

Abstract
Background
NADPH oxidase 4 (Nox4) has been implicated in cardiac remodeling, but its precise role in cardiac injury remains controversial. Furthermore, little is known about the downstream effector signaling pathways activated by Nox4-derived ROS in the myocardium. We investigated the role of Nox4 and Nox4 associated signaling pathways in the development of cardiac remodeling.
Methods and Results
Cardiac-specific human Nox4 transgenic mice (c-hNox4Tg) were generated. Four groups of mice were studied: 1) control mice (CTL): littermates that are negative for hNox4 transgene but Cre positive; 2) c-hNox4 Tg mice; 3) angiotensin II (AngII)-infused CTL mice and 4) c-hNox4Tg mice infused with AngII. The c-hNox4Tg mice exhibited approximately 10-fold increase in Nox4 protein expression and 8-fold increase in the production of reactive oxygen species, and manifested cardiac interstitial fibrosis. AngII-infusion to CTL mice increased cardiac Nox4 expression and induced fibrosis and hypertrophy. The Tg mice receiving AngII exhibited more advanced cardiac remodeling and robust elevation in Nox4 expression, indicating that AngII worsens cardiac injury, at least partially by enhancing Nox4 expression. Moreover, hNox4 transgene and/or AngII-infusion induced the expression of cardiac fetal genes and activated the Akt-mTOR and NFκB signaling pathways. Treatment of AngII-infused c-hNox4Tg mice with GKT137831, a Nox4/Nox1 inhibitor, abolished the increase in oxidative stress, suppressed Akt-mTOR and NFκB signaling pathway and attenuated cardiac remodeling.
Conclusion
Upregulation of Nox4 in the myocardium causes cardiac remodeling through activating Akt-mTOR and NFκB signaling pathways. Inhibition of Nox4 has therapeutic potential to treat cardiac remodeling.
Keywords: Reactive oxygen species, NADPH oxidase, fibrosis, hypertrophy, signaling pathways
Introductioin
Left ventricle remodeling, characterized by interstitial fibrosis and cardiomyocyte hypertrophy, is a common response to acute and chronic cardiac injury observed in various heart diseases in humans and animal models, commencing with hypertension, valvular disease, coronary artery disease, and cardiomyopathy that frequently eventuates in heart failure.1, 2 Over the past decade, strong evidence has implicated a common denominator, reactive oxygen species (ROS), in the development of cardiovascular pathology including cardiac remodeling.3 However, the failure of clinical trials with antioxidant compounds has underscored the need for better antioxidant therapies and a more thorough understanding of the source(s) and contribution of ROS in these diseases.4 Thus, it is important to identify the enzymatic source(s) of ROS so that specific targeting of the enzyme system can be developed to efficiently abolish the deleterious effects of ROS in pathological conditions.
Among the various potential sources of ROS, NADPH oxidases have emerged as major enzymes responsible for production of superoxide (O2•-) and hydrogen peroxide. Thus far, seven homologs/isoforms of these enzymes have been found in humans and animals, and are referred to as the Nox family of NADPH oxidases: Nox1 - 5, Duox1, and Duox2. These enzymes share the capacity to transport electrons across the plasma membrane and to generate O2•- and other downstream ROS, such as hydrogen peroxide (H2O2). Nox enzymes are professional ROS-producing enzymes distinguishing themselves from other sources where ROS are produced as a byproduct. In addition to Nox2, Nox4 is a major source of ROS in the heart.5 Both enzymes share sequence homology, but have distinct characteristics. For example, while Nox2 requires cytosolic factors for its activation,6 Nox4 is constitutively active and is regulated primarily at the level of its expression.7 Nox2 localizes primarily in the plasma membrane whereas Nox4 is found in intracellular membranes such as mitochondria, endoplasmic reticulum and nuclear membranes.5, 8, 9 Emerging data suggest contradictory role of Nox4 in cardiac remodeling with considerable debate on whether Nox4 is protective or deleterious during cardiac response to injury. For example, upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in the myocardium.8 Also, increased oxidative stress in the nucleus caused by Nox4 mediates oxidation of histone deacetylase 4 resulting in cardiac hypertrophy.10 In contrast to these results, Zhang et al11 reported that Nox4-null mice develop exaggerated contractile dysfunction, hypertrophy, and cardiac dilatation in response to chronic pressure overload, whereas Nox4 transgenic mice are protected. Importantly, signaling molecules targeted by Nox4 in the myocardium are incompletely defined.
The aim of this study was to elucidate the role of Nox4 in the development of cardiac remodeling and explore potential signal transduction pathways that may mediate the effect of Nox4. We generated a cardiac-specific human Nox4-transgenic mouse model (c-hNox4Tg) and investigated 1) the effect of overexpression of Nox4 on cardiac fibrosis and hypertrophy, 2) the genes that are activated by Nox4 in the heart that modulate cardiac remodeling, 3) the effector signaling pathways regulated by Nox4, and 4) the therapeutic potential of inhibiting Nox4 on cardiac remodeling. As the renin angiotensin system is activated in cardiac disease, we also examined the role of Nox4 in mediating the effects of angiotensin II (AngII) in the myocardium.
Methods
Generation of inducible cardiac specific human Nox4 transgenic mice
Lox-Stop-lox-human Nox4 transgenic mice (LSL-hNox4Tg) on a mixed genetic background of C57BL/6 were created in collaboration with Taconic Farm, Inc. The transgene is driven by a beta-actin promoter. Cardiomyocyte-specific humanNox4 transgenic mice (c-hNox4Tg) were generated by crossing homozygote LSL-hNox4Tg mice to tamoxifen-inducible myocardial specific Cre (αMHC-Cre) mice with genetic background of C57B/6 (see Supplemental Figure S1A for details about the transgene construct). Experiments were conducted in four groups of 16-week-old male mice: 1) Control mice (CTL), the littermates that are hNox4 negative but Cre positive; 2) c-hNox4Tg mice, littermates that are positive for both hNox4 and Cre; 3) angiotensin II infused CTL mice (WT+AngII) and 4) angiotensin II infused c-hNox4 Tg (Tg+AngII). Six mice were used in each group (n = 6). All protocols concerning the use of animals were approved by the Institutional Animal Care and Use Committee at the University of Texas Health Science Center at San Antonio (UTHSCSA). An expanded Materials and Methods section is in the online-only Data Supplement.
Tamoxifen induction, angiotensin II infusion and GKT137831 treatment
All four groups of 16 weeks old mice described above were treated with tamoxifen at a dose of 1 mg/ml in the drinking water for 7 days. At the end of tamoxifen treatment, either AngII infusion or vehicle control (water) was infused intraperitoneally via Alzet micro-osmotic pumps (model 1004) as previously described.12 AngII was infused at a concentration of 1 μg/kg/min for 14 days. GKT137831was mixed in standard chow diet and treatment with a dose of 40 mg/kg/day was started at the same time as tamoxifen treatment and lasted for a total of 3 weeks. Mice were anaesthetized and tissues were harvested at the end of the treatment (Supplemental Figure S1B). To evaluate the potential influence of tamoxifen or Cre activity, multiple control mice including C57B/6J, αMHC-Cre, LSL-hNox4 were treated with vehicle or tamoxifen in the drinking water as for the experimental groups above. Cardiac characteristics were compared. No difference was observed for all the parameters tested without or with tamoxifen treatment (Supplemental Table S4).
Statistical analysis
Data are expressed as mean ± SEM or mean ± SD. Normality analysis was performed before any test using the Shapiro-Wilk and Anderson-Darling statistics. When normality distributed, 2-tailed, unpaired or paired Student t tests were performed for 2-group comparisons and one-way ANOVA followed by the Tukey post hoc test for multi-group comparisons. When samples were not normally distributed, Mann-Whitney U test for 2-group comparisons and the Kruskal-Wallis test for multi-group comparisons followed by the Dunn test were performed. Differences with P<0.05 were considered as statistically significant. The “n” numbers for each group are indicated in the Figure legends.
Results
Characterization of cardiac-specific human Nox4 transgenic mice
To characterize cardiac-specific hNox4 transgenic mice, RT-PCR analysis was performed using total RNA prepared from the left ventricles. The human Nox4 (hNox4) transgene mRNA is exclusively expressed in cardiac-specific human Nox4 transgenic mice (c-hNox4 Tg) and levels of hNox4 mRNA were approximately 8-fold higher compared to endogenous mouse Nox4 mRNA (Figure 1A). The specificity of c-hNox4 expression was confirmed using liver tissue, as shown in Figure 1B, no expression of hNox4 mRNA was detected. The transgene was not detected in other tissues of the transgenic mice (data not shown). Western blot analysis shows that total expression of Nox4 protein was approximately 10-fold higher in c-hNox4 Tg mice than in control (CTL) mice (Figure 1C - E). These results indicate significant tissue-specific expression of the transgene. In addition, cardiac Nox activity is consistently increased in the Tg mice compared to control mice (Supplemental Figure S7C).
Figure 1. Myocardial specific expression of human Nox4 transgene (hNox4-Tg).
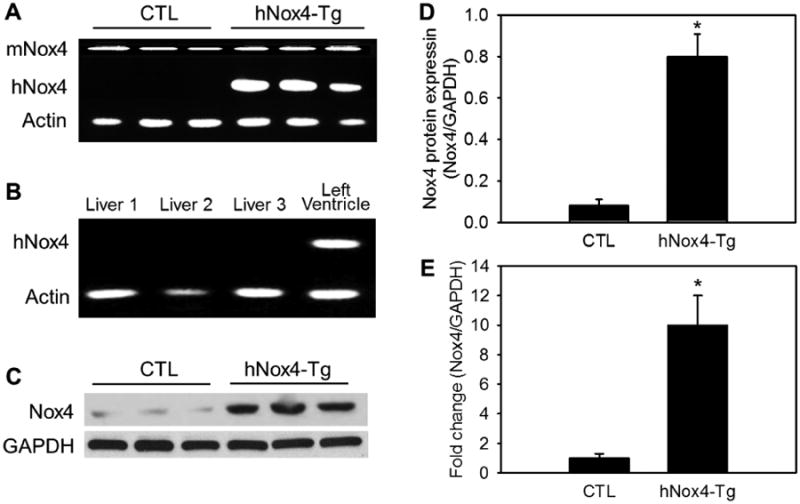
Expression of mouse (mNox4) and human Nox4 mRNAs in LV (A) and livers (B) was analyzed by RT-PCR. Protein expression of Nox4 in LV tissues was analyzed by Western blotting (C), and quantitative analyses are shown in panels D and E (the fold changes). GAPDH was used as internal control. The data shown are from three of six mice in each group. P < 0.001 hNox4-Tg vs CTL. n = 6.
Next, we examined the phenotype of the Tg mice infused with AngII or vehicle. Both Tg and CTL littermates exhibited similar body weight and size. No obvious behavioral abnormalities were observed after one week of tamoxifen treatment and following 2 weeks of angiotensin II (AngII) or vehicle (saline) infusion. However, AngII-infused mice displayed a significant increase in heart weight. Heart weight to body weight ratio and mean blood pressure were markedly augmented in AngII-infused mice compared to the CTL mice. Heart weight and heart weight to body weight ratio were significantly higher in Tg+AngII mice than in CTL+AngII mice. Mean blood pressure of Tg+AngII mice was slightly elevated compared to CTL+AngII mice, but the difference did not reach statistical significance. There was no significant difference in heart rate between all groups (Supplemental Table S1).
AngII-infusion increases cardiac Nox4 expression in CTL and Tg mice, but does not alter the expression of other Nox enzymes
We first examined the effect of AngII on cardiac Nox4 expression in CTL and in Tg mice. Infusion of AngII into CTL mice significantly increased Nox4 protein levels in the LV (Figure 2A, C and D). This increase was similar to that observed in Nox4 Tg mice. AngII infusion into Tg mice resulted in further increase in Nox4 expression in the LV likely representing the additive effect of the mouse endogenous Nox4 (induced by AngII) and the human Nox4 introduced by the transgene. Note that two Nox4 antibodies recognizing different epitopes of the protein showed similar results (Figure 2A). The first antibody is a rabbit polyclonal IgG against an epitope corresponding to amino acids 201-300 mapping within an internal region of Nox4 of human Nox4 (sc-30141, Santa Cruz). The second antibody is a rabbit monoclonal IgG against a peptide sequence within the NADPH binding domain of Nox4 that is conserved between human and mouse Nox4 protein sequences (Epitomics, Burlingame, Calif).13, 14 Both antibodies recognize human and mouse Nox4. The mechanism by which AngII upregulates Nox4 levels remains to be determined. Nox4 is regulated via both transcriptional as well as translational mechanisms. It is well established that the constitutive activity of Nox4 catalytic unit is potentiated by p22phox. We have found that AngII not only increases the levels of Nox4 but also the regulatory subunit p22phox. Analysis using qPCR with the LV tissues from AngII infused mice and control mice revealed that the level of p22phox mRNA is increased by AngII. Furthermore, AngII increased level of p22phox protein associated with Nox4 as demonstrated by immunoprecipitation/immunoblot, which may represent a means whereby AngII contributes to Nox4 activation (Supplemental Figure S10A - C).
Figure 2. Expression of Nox4 and other Nox isoforms in LV tissues.
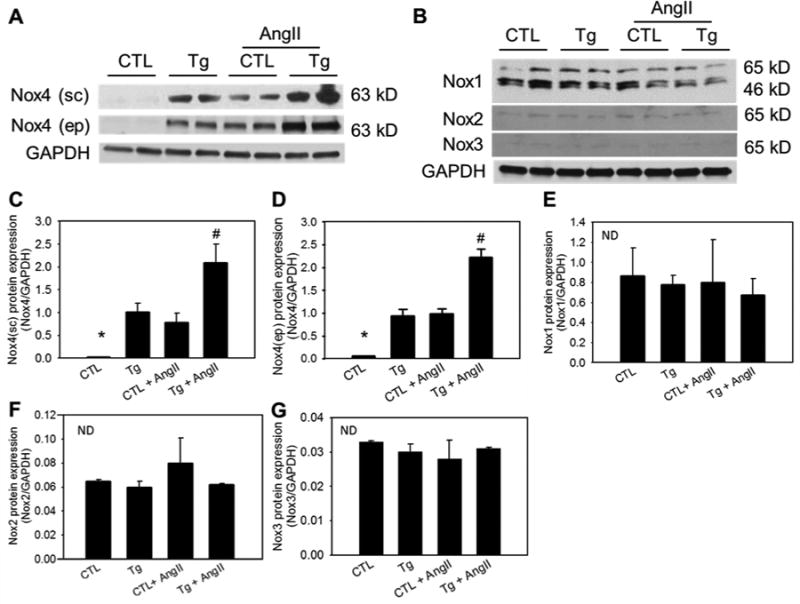
LV tissue of the four groups was harvested at the end of angiotensin II or vehicle infusion and subjected to Western blotting. A, Representative immunoblot showing Nox4 expression analyzed using Nox4 antibody form Santa Cruz (sc) or Epitomics (ep). B, Representative immunoblot showing protein expression of Nox1, Nox2 and Nox3. C - G, Quantitative densitometric analyses of the immunoblot data. *P < 0.01 vs the rest groups. #P < 0.01 vs the Tg or CTL+AngII. ND indicates no significant difference among all groups. GAPDH was used as internal control. n = 6.
Next, we determined the effect of AngII and the transgene on the levels of other Nox isoforms. As shown in Fig. 2B, LV tissues express abundant levels of Nox1. Overexpression of Nox4 and administration of AngII to CTL and Tg mice did not have any effect on Nox1 levels (Figure 2B and E). Similarly, LV tissues from Tg mice or from AngII-infused control or Tg mice showed no change in the levels of Nox2 and Nox3 (Figure. 2B, F and G). These results indicate that AngII treatment enhances the levels of Nox4 and amplifies the pathological effect of Nox4 on cardiac remodeling. Furthermore, overexpression of hNox4 does not alter expression of other Nox isoforms in the left ventricles.
Transgenic Nox4 and AngII additively promote ROS production in LV
Role of AngII-induced reactive oxygen species in the pathogenesis of cardiac injury is established. Furthermore, our results above demonstrate a potentiating effect of AngII in the Nox4 Tg mice on the expression of Nox4 in the LV. The enhanced expression of Nox4 was associated with increased NADPH oxidase activity (Supplemental Figure S7C) and increase in ROS production. DHE staining was used to detect ROS production in frozen sections of LV tissue. In this assay, the fluorescent intensity of DHE represents production of super oxide. DHE positive staining was significantly increased in the LV of Tg mice and CTL+AngII mice. Moreover, a robust increase in DHE positive staining was observed in LV of Tg+AngII mice compared to CTL mice, indicating that AngII enhances the effect of the transgene to increase ROS generation (Supplemental Figure S3). ROS production was confirmed using a fluorescent sensor-HyPer (Supplemental Figure S7B).
Overexpression of Nox4 leads to cardiac interstitial fibrosis
To investigate the role of Nox4 in cardiac remodeling, we examined fibrotic changes induced by transgenic hNox4 and/or AngII treatment using Masson's trichrome staining of the left ventricle. Interestingly, overexpression of hNox4 alone significantly induced interstitial fibrosis with 3-fold increase in collagen deposition (Figure 3A, blue area) over CTL mice. AngII-infusion tended to induce more fibrosis than the hNox4 transgene alone, but the difference between the both groups was not significant. The Tg+AngII mice exhibited more severe interstitial fibrosis, approximately 8-fold higher than in CTL mice (Figure 3A and B). Cardiac fibrosis is induced due to changes in the expression of specific markers.15-17 Thus, expression of several molecular markers of cardiac fibrosis was examined at the mRNA level. As shown in Figure 3C and 3D, expression of the fibrotic cytokines, transforming growth factor-beta1 (TGF-β1) and connective tissue growth factor (CTGF) mRNAs was significantly increased in the LV of Tg mice similar to that in AngII-infused control mice. Notably, AngII enhanced the expression of TGF-β1 and CTGF in the Tg mice as compared to that in control or Tg mice alone. Similar pattern of increased expression of fibrotic markers, fibronectin (FN), collagen I(a1) and collagen III (a1) was observed in AngII infused CTL and Tg mice and in Tg mice alone (Figure 3E - H). No change in PAI-1 expression was observed (Figure 3I). These results demonstrate that AngII as well as its downstream target Nox4 induce fibrotic responses in the LV.
Figure 3. Overexpression of Nox4 and or angiotensin II induce myocardial fibrosis.
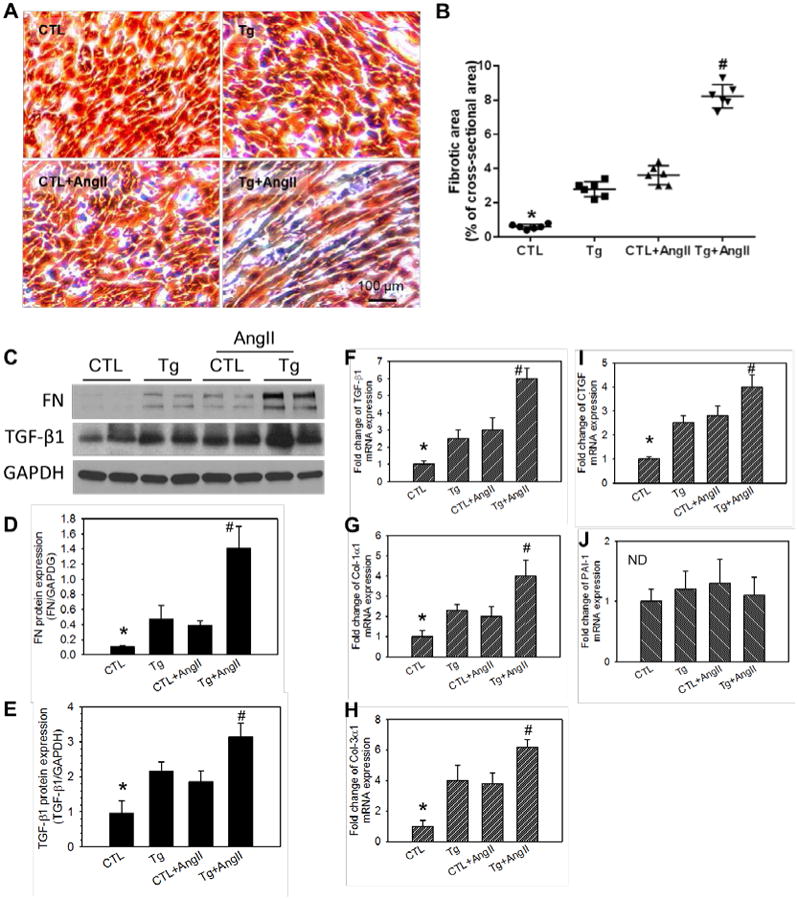
A, Representative microphotographs of LV tissue with Masson's trichrome staining. Collagen stain in blue, muscle in red, and nuclei in black. B, The quantification of fibrotic areas (blue stained collagen). Data are shown as % of section areas. C - E, Protein expression of markers of fibrosis, fibronectin and TGF-β1 detected by Western blotting (C) and the quantification analyses (D, E). F - J, mRNA expression of markers of fibrosis including TGF-β1 (F), collagen-1α1 (G), collagen-3α1 (H), connective tissue growth factor (I), and plasminogen activator inhibitor-1 (J). n = 6. *P < 0.01 vs the rest groups. #P < 0.01 vs the Tg or CTL+AngII. No significant difference between Tg and CTL+AngII. ND denotes no significant difference.
The fact that fibrosis develops in the LV of c-hNox4Tg mice suggests that overexpression of Nox4 in the myocardium induces interstitial collagen deposition by producing and or secreting FN, TGF-β1 or cytokines/growth factors that may act in a paracrine manner to prime differentiation / proliferation of interstitial mesenchymocytes/myofibroblasts. Analysis of TUNEL fluorescent staining shows that the number of apoptotic cardiac myocytes was markedly increased in Tg mice (P < 0.001 vs CTL), and that AngII alone induces myocardial apoptosis in CTL mice and potentiates the effect of the hNox4 transgene to enhance cell death (Supplemental Figure S4). We further performed histological analysis to determine the relationship of cell death and the development of fibrosis. Results suggest that increased expression of Nox4 in the myocardium causes ROS injury including myocyte death, thereby inducing paracrine and autocrine cytokines and growth factors leading to differentiation of interstitial fibroblasts into myofibroblasts and deposition of extracellular matrix/collagens in the LV (Supplemental Figure S5 and S6). Since cardiomyocyte apoptosis mainly induces reparative fibrosis, we quantified reparative fibrosis and perivascular fibrosis. The results showed that overexpression or AngII induced reparative fibrosis 2 to 3 folds more than perivascular fibrosis, suggesting an important effect of myocyte death on cardiac fibrosis (Supplemental Figure S12D).
To confirm the role of the Nox4 transgene in inducing cardiac fibrosis, we examined another line of the myocardial specific hNox4 transgenic mice. This line displayed approximately a 3.5-fold increase in Nox4 protein expression and increased fibrosis but not hypertrophy of the left ventricle compared to control mice (Supplemental Figure S13). Collectively, the data indicate that even modest increase in Nox4 induces cardiac remodeling.
Nox4 mediates angiotensin II-induced cardiac hypertrophy
Cardiac specific hNox4 overexpression alone did not change LV weight or the ratio of LV to body weight (Supplemental Table S1 and Figure 4B). To determine the effect of Nox4 over expression on cardiomyocyte hypertrophy, we performed morphometric analysis using H & E and WGA staining of the LV cross-sections. There was no significant difference in the size of cardiomyocytes between Tg and CTL mice (Figure 4, panels A and C; Supplemental Figure S12B and C), while AngII infusion increased myocyte size by about 32% in CTL+AngII (P < 0.05) and and 58% in Tg+AngII mice (P < 0.001) compared to CTL mice receiving vehicle. Myocyte size was significantly larger in AngII infused Tg mice than AngII infused CTL mice (P < 0.05). Furthermore, immunoblot analysis showed that protein expression of atrial natriuretic peptide (ANP), myocardin, beta myosin heavy chain (MyH7 or βMHC) and tropomyosin were increased by Nox4 transgene or AngII infusion (Figure 4D - H). AngII and the transgene had an additive effect on inducing the expression of all these hypertrophic markers (Figure 4D - H). These data suggest that Nox4 mediates AngII-induced cardiac hypertrophy by reactivating a set of fetal cardiac genes that are normally expressed in the heart only before birth.18, 19
Figure 4. Effect of Nox4 overexpression on myocardial hypertrophy.
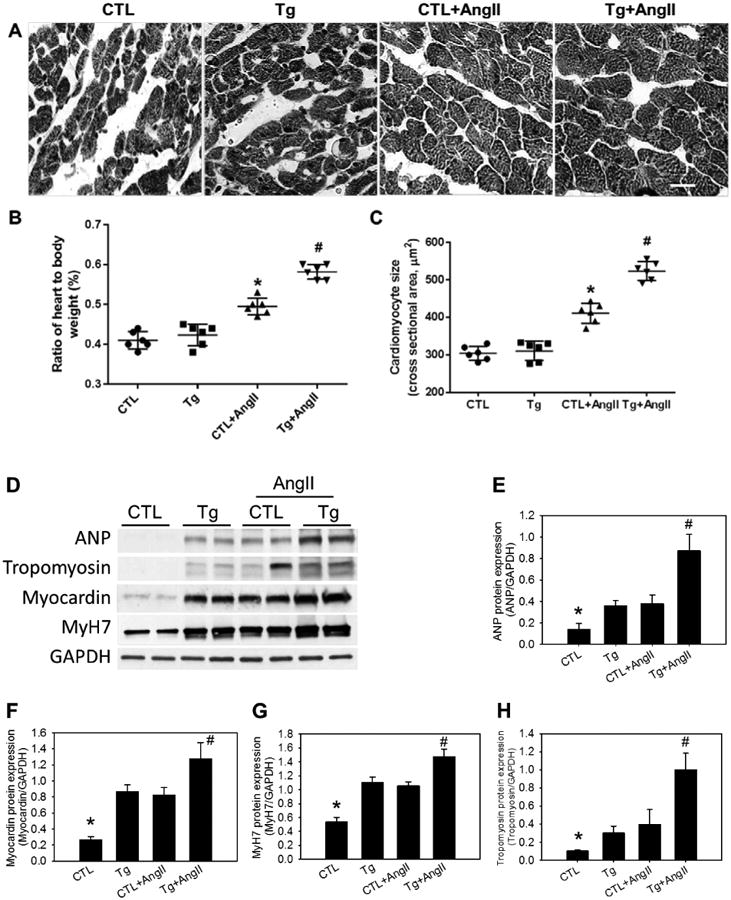
A, Representative microphotographs of cross-sectional views of the LV with Hematoxylin and Eosin staining (see Online Supplemental Figure 12A for a color version of Figure 4A). the white bar indicates 20 μm. B, The ratio of heart weight to body weight (%). C, Mean cross-sectional areas of cardiomyocytes. D - H, Protein expression of markers of cardiac hypertrophy including ANP, myocardin, MyH7, and tropomyosin in LV detected by Western blotting (D), and quantification analyses (E - H). In B and C, *P < 0.05 vs the rest of the groups, #P < 0.01 vs the CTL and the Tg. In E - H, *P < 0.01 vs the rest of the groups. #P < 0.01 vs the Tg or CTL+AngII, no significant difference between Tg and CTL+AngII. GAPGH was used as internal control. n = 6.
Nox4 activates Akt/mTOR/NFκB signaling
Signaling pathways that mediate the reactivation of cardiac fetal genes in postnatal hearts, in response to fibrotic/hypertrophic stimuli remain under investigation. Furthermore, little is known about the role of Nox4 in the amplication of these potential downstream signaling pathways. We explored whether Nox4 activates Akt signaling pathways. LV tissue from Tg mice showed significant increase in activating phosphorylation of Akt similar to that found in AngII infused control mice (Figure 5A - C). Note that AngII and hNox4 had additive effects on Akt phosphorylation at S473, while Akt phosphorylation at T308 seemed to be already maximal with AngII or hNox4 alone (Figure 5A and B). Next, we examined activation of mTOR as this enzyme is a downstream effector of activated Akt. Phosphorylation of S6 kinase and 4EBP-1 were used as indices of mTORC1 activation. Tg mice showed increased phosphorylation of S6K and 4EBP-1 that was enhanced by AngII infusion (Figure 5A, D - F). Note that increased phosphorylation of Akt at S473 indicates activation of mTORC2. Similarly, phosphorylation of mTOR was enhanced in AngII-infused control and Tg+ AngII mice. These results demonstrate activation of mTOR signal transduction downstream of Akt kinase in the LV of Nox4 overexpressing mice.
Figure 5. Overexpression of hNox4 transgene and angiotensin II activate Akt-mTOR and NFκB signaling by enhancing phosphorylation of components of the signaling pathways.
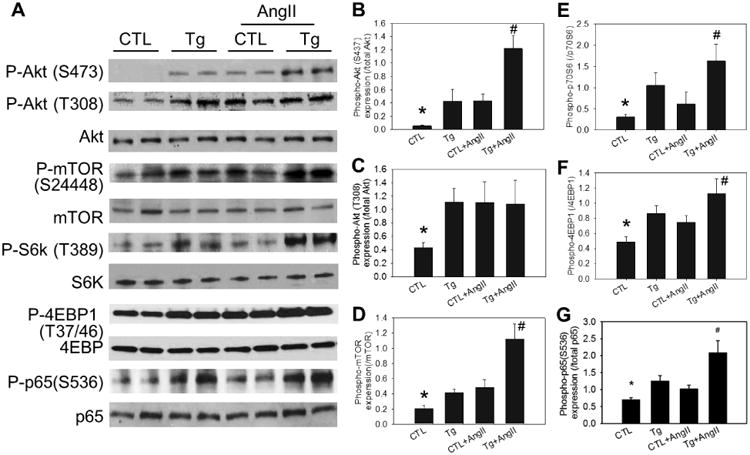
A, Representative immunoblot showing phosphorylation of Akt, mTOR complex 1, S6K, 4EBP1 and NFκB in LV tissue. B - E, Quantitative densitometric analyses of the immunoblot data. GAPDH was used as the internal control. n = 6. *P < 0.01 vs the rest groups. #P < 0.01 vs the Tg or CTL+AngII. No significant difference between Tg and CTL+AngII.
Next, we determined stimulation of another effector of Akt activation, the transcription factor NFkB. The p65 subunit of NFkB undergoes activating phosphorylation at S536 downstream of Akt activation. Therefore, we examined phosphorylation of p65 in the transgenic mice. As shown in Figure 5A and G, marked increase in p65 phosphorylation was found in hNox4 overexpressing mice. AngII infusion into the Tg mice significantly increased this phosphorylation as compared to control or Tg mice alone.
Effect of Nox4 inhibitor GKT137831 on cardiac remodeling
Our results described above demonstrate that overexpression of hNox4 in mouse heart leads to ROS production, cardiomyocyte apoptosis, fibrosis, and amplifies the same changes induced by AngII infusion. We subsequently verified that remodeling changes induced by hNox4 were indeed due to increased Nox4 activity, and more importantly we sought to determine whether Nox4 mediates AngII effects. To this end, we utilized a pharmacological inhibition strategy in our Nox4 overexpressing Tg mice. GKT137831 (Genkyotex, Geneva, Switzerland) is the first-in-class selective and potent Nox4 and Nox1 small molecule inhibitor with good oral bioavailability that has been shown to be well tolerated in healthy human subjects.20, 21 GKT137831 inhibits human and mouse NOX4 with similar potency, and should therefore inhibit hNox4 activity in Tg mice as well as endogenous mouse Nox4 in CTL mice infused with AngII. Treatment of AngII-infused Nox4 Tg mice with GKT137831 decreased ROS production and Nox activity in the LV (Supplemental Figure S7A - C). GKT137831 significantly prevented Nox4 and AngII-induced cardiac fibrosis and expression of fibrotic markers FN and TGF-β1 (Figure 6A - E). Additionally, inhibition of Nox4 by GKT137831 attenuated myocardial hypertrophy (Figure 7A, C and D). The increased expression of hypertrophic markers ANP, tropomyosin and myocardin was also prevented (P < 0.01) whereas MyH7 was not altered (Figure 7E - H). Myocardin is a member of the fetal cardiac genes and an essential component of a molecular switch for the expression of fetal contractile genes in cardiac muscle cells.22 This protein was reported to induce cardiac hypertrophy.19 Immunoblotting analysis showed that GKT137831 treatment decreased expression of myocardin protein (Figure 7B). In addition, GKT137831 robustly decreased the phosphorylation of Akt at THhr308, and the activation of mTORC1 (phosphorylation of S6K and 4EBP1), and mTORC2 (phosphorylation of Akt at S473) in AngII-treated Nox4 Tg mice (Figure 8A - E). Similarly, activating phosphorylation of p65 subunit of NFkB transcription factor was decreased by GKT137831 (Figure 8A and 8F). Treatment with the GKT compound had no effect on cardiac ROS production and did not alter pro-fibrotic markers and cardiac fetal genes in control mice (Supplemental Figure S7B and D). These data confirm that increased Nox4 activity is responsible for the LV changes observed in Tg mice, and identify Nox4 as a key mediator of AngII effects in the heart. Additionally, these results suggest that Akt-mTOR and NFκB are downstream of Nox4-derived ROS in mediating cardiac injury and remodeling.
Figure 6. Inhibition of Nox4 attenuates fibrotic change induced by hNox4 overexpression or/and angiotensin II.

A, Representative microphotographs of LV tissue with Masson's trichrome staining. Collagens stain in blue, muscle in red, and nuclei in black. B, The quantification of fibrotic area (blue stained collagen). Data are shown as % of section areas. C -E, Protein expression of markers of fibrosis fibronectin and TGF-β1 detected by the Western blotting (C) and the quantification analyses (D, E). n = 6. *P < 0.001.
Figure 7. Inhibition of Nox4 attenuates myocardial hypertrophy.
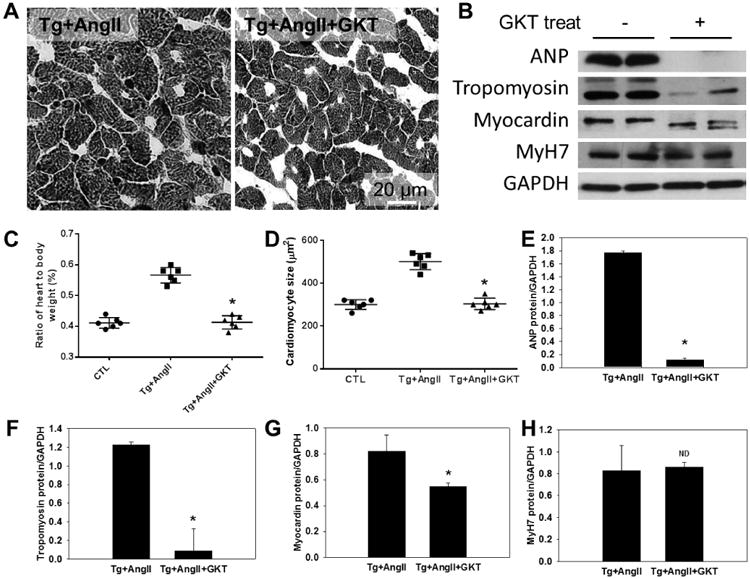
A, Representative microphotographs of cross-sectional views of the LV with Hematoxylin and Eosin staining, the white bar indicates 20 μm. B, Representative immunoblot showing that GKT treatment downregulates the expression of hypertrophic proteins. C. The ratio of heart weight to body weight (%). D, Mean cross-sectional size of cardiomyocyte. E - H, Quantification analyses of hypertrophic proteins shown in panel B. GAPDH was used as the internal control. *P < 0.01 vs Tg+AngII. ND, No significant difference between the two groups. n = 6.
Figure 8. Inhibition of Nox4 suppresses activation of Akt-mTOR and NFκB signaling.
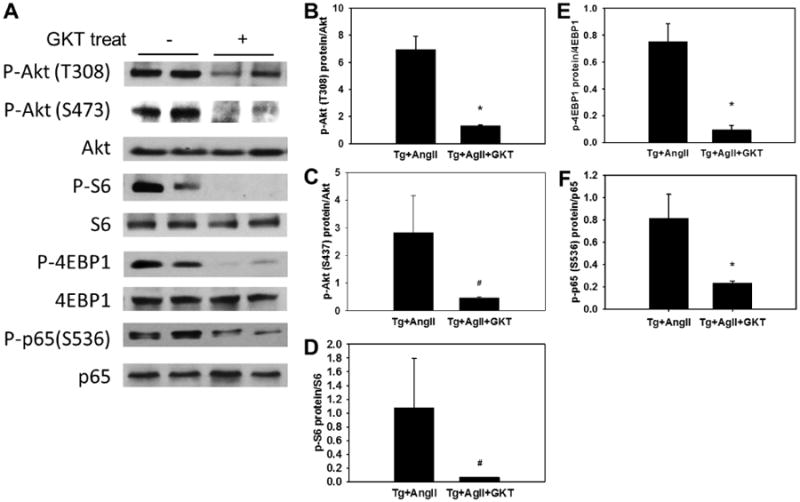
A, Representative immunoblot showing phosphorylation of Akt, mTOR complex 1 activity readout, and p65 in LV tissue. B - F, Quantitative densitometric analyses of the immunoblot data. *P < 0.001 between the two groups. #P < 0.05 between the two groups. n = 6.
Inhibition of mTOR or NFκB suppresses expression of markers of cardiac remodeling
To verify the implication of mTOR or NFκB signaling in our animal model, we performed tissue explant culture of left ventricles from hNox4 Tg mice infused with AngII and treated with 0.5 μM rapamycin, an inhibitor of mTOR, or μM pyrrolidine dithiocarbamate (PDTC), an inhibitor of NFκB, for 24 hours. Western bot analysis showed that these two inhibitors significantly suppress expression of FN, TGF-β, ANP and tropomyosin but not MyH7. Interestingly, myocardin was downregulated by rapamycin but not PDTC indicating that mTOR signaling but not NFκB regulates myocardin expression enhanced by Nox4 and AngII in our animal model (Supplemental Figure S8). To validate the explant culture approach, cell death and Nox4 expression in the tissue explants were measure after 24 hrs of culture and compared to freshly isolated tissues. Left ventricle explant cultures are histologically similar to freshly isolated tissue, and cell death rate in the cultured explants is slightly increased but this increase does not achieve statistical significance compared to freshly isolated tissue (Supplemental Figure S9).
Discussion
This study provides strong evidence demonstrating that increased expression/activation of Nox4 in the heart, either through transgenic overexpression or through the induction of endogenous Nox4 by AngII, induces cardiac remodeling including fibrosis and cardiomyocyte apoptosis. These pathological changes were associated with increased ROS production, expression of profibrotic cytokines, enhanced matrix accumulation and reactivation of the fetal gene program. Overexpression of hNox4 in the heart was not sufficient to cause the LV and cardiomyocyte hypertrophy. However, cardiac hNox4 overexpression was associated with increased expression of fetal genes generally associated with cardiomyocyte hypertrophy, and with the activation of the Akt/mTOR and NFκB signal transduction pathways that have been showed to be implicated in cardiac remodeling. The reasons for this discrepancy are not fully understood, but it is important to note that hNox4 Tg mice were normotensive. In contrast, AngII exerts similar effects in the heart but also increased blood pressure and LV hypertrophy. It is therefore possible that pressure overload is necessary for the development of frank LV hypertrophy. Alternatively, expression of the transgene for longer than 14 days may be required for the development of LV hypertrophy. AngII infusion markedly potentiated the effects of the Nox4 transgene, hNox4 overexpression conversely enhanced cardiac hypertrophy induced by AngII (Figure 2), suggesting that AngII plays a permissive role for Nox4 effects on these endpoints. Inhibition of Nox4 activity by GKT137831 decreases ROS levels, prevents the activation of the Akt-mTOR and NFκB and reactivation of the fetal gene program and markedly attenuates cardiac remodeling.
Nox4 is ubiquitously expressed in various tissues and cell types including renal cells, cardiac/smooth muscle, adipocytes and macrophages. Distinct from other members of the Nox family, Nox4 is constitutively active and increased expression invariably results in enhanced activity and increased ROS production. Nox4 is upregulated by vasoactive agonists such as AngII, endothelin-1, and phenylephrine and in response to cardiac injury and pathological conditions that result in cardiac remodeling.13, 14 Most recently, whole body and cardiac specific mouse Nox4 Tg and knockout (KO) mice were generated and used to investigate the role of Nox4 in cardiovascular diseases. Collectively, the data derived from such gene-manipulated mouse models raised a growing debate on whether Nox4 exerts adverse or protective actions in cardiovascular disease. To add to the complexity, discrepant results were reported in seemingly very similar experimental settings. For instance, transgenic mice overexpressing mouse Nox4 in a cardiac specific manner demonstrated that Nox4 is either protective11 or deleterious8 in models of the LV pressure overload. Interestingly, Matsushima et al. 10 recently showed that Nox4 global knockout and Nox4 tissue-specific knockout animals give rise to two opposite phenotypes when assessed in the same model. Cardiac-specific knockout of Nox4 protects against pressure overload-induced cardiac injury, whereas the global Nox4 knockout showed no protection when subjected to pressure overload10. This suggests that the group of proteins or signaling intermediates affected by Nox4 may have cell specific effects. Also, some of these pathways may protect cell function basally or in pathological conditions. Thus, global deletion of Nox4 may be deleterious independent of the disease induced. We generated tamoxifen-inducible myocardium-specific hNox4 Tg mice and examined the cardiac phenotype of these mice. Since the systemic and local renin angiotensin system is activated in cardiac disease, we also evaluated the role of Nox4 in mediating the effects of AngII in the myocardium. We also deployed a pharmacological inhibition strategy using the Nox4 and Nox1 inhibitor GKT137831 to establish direct effect of Nox4 on cardiac remodeling in transgenic mice overexpressing human Nox4 and infused with AngII. Nox4 was expressed, albeit at a relatively low level, in control mice suggesting that Nox4 may be required for normal cellular physiological processes (Figure 1A and Figure 2A). Interestingly, a 10-fold increase in Nox4 expression and interstitial fibrosis was observed in the LV of hNox4 Tg mice 14 days after induction of the transgene by tamoxifen. While markers of cardiomyocyte hypertrophy were also induced, no significant hypertrophy was present. Whether the prolonged overexpression of Nox4 results in hypertrophy remains to be determined. Studies by Ago et al. showed that overexpression of mouse Nox4 in Tg mice does not induce cardiac hypertrophy in young mice, but does cause cardiac myocyte hypertrophy as the transgenic mice advance to middle age, suggesting that long-term overexpression of Nox4 may be required for the development of cardiomyocyte hypertrophy.8
Control mice infused with AngII for 2 weeks exhibited similar levels of Nox4 expression in the LV as the hNox4 Tg mice and displayed cardiac fibrosis as well as hypertrophy, suggesting that AngII-induced cardiac hypertrophy involves additional mechanisms besides Nox4 expression (Figure 2A, C, D). For instance, the presence of high blood pressure in these mice may have contributed in the development of hypertrophy. The hNox4 Tg mice infused with AngII exhibited the highest expression of Nox4 and severe cardiac remodeling including both fibrosis and hypertrophy. It should be noted that the infusion of AngII in hNox4 Tg mice did not alter the expression of other Nox enzymes, (Figure 2B, E - I). However, in some conditions, manipulation of Nox4 gene may result in alteration of other Nox enzymes. For example, cardiac specific Nox4 KO mice have reduced cardiac hypertrophy/dysfunction in a mouse model of transverse aortic constriction (TAC), whereas systemic Nox4 KO mice exhibited more cardiac fibrosis and diastolic dysfunction than WT mice.10 Systemic deletion of Nox4 may change the status of other Nox enzymes not only in myocytes but also in other cardiac cells. In addition, global knockout of Nox4 may induce as-yet-unknown compensatory mechanisms during embryogenesis and development, resulting in changes in the expression of other Nox isoforms. Also, paracrine and endocrine metabolism may be regulated by whole body Nox4 deletion to affect the physiology/pathology of the disease process. Therefore, results from tissue-specific KO and systemic KO mice are not always comparable.
Cardiac myocytes proliferate rapidly during embryogenesis but lose their proliferative capacity soon after birth.23, 24 However, adult cardiac myocytes retain the ability to respond to mechanical, hemodynamic, hormonal, and pathologic stimuli and contribute to cardiac remodeling, including hypertrophic growth and interstitial expansion.25 Cardiac remodeling is accompanied by the activation of a set of fetal cardiac genes that are normally expressed in the embryonic heart.18, 26 The reactivation of cardiac fetal genes in postnatal cardiomyocytes in response to stress signals is useful markers for cardiac fibrosis and hypertrophy. The administration of GKT 137831 to hNox4 Tg mice infused with AngII, blocked the expression of cardiac fetal genes providing direct evidence that Nox4 mediates the enhanced expression of the fetal gene program (Figure 3, 4, 6, 7 and Supplemental Figure S7). Myocardin is a member of the fetal gene program and also represents a molecular switch that acts as co-activator of the transcription factor SRF (serum response factor) which regulates contractile genes in cardiac muscle cells.27, 28 This protein binds to the serum response element (SRE) in the promoter region of target genes. It controls the activity of many immediate-early genes, thereby regulating cell cycle, apoptosis, cell growth, and cell differentiation. Myocardin transactivates CArG box of ANP gene and induces expression of ANP, one of the most sensitive markers of hypertrophy.29, 30 Furthermore, myocardin has been implicated in the differentiation of myofibroblasts.31 Mouse myocardin gene encodes two alternatively spliced protein isoforms of 935 and 856 amino acids.32, 33 Inhibition of Nox4 by GKT137831 not only downregulated myocardin protein expression but also altered its molecular weight (Figure 7B, G), likely due to alternatively spliced transcript variants of myocardin.
Compared to the LVs from wild type mice, the LVs from hNox4 Tg mice and mice infused with AngII exhibited increase in phosphorylation of Akt at S473 and T308, as well as activation of mTOR and S6K pathway as evident by enhanced phosphorylation of 4EBP1 and S6 ribosomal protein. Both kinases were further activated in the LV of Nox4 Tg mice that received AngII infusion. Treatment with GKT137831 blocked the activation of Akt and mTOR/S6K pathway (Figure 5A - C, and 8A - C). Akt has been shown to be directly involved in physiological adaptive cardiac hypertrophy in response to IGF-I and exercise.34 Moreover, transgenic mice that overexpress Akt display spontaneous cardiac hypertrophy and pro-longed Akt activation in the myocardium results in dilatation and cardiac dysfunction.35 mTOR is a highly conserved serine/threonine kinase involved in vital cellular processes, including growth, gene transcription, and protein synthesis.36 Among the best studied properties of mTOR kinase is its involvement in protein translation and cell growth through phosphorylation of S6K and 4E-BP1. Although cardiac-specific mTOR-deficient mice develop a fatal, dilated cardiomyopathy,37 partial loss of mTOR activity37 and inhibition of mTOR by rapamycin38 significantly prevents cardiac hypertrophy. Administration of everolimus, a rapamycin analogue, decreases ventricle hypertrophy in kidney transplant patients.39 The activation of Akt-mTOR signaling likely contributes to cardiac fibrosis and hypertrophy by enhancing protein synthesis. We provide in vivo evidence of Nox4-derived ROS generation in mTOR activation in the course of cardiac remodeling.
Overexpression of Nox4 also results in activation of NFκB (Figure 5 and 8F-G). NFκB exists as an inactive dimeric complex, comprising p65 and p50 protein subunits bound to the inhibitor protein IκB. Activation of NFκB requires the phosphorylation-dependent degradation of IκBα, which is mediated by the IκBα kinase complex (IKK), IKKα, IKKβ, and IKKγ (NFκB essential modulator) subunits. The loss of IκBα exposes the nuclear localization motif on p65 NFκB subunit permitting its phosphorylation and nuclear targeting. NFκB activation is involved in the hypertrophic response of cultured cardiomyocytes40, 41 and is required for the development of cardiac hypertrophy in vivo42, 43 Moreover, there is evidence that NFκB and mTOR interact in a positive feedback loop.44, 45 Our in vitro results further verified that inhibition of mTOR or NFκB signaling significantly suppresses expression of cardiac remodeling markers enhanced by overexpressed Nox4 in our mouse models (Supplemental Figure S8). It is likely that Akt-mTOR-NFκB axis might work as a central signaling pathway and bi-directionally mediates cardiac remodeling. However, little is known about the role of Nox4 or Nox4-derived ROS in activation of Akt/mTOR and NFκB signaling pathways. While it is known that Nox4 is required for Akt activation in renal cells,46, 47 the data in the heart are very sparse. To the best of our knowledge, the present study is the first to demonstrate that Nox4 directly targets and activates mTOR, particularly mTORC1 complex, signaling. Interestingly, our observation that Nox4 overexpression increases phosphorylation of Akt on S473, an activating site targeted by mTORC2,48 implies that the ROS generated by Nox4 may also modulate mTORC2 complex. While numerous studies have described NFκB as an upstream regulator of Nox4 expression in vascular cells, few reports positioning Nox4 upstream of NFκB are available and are all related to toll like receptor-4 signaling.49, 50 Together, our findings suggest that mTOR and NFκB pathways may integrate Nox4-dependent ROS generation to the initiation of cardiac remodeling.
Data in mice treated with GKT137831 confirm the role of Nox4 in the development of cardiac remodeling. The fact that Nox4/Nox1 inhibitor GKT137831 attenuated cardiac damage in cardiac-specific Nox4 overexpressing mice treated with AngII strongly supports the idea that Nox4 is responsible for the pathologies observed. Although Nox2, Nox1 and Nox3 are also detected in the LV, with Nox1 being apparently expressed at a higher level than Nox2 and Nox3 in LV of our mouse models, Nox4 overexpression and/or AngII treatment did not change their basal expression. However, since the Nox2 or Nox1 activity is regulated by recruitment of cytosolic factors not via the control of its expression levels like Nox4, the role of Nox2 or Nox1 cannot be excluded. Thus, the protective effect of GKT137831 treatment most likely is resulted largely from the inhibition of Nox4 activity, but do not definitively exclude a partial contribution of Nox1. In addition to its primary localization in plasma membrane, Nox4 localizes to other subcellular compartments. In hNox4 Tg mice compared to control mice, we find that Nox4 is highly expressed in ER, mitochondria and nuclei (Supplemental Figure S11). These results may suggest the importance of subcellular translocation and localization in the regulation of Nox4 action in myocardium.
The dose of AngII for infusion used in this study is based on our previous studies.12, 51 Smaller doses of AngII infusion were tested; however were not sufficient to induce detectable cardiac morphological changes within the short-term (two week) treatment of these mice, may suggesting a difference of sensitivity to AngII either among different mouse species or between human subjects and mice. The renin-angiotensin system is upregulated or activated in diabetes and heart failure.52 Actually, AngII is synthesized locally in the heart and the concentrations of AngII in the cardiac myocytes that are available to bind to AngII receptors far exceeds blood circulating concentrations.53 Even in studies of cultured cardiomyocytes, concentrations of AngII in the cells are two orders of magnitude higher than the concentrations in the medium.53 Furthermore, various AngII levels in the myocardium may be induced in many different forms of cardiac injury. Therefore, the concentrations of AngII that may be achieved locally in pathological states may far exceed its circulating levels, and this may explain why we did not find detectable morphological pathological changes in the left ventricle of our mice when using lower doses of AngII infusion for two weeks.
Results from our second line of cardiac specific Nox4 transgenic mice showed that the transgenic mice displayed a 3.5 fold Nox4 protein expression and obvious fibrosis in the left ventricle versus control mice, but no myocardial hypertrophy, indicating a similarity in cardiac pathology between the two lines of Nox4 transgenic mice we generated using the same protocol, in spite of differences in Nox4 expression level (Supplemental Figure S13). Our data has shown that Nox4 expression in the left ventricle of transgenic mice predominantly was located in the endoplasmic reticulum, mitochondria and nuclei (Supplemental Figure S11), but we cannot exclude the possibility of expression of the transgene, if any, in un-physiological subcellular locations.
In summary, specific local upregulation of Nox4 in the heart enhances oxidative stress and induces cardiac remodeling, associated with activation of the Akt-mTOR and NFκB signaling pathways as well as activation of cardiac fetal contractile genes. Inhibition of Nox4 oxidase blocks the activation of these signaling pathways and attenuates cardiac remodeling, suggesting that NADPH oxidase inhibitors have the therapeutic potential to prevent/treat cardiac remodeling and potentially delay the development and reduce the severity of established heart failure (Supplemental Figure S14).
Supplementary Material
Acknowledgments
We thank Drs. Goutam Ghosh-Choudhury and Yves Gorin, at the UTHSCSA, and Philippe Wiesel at GenkyoTex for their valuable comments on the manuscript. We thank Dr. Yidong Chen, Professor of Epidemiology and Biostatistics at the UTHSCSA for a review on the statistical data and processes.
Sources of Funding: This work was supported in part by grants from JDRF (Multi-Project grant 4-2010-511), AHA (11SDG5380002) and RO1DK033665.
Footnotes
Disclosures: None
References
- 1.Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling--concepts and clinical implications: A consensus paper from an international forum on cardiac remodeling. Behalf of an international forum on cardiac remodeling. Journal of the American College of Cardiology. 2000;35:569–582. doi: 10.1016/s0735-1097(99)00630-0. [DOI] [PubMed] [Google Scholar]
- 2.Frey N, Katus HA, Olson EN, Hill JA. Hypertrophy of the heart: A new therapeutic target? Circulation. 2004;109:1580–1589. doi: 10.1161/01.CIR.0000120390.68287.BB. [DOI] [PubMed] [Google Scholar]
- 3.Tsutsui H, Kinugawa S, Matsushima S. Oxidative stress and heart failure. American journal of physiology Heart and circulatory physiology. 2011;301:H2181–2190. doi: 10.1152/ajpheart.00554.2011. [DOI] [PubMed] [Google Scholar]
- 4.Papaharalambus CA, Griendling KK. Basic mechanisms of oxidative stress and reactive oxygen species in cardiovascular injury. Trends in cardiovascular medicine. 2007;17:48–54. doi: 10.1016/j.tcm.2006.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuroda J, Ago T, Matsushima S, Zhai P, Schneider MD, Sadoshima J. Nadph oxidase 4 (nox4) is a major source of oxidative stress in the failing heart. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:15565–15570. doi: 10.1073/pnas.1002178107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takeya R, Ueno N, Kami K, Taura M, Kohjima M, Izaki T, Nunoi H, Sumimoto H. Novel human homologues of p47phox and p67phox participate in activation of superoxide-producing nadph oxidases. The Journal of biological chemistry. 2003;278:25234–25246. doi: 10.1074/jbc.M212856200. [DOI] [PubMed] [Google Scholar]
- 7.Dikalov SI, Dikalova AE, Bikineyeva AT, Schmidt HH, Harrison DG, Griendling KK. Distinct roles of nox1 and nox4 in basal and angiotensin ii-stimulated superoxide and hydrogen peroxide production. Free radical biology & medicine. 2008;45:1340–1351. doi: 10.1016/j.freeradbiomed.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ago T, Kuroda J, Pain J, Fu C, Li H, Sadoshima J. Upregulation of nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circulation research. 2010;106:1253–1264. doi: 10.1161/CIRCRESAHA.109.213116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Block K, Gorin Y, Abboud HE. Subcellular localization of nox4 and regulation in diabetes. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:14385–14390. doi: 10.1073/pnas.0906805106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matsushima S, Kuroda J, Ago T, Zhai P, Park JY, Xie LH, Tian B, Sadoshima J. Increase doxidative stress in the nucleus caused by nox4 mediates oxidation of hdac4 and cardiac hypertrophy. Circulation research. 2013;112:651–663. doi: 10.1161/CIRCRESAHA.112.279760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang M, Brewer AC, Schroder K, Santos CX, Grieve DJ, Wang M, Anilkumar N, Yu B, Dong X, Walker SJ, Brandes RP, Shah AM. Nadph oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:18121–18126. doi: 10.1073/pnas.1009700107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao Q, Ishibashi M, Hiasa K, Tan C, Takeshita A, Egashira K. Essential role of vascular endothelial growth factor in angiotensin ii-induced vascular inflammation and remodeling. Hypertension. 2004;44:264–270. doi: 10.1161/01.HYP.0000138688.78906.6b. [DOI] [PubMed] [Google Scholar]
- 13.Lee CF, Qiao M, Schroder K, Zhao Q, Asmis R. Nox4 is a novel inducible source of reactive oxygen species in monocytes and macrophages and mediates oxidized low density lipoprotein-induced macrophage death. Circulation research. 2010;106:1489–1497. doi: 10.1161/CIRCRESAHA.109.215392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ullevig S, Zhao Q, Lee CF, Seok Kim H, Zamora D, Asmis R. Nadph oxidase 4 mediates monocyte priming and accelerated chemotaxis induced by metabolic stress. Arteriosclerosis, thrombosis, and vascular biology. 2012;32:415–426. doi: 10.1161/ATVBAHA.111.238899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maalouf RM, Eid AA, Gorin YC, Block K, Escobar GP, Bailey S, Abboud HE. Nox4-derived reactive oxygen species mediate cardiomyocyte injury in early type 1 diabetes. American journal of physiology Cell physiology. 2012;302:C597–604. doi: 10.1152/ajpcell.00331.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuwahara F, Kai H, Tokuda K, Kai M, Takeshita A, Egashira K, Imaizumi T. Transforming growth factor-beta function blocking prevents myocardial fibrosis and diastolic dysfunction in pressure-overloaded rats. Circulation. 2002;106:130–135. doi: 10.1161/01.cir.0000020689.12472.e0. [DOI] [PubMed] [Google Scholar]
- 17.Hou J, Kato H, Cohen RA, Chobanian AV, Brecher P. Angiotensin ii-induced cardiac fibrosis in the rat is increased by chronic inhibition of nitric oxide synthase. The Journal of clinical investigation. 1995;96:2469–2477. doi: 10.1172/JCI118305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.MacLellan WR, Schneider MD. Genetic dissection of cardiac growth control pathways. Annual review of physiology. 2000;62:289–319. doi: 10.1146/annurev.physiol.62.1.289. [DOI] [PubMed] [Google Scholar]
- 19.Xing W, Zhang TC, Cao D, Wang Z, Antos CL, Li S, Wang Y, Olson EN, Wang DZ. Myocardin induces cardiomyocyte hypertrophy. Circulation research. 2006;98:1089–1097. doi: 10.1161/01.RES.0000218781.23144.3e. [DOI] [PubMed] [Google Scholar]
- 20.Aoyama T, Paik YH, Watanabe S, Laleu B, Gaggini F, Fioraso-Cartier L, Molango S, Heitz F, Merlot C, Szyndralewiez C, Page P, Brenner DA. Nicotinamide adenine dinucleotide phosphate oxidase in experimental liver fibrosis: Gkt137831 as a novel potential therapeutic agent. Hepatology. 2012;56:2316–2327. doi: 10.1002/hep.25938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Green DE, Murphy TC, Kang BY, Kleinhenz JM, Szyndralewiez C, Page P, Sutliff RL, Hart CM. The nox4 inhibitor gkt137831 attenuates hypoxia-induced pulmonary vascular cell proliferation. American journal of respiratory cell and molecular biology. 2012;47:718–726. doi: 10.1165/rcmb.2011-0418OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen JF, Wang S, Wu Q, Cao D, Nguyen T, Chen Y, Wang DZ. Myocardin marks the earliest cardiac gene expression and plays an important role in heart development. Anat Rec (Hoboken) 2008;291:1200–1211. doi: 10.1002/ar.20756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li F, Wang X, Gerdes AM. Formation of binucleated cardiac myocytes in rat heart: Ii. Cytoskeletal organisation. Journal of molecular and cellular cardiology. 1997;29:1553–1565. doi: 10.1006/jmcc.1997.0403. [DOI] [PubMed] [Google Scholar]
- 24.Li F, Wang X, Bunger PC, Gerdes AM. Formation of binucleated cardiac myocytes in rat heart: I. Role of actin-myosin contractile ring. Journal of molecular and cellular cardiology. 1997;29:1541–1551. doi: 10.1006/jmcc.1997.0381. [DOI] [PubMed] [Google Scholar]
- 25.Ahuja P, Sdek P, MacLellan WR. Cardiac myocyte cell cycle control in development, disease, and regeneration. Physiological reviews. 2007;87:521–544. doi: 10.1152/physrev.00032.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Olson EN, Schneider MD. Sizing up the heart: Development redux in disease. Genes & development. 2003;17:1937–1956. doi: 10.1101/gad.1110103. [DOI] [PubMed] [Google Scholar]
- 27.Wang D, Chang PS, Wang Z, Sutherland L, Richardson JA, Small E, Krieg PA, Olson EN. Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell. 2001;105:851–862. doi: 10.1016/s0092-8674(01)00404-4. [DOI] [PubMed] [Google Scholar]
- 28.Small EM, Thatcher JE, Sutherland LB, Kinoshita H, Gerard RD, Richardson JA, Dimaio JM, Sadek H, Kuwahara K, Olson EN. Myocardin-related transcription factor-a controls myofibroblast activation and fibrosis in response to myocardial infarction. Circulation research. 2010;107:294–304. doi: 10.1161/CIRCRESAHA.110.223172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Badorff C, Seeger FH, Zeiher AM, Dimmeler S. Glycogen synthase kinase 3beta inhibits myocardin-dependent transcription and hypertrophy induction through site-specific phosphorylation. Circulation research. 2005;97:645–654. doi: 10.1161/01.RES.0000184684.88750.FE. [DOI] [PubMed] [Google Scholar]
- 30.Boluyt MO, Zheng JS, Younes A, Long X, O'Neill L, Silverman H, Lakatta EG, Crow MT. Rapamycin inhibits alpha 1-adrenergic receptor-stimulated cardiac myocyte hypertrophy but not activation of hypertrophy-associated genes. Evidence for involvement of p70 s6 kinase. Circulation research. 1997;81:176–186. doi: 10.1161/01.res.81.2.176. [DOI] [PubMed] [Google Scholar]
- 31.Crider BJ, Risinger GM, Jr, Haaksma CJ, Howard EW, Tomasek JJ. Myocardin-related transcription factors a and b are key regulators of tgf-beta1-induced fibroblast to myofibroblast differentiation. The Journal of investigative dermatology. 2011;131:2378–2385. doi: 10.1038/jid.2011.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parmacek MS. Myocardin-related transcription factors: Critical coactivators regulating cardiovascular development and adaptation. Circulation research. 2007;100:633–644. doi: 10.1161/01.RES.0000259563.61091.e8. [DOI] [PubMed] [Google Scholar]
- 33.Imamura M, Long X, Nanda V, Miano JM. Expression and functional activity of four myocardin isoforms. Gene. 2010;464:1–10. doi: 10.1016/j.gene.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 34.Kim J, Wende AR, Sena S, Theobald HA, Soto J, Sloan C, Wayment BE, Litwin SE, Holzenberger M, LeRoith D, Abel ED. Insulin-like growth factor i receptor signaling is required for exercise-induced cardiac hypertrophy. Mol Endocrinol. 2008;22:2531–2543. doi: 10.1210/me.2008-0265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matsui T, Li L, Wu JC, Cook SA, Nagoshi T, Picard MH, Liao R, Rosenzweig A. Phenotypic spectrum caused by transgenic overexpression of activated akt in the heart. The Journal of biological chemistry. 2002;277:22896–22901. doi: 10.1074/jbc.M200347200. [DOI] [PubMed] [Google Scholar]
- 36.Shaw RJ, Cantley LC. Ras, pi(3)k and mtor signalling controls tumour cell growth. Nature. 2006;441:424–430. doi: 10.1038/nature04869. [DOI] [PubMed] [Google Scholar]
- 37.Zhang D, Contu R, Latronico MV, Zhang J, Rizzi R, Catalucci D, Miyamoto S, Huang K, Ceci M, Gu Y, Dalton ND, Peterson KL, Guan KL, Brown JH, Chen J, Sonenberg N, Condorelli G. Mtorc1 regulates cardiac function and myocyte survival through 4e-bp1 inhibition in mice. The Journal of clinical investigation. 2010;120:2805–2816. doi: 10.1172/JCI43008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McMullen JR, Sherwood MC, Tarnavski O, Zhang L, Dorfman AL, Shioi T, Izumo S. Inhibition of mtor signaling with rapamycin regresses established cardiac hypertrophy induced by pressure overload. Circulation. 2004;109:3050–3055. doi: 10.1161/01.CIR.0000130641.08705.45. [DOI] [PubMed] [Google Scholar]
- 39.Paoletti E, Marsano L, Bellino D, Cassottana P, Cannella G. Effect of everolimus on left ventricular hypertrophy of de novo kidney transplant recipients: A 1 year, randomized, controlled trial. Transplantation. 2012;93:503–508. doi: 10.1097/TP.0b013e318242be28. [DOI] [PubMed] [Google Scholar]
- 40.Yamamoto K, Ohki R, Lee RT, Ikeda U, Shimada K. Peroxisome proliferator-activated receptor gamma activators inhibit cardiac hypertrophy in cardiac myocytes. Circulation. 2001;104:1670–1675. doi: 10.1161/hc4001.097186. [DOI] [PubMed] [Google Scholar]
- 41.Pagani FD, DerSimonian H, Zawadzka A, Wetzel K, Edge AS, Jacoby DB, Dinsmore JH, Wright S, Aretz TH, Eisen HJ, Aaronson KD. Autologous skeletal myoblasts transplanted to ischemia-damaged myocardium in humans. Histological analysis of cell survival and differentiation. Journal of the American College of Cardiology. 2003;41:879–888. doi: 10.1016/s0735-1097(03)00081-0. [DOI] [PubMed] [Google Scholar]
- 42.Li Y, Ha T, Gao X, Kelley J, Williams DL, Browder IW, Kao RL, Li C. Nf-kappab activation is required for the development of cardiac hypertrophy in vivo. American journal of physiology Heart and circulatory physiology. 2004;287:H1712–1720. doi: 10.1152/ajpheart.00124.2004. [DOI] [PubMed] [Google Scholar]
- 43.Ha T, Li Y, Gao X, McMullen JR, Shioi T, Izumo S, Kelley JL, Zhao A, Haddad GE, Williams DL, Browder IW, Kao RL, Li C. Attenuation of cardiac hypertrophy by inhibiting both mtor and nfkappab activation in vivo. Free radical biology & medicine. 2005;39:1570–1580. doi: 10.1016/j.freeradbiomed.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 44.Dan HC, Cooper MJ, Cogswell PC, Duncan JA, Ting JP, Baldwin AS. Akt-dependent regulation of nf-{kappa}b is controlled by mtor and raptor in association with ikk. Genes & development. 2008;22:1490–1500. doi: 10.1101/gad.1662308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dhingra R, Gang H, Wang Y, Biala AK, Aviv Y, Margulets V, Tee A, Kirshenbaum LA. Bidirectional regulation of nuclear factor-kappab and mammalian target of rapamycin signaling functionally links bnip3 gene repression and cell survival of ventricular myocytes. Circulation Heart failure. 2013;6:335–343. doi: 10.1161/CIRCHEARTFAILURE.112.000061. [DOI] [PubMed] [Google Scholar]
- 46.Gorin Y, Ricono JM, Kim NH, Bhandari B, Choudhury GG, Abboud HE. Nox4 mediates angiotensin ii-induced activation of akt/protein kinase b in mesangial cells. American journal of physiology Renal physiology. 2003;285:F219–229. doi: 10.1152/ajprenal.00414.2002. [DOI] [PubMed] [Google Scholar]
- 47.New DD, Block K, Bhandhari B, Gorin Y, Abboud HE. Igf-i increases the expression of fibronectin by nox4-dependent akt phosphorylation in renal tubular epithelial cells. American journal of physiology Cell physiology. 2012;302:C122–130. doi: 10.1152/ajpcell.00141.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zoncu R, Efeyan A, Sabatini DM. Mtor: From growth signal integration to cancer, diabetes and ageing. Nature reviews Molecular cell biology. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Williams CR, Lu X, Sutliff RL, Hart CM. Rosiglitazone attenuates nf-kappab-mediated nox4 upregulation in hyperglycemia-activated endothelial cells. American journal of physiology Cell physiology. 2012;303:C213–223. doi: 10.1152/ajpcell.00227.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Manea A, Tanase LI, Raicu M, Simionescu M. Transcriptional regulation of nadph oxidase isoforms, nox1 and nox4, by nuclear factor-kappab in human aortic smooth muscle cells. Biochemical and biophysical research communications. 2010;396:901–907. doi: 10.1016/j.bbrc.2010.05.019. [DOI] [PubMed] [Google Scholar]
- 51.Ishibashi M, Egashira K, Zhao Q, Hiasa K, Ohtani K, Ihara Y, Charo IF, Kura S, Tsuzuki T, Takeshita A, Sunagawa K. Bone marrow-derived monocyte chemoattractant protein-1 receptor ccr2 is critical in angiotensin ii-induced acceleration of atherosclerosis and aneurysm formation in hypercholesterolemic mice. Arteriosclerosis, thrombosis, and vascular biology. 2004;24:e174–178. doi: 10.1161/01.ATV.0000143384.69170.2d. [DOI] [PubMed] [Google Scholar]
- 52.Asghar O, Al-Sunni A, Khavandi K, Khavandi A, Withers S, Greenstein A, Heagerty AM, Malik RA. Diabetic cardiomyopathy. Clin Sci (Lond) 2009;116:741–760. doi: 10.1042/CS20080500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Paul M, Poyan Mehr A, Kreutz R. Physiology of local renin-angiotensin systems. Physiological reviews. 2006;86:747–803. doi: 10.1152/physrev.00036.2005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.