

Abstract
Chronic lymphocytic leukemia (CLL) is a highly heterogeneous hematologic malignancy and characterized by dysregulation of cell death pathways. Apoptosis and necroptosis are the two major cell death processes, and substantial evidence showed up-regulation of several pro-survival factors in CLL cells. Autophagy, as a dual player in mediating cell death and survival, is largely regarded to be an alternative target in the treatment of CLL. Numerous novel drugs have been developed and are being investigated in clinical trials. It is necessary to depict the impaired cell death pathways in CLL and the pro-survival factors targeted by noncytotoxic drugs directly or indirectly. Here we summarize three dysregulated cell death mechanisms in CLL, and present the current knowledge of drugs that orchestrate cell death via targeting pro-survival factors and the clinical effects as well.
Keywords: Chronic lymphocytic leukemia, apoptosis, necroptosis, autophagy, therapy
Introduction
Chronic lymphocytic leukemia (CLL) is one of the most prevalent types of hematologic malignancies in Western countries. The clinical manifestations of CLL are highly heterogeneous, ranging from an indolent and asymptomatic period to a progressive symptom-causing phase [1]. CLL cells fail in overcoming the differentiation hurdle and are arrested in G0/G1 of the cell cycle. The progressive clonal accumulation of a CD5-positive subpopulation of B lymphocytes with mature appearance in the bone marrow, lymph nodes and peripheral blood will ultimately lead to occurrence and development of the disease [2]. Multiple external (microenvironmental stimuli and antigenic drive) and internal (genetic and epigenetic) events have been elucidated to play pivotal roles in the transformation, progression and evolution of CLL [3]. Despite the substantial progress in pathobiology research and the development of effective treatment regimens, CLL is still largely an incurable disease [4]. A personalized therapeutic approach based on genetic and molecular status could be preferable to the comprehensive treatment of CLL [4].
In-depth understanding of the existing cellular pathways by which CLL cells undergo metabolic cell death is required to determine the efficacy of therapeutic treatments as well as the exploration of novel strategies. Moreover, the expression levels of regulatory mediators involved could be altered in the context of CLL which could contribute to chemotherapy resistance. Therefore it is necessary to depict the different cell death pathways, particularly highlight the key mediators and interconnections in CLL cells. Here we focus on three cell death processes in CLL, namely apoptosis, necroptosis and autophagy, and we summarize the current pharmacological approaches to induce cell death through targeting these pathways.
Cell death pathways in apoptosis, necroptosis, and autophagy
Dying the right way, particularly the apoptotic, necrotic or autophagic pathways, is the end of a cell’s finite life. Once the deficiency occurred in one of the cell death pathways, the alternative pathways burdened. Although a number of investigations have depicted the landscape of molecular mechanisms through which cells undergo death pathways, the detailed signals involved are still not fully understood (Figure 1).
Figure 1.
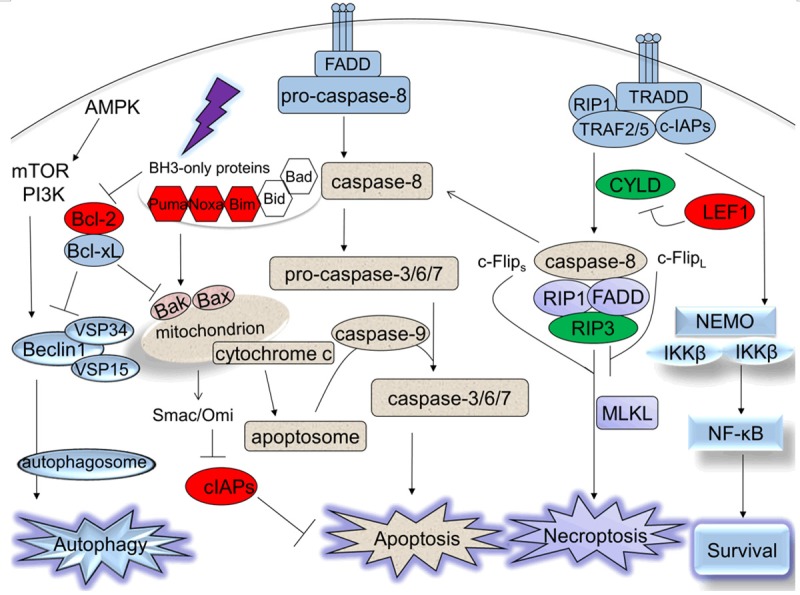
Schematic representation of different cell death pathways and their interactions. Apoptosis, necroptosis and autophagy are three major types of cell death, characterized by different factors involved in. The impaired factors which are up-regulated in CLL are colored in red and down-regulated in CLL are colored in green.
Pathways in apoptosis
Apoptosis is considered to be the principal death modality. It can be triggered by two main pathways (the extrinsic and intrinsic pathways) and eventually converge at the phase executed by caspases, leading to the cell destruction [5].
The extrinsic apoptotic pathway (also known as the death receptor pathway) is triggered by extrinsic ligands through activation of death receptors, such as CD95 (Fas), tumor necrosis factor receptor 1 (TNFR1) and tumor necrosis factor (TNF)-related apoptosis-inducing ligand receptor (TRAILR) [6]. The recruitment of several proteins, including Fas-associated death domain (FADD), TNFR1-associated death domain (TRADD) and pro-caspase-8, leads to the formation of the death inducing signaling complex (DISC) and activation of caspase-8, thus activating downstream effector caspases-3, -6 and/or -7 [6].
The intrinsic apoptotic pathway (also known as the mitochondrial pathway) has been extensively used to indicate instances of apoptotic cell death that are triggered by intracellular death signals such as DNA damage, nutrient deprivation and oxidative stress. The intrinsic pathway is regulated by dynamic interplay between members of B cell lymphoma (Bcl)-2 family [7]. The anti-apoptotic Bcl-2 family members (Bcl-2, Bcl-xL, Bcl-w, Mcl-1 and A1) prevent the pro-apoptotic Bcl-2 family members Bax and Bak from inducing the formation of mitochondrial outer membrane permeabilization (MOMP). The Bcl-2-homology 3 (BH3)-only proteins (Bid, Bad, Bim, Bik, Bnip3, Bmf, Hrk, Noxa and Puma) relieve the inhibition of Bax and Bak by antagonizing the anti-apoptotic Bcl-2 family members or activating Bax and Bak directly. The occurrence of MOMP results in the release of cytochrome c, which subsequently binds with apoptotic peptidase activating factor 1 (Apaf-1) and then recruits pro-caspase-9, thus assembling the complex cytochrome c/Apaf-1/caspase-9 apoptosome [6]. The activated caspase-9 triggers the downstream activation of effector caspases-3, -6, and -7, which play important roles in apoptotic cell death. Additionally, other pro-apoptotic factors released from the mitochondria such as second mitochondria-derived activator of caspases (Smac) and the mature serine protease Omi/HtrA2 neutralize the cellular inhibitor of apoptosis proteins (cIAPs), which inactivate the caspases directly or indirectly [8].
Pathways in necroptosis
Necroptosis, a type of regulated necrotic cell death mechanism, maintains some apoptotic features like the formation of functional complexes and is characterized by necrotic morphology. Necroptosis prevails in the apoptotic-deficient conditions. Pan-caspase inhibitor such as benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone (zVAD) may suppress apoptosis and switch apoptotic response to necroptosis [9]. The execution of this form of cell death is associated with the mitochondrial reactive oxygen species (ROS) production, lysosomal membrane permeabilization (LMP) and Poly (ADP-ribose) polymerase (PARP) hyperactivation [10].
Necroptosis is described as a receptor-interacting protein 1 (RIP1) kinase/receptor-interacting protein 3 (RIP3) kinase complex-dependent programmed cell death in response to various extracellular and intracellular stimuli, such as TNF family, Fas ligand (FasL), toll-like receptors (TLRs), lipopolysaccharides (LPS) as well as genotoxic stress [11].
Upon the activation of tumor necrosis factor receptor (TNFR), the downstream signaling is triggered by the formation of complex I, which recruits proteins containing death domain, such as TRADD, FADD, TNF-α receptor associated factor (TRAF) 2/5, cIAP1/2 and RIP1. In complex I, RIP1 is polyubiquitinated by E3 ligase TRAF2/5 and cIAPs, and then is able to activate NF-kB essential modulator (NEMO) and IkB kinase (IKK) complex, which promotes the activation of NF-kB pathway, thereby inducing cell survival [12-14]. When RIP1 and TRADD are released from complex I following cylindromatosis (CYLD) deubiquitination, another complex called complex II (also known as DISC) is assembled by recruiting RIP1, TRADD, FADD, RIP3, and caspase-8. RIP3 is activated after phosphorylated by the serine/threonine kinase RIP1 [15].
The formation of complex II is thought to trigger cell death through apoptosis or necroptosis, determined by the functional status of the key mediator caspase-8 [15]. Studies showed that the caspase-8 homodimers or caspase-8/cellular FLICE (FADD-like IL-1β-converting enzyme)-inhibitory protein (c-FLIP) long isoform (c-FlipL) heterodimers negatively regulate necroptosis by cleaving and inactivating both RIP1 and RIP3, and the homodimers can drive the process to the pro-apoptotic caspase activation mode, while the presence of caspase-8/c-FLIP heterodimers short isoform (c-Flips) or the absence of functional caspase-8 may promote RIP1/RIP3-dependent necroptosis [16]. Also the mixed lineage kinase domain like protein (MLKL), which is a pseudokinase, acts as an important substrate of RIP3 to mediate downstream necroptosis signaling pathways [16].
Pathways in autophagy
Autophagy has been extensively reported in the past few decades. Autophagy is an adaptive cellular metabolism, including macroautophagy, microautophagy and chaperone-mediated autophagy, in an attempt to mitigate various cell stress stimuli such as starvation, hypoxia, endoplasmic reticulum (ER) stress, and unfolded protein. The role autophagy plays in the cell death process remains controversial, most likely depending on different contexts. Autophagy may be regarded as a dynamic pro-survival homeostatic mechanism by which excessive or unnecessary constituents and damaged organelles are fused with lysosomes for degradation and recycling; however, it may be regarded as a mechanism that eventually promotes autophagic cell death if the massive stress occurs [17].
In response to starvation or energy depletion, the mammalian target of rapamycin (mTOR) complex 1 (mTORC1) is inhibited, which permits activation of UNC-51-like kinase 1 (ULK1)-autophagy-related genes (Atg) 13-Atg101-FIP200 complex, initiating the formation of autophagosomes. The stimulation of class I phosphatidylinositol 3-kinase (PI3K)-AKT pathway enables mTORC1 to negatively regulate the ULK-Atg13-Atg101-FIP200 complex [17]. Another complex, the class III PI3K (Vps34) complex composed of Vps34, Vps15, Beclin1 and Barkor is required to generate phosphatidylinositol 3-phosphate during the autophagosomal membrane nucleation [17].
The autophagosomal elongation process requires two ubiquitin-like conjugation systems. First, the ubiquitin-like proteins Atg12 is conjugated to an internal lysine of Atg5 via Atg7 and Atg10 enzymes. Subsequently the complex Atg5-Atg12-Atg16L1 is formed, which is essential for the elongation and the closure of the autophagosomal membranes. The ubiquitin-like protein microtubule-associated protein 1 light chain 3 (LC3) is implicated in a second conjugation system. Free cytosolic LC3 (referred to as LC3-I) is lipidated by phosphatidyl-ethanolamine (PE), leading to the conversion to LC3-II (PE-conjugated form). LC3-II is a key regulator which binds to the autophagic membranes. Finally, the lysosomal membrane protein 2 (Lamp2) and the GTPase Rab7 are required for the fusion of autophagosomes with the lysosomes [18,19]. The contents are degraded by lysosomal acid hydrolases after autophagosomes fusion with lysosomes [19].
Bcl-2 anti-apoptotic family members Bcl-2 and Bcl-xL may inhibit autophagy by binding to the essential component Beclin-1, preventing the formation of the class III PI3K (Vps34) complex. Conversely, the pro-apoptotic BH3-only proteins could induce autophagy by interfering with the interaction between Bcl-2/Bcl-xL and Beclin-1 [20,21]. Furthermore, it has been demonstrated that only Bcl-2/Bcl-xL and Beclin-1 complex located in the endoplasmic reticulum (ER) inhibit autophagy, suggesting subcellular localization matters in the regulation of autophagy. Similarly, nuclear p53 could induce autophagy while cytoplasmic p53 may repress autophagy [22,23].
Defective cell death pathways in CLL
Impaired cell death processes rather than excessive cellular proliferation in CLL have been demonstrated as the key event that contributes to the accumulation of malignant lymphocytes and the resistance to chemotherapy as well. Moreover, microenvironment supports CLL cells via interaction and chemokines, constantly stimulating CLL cells and promoting the resistance to cell death [24,25] (Figure 1).
Dysregulation of apoptosis may account for the development of malignant B cells in CLL. Several factors may be implicated in the defective apoptotic cell death, including the impaired apoptotic regulators induced by survival signals from the microenvironment and the genetic defects in CLL cells such as p53 mutation or deletion [26]. B-cell receptor (BCR)-dependent antigen stimulation and/or BCR-independent interactions with the microenvironment favor the activation of CLL B cells [27]. The activated survival pathways such as NF-kB or PI3K/AKT pathway in CLL cells lead to the upregulation of key anti-apoptotic Bcl-2 family members as well as cIAPs, which contributes to the chemotherapy resistance, disease progression and poor clinical prognosis [25,28]. We detected the elevated expression of Noxa, Bim and Bcl-2 occurs in CLL patients compared with normal controls, while the low expression of Puma is associated with variant clinical characteristics indicating poor prognosis [29]. Another Bcl-2 family protein, Mcl-1, has been shown to be strongly related with VH gene mutation status, CD38 expression, and ZAP-70 expression. Moreover, the higher Mcl-1/Bax ratios and/or Bcl-2/Bax ratios identify CLL patients with a significantly shorter overall survival (OS) and resistance to fludarabine [30-34].
It has been reported that the expression of anti-apoptotic proteins cIAP1 and cIAP2 in CLL cells is significantly higher than in lymphocytes from healthy donors, whereas Smac, which is the antagonist of cIAPs, is down-regulated in CLL. In comparison to stable CLL, higher expression of cIAPs (cIAP1, cIAP2, XIAP, and survivin) and lower levels of Smac are observed in patients with progressive CLL. Moreover, high concurrent expression of cIAP1 and survivin appears to be associated with shorter OS [35]. In response to cellular stress, p53 expression is upregulated and it transactivates pro-apoptotic target genes, which leads to the release of cytochrome c and apoptosis of the cells [36]. Therefore, the p53-dependent apoptotic response is not effective in CLL patients with p53 dysfunction.
Our research group has discovered that CLL cells have defects not only in apoptotic program but also in necroptotic signaling. We found that CLL cells exhibit impaired necroptosis following treatment with TNFα or TNFα plus zVAD. In addition, we detected two crucial necroptotic response regulators RIP3 and CYLD are frequently down-regulated in CLL cells, while no difference in RIP1 expression compared with healthy controls [9]. Lymphoid enhancer-binding factor 1 (LEF1), a downstream effector of the Wnt/β-catenin pathway, is reported to be highly expressed in CLL cells [37]. We identified the higher level of LEF1 in CLL and it may mediate as a transcription repressor of CYLD independent of β-catenin in CLL. LEF1 knockdown sensitized CLL cells to death receptor ligation induced necroptotic cell death, indicating the necroptosis resistance in CLL cells partly attributed to the defective regulators [9].
Activation of ER stress resulted from perturbed protein folding is associated with apoptotic cell death pathway, and is also accompanied the induction of autophagy in some situations [38]. Rosati et al. found that induced ER stress triggers CLL cell apoptosis and Mahoney et al. showed ER stress-inducing agents may induce autophagy in CLL cells [39]. Moreover, inhibition of autophagy increases the effects of drugs on CLL, suggesting autophagy participates in the pro-survival mechanism following ER stress and may lead to drug resistance [40,41]. However, several studies argue that the ER stress response facilitates CLL progression, which should be inhibited rather than be induced. Kriss and colleagues revealed aberrantly activated ER stress response including the IRE-1/XBP-1 pathway in Eμ-TCL1 CLL mouse model and in human CLL as well [42]. In addition, the increased cell apoptosis could be observed by blocking IRE-1/XBP-1 pathway [42,43].
Therapeutic targeting of cell death pathways in CLL
The major cell death processes are interlinked and contain many common regulatory mechanisms [44]. Exploration of our understanding of the major pathways which regulate cell viability could provide a plethora of opportunities for effective anti-cancer therapeutics. With so many drugs developed now, it is critical to determine which one or combination is the most appropriate for a patient based on the death pathways perturbed by the agents, especially when the genetic abnormalities and other factors which may elicit resistance to some kind of drugs exist. Accordingly, several agents targeting pro-survival molecules have entered clinical trials and the prospects for these drugs in clinical practice are discussed (Table 1).
Table 1.
Summary of different cell death-inducing agents in the era of targeting treatment in CLL
| Type of cell death | Targets | Agents | Reference |
|---|---|---|---|
| Apoptosis/necroptosis/autophagy | Pro-survival Bcl-2 mRNA or Bcl-2 family proteins | Oblimersen sodium, Obatoclax mesylate, ABTs | [45-47], [48-53], [54-58] |
| Apoptosis/autophagy | Histone deacetylase | MGCD0103, Valproic acid | [64-67], [103,104] |
| Apoptosis/autophagy | Cyclin-dependent kinase | Flavopiridol, Dinaciclib | [40,68-72], [73,74] |
| Apoptosis/necroptosis/autophagy | Proteasome | Bortezomib, Carfizomib | [75-84], [85,86] |
| Apoptosis/necroptosis | cIAP proteins | Smac-mimetics, XIAP inhibitor | [25], [87] |
| Apoptosis | B cell receptor signaling pathway kinases | Dasatinib, Bafetinib, Fostamatinib, Entospletinib, Idelalisib, Ibrutinib | [90-93], [94], [95] , [96-98], [99-102] |
Inhibitors of pro-survival Bcl-2 family proteins
Oblimersen sodium (G3139) is a CpG-containing oligonucleotide antisense drug specifically binding to the Bcl-2 mRNA sequence, resulting in mRNA degradation [45]. An early non-randomized phase I/II study conducted by O’Brien and colleagues showed that oblimersen sodium had dose-limiting reactions in patients with advanced CLL [46]. Hence, a phase III randomized clinical trial of fludarabine and cyclophosphamide with or without oblimersen was investigated in patients with relapsed or refractory CLL based on promising early phase trials. The complete response or nodular partial response (CR/nPR) rate was 7% with chemotherapy (fludarabine plus cyclophosphamide, FC) only group and 17% with oblimersen plus chemotherapy (FCO) group. The maximum benefit from oblimersen-containing regimen was observed in fludarabine-sensitive (relapsed) patients with the rate of CR/nPR being 25% in comparison to 6% in the chemotherapy-only group, and had an attendant 50% reduction in the risk of death [45]. Although the 5-year long term survival rate showed no significant difference between FCO group and FC group, patients who achieved CR/nPR from the oblimersen-containing group had a significant 5-year survival benefit. Clinical trials have identified the modest efficacy of oblimersen combined with FC in relapsed/refractory CLL, especially in a subset of fludarabine-sensitive patients [47].
Obatoclax (GX15-070) is a pan-BCL-2 family antagonist, mainly interacting with Bcl-2, Bcl-xL, Bax, and Mcl-1 with low binding affinity. It has been reported to enhance cell death via mitochondrial apoptosis, and Beclin-1 dependent autophagy in primary CLL cells as well [48,49]. Recently, Basit et al. showed that obatoclax triggered RIP1 and RIP3 mediated necroptosis by promoting the assembly of the necrosome on autophagosomal membranes [50]; however, it has not been demonstrated whether this scenario occurs in CLL cells. The phase I study of single-agent obatoclax indicated biologic and limited clinical activity in heavily pretreated patients with advanced CLL patients and among whom only 4% achieved partial response (PR) [51]. In a phase I/II study revealed that combined obatoclax plus bortezomib was feasible in patients with mantle cell lymphoma (MCL) [52], suggesting obatoclax might enhance the cytotoxicity of treatment in combination with other drugs [53].
ABT-737 is the first effective BH3 mimetic inhibitor selectively targets the pro-survival proteins Bcl-2, Bcl-xL and Bcl-w (but not Mcl-1 or A1), thereby releasing the pro-apoptosis proteins [54]. It has been developed into orally-available agent ABT-263, and in a phase II study, previously untreated CLL patients treated with ABT-263 combined with rituximab achieved higher CR/PR rates than treated with rituximab alone. The significantly better treatment outcome with ABT-263 was also seen in CLL patients with del (17p) or high levels of Bcl-2. Maintenance with ABT-263 until disease progression or unacceptable toxicity appeared to yield the highest CR/PR rates and prolonged progression-free survival (PFS) [55].
ABT-199 is a second generation orally bioavailable Bcl-2-selective inhibitor, specifically leading to Bax/Bak-mediated apoptosis and may spare platelets in order to avoid clinically significant thrombocytopenia caused by predecessor ABT-263 [56]. A related study reported the combination of ABT-199 and rituximab was feasible and achieved an ORR of 86% with 31% CR in patients with CLL/SLL [57]. Choudhary et al. found that high intrinsic Mcl-1 and Bcl-xL expression and activity accounted for ABT-199 resistance, and ABT-199 was more effective when combined with PI3K/AKT/mTOR inhibitors to sequester AKT activation and release pro-apoptotic protein Bim, and thus led to cell death in this setting [58]. Combinations with other drugs such as obinutuzumab, and bendamustine are currently under clinical trials.
Histone deacetylase inhibitors
Histone deacetylases (HDACs) play a crucial role in chromatin conformation and gene expression [59]. Deregulation of HDACs has been observed in several cancers including CLL. Some HDACs (HDAC 6, 7, 11, sirtuin (SIRT) 3, SIRT 6 and 7) are overexpressed while some HDACs (HDAC 2 and SIRT 4) are under-expressed in CLL patients [60]. In addition, elevated HDACs activity is observed in CLL patients with shorter TFS and OS [61]. It has been showed that upregulated HDACs mediated the epigenetic silencing of tumor suppressor genes encoding microRNAs such as miR-15a, miR-16 and miR-29b in CLL. HDAC inhibitors (HDACi) may partially restore the expression of these microRNAs which elicit the loss of Mcl-1 expression and mitochondrial dysfunction [62]. Additionally, HDACi could induce apoptosis by increasing the expression of Bim and Noxa which bound to anti-apoptotic Bcl-2 members [63]. These results suggest that HDAC inhibitors are emerging as potent anticancer agents.
MGCD0103 is an orally available HDAC inhibitor which selectively targets class I (HDAC 1, 2, and 3) and class IV (HDAC 11) enzymes [64]. It has been identified as not only an apoptosis inducer but also an autophagy suppressor. El-Khoury et al. showed that MGCD0103 mainly activated the intrinsic caspase-dependent apoptotic cell death pathway through declining Mcl-1 expression, inducing the translocation of Bax to the mitochondria, and releasing cytochrome c [65]. Besides, MGCD0103 inhibits autophagy in primary CLL cells through transcriptional downregulation of autophagy genes [66]. A phase II study of MGCD0103 was conducted in patients with relapsed/refractory CLL. The efficacy of single agent MGCD103 was limited and the majority of patients could only tolerate up to 2 cycles of MGCD0103 [67]. Further investigation will be necessary to evaluate whether the combination of HDACi with autophagy inhibitors or other drugs potentiate CLL cell death.
Cyclin-dependent kinase inhibitors
Flavopiridol (HMR-1275, L-868275) is a semisynthetic flavone derived from rohitukine which is an alkaloid extracts from India plants. It is a broad spectrum cyclin-dependent kinase (CDK) inhibitor that could effectively induce apoptosis in CLL cells by inactivating RNA polymerase II, thus inhibiting Mcl-1 expression but not Bcl-2 in transcriptional level and activating caspase-3, regardless of genic alterations of del (17p) or p53 mutation [68]. Flavopiridol also induces ER stress in CLL cells, which contributes to cell death through IRE1-mediated activation of ASK1 and caspase-4. Autophagy induced by flavopiridol has been demonstrated to be a protective process in CLL cells [40]. Clinical responses from a phase II study revealed that 57% patients with del (17p) and 50% patients with del (11q) responded to flavopiridol treatment [69]. However, tumor lysis syndrome (TLS) was observed in over 40% of patients with CLL receiving flavopiridol [70]. Stephens and colleagues combined cyclophosphamide, alvocidib (flavopiridol) and rituximab (CAR) to alleviate TLS, and the regimen was tolerable and beneficial without TLS in high-risk CLL patients [71,72].
Developed from broad inhibitor flavopiridol, dinaciclib (SCH727965) is a selective inhibitor of CDK 1, 2, 5 and 9. Five patients with relapsed/refractory CLL were enrolled in a phase I study, and were treated with dinaciclib in combination with rituximab. Four patients achieved stable disease (SD), and one achieved CR [73]. In a phase II study, 54% patients with del (17p) responded to dinaciclib treatment and presented with a median PFS of 481 days [74]. These clinical data suggests CDK inhibitor as a promising agent particularly in high-risk CLL patients.
Proteasome inhibitors
Bortezomib (PS-341) is a proteasome inhibitor approved for the treatment of multiple myeloma and MCL [75]. The antineoplastic effect of bortezomib includes inhibition of cell growth signaling pathways, induction of apoptosis, and inhibition of cellular adhesion molecule expression [76]. Proteasome inhibitors can lead to accumulation of a variety of pro-apoptotic proteins in malignant cells rather than normal cells [77]. Bortezomib, represented as the first proteasome inhibitor, has shown positive clinical benefits toward hematological malignancies through upregulating pro-apoptotic protein Noxa expression, which may interact with anti-apoptotic proteins Bcl-xL and Bcl-2, resulting in the activation of the classic mitochondria-dependent intrinsic apoptotic pathway [78,79]. Bortezomib has also been identified as the suppresser of NF-kB signaling pathway through preventing the degradation of IκB [80]. Nevertheless, an early randomized phase II study presented evidence that patients with fludarabine-refractory CLL treated with this single agent failed to achieve objective responses [76,80]. The hampered therapeutic efficacy of bortezomib has been partly attributed to flavonoids presented in plasma and some dietary natural compounds, as well as the accumulation of Mcl-1, all of which may compromise the ability of bortezomib-induced apoptosis [79,81,82]. The clinical benefit of bortezomib is not satisfied due to the emergence of resistance associated with several underlying molecular mechanisms as well [83]. The disappointing results indicate that bortezomib in combination with other agents is required in the treatment of CLL. Clinical data from a combination of rituximab and bortezomib in patients with refractory or relapsed indolent lymphoma (CLL included) revealed an ORR of 70% and a PFS rate of 41% at 2 years. The regimen was well-tolerated with low toxicity [84].
Carfilzomib (PR-171) is a second-generation proteasome inhibitor that irreversibly inhibits the chymotrypsin-like subunit activity of the proteasome. Unlike bortezomib, carfilzomib is equally effective in the presence or absence of human serum [85]. Carfilzomib functions irrespective of del (17p) or p53 mutation in CLL cells, generating caspase dependent cytotoxicity. Moreover, carfilzomib promotes an atypical NF-kB response not only by decreasing IκBα expression but also increasing the phosphorylation of IκBα, and importantly, without inducing the transcription of some canonical NF-kB related cytokines and survival factors [85]. Preliminary outcomes from a phase I dose-escalation trial of carfilzomib revealed acceptable tolerability in relapsed and refractory CLL/SLL patients; however, no patient was sufficient to be evaluated as CR/PR, although minimal efficacy was observed [86].
Death receptor pathway activators
cIAPs antagonist
Over the decades, a variety of small molecules have been developed to neutralize overexpressed cIAP proteins, and thereby promote caspase activation and apoptosis. Scavullo et al. found Smac-mimetic compound Smac66 had the ability to antagonize members of the cIAP family and upregulated the expression of activated caspase-3 and PARP, indicating the potential therapeutic use as monotherapy rather than in combination against CLL cells [87]. XIAP inhibitors synergized with TRAIL could sensitize CLL cells with caspase-3 induced and caspase-dependent apoptosis, even in CLL patients with unfavorable prognosis [25].
The recent evidence showed that, similar to the previous studies, the degradation of cIAP1/2 and the production of TNFα were observed in CLL cells treated with Smac-mimetic. However, after the treatment, CD40-stimulated CLL cells were unable to form the ripoptosome complex and be killed by apoptosis or necroptosis [88]. Thus, further studies are required to elucidate the availability of cIAP antagonist induced apoptosis-based therapy in CLL.
B-cell receptor signaling inhibitors
The increased activation of downstream pathway of the BCR plays a pivotal role in the pathogenesis of CLL and is accountable for providing growth signals to prolonged survival of CLL cells. BCR-associated components including Lyn tyrosine kinase, Spleen tyrosine kinase (Syk), Bruton’s tyrosine kinase (Btk) and PI3K have been regarded as the focus of novel targets for CLL treatment with obvious clinical benefits [89].
Lyn tyrosine kinase inhibitor
Dasatinib (BMS-354825), a dual c-abl and pan-Src family kinases inhibitor, especially as a sustained inhibitor of Lyn tyrosine kinase in CLL cells, has been widely studied with the ability of triggering ER stress and reducing the expression of Mcl-1 and Bcl-xL [90]. Aside from the death-inducing process mediated by dasatinib, the protective autophagy is also observed in the wide type p53 CLL lymphocytes [91]. In a phase II clinical trial in patients with relapsed/refractory CLL, dasatinib was well tolerated and a great reduction in lymph nodes or extranodal masses was observed in the majority of patients [92]. Kater et al. conducted a phase II trial of dasatinib in combination with fludarabine in refractory patients with CLL. The modest clinical efficacy was reported with an ORR of 18% and a median PFS of 6.3 months [93].
Bafetinib (INNO-406) is a potent second generation Lyn tyrosine kinase inhibitor. A phase II study of bafetinib as treatment in patients with relapsed or refractory CLL has been completed, yet no related results provided.
Spleen tyrosine kinase inhibitor
Fostamatinib (R788) is the first oral Syk inhibitor which shows significant clinical activity in patients with mature B-cell malignancies. In a phase I/II clinical trial investigating the effect of fostamatinib in recurrent B-cell NHL and CLL, the ORR in patients with SLL/CLL showed the highest response rate of 55% and the median PFS was 6.4 months [94].
Entospletinib (GS-9973) is a selective inhibitor of Syk. In a phase II study performed by Friedberg et al., the ORR was 61% with median PFS of 13.8 months in patients with relapsed or refractory CLL, albeit no subject achieved a CR [95].
Phosphatidylinositol-3 kinase inhibitor
Idela-lisib (GS-1101, CAL-101) is a potent inhibitor of the delta isoform of PI3K. Idelalisib was given to patients with relapsed/refractory CLL in a phase I study. Among all patients in this study, the ORR was 72% with the median PFS of 15.8 months, and the nodal responses rate was 81%. Moreover, patients with del (17p) and/or TP53 mutation had an inferior PFS of only 5 months, while patients without this abnormality had a PFS of 41 months [96]. In a phase 3 study, the efficacy of idelalisib was compared between rituximab combined with idelalisib and with placebo in relapsed CLL patients. The ORR was 13%, and the median duration of PFS was 5.5 months in the placebo group, whereas the ORR was 81% and the median PFS was not reached in the idelalisib group [97]. Based on the current results from the trials of idelalisib in relapsed/refractory CLL, idelalisib was found to have an acceptable safety profile, and was associated with significant improvement in the clinical responses. Otherwise, the attempts to combine entospletinib with idelalisib enhanced the inhibition of BCR-mediated signaling, thereby synergistically decreasing the viability of CLL cells and disrupting chemokine expression in CLL cells [98].
Bruton’s tyrosine kinase inhibitor
Ibrutinib (PCI-32765) is a first-in-class oral bioavailable, covalent inhibitor of Bruton’s tyrosine kinase (Btk). In early phase studies, Ibrutinib was associated with a high frequency of durable remissions among patients with previously treated CLL [99]. Significantly, improved PFS, OS, and ORR were showed in a randomised comparison of ibrutinib versus the anti-CD20 antibody ofatumumab in previously treated CLL [100]. Ibrutinib acts through a p53-independent mechanism whereby high-risk CLL patients responded equally as well as low-risk patients to ibrutinib [101]. The phase II clinical trial of ibrutinib has demonstrated its efficacy in TP53-aberrant CLL patients. At the time of assessment, 97% of previously untreated patients and 80% of patients with relapsed/refractory patients achieved an objective response [101]. A median 3-year follow-up of single-agent ibrutinib administrated in 132 CLL/SLL patients demonstrated the durable responses and the diminished toxicity over time [102]. The encouraging results warrant the further studies in ibrutinib and there are more than thirty ongoing clinical trials with regard to ibrutinib monotherapy or in cooperation with other drugs in CLL.
Conclusion
The landscape of pharmaceutical approaches in CLL is shifting gradually from traditional chemotherapy to targeted agents, focusing on the precise targeting effect rather than the increase of cytotoxicity. Over the decades, mounting evidence has delineated the abnormal characteristics as stumbling blocks to cell death in CLL cells. Understanding the abnormal death processes by which cells are trapped in the prolonged survival is the first key to patient survival. The second key is the in-depth studies of novel drugs both in vivo and in vitro, given the vague mechanisms whereby the cells execute death pathways. Further, apart from the sustained investigations for the optimal use of novel drugs in clinical trials, the design of the appropriate therapeutic combination of agents may be of pivotal importance as the third key for improved effects of therapies generally and even individually.
Acknowledgements
This study was supported by National Natural Science Foundation of China (30971296, 81170485, 81170488, 81370657, 81470328), Key Projects of Health Department of Jiangsu Province (K201108), Jiangsu Province’s Medical Elite Program (RC2011169), National Public Health Grand Research Foundation (201202017), Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institute (JX10231801), Program for Development of Innovative Research Teams in the First Affiliated Hospital of Nanjing Medical University, Project of National Key Clinical Specialty, Jiangsu Province Graduate Students Research and Innovation Plan (JX22013282), National Science & Technology Pillar Program (2014BAI09B12), and Project funded by Jiangsu Provincial Special Program of Medical Science (BL2014086).
Disclosure of conflict of interest
None.
References
- 1.Decker T, Sandherr M, Goetze K, Oelsner M, Ringshausen I, Peschel C. A pilot trial of the mTOR (mammalian target of rapamycin) inhibitor RAD001 in patients with advanced B-CLL. Ann Hematol. 2009;88:221–227. doi: 10.1007/s00277-008-0582-9. [DOI] [PubMed] [Google Scholar]
- 2.Rai KR, Chiorazzi N. Determining the clinical course and outcome in chronic lymphocytic leukemia. N Engl J Med. 2003;348:1797–1799. doi: 10.1056/NEJMe030032. [DOI] [PubMed] [Google Scholar]
- 3.Zenz T, Mertens D, Kuppers R, Dohner H, Stilgenbauer S. From pathogenesis to treatment of chronic lymphocytic leukaemia. Nat Rev Cancer. 2010;10:37–50. doi: 10.1038/nrc2764. [DOI] [PubMed] [Google Scholar]
- 4.Rozovski U, Hazan-Halevy I, Keating MJ, Estrov Z. Personalized medicine in CLL: current status and future perspectives. Cancer Lett. 2014;352:4–14. doi: 10.1016/j.canlet.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Radogna F, Dicato M, Diederich M. Cancertype-specific crosstalk between autophagy, necroptosis and apoptosis as a pharmacological target. Biochem Pharmacol. 2015;94:1–11. doi: 10.1016/j.bcp.2014.12.018. [DOI] [PubMed] [Google Scholar]
- 6.Marino G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15:81–94. doi: 10.1038/nrm3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eimon PM, Kratz E, Varfolomeev E, Hymowitz SG, Stern H, Zha J, Ashkenazi A. Delineation of the cell-extrinsic apoptosis pathway in the zebrafish. Cell Death Differ. 2006;13:1619–1630. doi: 10.1038/sj.cdd.4402015. [DOI] [PubMed] [Google Scholar]
- 8.Long JS, Ryan KM. New frontiers in promoting tumour cell death: targeting apoptosis, necroptosis and autophagy. Oncogene. 2012;31:5045–5060. doi: 10.1038/onc.2012.7. [DOI] [PubMed] [Google Scholar]
- 9.Liu P, Xu B, Shen W, Zhu H, Wu W, Fu Y, Chen H, Dong H, Zhu Y, Miao K, Xu W, Li J. Dysregulation of TNFalpha-induced necroptotic signaling in chronic lymphocytic leukemia: suppression of CYLD gene by LEF1. Leukemia. 2012;26:1293–1300. doi: 10.1038/leu.2011.357. [DOI] [PubMed] [Google Scholar]
- 10.Repnik U, Hafner CM, Turk B. Lysosomal membrane permeabilization in cell death: concepts and challenges. Mitochondrion. 2014;19:49–57. doi: 10.1016/j.mito.2014.06.006. [DOI] [PubMed] [Google Scholar]
- 11.Giampietri C, Starace D, Petrungaro S, Filippini A, Ziparo E. Necroptosis: molecular signalling and translational implications. Int J Cell Biol. 2014;2014:490275. doi: 10.1155/2014/490275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Devin A, Cook A, Lin Y, Rodriguez Y, Kelliher M, Liu Z. The distinct roles of TRAF2 and RIP in IKK activation by TNF-R1: TRAF2 recruits IKK to TNF-R1 while RIP mediates IKK activation. Immunity. 2000;12:419–429. doi: 10.1016/s1074-7613(00)80194-6. [DOI] [PubMed] [Google Scholar]
- 13.Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J, Gillard JW, Jaquith JB, Morris SJ, Barker PA. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell. 2008;30:689–700. doi: 10.1016/j.molcel.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 14.Varfolomeev E, Goncharov T, Fedorova AV, Dynek JN, Zobel K, Deshayes K, Fairbrother WJ, Vucic D. c-IAP1 and c-IAP2 are critical mediators of tumor necrosis factor alpha (TNFalpha)-induced NF-kappaB activation. J Biol Chem. 2008;283:24295–24299. doi: 10.1074/jbc.C800128200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Christofferson DE, Yuan J. Necroptosis as an alternative form of programmed cell death. Curr Opin Cell Biol. 2010;22:263–268. doi: 10.1016/j.ceb.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M, Cain K, MacFarlane M, Hacker G, Leverkus M. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell. 2011;43:449–463. doi: 10.1016/j.molcel.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:1845–1846. doi: 10.1056/NEJMc1303158. [DOI] [PubMed] [Google Scholar]
- 18.Trocoli A, Djavaheri-Mergny M. The complex interplay between autophagy and NF-kappaB signaling pathways in cancer cells. Am J Cancer Res. 2011;1:629–649. [PMC free article] [PubMed] [Google Scholar]
- 19.Jin M, Klionsky DJ. Regulation of autophagy: modulation of the size and number of autophagosomes. FEBS Lett. 2014;588:2457–2463. doi: 10.1016/j.febslet.2014.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 21.Levine B, Sinha S, Kroemer G. Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy. 2008;4:600–606. doi: 10.4161/auto.6260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maiuri MC, Le TG, Criollo A, Rain JC, Gautier F, Juin P, Tasdemir E, Pierron G, Troulinaki K, Tavernarakis N, Hickman JA, Geneste O, Kroemer G. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007;26:2527–2539. doi: 10.1038/sj.emboj.7601689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maiuri MC, Galluzzi L, Morselli E, Kepp O, Malik SA, Kroemer G. Autophagy regulation by p53. Curr Opin Cell Biol. 2010;22:181–185. doi: 10.1016/j.ceb.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 24.Danilov AV, Danilova OV, Klein AK, Huber BT. Molecular pathogenesis of chronic lymphocytic leukemia. Curr Mol Med. 2006;6:665–675. doi: 10.2174/156652406778195008. [DOI] [PubMed] [Google Scholar]
- 25.Loeder S, Zenz T, Schnaiter A, Mertens D, Winkler D, Dohner H, Debatin KM, Stilgenbauer S, Fulda S. A novel paradigm to trigger apoptosis in chronic lymphocytic leukemia. Cancer Res. 2009;69:8977–8986. doi: 10.1158/0008-5472.CAN-09-2604. [DOI] [PubMed] [Google Scholar]
- 26.Dighiero G, Hamblin TJ. Chronic lymphocytic leukaemia. Lancet. 2008;371:1017–1029. doi: 10.1016/S0140-6736(08)60456-0. [DOI] [PubMed] [Google Scholar]
- 27.Ghia P, Chiorazzi N, Stamatopoulos K. Microenvironmental influences in chronic lymphocytic leukaemia: the role of antigen stimulation. J Intern Med. 2008;264:549–562. doi: 10.1111/j.1365-2796.2008.02030.x. [DOI] [PubMed] [Google Scholar]
- 28.Buggins AG, Pepper CJ. The role of Bcl-2 family proteins in chronic lymphocytic leukaemia. Leuk Res. 2010;34:837–842. doi: 10.1016/j.leukres.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 29.Zhang LN, Li JY, Xu W. A review of the role of Puma, Noxa and Bim in the tumorigenesis, therapy and drug resistance of chronic lymphocytic leukemia. Cancer Gene Ther. 2013;20:1–7. doi: 10.1038/cgt.2012.84. [DOI] [PubMed] [Google Scholar]
- 30.Pepper C, Hoy T, Bentley DP. Bcl-2/Bax ratios in chronic lymphocytic leukaemia and their correlation with in vitro apoptosis and clinical resistance. Br J Cancer. 1997;76:935–938. doi: 10.1038/bjc.1997.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Molica S, Dattilo A, Giulino C, Levato D, Levato L. Increased bcl-2/bax ratio in B-cell chronic lymphocytic leukemia is associated with a progressive pattern of disease. Haematologica. 1998;83:1122–1124. [PubMed] [Google Scholar]
- 32.Pepper C, Thomas A, Hoy T, Bentley P. Chlorambucil resistance in B-cell chronic lymphocytic leukaemia is mediated through failed Bax induction and selection of high Bcl-2-expressing subclones. Br J Haematol. 1999;104:581–588. doi: 10.1046/j.1365-2141.1999.01210.x. [DOI] [PubMed] [Google Scholar]
- 33.Starczynski J, Pepper C, Pratt G, Hooper L, Thomas A, Milligan D, Bentley P, Fegan C. Common polymorphism G(-248)A in the promoter region of the bax gene results in significantly shorter survival in patients with chronic lymphocytic Leukemia once treatment is initiated. J. Clin. Oncol. 2005;23:1514–1521. doi: 10.1200/JCO.2005.02.192. [DOI] [PubMed] [Google Scholar]
- 34.Pepper C, Lin TT, Pratt G, Hewamana S, Brennan P, Hiller L, Hills R, Ward R, Starczynski J, Austen B, Hooper L, Stankovic T, Fegan C. Mcl-1 expression has in vitro and in vivo significance in chronic lymphocytic leukemia and is associated with other poor prognostic markers. Blood. 2008;112:3807–3817. doi: 10.1182/blood-2008-05-157131. [DOI] [PubMed] [Google Scholar]
- 35.Grzybowska-Izydorczyk O, Cebula B, Robak T, Smolewski P. Expression and prognostic significance of the inhibitor of apoptosis protein (IAP) family and its antagonists in chronic lymphocytic leukaemia. Eur J Cancer. 2010;46:800–810. doi: 10.1016/j.ejca.2009.11.023. [DOI] [PubMed] [Google Scholar]
- 36.Erster S, Mihara M, Kim RH, Petrenko O, Moll UM. In vivo mitochondrial p53 translocation triggers a rapid first wave of cell death in response to DNA damage that can precede p53 target gene activation. Mol Cell Biol. 2004;24:6728–6741. doi: 10.1128/MCB.24.15.6728-6741.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gutierrez NC, Ocio EM, de Las Rivas J, Maiso P, Delgado M, Ferminan E, Arcos MJ, Sanchez ML, Hernandez JM, San MJF. Gene expression profiling of B lymphocytes and plasma cells from Waldenstrom’s macroglobulinemia: comparison with expression patterns of the same cell counterparts from chronic lymphocytic leukemia, multiple myeloma and normal individuals. Leukemia. 2007;21:541–549. doi: 10.1038/sj.leu.2404520. [DOI] [PubMed] [Google Scholar]
- 38.Verfaillie T, Salazar M, Velasco G, Agostinis P. Linking ER Stress to Autophagy: Potential Implications for Cancer Therapy. Int J Cell Biol. 2010;2010:930509. doi: 10.1155/2010/930509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rosati E, Sabatini R, Rampino G, Tabilio A, Di IM, Fettucciari K, Bartoli A, Coaccioli S, Screpanti I, Marconi P. Constitutively activated Notch signaling is involved in survival and apoptosis resistance of B-CLL cells. Blood. 2009;113:856–865. doi: 10.1182/blood-2008-02-139725. [DOI] [PubMed] [Google Scholar]
- 40.Mahoney E, Lucas DM, Gupta SV, Wagner AJ, Herman SE, Smith LL, Yeh YY, Andritsos L, Jones JA, Flynn JM, Blum KA, Zhang X, Lehman A, Kong H, Gurcan M, Grever MR, Johnson AJ, Byrd JC. ER stress and autophagy: new discoveries in the mechanism of action and drug resistance of the cyclin-dependent kinase inhibitor flavopiridol. Blood. 2012;120:1262–1273. doi: 10.1182/blood-2011-12-400184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mahoney E, Maddocks K, Flynn J, Jones J, Cole SL, Zhang X, Byrd JC, Johnson AJ. Identification of endoplasmic reticulum stress-inducing agents by antagonizing autophagy: a new potential strategy for identification of anti-cancer therapeutics in B-cell malignancies. Leuk Lymphoma. 2013;54:2685–2692. doi: 10.3109/10428194.2013.781168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kriss CL, Pinilla-Ibarz JA, Mailloux AW, Powers JJ, Tang CH, Kang CW, Zanesi N, Epling-Burnette PK, Sotomayor EM, Croce CM, Del VJR, Hu CC. Overexpression of TCL1 activates the endoplasmic reticulum stress response: a novel mechanism of leukemic progression in mice. Blood. 2012;120:1027–1038. doi: 10.1182/blood-2011-11-394346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tang CH, Ranatunga S, Kriss CL, Cubitt CL, Tao J, Pinilla-Ibarz JA, Del VJR, Hu CC. Inhibition of ER stress-associated IRE-1/XBP-1 pathway reduces leukemic cell survival. J Clin Invest. 2014;124:2585–2598. doi: 10.1172/JCI73448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fayaz SM, Suvanish KVS, Rajanikant GK. Necroptosis: who knew there were so many interesting ways to die. CNS Neurol Disord Drug Targets. 2014;13:42–51. doi: 10.2174/18715273113126660189. [DOI] [PubMed] [Google Scholar]
- 45.O’Brien S, Moore JO, Boyd TE, Larratt LM, Skotnicki A, Koziner B, Chanan-Khan AA, Seymour JF, Bociek RG, Pavletic S, Rai KR. Randomized phase III trial of fludarabine plus cyclophosphamide with or without oblimersen sodium (Bcl-2 antisense) in patients with relapsed or refractory chronic lymphocytic leukemia. J. Clin. Oncol. 2007;25:1114–1120. doi: 10.1200/JCO.2006.07.1191. [DOI] [PubMed] [Google Scholar]
- 46.O’Brien SM, Cunningham CC, Golenkov AK, Turkina AG, Novick SC, Rai KR. Phase I to II multicenter study of oblimersen sodium, a Bcl-2 antisense oligonucleotide, in patients with advanced chronic lymphocytic leukemia. J. Clin. Oncol. 2005;23:7697–7702. doi: 10.1200/JCO.2005.02.4364. [DOI] [PubMed] [Google Scholar]
- 47.O’Brien S, Moore JO, Boyd TE, Larratt LM, Skotnicki AB, Koziner B, Chanan-Khan AA, Seymour JF, Gribben J, Itri LM, Rai KR. 5-year survival in patients with relapsed or refractory chronic lymphocytic leukemia in a randomized, phase III trial of fludarabine plus cyclophosphamide with or without oblimersen. J. Clin. Oncol. 2009;27:5208–5212. doi: 10.1200/JCO.2009.22.5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Perez-Galan P, Roue G, Lopez-Guerra M, Nguyen M, Villamor N, Montserrat E, Shore GC, Campo E, Colomer D. BCL-2 phosphorylation modulates sensitivity to the BH3 mimetic GX15-070 (Obatoclax) and reduces its synergistic interaction with bortezomib in chronic lymphocytic leukemia cells. Leukemia. 2008;22:1712–1720. doi: 10.1038/leu.2008.175. [DOI] [PubMed] [Google Scholar]
- 49.Sharma A, Singh K, Mazumder S, Hill BT, Kalaycio M, Almasan A. BECN1 and BIM interactions with MCL-1 determine fludarabine resistance in leukemic B cells. Cell Death Dis. 2013;4:e628. doi: 10.1038/cddis.2013.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Basit F, Cristofanon S, Fulda S. Obatoclax (GX15-070) triggers necroptosis by promoting the assembly of the necrosome on autophagosomal membranes. Cell Death Differ. 2013;20:1161–1173. doi: 10.1038/cdd.2013.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.O’Brien SM, Claxton DF, Crump M, Faderl S, Kipps T, Keating MJ, Viallet J, Cheson BD. Phase I study of obatoclax mesylate (GX15-070), a small molecule pan-Bcl-2 family antagonist, in patients with advanced chronic lymphocytic leukemia. Blood. 2009;113:299–305. doi: 10.1182/blood-2008-02-137943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goy A, Hernandez-Ilzaliturri FJ, Kahl B, Ford P, Protomastro E, Berger M. A phase I/II study of the pan Bcl-2 inhibitor obatoclax mesylate plus bortezomib for relapsed or refractory mantle cell lymphoma. Leuk Lymphoma. 2014;55:2761–2768. doi: 10.3109/10428194.2014.907891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Joudeh J, Claxton D. Obatoclax mesylate: pharmacology and potential for therapy of hematological neoplasms. Expert Opin Investig Drugs. 2012;21:363–373. doi: 10.1517/13543784.2012.652302. [DOI] [PubMed] [Google Scholar]
- 54.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O’Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 55.Kipps TJ, Eradat H, Grosicki S, Catalano J, Cosolo W, Dyagil I, Yalamanchili S, Chai A, Saharanaman S, Punnoose E, Hurst D, Pylypenko H. A phase 2 study of the BH3 mimetic BCL2 inhibitor navitoclax (ABT-263) with or without rituximab, in previously untreated B-cell chronic lymphocytic leukemia. Leuk Lymphoma. 2015;3:1–30. doi: 10.3109/10428194.2015.1030638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, Huang DC, Hymowitz SG, Jin S, Khaw SL, Kovar PJ, Lam LT, Lee J, Maecker HL, Marsh KC, Mason KD, Mitten MJ, Nimmer PM, Oleksijew A, Park CH, Park CM, Phillips DC, Roberts AW, Sampath D, Seymour JF, Smith ML. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19:202–208. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 57.Roberts AW, Ma S, Brander DM, Kipps TJ, Barrientos JC, Davids MS, Anderson MA, Tam C, Mason-Bright T, Rudersdorf NK, Gressick L, Yang JN, Munasinghe W, Zhu M, Cerri E, Enschede SH, Humerickhouse RA, Seymour JF. Determination of Recommended Phase 2 Dose of ABT-199 (GDC-0199) Combined with Rituximab (R) in Patients with Relapsed/Refractory (R/R) Chronic Lymphocytic Leukemia (CLL) American Society of Hematology Annual Meeting. 2014 Abstract #325. [Google Scholar]
- 58.Choudhary GS, Al-Harbi S, Mazumder S, Hill BT, Smith MR, Bodo J, Hsi ED, Almasan A. MCL-1 and BCL-xL-dependent resistance to the BCL-2 inhibitor ABT-199 can be overcome by preventing PI3K/AKT/mTOR activation in lymphoid malignancies. Cell Death Dis. 2015;6:e1593. doi: 10.1038/cddis.2014.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gallinari P, Di MS, Jones P, Pallaoro M, Steinkuhler C. HDACs, histone deacetylation and gene transcription: from molecular biology to cancer therapeutics. Cell Res. 2007;17:195–211. doi: 10.1038/sj.cr.7310149. [DOI] [PubMed] [Google Scholar]
- 60.Van Damme M, Crompot E, Meuleman N, Mineur P, Bron D, Lagneaux L, Stamatopoulos B. HDAC isoenzyme expression is deregulated in chronic lymphocytic leukemia B-cells and has a complex prognostic significance. Epigenetics. 2012;7:1403–1412. doi: 10.4161/epi.22674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Van Damme M, Crompot E, Meuleman N, Mineur P, Dessars B, El HH, Bron D, Lagneaux L, Stamatopoulos B. Global histone deacetylase enzymatic activity is an independent prognostic marker associated with a shorter overall survival in chronic lymphocytic leukemia patients. Epigenetics. 2014;9:1374–1381. doi: 10.4161/15592294.2014.969628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sampath D, Liu C, Vasan K, Sulda M, Puduvalli VK, Wierda WG, Keating MJ. Histone deacetylases mediate the silencing of miR-15a, miR-16, and miR-29b in chronic lymphocytic leukemia. Blood. 2012;119:1162–1172. doi: 10.1182/blood-2011-05-351510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Inoue S, Riley J, Gant TW, Dyer MJ, Cohen GM. Apoptosis induced by histone deacetylase inhibitors in leukemic cells is mediated by Bim and Noxa. Leukemia. 2007;21:1773–1782. doi: 10.1038/sj.leu.2404760. [DOI] [PubMed] [Google Scholar]
- 64.Zhou N, Moradei O, Raeppel S, Leit S, Frechette S, Gaudette F, Paquin I, Bernstein N, Bouchain G, Vaisburg A, Jin Z, Gillespie J, Wang J, Fournel M, Yan PT, Trachy-Bourget MC, Kalita A, Lu A, Rahil J, MacLeod AR, Li Z, Besterman JM, Delorme D. Discovery of N-(2-aminophenyl)-4-[(4-pyridin-3-ylpyrimidin-2-ylamino) methyl] benzamide (MGCD0103), an orally active histone deacetylase inhibitor. J Med Chem. 2008;51:4072–4075. doi: 10.1021/jm800251w. [DOI] [PubMed] [Google Scholar]
- 65.El-Khoury V, Moussay E, Janji B, Palissot V, Aouali N, Brons NH, Van Moer K, Pierson S, Van Dyck E, Berchem G. The histone deacetylase inhibitor MGCD0103 induces apoptosis in B-cell chronic lymphocytic leukemia cells through a mitochondria-mediated caspase activation cascade. Mol Cancer Ther. 2010;9:1349–1360. doi: 10.1158/1535-7163.MCT-09-1000. [DOI] [PubMed] [Google Scholar]
- 66.El-Khoury V, Pierson S, Szwarcbart E, Brons NH, Roland O, Cherrier-De WS, Plawny L, Van Dyck E, Berchem G. Disruption of autophagy by the histone deacetylase inhibitor MGCD0103 and its therapeutic implication in B-cell chronic lymphocytic leukemia. Leukemia. 2014;28:1636–1646. doi: 10.1038/leu.2014.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Blum KA, Advani A, Fernandez L, Van Der Jagt R, Brandwein J, Kambhampati S, Kassis J, Davis M, Bonfils C, Dubay M, Dumouchel J, Drouin M, Lucas DM, Martell RE, Byrd JC. Phase II study of the histone deacetylase inhibitor MGCD0103 in patients with previously treated chronic lymphocytic leukaemia. Br J Haematol. 2009;147:507–514. doi: 10.1111/j.1365-2141.2009.07881.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Byrd JC, Shinn C, Waselenko JK, Fuchs EJ, Lehman TA, Nguyen PL, Flinn IW, Diehl LF, Sausville E, Grever MR. Flavopiridol induces apoptosis in chronic lymphocytic leukemia cells via activation of caspase-3 without evidence of bcl-2 modulation or dependence on functional p53. Blood. 1998;92:3804–3816. [PubMed] [Google Scholar]
- 69.Lin TS, Ruppert AS, Johnson AJ, Fischer B, Heerema NA, Andritsos LA, Blum KA, Flynn JM, Jones JA, Hu W, Moran ME, Mitchell SM, Smith LL, Wagner AJ, Raymond CA, Schaaf LJ, Phelps MA, Villalona-Calero MA, Grever MR, Byrd JC. Phase II study of flavopiridol in relapsed chronic lymphocytic leukemia demonstrating high response rates in genetically high-risk disease. J. Clin. Oncol. 2009;27:6012–6018. doi: 10.1200/JCO.2009.22.6944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Blum KA, Ruppert AS, Woyach JA, Jones JA, Andritsos L, Flynn JM, Rovin B, Villalona-Calero M, Ji J, Phelps M, Johnson AJ, Grever MR, Byrd JC. Risk factors for tumor lysis syndrome in patients with chronic lymphocytic leukemia treated with the cyclin-dependent kinase inhibitor, flavopiridol. Leukemia. 2011;25:1444–1451. doi: 10.1038/leu.2011.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hallek M, Fischer K, Fingerle-Rowson G, Fink AM, Busch R, Mayer J, Hensel M, Hopfinger G, Hess G, von GU, Bergmann M, Catalano J, Zinzani PL, Caligaris-Cappio F, Seymour JF, Berrebi A, Jager U, Cazin B, Trneny M, Westermann A, Wendtner CM, Eichhorst BF, Staib P, Buhler A, Winkler D, Zenz T, Bottcher S, Ritgen M, Mendila M, Kneba M. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet. 2010;376:1164–1174. doi: 10.1016/S0140-6736(10)61381-5. [DOI] [PubMed] [Google Scholar]
- 72.Stephens DM, Ruppert AS, Maddocks K, Andritsos L, Baiocchi R, Jones J, Johnson AJ, Smith LL, Zhao Y, Ling Y, Li J, Phelps MA, Grever MR, Byrd JC, Flynn JM. Cyclophosphamide, alvocidib (flavopiridol), and rituximab, a novel feasible chemoimmunotherapy regimen for patients with high-risk chronic lymphocytic leukemia. Leuk Res. 2013;37:1195–1199. doi: 10.1016/j.leukres.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fabre C, Gobbi M, Ezzili C, Zoubir M, Sablin MP, Small K, Im E, Shinwari N, Zhang D, Zhou H, Le TC. Clinical study of the novel cyclin-dependent kinase inhibitor dinaciclib in combination with rituximab in relapsed/refractory chronic lymphocytic leukemia patients. Cancer Chemother Pharmacol. 2014;74:1057–1064. doi: 10.1007/s00280-014-2583-9. [DOI] [PubMed] [Google Scholar]
- 74.Flynn J, Jones J, Johnson AJ, Andritsos L, Maddocks K, Jaglowski S, Hessler J, Grever MR, Im E, Zhou H, Zhu Y, Zhang D, Small K, Bannerji R, Byrd JC. Dinaciclib is a novel cyclin-dependent kinase inhibitor with significant clinical activity in relapsed and refractory chronic lymphocytic leukemia. Leukemia. 2015;29:1524–9. doi: 10.1038/leu.2015.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu FT, Agrawal SG, Gribben JG, Ye H, Du MQ, Newland AC, Jia L. Bortezomib blocks Bax degradation in malignant B cells during treatment with TRAIL. Blood. 2008;111:2797–2805. doi: 10.1182/blood-2007-08-110445. [DOI] [PubMed] [Google Scholar]
- 76.Faderl S, Rai K, Gribben J, Byrd JC, Flinn IW, O’Brien S, Sheng S, Esseltine DL, Keating MJ. Phase II study of single-agent bortezomib for the treatment of patients with fludarabine-refractory B-cell chronic lymphocytic leukemia. Cancer. 2006;107:916–924. doi: 10.1002/cncr.22097. [DOI] [PubMed] [Google Scholar]
- 77.Buac D, Shen M, Schmitt S, Kona FR, Deshmukh R, Zhang Z, Neslund-Dudas C, Mitra B, Dou QP. From bortezomib to other inhibitors of the proteasome and beyond. Curr Pharm Des. 2013;19:4025–4038. doi: 10.2174/1381612811319220012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Baou M, Kohlhaas SL, Butterworth M, Vogler M, Dinsdale D, Walewska R, Majid A, Eldering E, Dyer MJ, Cohen GM. Role of NOXA and its ubiquitination in proteasome inhibitor-induced apoptosis in chronic lymphocytic leukemia cells. Haematologica. 2010;95:1510–1518. doi: 10.3324/haematol.2010.022368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen D, Frezza M, Schmitt S, Kanwar J, Dou QP. Bortezomib as the first proteasome inhibitor anticancer drug: current status and future perspectives. Curr Cancer Drug Targets. 2011;11:239–253. doi: 10.2174/156800911794519752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Panwalkar A, Verstovsek S, Giles F. Nuclear factor-kappaB modulation as a therapeutic approach in hematologic malignancies. Cancer. 2004;100:1578–1589. doi: 10.1002/cncr.20182. [DOI] [PubMed] [Google Scholar]
- 81.Nencioni A, Hua F, Dillon CP, Yokoo R, Scheiermann C, Cardone MH, Barbieri E, Rocco I, Garuti A, Wesselborg S, Belka C, Brossart P, Patrone F, Ballestrero A. Evidence for a protective role of Mcl-1 in proteasome inhibitor-induced apoptosis. Blood. 2005;105:3255–3262. doi: 10.1182/blood-2004-10-3984. [DOI] [PubMed] [Google Scholar]
- 82.Liu FT, Agrawal SG, Movasaghi Z, Wyatt PB, Rehman IU, Gribben JG, Newland AC, Jia L. Dietary flavonoids inhibit the anticancer effects of the proteasome inhibitor bortezomib. Blood. 2008;112:3835–3846. doi: 10.1182/blood-2008-04-150227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Niewerth D, Jansen G, Assaraf YG, Zweegman S, Kaspers GJ, Cloos J. Molecular basis of resistance to proteasome inhibitors in hematological malignancies. Drug Resist Updat. 2015;18:18–35. doi: 10.1016/j.drup.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 84.Yun H, Zhang HL, Wang HQ. Rituximab and bortezomib (RB): a new effective regimen for refractory or relapsed indolent lymphomas. Med Oncol. 2015;32:353. doi: 10.1007/s12032-014-0353-5. [DOI] [PubMed] [Google Scholar]
- 85.Gupta SV, Hertlein E, Lu Y, Sass EJ, Lapalombella R, Chen TL, Davis ME, Woyach JA, Lehman A, Jarjoura D, Byrd JC, Lucas DM. The proteasome inhibitor carfilzomib functions independently of p53 to induce cytotoxicity and an atypical NF-kappaB response in chronic lymphocytic leukemia cells. Clin Cancer Res. 2013;19:2406–2419. doi: 10.1158/1078-0432.CCR-12-2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Awan FT, Flynn JM, Jones JA, Andritsos LA, Maddocks KJ, Sass EJ, Lucas MS, Chase W, Waymer S, Ling Y, Jiang Y, Phelps MA, Byrd JC, Lucas DM, Woyach JA. Phase I dose escalation trial of the novel proteasome inhibitor carfilzomib in patients with relapsed chronic lymphocytic leukemia and small lymphocytic lymphoma. Leuk Lymphoma. 2015;3:1–7. doi: 10.3109/10428194.2015.1014368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Scavullo C, Servida F, Lecis D, Onida F, Drago C, Ferrante L, Seneci P, Barcellini W, Lionetti M, Todoerti K, Neri A, Delia D, Deliliers GL. Singleagent Smac-mimetic compounds induce apoptosis in B chronic lymphocytic leukaemia (BCLL) Leuk Res. 2013;37:809–815. doi: 10.1016/j.leukres.2013.03.016. [DOI] [PubMed] [Google Scholar]
- 88.Maas C, Tromp JM, van Laar J, Thijssen R, Elias JA, Malara A, Krippner-Heidenreich A, Silke J, van Oers MH, Eldering E. CLL cells are resistant to smac mimetics because of an inability to form a ripoptosome complex. Cell Death Dis. 2013;4:e782. doi: 10.1038/cddis.2013.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Robak T, Robak P. BCR signaling in chronic lymphocytic leukemia and related inhibitors currently in clinical studies. Int Rev Immunol. 2013;32:358–376. doi: 10.3109/08830185.2013.786711. [DOI] [PubMed] [Google Scholar]
- 90.Veldurthy A, Patz M, Hagist S, Pallasch CP, Wendtner CM, Hallek M, Krause G. The kinase inhibitor dasatinib induces apoptosis in chronic lymphocytic leukemia cells in vitro with preference for a subgroup of patients with unmutated IgVH genes. Blood. 2008;112:1443–1452. doi: 10.1182/blood-2007-11-123984. [DOI] [PubMed] [Google Scholar]
- 91.Amrein L, Soulieres D, Johnston JB, Aloyz R. p53 and autophagy contribute to dasatinib resistance in primary CLL lymphocytes. Leuk Res. 2011;35:99–102. doi: 10.1016/j.leukres.2010.05.029. [DOI] [PubMed] [Google Scholar]
- 92.Amrein PC, Attar EC, Takvorian T, Hochberg EP, Ballen KK, Leahy KM, Fisher DC, Lacasce AS, Jacobsen ED, Armand P, Hasserjian RP, Werner L, Neuberg D, Brown JR. Phase II study of dasatinib in relapsed or refractory chronic lymphocytic leukemia. Clin Cancer Res. 2011;17:2977–2986. doi: 10.1158/1078-0432.CCR-10-2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kater AP, Spiering M, Liu RD, Doreen TRG, Slinger E, Tonino SH, Beckers MM, Daenen S, Doorduijn JK, Lankheet NA, Luijks DM, Eldering E, van Oers MH. Dasatinib in combination with fludarabine in patients with refractory chronic lymphocytic leukemia: a multicenter phase 2 study. Leuk Res. 2014;38:34–41. doi: 10.1016/j.leukres.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 94.Friedberg JW, Sharman J, Sweetenham J, Johnston PB, Vose JM, Lacasce A, Schaefer-Cutillo J, De Vos S, Sinha R, Leonard JP, Cripe LD, Gregory SA, Sterba MP, Lowe AM, Levy R, Shipp MA. Inhibition of Syk with fostamatinib disodium has significant clinical activity in non-Hodgkin lymphoma and chronic lymphocytic leukemia. Blood. 2010;115:2578–2585. doi: 10.1182/blood-2009-08-236471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sharman J, Hawkins M, Kolibaba K, Boxer M, Klein L, Wu M, Hu J, Abella S, Yasenchak C. An open-label phase 2 trial of entospletinib (GS-9973), a selective spleen tyrosine kinase inhibitor, in chronic lymphocytic leukemia. Blood. 2015;125:2336–2343. doi: 10.1182/blood-2014-08-595934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Brown JR, Byrd JC, Coutre SE, Benson DM, Flinn IW, Wagner-Johnston ND, Spurgeon SE, Kahl BS, Bello C, Webb HK, Johnson DM, Peterman S, Li D, Jahn TM, Lannutti BJ, Ulrich RG, Yu AS, Miller LL, Furman RR. Idelalisib, an inhibitor of phosphatidylinositol 3-kinase p110delta, for relapsed/refractory chronic lymphocytic leukemia. Blood. 2014;123:3390–3397. doi: 10.1182/blood-2013-11-535047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Furman RR, Sharman JP, Coutre SE, Cheson BD, Pagel JM, Hillmen P, Barrientos JC, Zelenetz AD, Kipps TJ, Flinn I, Ghia P, Eradat H, Ervin T, Lamanna N, Coiffier B, Pettitt AR, Ma S, Stilgenbauer S, Cramer P, Aiello M, Johnson DM, Miller LL, Li D, Jahn TM, Dansey RD, Hallek M, O’Brien SM. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med. 2014;370:997–1007. doi: 10.1056/NEJMoa1315226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Burke RT, Meadows S, Loriaux MM, Currie KS, Mitchell SA, Maciejewski P, Clarke AS, Dipaolo JA, Druker BJ, Lannutti BJ, Spurgeon SE. A potential therapeutic strategy for chronic lymphocytic leukemia by combining Idelalisib and GS-9973, a novel spleen tyrosine kinase (Syk) inhibitor. Oncotarget. 2014;5:908–915. doi: 10.18632/oncotarget.1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, Grant B, Sharman JP, Coleman M, Wierda WG, Jones JA, Zhao W, Heerema NA, Johnson AJ, Sukbuntherng J, Chang BY, Clow F, Hedrick E, Buggy JJ, James DF, O’Brien S. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369:32–42. doi: 10.1056/NEJMoa1215637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Byrd JC, Brown JR, O’Brien S, Barrientos JC, Kay NE, Reddy NM, Coutre S, Tam CS, Mulligan SP, Jaeger U, Devereux S, Barr PM, Furman RR, Kipps TJ, Cymbalista F, Pocock C, Thornton P, Caligaris-Cappio F, Robak T, Delgado J, Schuster SJ, Montillo M, Schuh A, de Vos S, Gill D, Bloor A, Dearden C, Moreno C, Jones JJ, Chu AD. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med. 2014;371:213–223. doi: 10.1056/NEJMoa1400376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Farooqui MZ, Valdez J, Martyr S, Aue G, Saba N, Niemann CU, Herman SE, Tian X, Marti G, Soto S, Hughes TE, Jones J, Lipsky A, Pittaluga S, Stetler-Stevenson M, Yuan C, Lee YS, Pedersen LB, Geisler CH, Calvo KR, Arthur DC, Maric I, Childs R, Young NS, Wiestner A. Ibrutinib for previously untreated and relapsed or refractory chronic lymphocytic leukaemia with TP53 aberrations: a phase 2, single-arm trial. Lancet Oncol. 2015;16:169–176. doi: 10.1016/S1470-2045(14)71182-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Byrd JC, Furman RR, Coutre SE, Burger JA, Blum KA, Coleman M, Wierda WG, Jones JA, Zhao W, Heerema NA, Johnson AJ, Shaw Y, Bilotti E, Zhou C, James DF, O’Brien S. Three-year follow-up of treatment-naive and previously treated patients with CLL and SLL receiving single-agent ibrutinib. Blood. 2015;125:2497–2506. doi: 10.1182/blood-2014-10-606038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yoon JY, Szwajcer D, Ishdorj G, Benjaminson P, Xiao W, Kumar R, Johnston JB, Gibson SB. Synergistic apoptotic response between valproic acid and fludarabine in chronic lymphocytic leukaemia (CLL) cells involves the lysosomal protease cathepsin B. Blood Cancer J. 2013;3:e153. doi: 10.1038/bcj.2013.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Karp M, Kosior K, Karczmarczyk A, Zajac M, Zaleska J, Tomczak W, Chocholska S, Hus M, Dmoszynska A, Giannopoulos K. Cytotoxic activity of valproic Acid on primary chronic lymphocytic leukemia cells. Adv Clin Exp Med. 2015;24:55–62. doi: 10.17219/acem/29264. [DOI] [PubMed] [Google Scholar]