.05 (two-way ANOVA followed by Bonferroni post hoc test; n = 5 or 6 per group). (D) The phosphorylation of CREB at S133 is also reduced in NAc of Rgs9KO mice at the time point these mice show an antiallodynic response. *P < 0.05 (two-way ANOVA followed by Bonferroni post hoc test; n = 3 per group). (E) Co-IP assays at 2 wk after DMI treatment show that DMI promotes complexes between GRK2 and Gβ subunits in Rgs9KO, but not in Rgs9WT, ventral striatal tissue. *P < 0.05 (t test; n = 4 per group). (F) DMI also promotes complexes between G protein β subunits and pHDAC5 S498 (the phosphorylated, cytoplasmic form of the protein). Interestingly, this interaction is abolished in Rgs9KO mice, where these complexes are undetectable. *P < 0.05 (t test; n = 4 per group). IB, immunoblotting; OD, optical density. Data are presented as mean ± SEM.
We next investigated the effect of DMI on Gβγ-related signal-transduction events. GPCR activation is known to promote the formation of complexes between Gβγ subunits and GRK kinases (20, 21). We hypothesized that the antiallodynic response observed with low doses of DMI in Rgs9KO mice is associated with increased levels of GRK2/Gβ complexes, and we used co-IP assays to monitor these complexes in ventral striatal tissue. As shown in Fig. 3E, DMI treatment promotes the formation of complexes between GRK2 and Gβ subunits in the Rgs9KO group. GPCR activation also promotes complexes between Gβ subunits and the epigenetic modifier HDAC5 (22, 23). HDAC5 is expressed in several brain regions, mediating sensory and affective responses and TCA actions (10, 24). Here we show that chronic DMI promotes complexes between Gβ subunits and a phosphorylated (cytoplasmic) form of HDAC5 (S498), whereas these complexes are markedly decreased in the NAc of Rgs9KO mice (Fig. 3F). HDAC5 can be found in both cytoplasmic and nuclear compartments, and the nuclear translocation of this protein is controlled by phosphorylation (25, 26), as well as by complexes with Gβγ subunits (22, 23). Because DMI promotes complexes between Gβ subunits and HDAC5, we hypothesized that upon DMI administration, HDAC5 translocates to the nucleus and binds to chromatin complexes to suppress gene expression (an effect that delays the onset of drug action). When we used a nuclear/cytoplasmic fractionation protocol and monitored HDAC5 and Gβ levels in nuclear and cytoplasmic compartments, we found that acute DMI application (15 mg/kg, i.p.) increases the levels of both HDAC5 and Gβ proteins in the nucleus without affecting the nuclear/cytoplasmic distribution of Gβ5 and RGS9-2 (Fig. 4A). The acute DMI stimulation of HDAC5 nuclear translocation was absent in Rgs9KO mice (Fig. 4B). We also found that after chronic DMI administration (3 wk, a time point when both genotypes respond to DMI), no nuclear translocation of HDAC5 is observed in Rgs9WT–SNI or Rgs9KO–SNI mice (Fig. 4C). Notably, at the 2-wk time point at which only Rgs9KO animals show a response to DMI, no changes in HDAC5 phosphorylation at site Ser-498 were observed, further supporting the hypothesis that there is no nuclear translocation under these conditions (Fig. S2).
Fig. 4.

DMI affects the nuclear shuttling of HDAC5. We used nuclear/cytoplasmic fractionation assays to monitor the expression of HDAC5, Gβγ subunits, RGS9-2, and Gβ5 in the nucleus and cytoplasm of ventral striatal tissue of Rgs9WT and Rgs9KO mice after acute or chronic DMI treatment. (A) Acute DMI administration promotes the nuclear translocation of HDAC5 and Gβ subunits in tissue from Rgs9WT mice, but not in Rgs9KO mice. There is no change in the expression of RGS9-2 or Gβ5 in nuclear vs. cytoplasmic fractions after acute DMI treatment. *P < 0.05 (t test; n = 4 or 5 per group). This antibody detects only the nonphosphorylated nuclear form of the protein. (B) Acute DMI does not promote HDAC5 or Gβ5 levels in the nucleus of Rgs9KO mice. (C) The nuclear and cytoplasmic levels of HDAC5 and PanGβ were monitored in the ventral striata of Rgs9WT and Rgs9KO mice after 3 wk of DMI treatment (a time point at which both phenotypes respond). No differences in nuclear or cytoplasmic levels of these proteins were observed between chronic saline or chronic DMI groups. (D) Hdac5KO mice develop an antiallodynic response to DMI in the Von Frey assay (5 mg/kg, i.p., twice a day) within 2 wk of treatment. *P < 0.05 (two-way ANOVA followed by Bonferroni post hoc test; n = 7–9 per group. (E) Bilateral overexpression of Hdac5 in the NAc of C57BL/6 mice prevents the antiallodynic effects of DMI (10 mg/kg, i.p., twice a day). Viral infections were performed on day 5 of the DMI treatment. HSV–LacZ-infected animals show a significant antiallodynic response in the Von Frey assay after 18 d of DMI treatment, which is not observed in the Hdac5-overexpressing mice. *P < 0.05 (two-way ANOVA followed by Bonferroni post hoc test; n = 9 per group). BL, baseline; Cyt, cytoplasmic; Nucl, nuclear. Data are presented as mean ± SEM.
Fig. S2.

Regulation of HDAC5 phosphorylation sites in NAc under SNI and chronic DMI treatment. (A) At 2 wk after SNI (or sham) surgery, C57BL/6 mice were chronically treated with DMI (15 mg/kg, i.p) or saline. NAc tissue was collected at the time point where the Von Frey antiallodynic response was evident. HDAC5 phosphorylation of S498 (Left) and S279 (Right) sites was decreased in SNI groups treated with DMI. *P < 0.05 (n = 5 or 6; two-way ANOVA). (B) Rgs9WT–SNI and Rgs9KO–SNI mice were chronically treated with saline or DMI (15 mg/kg, i.p., twice a day), and NAc tissue was collected at the time point at which only the mutant mice developed an antiallodynic response to DMI. HDAC5 phosphorylation at site S498 remained unchanged (Left), and HDAC5 phosphorylation at S279 (Right) was decreased in the mutant DMI-treated mice. *P < 0.05 (n = 5 or 6; two-way ANOVA). Data are presented as mean ± SEM.
We then investigated the role of HDAC5 in antiallodynic responses to DMI. Genetic ablation of Hdac5 shifted the DMI dose–response to the left, because Hdac5 knockout mice, but not their wild-type littermates, showed antiallodynic responses to a low-dose regimen of DMI (5 mg/kg, i.p., twice a day; Fig. 4D). To determine whether HDAC5 actions in the NAc contribute to this phenotype, we used HSV-mediated gene transfer to overexpress Hdac5 in adult C57BL/6 mice after 2 wk of SNI and 5 d of DMI treatment. As shown in Fig. 4E, HSV–Hdac5 infection in the NAc prevented the antiallodynic effect of DMI (10 mg/kg, i.p., twice a day) in the Von Frey test.
The first set of our studies demonstrated that ablation of the Rgs9 gene affects several protein–protein interactions, phosphorylation events, and epigenetic mechanisms that are associated with DMI responses. Because the therapeutic effects of TCAs are associated with changes in gene expression (8, 27), the last part of this study used RNA-seq analysis to gain an understanding of the adaptations in gene expression associated with knockout of Rgs9, nerve injury, and DMI treatment in the NAc. For the RNA-seq studies, we used six different groups of mice: naive Rgs9WT and Rgs9KO, SNI-operated Rgs9WT and Rgs9KO mice treated with saline, and SNI-operated Rgs9WT and Rgs9KO mice treated with DMI. We used similar time points of DMI treatment (15 mg/kg, i.p., twice a day) as in our behavioral assays. NAc tissue from wild-type and mutant mice was collected at the time point of 2 wk, when only knockout mice show an antiallodynic response (Figs. 1E and 5A). Results were analyzed as pairwise comparisons between the groups, and gene regulation is inferred by fold change. Information regarding gene ontology (GO) and number of genes in each experimental group is provided in Fig. S3. To further illustrate a role of Rgs9 on gene adaptations in the NAc, we constructed Venn diagrams of data from wild-type and mutant mice compared with their corresponding SNI–DMI groups (Fig. 5B). As expected, the impact of drug treatment was evident on the Rgs9KO SNI–DMI mice, because this group demonstrates an antiallodynic response at the time point examined. However, the changes between saline-treated and naive Rgs9KO mice were almost one-third of those observed in the wild-type groups, although neuropathic pain affects the expression of a large number of genes in the NAc of wild-type mice (Fig. 5C). Even the 10 overlapping genes changed in the opposite direction, as illustrated in the embedded heatmap and in Table S1. Interestingly, when including the differences in gene adaptations between the naive wild-type and knockout groups, we found that more than half of the genes affected by Rgs9 deletion overlap with the genes altered after nerve injury induction in Rgs9WT mice, suggesting that naive Rgs9KO mice possess adaptive changes in the expression of genes involved in neuropathic pain.
Fig. 5.

NAc-specific RNA-seq profiling of Rgs9 knockout in naive and neuropathic pain mice, treated either with saline or DMI. (A) Timeline of the RNA-seq experiments. Briefly, NAc tissue was collected at the time point when only the mutant mice responded to the chronic DMI treatment. (B) Venn diagram depiction of the effect of drug treatment on gene expression. As expected, gene expression was slightly affected in the NAc of Rgs9WT SNI–DMI mice compared with saline-treated controls, because tissue collection was performed at the time point at which only the mutant mice developed antiallodynic response. (C) Neuropathic pain effect on gene expression in drug-naive mice. Venn diagram between the groups of naive and neuropathic pain-treated Rgs9WT and Rgs9KO mice suggests that the effect of SNI on gene expression was less potent in the NAc of mutant mice, whereas SNI affected different genes in the wild-type vs. mutant mice. The small embedded heatmap shows that even most of the 10 overlapping genes change to opposite direction. When differentially expressed genes between naive Rgs9WT and Rgs9KO mice were added to the Venn diagram, it was revealed that half of the adaptations induced after SNI surgery in wild-type mice are already present in the NAc of the naive mutant mice, possibly explaining the small effect in the Rgs9KO NAc after SNI. (D) Heatmap analysis reveals that the gene regulation pattern in the NAc of Rgs9KO naive mice is very similar to the gene regulation induced by DMI treatment, suggesting that NAc-specific gene adaptations in the absence of Rgs9 promote DMI’s actions. (E, Left) Venn diagram of the genes from naive and SNI–DMI groups of Rgs9WT and Rgs9KO mice reveals an overlap of 53 genes with very similar regulation, as indicated by the embedded heatmap. (E, Right) The table lists some of these genes with the corresponding fold changes. Importantly, the direction of the change is similar between the groups of naive and SNI–DMI-treated mice, suggesting that this adaptive gene regulation in the Rgs9KO naive mice facilitates DMI’s antiallodynic effect.
Fig. S3.

GO and experimental groups of the RNA-seq study. (A) To gain an overview of the pathways affected by our genetic or pharmacological manipulations, GO analysis was performed for all of the significantly altered genes identified from the RNA-seq analysis, regardless of experimental group. The analysis revealed changes in the expression of genes that play a role in antidepressant drug responses or mood or pain disorders, including Wingless-related integration site signaling pathway (Wnt) signaling, alpha-adrenergic receptor, and heterotrimeric G-protein–signaling pathways. Only pathways that reached statistical significance are shown. (B) The same approach was followed to unravel which biological processes were most represented in the RNA-seq results. The enriched biological processes, along with the assigned P value and the number of genes for every GO term, are shown on the graph. (C) Summary of the main group comparisons of the study, including the total number of significant up- and down-regulated genes within each group. As expected, the least change was observed in the comparison between Rgs9WT SNI–Sal and Rgs9WT SNI–DMI groups, because at this time point, wild-type mice fail to show an antiallodynic response. On the contrary, most gene expression changes were observed between naive Rgs9WT and naive Rgs9KO mice and between naive Rgs9WT and Rgs9WT SNI–Sal groups.
Table S1.
Table of overlapping genes between the group comparisons of Rgs9WT naïve vs. Rgs9WT SNI–Sal and Rgs9KO naïve vs. Rgs9KO SNI–Sal
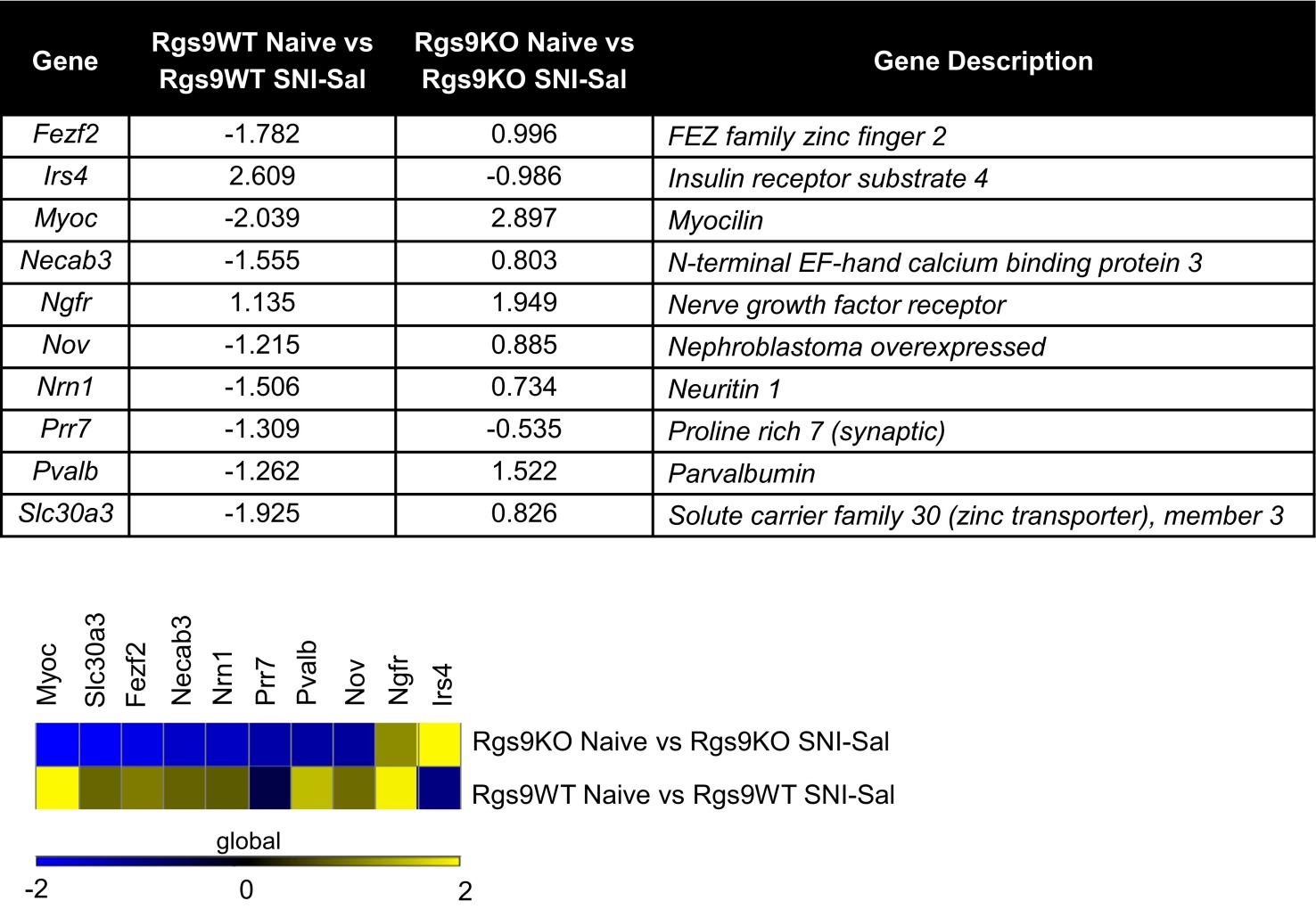 |
Venn diagram between Rgs9WT naive vs. Rgs9WT SNI–Sal and Rgs9KO naive vs. Rgs9KO SNI–Sal mice (Fig. 5E) revealed 10 overlapping genes, listed in the table in Upper, with corresponding fold changes. Notably, the majority of the genes change to the opposite direction between the two groups, suggesting that neuropathic pain effect on NAc gene expression, even when affecting the same genes, is very different in the mutant mice compared with wild-type controls. (Lower) The latter difference is further illustrated with heatmap analysis.
Next, we used heatmap analysis to depict the significant fold changes in gene expression between Rgs9WT and Rgs9KO mice, from the naive, SNI–Sal-, and SNI–DMI-treated groups. Using as reference the significantly changed genes from the comparison of naive mice groups, the heatmap reveals that gene regulation pattern in the NAc of Rgs9KO naive mice is very similar to the gene regulation induced by DMI treatment (Fig. 5D). Conversely, when genes from the SNI–DMI condition were used as reference, the resulting heatmap produced a very similar pattern, suggesting that DMI treatment reverses the effect of nerve injury on gene expression, which now resembles gene regulation in naive animals (Fig. S4A). Ontology analysis revealed that both naive and SNI–DMI groups are involved in similar cellular processes and pathways (Fig. S4B). Annotation of the genes from these groups to specific categories using keyword terms shows that most of the genes are related to signal transduction events (Fig. S5A). Moreover, 53 of the genes overlap with those changed between the naive and DMI-treated SNI groups of Rgs9WT and Rgs9KO mice (Fig. 5E). Representative genes are included in the table in Fig. 5E. Specifically, in Rgs9KO NAc, the early response to DMI induces the expression of genes encoding ion channels, transcription factors, neuronal guidance/differentiation, and signal transduction molecules known to be involved in several forms of pain and/or antidepressant drug actions. For example, mice lacking the adenylate cyclase-activating polypeptide 1 (Adcyap1) do not develop inflammatory or neuropathic pain and exhibit depression-like behaviors (28, 57). We show a reduction of Adcyap1 expression in the NAc of Rgs9KO mice that respond to DMI treatment, supporting the hypothesis that reduction of ADCYAP1 activity prevents pain-like responses (Fig. 5E). Moreover, the negative regulation of brain-derived neurotrophic factor (Bdnf) in the Rgs9KO SNI–DMI group correlates with reports on a role of Bdnf in neuropathic pain (29). Notably, the frizzled-B (FrzB) and dickkopf-like 1 (Dkkl1) genes (Fig. 5E) are members of the Wnt signaling pathway, which has an emerging role in neuropathic pain pathogenesis, depression, and antidepressant drug actions (11, 30).
Fig. S4.

Rgs9KO naïve and SNI–DMI mice share similar expression profiles and biological processes. (A) Heatmap of the genes regulated by chronic DMI in the NAc of neuropathic pain–RGS9KO group reveals similar expression profiling in the RGS9KO naive group, suggesting that ablation of RGS9 promotes adaptive changes that facilitate DMI’s actions. (B) GO analysis revealed similar biological processes (black) and KEGG/PANTHER pathways (gray) enriched in the naive and SNI–DMI groups of mice.
Fig. S5.

GO and protein interaction network for the group comparison Rgs9WT SNI–DMI vs. Rgs9KO SNI–DMI. (A) The statistically significant genes between the comparison of NAc from Rgs9WT SNI–DMI and Rgs9KO SNI–DMI groups from the RNA-seq study were submitted for gene ontology analysis, using the GO term SP_PIR_KEYWORDS (Swiss-Prot and Protein Information Resource databases), which annotates genes to specific categories by using keyword terms. The resulting categories with P < 0.05 and the corresponding number of genes are indicated on the pie graph. (B) The list of genes from the comparison of Rgs9WT SNI–DMI vs. Rgs9KO SNI–DMI was also used to construct a possible protein interaction network using the STRING database and the high-confidence mode 0.07. Although the network contains numerous individual nodes, there is a highly intercorrelated network in the center, involving genes abundant in the NAc and also relevant to chronic pain as well as antidepressant treatment.
Heatmaps of the genes between group comparisons of Rgs9KO mice are shown in Fig. 6A. Chronic DMI treatment completely reverses the gene signature established after SNI induction in the knockout mice. GO reveals that the majority of these genes regulate cell adhesion, neuronal differentiation, and cell motion (Fig. S6). The table in Fig. 6B lists several of these genes, which are implicated in nociception, depression, and antidepressant drug actions. For example, the transcription factors Bhlhe22 and Npas4 have been implicated in nociceptive and depressive behaviors respectively (58, 59). Importantly, the direction of gene expression change in the SNI–DMI-treated Rgs9KO mice is the same as in the naive Rgs9KO mice, suggesting that the basal gene expression profile upon knockout of Rgs9 facilitates DMI actions. Some of the genes altered by DMI treatment in Rgs9KO mice with SNI encode proteins that are predicted via the STRING database to interact with each other (Fig. 6C). RNA-seq analysis findings were verified by quantitative PCR (qPCR). Fig. 6 D and E shows data from qPCR analysis of NAc cDNA from naive Rgs9WT and Rgs9KO mice. Changes in expression of mRNAs between Rgs9WT and Rgs9KO SNI mice after 2 wk of DMI treatment are shown in Fig. 6F. Adenylate cyclase 1 (Adcy1), along with the Adcyap1, Bdnf, and Cholecystokinin (Cck) genes, are part of protein-interaction networks that form between products of the genes belonging to the comparison of SNI–DMI groups of mice as shown in Fig. S5B.
Fig. 6.

Rgs9 gene knockout promotes adaptive changes that resemble DMI’s therapeutic effect. (A) Heatmap of the genes regulated by chronic DMI in the NAc of Rgs9KO SNI group reveals that chronic antidepressant treatment almost completely reversed the adaptive changes in the group of mutant mice under neuropathic pain, suggesting that ablation of Rgs9 promotes adaptive changes that facilitate DMI’s actions. (B) Table depicting fold changes for the genes between the aforementioned group comparison as well as the naive group of mice. Importantly, the direction of the change for many genes is similar between the first and the third group comparisons shown in the table, suggesting once again that adaptive changes in the NAc of Rgs9KO naive mice are beneficial for the action of DMI. (C) Moreover, by using the STRING database for protein–protein interactions, a highly interconnected network of gene products from the Rgs9KO SNI–DMI compared with Rgs9KO SNI–Sal group was revealed. (D and E) To validate the RNA-seq results, several genes from the groups of naive Rgs9WT vs. Rgs9KO and Rgs9WT SNI–DMI vs. Rgs9KO SNI–DMI mice were selected to be tested with qPCR. (D) Up-regulated genes. (E) Down-regulated genes. *P < 0.05; **P < 0.01; ***P < 0.001 (t test, unpaired two-tailed). (F) Similarly, 18 of 22 selected genes (19 genes shown here) from the group of mice that responded to DMI treatment were confirmed by qPCR (data expressed as log2 fold change). *P < 0.05 (t test, unpaired two-tailed). N.c., no change.
Fig. S6.

Biological processes enriched in SNI–Sal vs. SNI–DMI Rgs9KO groups of RNA-seq analysis. GO analysis for the genes affected by DMI revealed that in Rgs9KO groups, response to the drug’s antiallodynic effect is mediated partly by biological processes such as neuron differentiation, cell adhesion, and neuron development.
Discussion
Neuropathic pain is a complex chronic disorder that involves adaptations in the spinal cord, as well as in several brain regions controlling pain perception, mood, motivation, and learning. Recent reports implicate multiple forebrain regions, such as the amygdala, the anterior cingulate cortex, and the NAc, in the pathophysiology of chronic pain syndromes (31–33). The data presented here demonstrate that RGS9-2 complexes in the NAc negatively modulate a number of cytoplasmic and nuclear events, which ultimately regulate the onset of action and therapeutic efficacy of DMI and other antidepressants in a neuropathic pain state. Although it is clear that alleviation of chronic pain symptoms by TCAs involves adaptations in monoamine responses in several CNS regions, the data presented here indicate that modulation of GPCR signaling in the NAc by RGS9-2 contributes to their therapeutic effectiveness.
We focused on the investigation of RGS9-2 because this molecule is a potent modulator of GPCR activity in the striatum and affects behavioral responses to several psychoactive or monoamine-targeting compounds (2, 5). Several preclinical studies have shown that DMI has antiallodynic properties in models of neuropathic pain (34–37). The DMI dose–response may vary between genetic backgrounds, neuropathic pain models, and type of Von Frey hair, but overall, chronic DMI administration promotes antiallodynic responses in rodents. DMI treatment affects the activation of the transcription factor CREB and is associated with changes in phosphoprotein levels in the brain reward center (9, 18, 38, 39). By controlling the availability of Gα and βγ subunits to effector molecules, RGS9-2 negatively regulates protein interactions and levels of phosphoproteins involved in antidepressant responsiveness. Here, we show that chronic DMI administration reduces CREB (S133) and GluR1 (S845) phosphorylation in the NAc of Rgs9KO mice. Both of these phosphorylation sites are targets of PKA (40). Notably, a recent study demonstrated that this GluR1 phosphorylation in the insular cortex contributes to postsynaptic amplification of neuropathic pain (33).
We also show that ablation of Rgs9 affects the activation of Gβγ effectors such as GRK2 and HDAC5. Acute DMI administration promotes the formation of complexes between HDAC5 and Gβ subunits and transiently promotes nuclear translocation of HDAC5, which disappears with prolonged treatment concurrent with the manifestation of the antiallodynic response. The lack of DMI-induced HDAC5 nuclear translocation in Rgs9KO mice likely explains the accelerated antiallodynic phenotype observed in these mice. Consistent with these data, the onset and efficacy of DMI antiallodynic action are also altered by Hdac5 deletion. Because DMI treatment enhances complex formation between GRK2 and HDAC5, future work should elucidate the Gβ subunit specificity of these events, as well as the role of other HDACs in the NAc in DMI responsiveness. Nevertheless, these data suggest that compounds targeting HDAC5 may amplify effects of TCAs in the treatment of neuropathic pain. However, manipulation of RGS9-2 activity may be more advantageous than targeting HDAC5, because RGS9-2 is primarily expressed in the striatum. Although no pharmacologic tools for manipulating RGS9-2 activity are currently available, several efforts are directed toward targeting key protein interactions to antagonize the effects of RGS9-2 complexes.
We also used a more open-ended approach to identify the genes in the NAc regulated by DMI under neuropathic pain states. RNA-seq analyses reveal that chronic DMI alters expression of a number of genes, including those encoding GPCRs, ion channels, adenylyl cyclase targets, and components of the Wnt signaling pathway—several of which have been shown to play a prominent role in nociceptive transmission, mood disorders, and antidepressant drug actions. Differential gene-expression analysis revealed the down-regulation of many genes with DMI treatment after SNI in Rgs9KO mice compared with wild-type controls, including Adcyap1 and Bdnf, for which reduced function is associated with reduced neuropathic pain sensitivity (28, 29), and Adcy1, the deletion of which reduces mechanical allodynia and inflammatory pain sensitivity (41). The expression of regulator of neurite outgrowth neuritin 1 (Nrn1), a gene that is stimulated by Bdnf and is implicated in antidepressant actions (42, 43) and diabetic neuropathy (44), is also altered. Several other genes with documented roles in chronic pain and antidepressant actions that are down-regulated by chronic DMI treatment in the Rgs9KO SNI group, including Neuropeptide Y1 receptor (Npy1R) (Fig. 6B, 60), were also altered (45). Neuropeptide systems constitute major drug development targets for the treatment of anxiety and depression, which are often comorbid with chronic pain (46, 47). Our RNA-seq study also reveals that DMI down-regulates Cck and vasointestinal peptide (Vip) in Rgs9KO mice, similar to what has been reported with chronic imipramine treatment (43). Interestingly, Cck in medial prefrontal–NAc projections mediates depressive symptoms, and blockade of the Cck B receptor promotes resilience (48). Notably, the pattern of Cck and Vip regulation is similar between naive Rgs9KO and wild-type mice, suggesting that adaptive changes in the NAc of Rgs9KO mice establish a pattern/signature that facilitates the antiallodynic effect of DMI. Cck and Vip, as well as Adcyap1, have been proposed as promising therapeutic targets for the treatment of neuropathic pain (49) and have been listed in other published microarray datasets from stress and depression studies (10, 43, 50) (Table S2). Also regulated by DMI in Rgs9KO mice were the peptides oxytocin, which has been implicated in antinociceptive mechanisms (51), and galanin (Gal), which has a profound role in nerve injury (52, 53) and neuropathic pain-induced motivational changes (54).
Table S2.
Table of common genes between our RNA-seq study and other genome-wide studies
| HDAC5 WT vs. HDAC5 KO (10) | MEF2 target genes (50) | Imipramine (43) |
| Acta2 | Adora2a | Adora2a |
| Adcyap1 | Asb4 | Agt |
| Agt | C1qtnf6 | Basp1 |
| Cck | Cckbr | Bhlhb5 |
| Cckbr | Clic4 | Calcr |
| Celsr3 | Col5a1 | Cck |
| Col6a1 | Coro6 | Ccnd2 |
| Crhbp | Emx1 | Chrdl1 |
| Dio2 | Esrrb | Cib2 |
| Dlgap1 | Galnt9 | Col11a1 |
| E2f1 | Gdf10 | Crtac1 |
| Gdf10 | Gnrh1 | Dlgap1 |
| Gucy2g | Gprin3 | Drd1a |
| Hs3st2 | Isoc1 | Fdps |
| Kbtbd5 | Kl | Frzb |
| Kcnc4 | Lrrc55 | Hs3st2 |
| Kcnh8 | Mapk11 | Htr4 |
| Kcnk4 | Musk | Khdrbs3 |
| Lgi2 | Otof | LOC302671 |
| Mef2c | Peg10 | Neurod1 |
| Nefh | Plch2 | Nr2f1 |
| Nr2f1 | Pnoc | Nrn1 |
| Nts | Scn1b | Nts |
| Olfm1 | Sec14l3 | Nxph3 |
| Pvalb | Serinc2 | Olfm1 |
| Ramp3 | Slc2a13 | Pdzrn3 |
| Slc17a6 | Sostdc1 | Pvalb |
| Slc18a2 | Stk32a | Rtn4r |
| Tmem25 | Stk32c | Slc5a7 |
| Wnt9a | Sytl2 | Stx1a |
| Tesc | Sv2c | |
| Vip | ||
| Zfpm2 |
Gene lists retrieved from NAc microarrays in HDAC5 WT and HDAC5KO acutely treated with saline (10), ChIP-seq analysis to identify MEF2 target genes in the NAc (50) (MEF2 is a major target of HDAC5), and microarrays in the NAc of chronically imipramine-treated mice (43). In bold are the common genes between the three studies and our RNA-seq.
Our RNA-seq studies also identified a number of GPCRs known to be involved in antidepressant drug actions and chronic pain. This list includes the gene encoding dopamine receptor D1 (Drd1a), which is potently regulated by RGS9-2 (1). This receptor positively modulates nociceptive responses (55), and its expression is decreased in the NAc 28 d after the induction of nerve injury (56). Moreover, chronic TCA treatment leads to an increase in Drd1a expression in the rodent NAc (43). Our findings suggest that the up-regulation of Drd1a expression in the NAc of naive Rgs9KO mice facilitates the actions of DMI. Finally, our results support a major role of the Wnt signaling pathway in DMI responses (Fig. 6E and Figs. S3A and S4B). This pathway contributes to the development of hyperalgesia and allodynia after nerve injury (30) and is involved in antidepressant drug actions (43).
In summary, this study provides information on NAc-specific intracellular mechanisms modulating the antiallodynic and antidepressant actions of TCAs in neuropathic pain states. RGS9-2 modulates a wide range of cytoplasmic and nuclear functions that both enhance DMI efficacy and accelerate the onset of action. RGS9-2 acts via a complex mechanism involving protein interactions, phosphorylation events, and control of HDAC5 repressor function. Our work provides additional evidence on the interrelationship between chronic pain and depression, showing that neuropathic pain promotes adaptations in networks associated with mood and motivation several weeks before the development of mood disorders. Pharmacological interventions that target some of these adaptations in the NAc not only reverse depression-like behaviors but also alleviate sensory deficits such as mechanical allodynia, most likely by altering the perception of noxious stimuli. As the NAc emerges as a key regulatory region for both sensory and affective symptoms of neuropathic pain, future studies will use information from proteomic analysis and the RNA-seq studies to further understand the GPCRs and pathways affected by RGS9-2 to identify novel drug targets. We will also evaluate the role of RGS9-2 in the therapeutic actions of other categories of monoamine targeting antidepressants that are used for the treatment of chronic pain (such as venlafaxine and tapentadol) to determine the role of specific monoamine receptors in drug responsiveness as well as the role of specific Gβ subunits and epigenetic molecules. We now aim to develop novel pharmacologic strategies to target RGS9-2 complexes to accelerate the onset of action and to enhance the therapeutic efficacy of antidepressants in models of chronic pain and depression.
Materials and Methods
Animals and Drug Treatments.
For all behavioral experiments, 2- to 3-mo-old Rgs9 or Hdac5 knockout mice and their wild-type littermates or adult C57BL/6 mice (The Jackson Laboratory) were used. All behavioral studies were performed in adult male mice, except for the NTL and DLX Von Frey studies, which were performed in adult female mice. Mice were group-housed (four or five per cage) on a 12-h light/dark cycle, provided with food and water ad libitum. Animal handling was in accordance to the animal care and use committees of Icahn School of Medicine at Mount Sinai and the University of Crete. DMI (Sigma Aldrich), NTL (Sigma Aldrich), and DLX (Tocris Bioscience) were dissolved in distilled water and further diluted in saline. Saline was also used as vehicle treatment. Injections were performed at 9:00 AM and at 9:00 PM. More details about drug injections for each of the experiments are provided in Results.
Stereotaxic Surgery and Viral Localization.
Stereotaxic coordinates for viral vector injections into the NAc were as follows: AP= +1.6, AL= +1.5, and DV= −4.4 at an angle of 10° from the midline (5). HSV–Rgs9-2, HSV–Hdac5, and HSV–LacZ vectors were generated by R.L.N. (2, 10). AAV–Rgs9-2 and AAV–GFP vectors were generated according to published methods (5). Immunohistochemistry was performed by using chicken polyclonal anti-GFP antibody (1:300; Aveslab).
SNI Model and Von Frey Test for Mechanical Allodynia.
The SNI operation was performed under Avertine (2,2,2-tribromoethanol; Sigma-Aldrich) general anesthesia (18). With the help of a stereomicroscope, skin incision of the left hind-limb at mid-thigh level, followed by muscle layers separation, revealed the sciatic nerve and its three branches. The common peroneal and the sural nerves were carefully ligated with 6.0 silk suture (Ethicon; Johnson & Johnson Intl.) and transected, and 1- to 2-mm sections of these nerves were removed, while the tibial nerve was left intact. Skin was then closed with silk 4.0 sutures (Ethicon; Johnson & Johnson Intl). The same procedure was followed in sham-operated mice, but the nerves were left intact (18). For the assessment of mechanical allodynia, we used Von Frey (18, 19) with ascending forces expressed in grams (0.1–3.6 g; Electronic Von Frey Anesthesiometer; IITC). Each filament was applied five times in a row against the lateral area of the paw. Hindpaw withdrawal or licking induced by the filament was defined as positive pain response. A positive response in three of five repetitive stimuli was defined as the pain threshold. Von Frey measurements were performed at least 4 h after antidepressant drug injection. Mice were habituated to the Von Frey apparatus for 30 min every day for 10 d, before the SNI surgeries. Mice were monitored for Von Frey responses at least 4 h after the morning antidepressant drug injection.
FST.
For the FST, mice were placed in individual glass cylinders (46-cm height × 18-cm diameter) containing water at room temperature (25 °C) at a depth of 15 cm. DMI or vehicle (saline) was administered at 24 h (i.p.), 5 h (i.p), and 1 h (s.c.) before the assay (18).
Nucleo-Cytoplasmic Fractionation, Western Blotting, and Co-IP Assays.
Nucleo-cytoplasmic fractionation was performed as described with minor modifications (61). Western blotting and co-IP assays were performed as described (6). The following antibodies were used for co-IP and Western blot analysis: rabbit protein A-purified anti-RGS9 (1:1,000; ref. 6), rabbit anti-Gβ5 C terminus (1:20,000; W. Simonds, National Institute of Diabetes, Digestive and Kidney Diseases, Bethesda), rabbit anti-Gβ (1:40,000; Santa Cruz), rabbit anti–p-HDAC5 (S498; 1:1,000; Abcam), rabbit anti–p-CREB (S133; 1:1,000; Cell Signaling), rabbit anti-CREB (1:1,000; Cell Signaling), rabbit anti–p-GluR1 (S845; 1:1,000; Cell Signaling), rabbit anti-GluR1 (1:1,000; Cell Signaling), mouse anti-HDAC5 (1:1,000; Abcam), mouse anti–beta-tubulin (1:40,000; Sigma-Aldrich), and mouse anti-GRK2 (1:2,000; Santa Cruz). Rabbit anti–p-HDAC5 (S279; 1:1,000; ref. 24) was provided by C. W. Cowan (Harvard University).
RNA-Seq Studies and qPCR.
Six groups of mice with three biological replicates per group were used for the RNA-seq study. In brief, NAc punches were taken from individual mice (six animals per treatment group), and total RNA was isolated from pooled bilateral punches of two animals per replicate (three replicates per treatment group), as described above with TRIzol according to manufacturer’s protocol. RNA was purified with RNeasy Micro columns (Qiagen), and an Agilent 2100 Bioanalyzer confirmed that the RNA integrity numbers were >8.0. The poly-A–containing mRNA was purified by using poly-T oligo-attached magnetic beads, and the mRNA-seq library was prepared from each pooled RNA sample by using the TruSeq RNA Sample Preparation Kit v2 (no. RS-122-2002), according to the instructions of Illumina. RNA-seq was performed on the Illumina HiSeq2000 machine at Mount Sinai’s Genomic Core facility. The RNA-seq read alignment and differential analysis were performed by using the TopHat2 (62) and Cufflinks (63) packages. Significantly differentially expressed genes were determined by using a false discovery rate of <5% in this analysis. Details on RNA-seq quality control metrics are provided in Table S3. qPCR was performed by using SYBR green (Quanta Biosciences; catalog no. 95073) on an Applied Biosystems 7500 system. Reactions were run in triplicate and analyzed by using the ΔΔCt method and GAPDH as normalization control.
Table S3.
Quality control for the RNA-seq study
| Sample | Total reads | Mapped reads | Mitochondrial RNA reads | rRNA reads | Mapping rate | Mitochondrial RNA rate | rRNA rate |
| R9_KO_naive_1 | 63,587,606 | 59,686,286 | 6,186,111 | 11,417 | 93.8647 | 10.3644 | 0.0191 |
| R9_KO_naive_2 | 59,821,512 | 56,188,894 | 5,811,752 | 10,037 | 93.9276 | 10.3432 | 0.0179 |
| R9_KO_naive_3 | 57,791,560 | 53,991,111 | 4,554,330 | 10,618 | 93.4239 | 8.4353 | 0.0197 |
| R9_KO_SN1_DMI_1 | 62,734,095 | 58,959,395 | 5,581,930 | 11,830 | 93.9830 | 9.4674 | 0.0201 |
| R9_KO_SN1_DMI_2 | 58,354,570 | 54,372,413 | 5,345,565 | 10,122 | 93.1759 | 9.8314 | 0.0186 |
| R9_KO_SN1_DMI_3 | 60,916,951 | 57,277,403 | 5,022,732 | 11,680 | 94.0254 | 8.7691 | 0.0204 |
| R9_KO_SN1_Sal_1 | 59,309,317 | 55,514,417 | 5,923,850 | 10,404 | 93.6015 | 10.6708 | 0.0187 |
| R9_KO_SN1_Sal_2 | 55,182,236 | 51,793,845 | 5,327,948 | 9,803 | 93.8596 | 10.2868 | 0.0189 |
| R9_KO_SN1_Sal_3 | 65,201,565 | 61,188,398 | 5,127,970 | 12,224 | 93.8450 | 8.3806 | 0.0200 |
| R9_WT_naive_1 | 54,736,173 | 51,339,669 | 2,511,171 | 6,077 | 93.7948 | 4.8913 | 0.0118 |
| R9_WT_naive_2 | 61,383,196 | 57,636,097 | 4,954,274 | 11,606 | 93.8956 | 8.5958 | 0.0201 |
| R9_WT_naive_3 | 67,295,752 | 63,047,718 | 4,836,466 | 12,415 | 93.6875 | 7.6711 | 0.0197 |
| R9_WT_SN1_DMI_1 | 58,888,578 | 55,284,954 | 4,945,464 | 10,759 | 93.8806 | 8.9454 | 0.0195 |
| R9_WT_SN1_DMI_2 | 56,628,928 | 53,020,065 | 4,541,400 | 10,538 | 93.6272 | 8.5654 | 0.0199 |
| R9_WT_SN1_DMI_3 | 57,747,180 | 54,204,918 | 4,333,153 | 10,421 | 93.8659 | 7.9940 | 0.0192 |
| R9_WT_SN1_Sal_1 | 63,472,320 | 59,419,971 | 5,745,035 | 11,705 | 93.6156 | 9.6685 | 0.0197 |
| R9_WT_SN1_Sal_2 | 58,830,460 | 55,028,236 | 5,005,685 | 10,665 | 93.5370 | 9.0966 | 0.0194 |
| R9_WT_SN1_Sal_3 | 58,805,738 | 55,155,160 | 4,891,616 | 11,063 | 93.7921 | 8.8688 | 0.0201 |
Indicated are the number of reads and corresponding mapping rates regarding each of the samples used for sequencing.
Bioinformatic Analysis.
To identify pathway and gene functional annotations that were overrepresented in our RNA-seq results, GO analysis was performed by using the functional annotation and clustering software tool available through the online Database for Annotation, Visualization and Integrated Discovery (DAVID) Bioinformatics Resources (Version 6.7; https://david.ncifcrf.gov/. Three GO terms, biological process (BP_FAT), molecular function (MF_FAT), and Swiss-Prot and Protein Information Resource (SP_PIR) keywords were used to cluster genes. Only terms with P < 0.05 are reported. The Venn diagrams were generated by using the BioVenn-web application (www.cmbi.ru.nl/cdd/biovenn/index.php). To identify known and potential protein interactions between the products of the genes involved in our RNA-seq results, we used the STRING (Version 9.1) software and database (64).
Statistical Analysis.
For all behavioral experiments, data were analyzed by using two-way ANOVA followed by the Bonferroni post hoc test, as indicated in each figure. For Western blot and co-IP assays, we used the t test or two-way ANOVA as indicated in each figure. For nuclear translocation assays and qPCR analysis, we used Student’s t test.
Acknowledgments
This work was supported by National Institute on Drug Abuse Grant PPG-POIDAO8227 (to V.Z.), National Institute of Neurological Disorders and Stroke Grant NS086444 (to V.Z. and L.S.), and the Greek Secretariat for Research and Technology and the 7th EU Framework (Aristia I; to V.Z., D.T., V.M., and S.G.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE71527).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1504283112/-/DCSupplemental.
References
- 1.Traynor JR, Terzi D, Caldarone BJ, Zachariou V. RGS9-2: Probing an intracellular modulator of behavior as a drug target. Trends Pharmacol Sci. 2009;30(3):105–111. doi: 10.1016/j.tips.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zachariou V, et al. Essential role for RGS9 in opiate action. Proc Natl Acad Sci USA. 2003;100(23):13656–13661. doi: 10.1073/pnas.2232594100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sondek J, Siderovski DP. Ggamma-like (GGL) domains: New frontiers in G-protein signaling and beta-propeller scaffolding. Biochem Pharmacol. 2001;61(11):1329–1337. doi: 10.1016/s0006-2952(01)00633-5. [DOI] [PubMed] [Google Scholar]
- 4.Jayaraman M, Zhou H, Jia L, Cain MD, Blumer KJ. R9AP and R7BP: Traffic cops for the RGS7 family in phototransduction and neuronal GPCR signaling. Trends Pharmacol Sci. 2009;30(1):17–24. doi: 10.1016/j.tips.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gaspari S, et al. Nucleus accumbens-specific interventions in RGS9-2 activity modulate responses to morphine. Neuropsychopharmacology. 2014;39(8):1968–1977. doi: 10.1038/npp.2014.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Psifogeorgou K, et al. A unique role of RGS9-2 in the striatum as a positive or negative regulator of opiate analgesia. J Neurosci. 2011;31(15):5617–5624. doi: 10.1523/JNEUROSCI.4146-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kovoor A, et al. D2 dopamine receptors colocalize regulator of G-protein signaling 9-2 (RGS9-2) via the RGS9 DEP domain, and RGS9 knock-out mice develop dyskinesias associated with dopamine pathways. J Neurosci. 2005;25(8):2157–2165. doi: 10.1523/JNEUROSCI.2840-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nestler EJ, Carlezon WA., Jr The mesolimbic dopamine reward circuit in depression. Biol Psychiatry. 2006;59(12):1151–1159. doi: 10.1016/j.biopsych.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 9.Chartoff EH, et al. Desipramine reduces stress-activated dynorphin expression and CREB phosphorylation in NAc tissue. Mol Pharmacol. 2009;75(3):704–712. doi: 10.1124/mol.108.051417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Renthal W, et al. Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron. 2007;56(3):517–529. doi: 10.1016/j.neuron.2007.09.032. [DOI] [PubMed] [Google Scholar]
- 11.Dias C, et al. β-catenin mediates stress resilience through Dicer1/microRNA regulation. Nature. 2014;516(7529):51–55. doi: 10.1038/nature13976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cruccu G. Treatment of painful neuropathy. Curr Opin Neurol. 2007;20(5):531–535. doi: 10.1097/WCO.0b013e328285dfd6. [DOI] [PubMed] [Google Scholar]
- 13.Max MB, et al. Efficacy of desipramine in painful diabetic neuropathy: A placebo-controlled trial. Pain. 1991;45(1):3–9. doi: 10.1016/0304-3959(91)90157-S. [DOI] [PubMed] [Google Scholar]
- 14.Radat F, Margot-Duclot A, Attal N. Psychiatric co-morbidities in patients with chronic peripheral neuropathic pain: A multicentre cohort study. Eur J Pain. 2013;17(10):1547–1557. doi: 10.1002/j.1532-2149.2013.00334.x. [DOI] [PubMed] [Google Scholar]
- 15.Cottingham C, Chen Y, Jiao K, Wang Q. The antidepressant desipramine is an arrestin-biased ligand at the α(2A)-adrenergic receptor driving receptor down-regulation in vitro and in vivo. J Biol Chem. 2011;286(41):36063–36075. doi: 10.1074/jbc.M111.261578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang HT, Whisler LR, Huang Y, Xiang Y, O’Donnell JM. Postsynaptic alpha-2 adrenergic receptors are critical for the antidepressant-like effects of desipramine on behavior. Neuropsychopharmacology. 2009;34(4):1067–1077. doi: 10.1038/npp.2008.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shields SD, Eckert WA, 3rd, Basbaum AI. Spared nerve injury model of neuropathic pain in the mouse: A behavioral and anatomic analysis. J Pain. 2003;4(8):465–470. doi: 10.1067/s1526-5900(03)00781-8. [DOI] [PubMed] [Google Scholar]
- 18.Stratinaki M, et al. Regulator of G protein signaling 4 [corrected] is a crucial modulator of antidepressant drug action in depression and neuropathic pain models. Proc Natl Acad Sci USA. 2013;110(20):8254–8259. doi: 10.1073/pnas.1214696110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Terzi D, et al. RGS9-2 modulates sensory and mood related symptoms of neuropathic pain. Neurobiol Learn Mem. 2014;115:43–48. doi: 10.1016/j.nlm.2014.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daaka Y, et al. Receptor and G betagamma isoform-specific interactions with G protein-coupled receptor kinases. Proc Natl Acad Sci USA. 1997;94(6):2180–2185. doi: 10.1073/pnas.94.6.2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Penela P, Murga C, Ribas C, Lafarga V, Mayor F., Jr The complex G protein-coupled receptor kinase 2 (GRK2) interactome unveils new physiopathological targets. Br J Pharmacol. 2010;160(4):821–832. doi: 10.1111/j.1476-5381.2010.00727.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bhatnagar A, et al. Interaction of G-protein βγ complex with chromatin modulates GPCR-dependent gene regulation. PLoS One. 2013;8(1):e52689. doi: 10.1371/journal.pone.0052689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spiegelberg BD, Hamm HE. G betagamma binds histone deacetylase 5 (HDAC5) and inhibits its transcriptional co-repression activity. J Biol Chem. 2005;280(50):41769–41776. doi: 10.1074/jbc.M504066200. [DOI] [PubMed] [Google Scholar]
- 24.Taniguchi M, et al. Histone deacetylase 5 limits cocaine reward through cAMP-induced nuclear import. Neuron. 2012;73(1):108–120. doi: 10.1016/j.neuron.2011.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chawla S, Vanhoutte P, Arnold FJ, Huang CL, Bading H. Neuronal activity-dependent nucleocytoplasmic shuttling of HDAC4 and HDAC5. J Neurochem. 2003;85(1):151–159. doi: 10.1046/j.1471-4159.2003.01648.x. [DOI] [PubMed] [Google Scholar]
- 26.McKinsey TA, Zhang CL, Olson EN. Identification of a signal-responsive nuclear export sequence in class II histone deacetylases. Mol Cell Biol. 2001;21(18):6312–6321. doi: 10.1128/MCB.21.18.6312-6321.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Malberg JE, Blendy JA. Antidepressant action: To the nucleus and beyond. Trends Pharmacol Sci. 2005;26(12):631–638. doi: 10.1016/j.tips.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 28.Mabuchi T, et al. Pituitary adenylate cyclase-activating polypeptide is required for the development of spinal sensitization and induction of neuropathic pain. J Neurosci. 2004;24(33):7283–7291. doi: 10.1523/JNEUROSCI.0983-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yajima Y, et al. Direct evidence for the involvement of brain-derived neurotrophic factor in the development of a neuropathic pain-like state in mice. J Neurochem. 2005;93(3):584–594. doi: 10.1111/j.1471-4159.2005.03045.x. [DOI] [PubMed] [Google Scholar]
- 30.Zhang YK, et al. WNT signaling underlies the pathogenesis of neuropathic pain in rodents. J Clin Invest. 2013;123(5):2268–2286. doi: 10.1172/JCI65364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Apkarian AV, Hashmi JA, Baliki MN. Pain and the brain: Specificity and plasticity of the brain in clinical chronic pain. Pain. 2011;152(3) Suppl:S49–S64. doi: 10.1016/j.pain.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.von Hehn CA, Baron R, Woolf CJ. Deconstructing the neuropathic pain phenotype to reveal neural mechanisms. Neuron. 2012;73(4):638–652. doi: 10.1016/j.neuron.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qiu S, et al. GluA1 phosphorylation contributes to postsynaptic amplification of neuropathic pain in the insular cortex. J Neurosci. 2014;34(40):13505–13515. doi: 10.1523/JNEUROSCI.1431-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sands SA, McCarson KE, Enna SJ. Relationship between the antinociceptive response to desipramine and changes in GABAB receptor function and subunit expression in the dorsal horn of the rat spinal cord. Biochem Pharmacol. 2004;67(4):743–749. doi: 10.1016/j.bcp.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 35.Alba-Delgado C, Mico JA, Sánchez-Blázquez P, Berrocoso E. Analgesic antidepressants promote the responsiveness of locus coeruleus neurons to noxious stimulation: Implications for neuropathic pain. Pain. 2012;153(7):1438–1449. doi: 10.1016/j.pain.2012.03.034. [DOI] [PubMed] [Google Scholar]
- 36.Farghaly HS, Abdel-Zaher AO, Mostafa MG, Kotb HI. Comparative evaluation of the effect of tricyclic antidepressants on inducible nitric oxide synthase expression in neuropathic pain model. Nitric Oxide. 2012;27(2):88–94. doi: 10.1016/j.niox.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 37.Yalcin I, et al. Beta2-adrenoceptors are essential for desipramine, venlafaxine or reboxetine action in neuropathic pain. Neurobiol Dis. 2009;33(3):386–394. doi: 10.1016/j.nbd.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 38.Becerra L, Navratilova E, Porreca F, Borsook D. Analogous responses in the nucleus accumbens and cingulate cortex to pain onset (aversion) and offset (relief) in rats and humans. J Neurophysiol. 2013;110(5):1221–1226. doi: 10.1152/jn.00284.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blendy JA. The role of CREB in depression and antidepressant treatment. Biol Psychiatry. 2006;59(12):1144–1150. doi: 10.1016/j.biopsych.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 40.Roche KW, O’Brien RJ, Mammen AL, Bernhardt J, Huganir RL. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron. 1996;16(6):1179–1188. doi: 10.1016/s0896-6273(00)80144-0. [DOI] [PubMed] [Google Scholar]
- 41.Wei F, et al. Genetic elimination of behavioral sensitization in mice lacking calmodulin-stimulated adenylyl cyclases. Neuron. 2002;36(4):713–726. doi: 10.1016/s0896-6273(02)01019-x. [DOI] [PubMed] [Google Scholar]
- 42.Son H, et al. Neuritin produces antidepressant actions and blocks the neuronal and behavioral deficits caused by chronic stress. Proc Natl Acad Sci USA. 2012;109(28):11378–11383. doi: 10.1073/pnas.1201191109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wallace DL, et al. CREB regulation of nucleus accumbens excitability mediates social isolation-induced behavioral deficits. Nat Neurosci. 2009;12(2):200–209. doi: 10.1038/nn.2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Karamoysoyli E, Burnand RC, Tomlinson DR, Gardiner NJ. Neuritin mediates nerve growth factor-induced axonal regeneration and is deficient in experimental diabetic neuropathy. Diabetes. 2008;57(1):181–189. doi: 10.2337/db07-0895. [DOI] [PubMed] [Google Scholar]
- 45.Naveilhan P, et al. Reduced antinociception and plasma extravasation in mice lacking a neuropeptide Y receptor. Nature. 2001;409(6819):513–517. doi: 10.1038/35054063. [DOI] [PubMed] [Google Scholar]
- 46.Bair MJ, Robinson RL, Katon W, Kroenke K. Depression and pain comorbidity: A literature review. Arch Intern Med. 2003;163(20):2433–2445. doi: 10.1001/archinte.163.20.2433. [DOI] [PubMed] [Google Scholar]
- 47.Yalcin I, Barrot M. The anxiodepressive comorbidity in chronic pain. Curr Opin Anaesthesiol. 2014;27(5):520–527. doi: 10.1097/ACO.0000000000000116. [DOI] [PubMed] [Google Scholar]
- 48.Vialou V, et al. Prefrontal cortical circuit for depression- and anxiety-related behaviors mediated by cholecystokinin: Role of ΔFosB. J Neurosci. 2014;34(11):3878–3887. doi: 10.1523/JNEUROSCI.1787-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Raju HB, Englander Z, Capobianco E, Tsinoremas NF, Lerch JK. Identification of potential therapeutic targets in a model of neuropathic pain. Front Genet. 2014;5:131. doi: 10.3389/fgene.2014.00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pulipparacharuvil S, et al. Cocaine regulates MEF2 to control synaptic and behavioral plasticity. Neuron. 2008;59(4):621–633. doi: 10.1016/j.neuron.2008.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Robinson DA, et al. Oxytocin mediates stress-induced analgesia in adult mice. J Physiol. 2002;540(Pt 2):593–606. doi: 10.1113/jphysiol.2001.013492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Holmes FE, et al. Transgenic overexpression of galanin in the dorsal root ganglia modulates pain-related behavior. Proc Natl Acad Sci USA. 2003;100(10):6180–6185. doi: 10.1073/pnas.0937087100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Holmes A, Picciotto MR. Galanin: A novel therapeutic target for depression, anxiety disorders and drug addiction? CNS Neurol Disord Drug Targets. 2006;5(2):225–232. doi: 10.2174/187152706776359600. [DOI] [PubMed] [Google Scholar]
- 54.Schwartz N, et al. Chronic pain. Decreased motivation during chronic pain requires long-term depression in the nucleus accumbens. Science. 2014;345(6196):535–542. doi: 10.1126/science.1253994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ortiz O, et al. Associative learning and CA3-CA1 synaptic plasticity are impaired in D1R null, Drd1a-/- mice and in hippocampal siRNA silenced Drd1a mice. J Neurosci. 2010;30(37):12288–12300. doi: 10.1523/JNEUROSCI.2655-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chang PC, et al. Role of nucleus accumbens in neuropathic pain: Linked multi-scale evidence in the rat transitioning to neuropathic pain. Pain. 2014;155(6):1128–1139. doi: 10.1016/j.pain.2014.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hashimoto H, et al. Depression-like behavior in the forced swimming test in PACAP-deficient mice: Amelioration by the atypical antipsychotic risperidone. J Neurochem. 2009;110(2):595–602. doi: 10.1111/j.1471-4159.2009.06168.x. [DOI] [PubMed] [Google Scholar]
- 58.Ross SE, et al. Loss of inhibitory interneurons in the dorsal spinal cord and elevated itch in Bhlhb5 mutant mice. Neuron. 2010;65(6):886–898. doi: 10.1016/j.neuron.2010.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Furukawa-Hibi Y, Yun J, Nagai T, Yamada K. Transcriptional suppression of the neuronal PAS domain 4 (Npas4) gene by stress via the binding of agonist-bound glucocorticoid receptor to its promoter. J Neurochem. 2012;123(5):866–875. doi: 10.1111/jnc.12034. [DOI] [PubMed] [Google Scholar]
- 60.Redrobe JP, Dumont Y, Fournier A, Quirion R. The neuropeptide Y (NPY) Y1 receptor subtype mediates NPY-induced antidepressant-like activity in the mouse forced swimming test. Neuropsychopharmacology. 2002;26(5):615–624. doi: 10.1016/S0893-133X(01)00403-1. [DOI] [PubMed] [Google Scholar]
- 61.Eliopoulos AG, et al. Epstein-Barr virus-encoded latent infection membrane protein 1 regulates the processing of p100 NF-kappaB2 to p52 via an IKKgamma/NEMO-independent signalling pathway. Oncogene. 2003;22(48):7557–7569. doi: 10.1038/sj.onc.1207120. [DOI] [PubMed] [Google Scholar]
- 62.Kim D, et al. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14(4):R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Trapnell C, et al. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31(1):46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Franceschini A, et al. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41(Database issue):D808–D815. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]