

Abstract
Arsenic (As), cadmium (Cd), and lead (Pb) in select combinations are proved to affect the viability of astrocyte. However, their role in glioma, an aggressive astroglial tumor, is unexplored. We analyzed the effect of As+Cd+Pb on C6-glioma cells derived from rat glioma. We determined the lethal concentration (LC) of individual metal, and then treated C6-glioma cells with As+Cd+Pb at LC-5 (As: 5 mM, Cd: 2.5 mM and Pb: 15 mM), and concentrations that were double or triple of LC5. As+Cd+Pb induced dose-dependent reduction in C6-glioma viability. Cell death was due to apoptotic DNA fragmentation, detected through terminal deoxynucleotidyl transferase-mediated dUTP-nick-end labeling. An enhanced cleavage of caspase-9 indicated the apoptosis to be mitochondria-mediated. An increase in pro-apoptotic Bcl-2-associated-X protein (Bax) and decrease in anti-apoptotic Bcl2 resulting in a Bax/Bcl2 ratio > 1.0 validated mitochondrial apoptosis. Exploring apoptotic regulatory mechanism revealed an alteration in glial cell morphology and augmentation of astroglial marker, glial fibrillary acidic protein (GFAP), that demonstrated co-localization with cleaved caspase-9. The glial activation was accompanied by inflammation, involving the up-regulation of interleukin-1 (IL-1) and IL-1-receptor. IL-1 also contributed to apoptosis, as evident from the attenuation of cleaved caspase-9 upon treatment with IL-1receptor antagonist. Investigating the involvement of Mitogen-activated protein kinases (MAPKs) revealed the activation of P38 as indicated by an increased phospho-p38 expression. p38-MAPK inhibitor, SB203580, prevented caspase-9 activation, which further suppoted the involvement of p38-MAPK in C6-glioma apoptosis. Overall our data demonstrate the toxic effect of As+Cd+Pb on C6-glioma, which is mediated by mitochondria-dependent apoptosis that requires astroglial activation, inflammation and p38-MAPK signaling. As+Cd+Pb combination treatment may have a potential therapeutic usage against glial tumors.
Keywords: As+Cd+Pb, caspase-9, Bax/Bcl2, GFAP, IL-1, phospho-P38
Introduction
Malignant gliomas are the most common and aggressive type of primary brain tumors, conferring poor prognosis [1]. Current available glioblastoma therapy claims a survival of 1-2 year post-diagnosis [2,3], where drug penetrability through the blood-brain barrier (BBB) poses a problem [4].
Dysregulation of normal apoptotic signaling mechanisms is important during the transformation process of cancer [5]. Likewise, glioma cells exhibit an impaired apoptosis, prompting growth and invasiveness of malignant tumor [6,7]. Thus, stimulation of glioma cell apoptosis is deemed as a promising treatment strategy to reduce glioma tumor invasion.
Apoptosis is a physiological cell suicide program prominently involving the mitochondria [8]. Caspases play an important role in apoptotic mitochondrial damage, where aspartic acid specific protease, caspase-9, is the initiator [9]. Pro-caspase-9 cleavage generates its active form that further triggers effector caspases [10]. Activated caspase cascade culminates in altered apoptotic phenotypes, of which nuclear fragmentation is a major one [11]. Another important component of mitochondria-mediated apoptosis is basal cell lymphoma protein-2 (Bcl2) family [12]. Proteins of Bcl2 family have pro- or anti-apoptotic activities that control mitochondrial membrane permeability and thereby cellular apoptosis [13].
Reduction in glioma tumor invasion and glial cell number has been proven to result from an increase in Bax/Bcl2 ratio and the caspase activation [14,15]. Of several mechanisms regulating this apoptotic machinery, astroglial activation, marked by glial fibrillary acidic protein (GFAP) expression, is an important one [16,17]. Inflammation and inflammatory cytokines modulate this astroglia-regulated apoptotic process resulting in altered growth of malignant glioma [18,19]. Mitogen activated protein kinases (MAPK) are also key contributors of glial apoptosis [20,21]. Extracellular signal-regulated kinase (ERK) [22], c-Jun N-terminal kinases (JNK) [23] and P38 [24,25], the three major MAPK pathways [26], regulate caspase activation and thereby cellular apoptosis in the malignant glioma.
It has been earlier demonstrated that exposure to a mixture of arsenic (As), cadmium (Cd) and lead (Pb) (i. e. As+Cd+Pb) disrupts blood-brain barrier (BBB) allowing dose-dependent penetration of metals in the rat brain [27]. The metals affected the expression levels of GFAP and viability of astrocytes through activation of ERK and JNK signaling [27]. Arsenic, Cd and Pb have proven anti-proliferative and pro-apoptotic effects in cancer cells as well [28-30]. We thus hypothesized that As+Cd+Pb, with the ability to penetrate BBB, may affect the viability of malignant glioma and serve as chemotherapeutic agent. We assumed a greater effect in mixture than the individual metals. We assessed the potential apoptotic effect of this heavy metal mixture on C6-glioma, a widely used astrocyte like cell line [31,32]. We examined the possibility of altered astroglial activation and inflammation, and scanned modulation in MAPK signaling. Overall, our study identified mechanism affecting viability of malignant glial cells upon As+Cd+Pb-exposure.
Materials and methods
Reagents
Dulbecco’s Modified Eagle medium (DMEM)-F12, fetal bovine serum (FBS), pennicilin-streptomycin (Pen-Strep), HEPES buffer, L-glutamine, PBS and protein markers were procured from Invitrogen (Carlsbad, CA). Na-arsenite, Cd-chloride, Pb-acetate, Ponceau S stain, Na-orthovanadate, NaF, PMSF, protease inhibitor cocktail, MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide], Hoechst 33258 stain and Cellytic mammalian cell lysis reagent was procured from Sigma Chemical Co. (St Louis, MO). Terminal deoxynucleotide-transferase (TdT)-mediated dUTP nick end labeling (TUNEL) kit was procured from Roche (Indianapolis, IN). Bicinchoninic acid (BCA) assay kit was procured from Merck Co. (Bangalore, India). Rabbit polyclonal antibodies to pro-caspase-9, cleaved caspase-9, Bax, Bcl2, p-P38 and P-38 were purchased from Cell Signaling Technology (Danvers, MA). Mouse monoclonal antibody to GFAP and polyvinylidene difluoride (PVDF) membrane was purchased from Millipore (Temecula, CA). Rabbit polyclonal antibody to IL-1 was obtained from Abcam (Cambridge, MA). Rabbit polyclonal antibody to interleukin-1 receptor-1 (IL-1R1) was purchased from Santa Cruz Biotechnology (Dallas, texas). Mouse monoclonal antibody to β-actin and horseradishperoxide (HRP)-conjugated secondary anti-rabbit IgG and antimouse IgG were purchased from Sigma Chemical Co. (St Louis, MO). Supersignal west femto maximum sensitivity substrate for western blotting was purchased from PIERCE Biotechnology (Rockford, IL). Alexa Fluor® 488 goat anti-mouse IgG, Alexa Fluor® 546 goat anti-rabbit IgG and Alexa Fluor® 594 goat anti-rabbit IgG was from Invitrogen (Carlsbad, CA).
Cell culture and MTT assay
C6 glioma cell line was incubated in DMEM-F12, HEPES, L-glutamine, 10% FBS and 1% Penn-Strep complete growth medium. To measure C6 cell viability, MTT was performed as described before [33]. Briefly, C6 cells were grown to 80% confluency and pre-incubated for 2 h in reduced serum medium (0.5% FBS) in tissue culture incubator at 37°C with 5% CO2-95% air and optimum humidity. Cells were then treated with As, Cd, Pb or As+Cd+Pb at a concentration range of 0.01 to 200 μM and incubated overnight in a reduced serum medium. For MTT assay, cells were treated with 10 μl MTT (10.4 mg/ml) and incubated for 4 h. This was followed by a treatment with 50% dimethylformamide-20% SDS and incubation for 12 h, to solubilize the formazan released from MTT. Absorbance was then measured at 595 nm with background subtraction at 655 nm. Lethal concentration (LC) of the metals was determined using GraphPad Prism 3.0 software.
DNA fragmentation-apoptosis assay
C6 cells were plated on cover slips and DNA fragmentation examined through TUNEL assay as described before [34]. Briefly, C6 cells were treated with As+Cd+Pb at LC-5 (As: 5 μM, Cd: 2.5 μM and Pb: 15 μM), LC-10 (As: 10 μM, Cd: 5 μM and Pb: 30 μM) and LC-20 (As: 20 μM, Cd: 10 μM and Pb: 60 μM) values or vehicle (water) overnight and fixed with freshly prepared 4% paraformaldehyde (PFA). Apoptosis assay was then performed with in situ cell death detection TUNEL kit following manufacturer’s protocol. Cells were then counterstained with Hoechst 33258 (0.2 mM) for 5 min, visualized and image captured using a fluorescence microscope (Nikon Instech Co. Ltd., Kawasaki, Kanagawa, Japan). TUNEL-positive cells were counted from five different fields using Image-Pro plus 5.1 software (Media Cybernetics Inc.). About 1000 cells in each cover slip were marked. Apoptosis was quantitated, and Apoptotic Index represented by the number of TUNEL-positive cells per 100 Hoechst stained nuclei [35].
Cell treatments
C6 cells were treated overnight with vehicle (water) or As+Cd+Pb at T1, T2 and T3, where T1 represents As+Cd+Pb at LC-5 (As: 5 μM, Cd: 2.5 μM and Pb: 15 μM); T2, LC-10 (As: 10 μM, Cd: 5 μM and Pb: 30 μM) and T3, LC-20 (As: 20 μM, Cd: 10 μM and Pb: 60 μM).
Combination index (CI)
To compare the effect of As+Cd+Pb with their calculated additive effects, a combination index (CI) was calculated using the software (Calcusyn; Biosoft, Manchester, United Kingdom). The CI values less than, equal to, or more than 1 indicates synergy, additivity, or antagonism, respectively [36].
Western blotting
Expression levels of pro-caspase-9, cleaved caspase-9, Bax, Bcl2, p-P38, p-38 and β-actin were determined from C6 cell lysates through western blotting as described earlier [33]. Briefly, C6 cells were first washed with ice-cold PBS. Ice-cold Cell Lysis Reagent supplemented with protease inhibitor cocktail (a mixture of 4-(2-aminoethyl) benzenesulfonyl fluoride, pepstatinA, E-64, bestatin, leupeptin, and aprotinin) was added to the cells that were then kept on ice for 20 min. To check for expression levels of p-p38 and p38, cells were co-supplemented with protein extraction reagent supplemented with 1 mM sodium-orthovandate and 25 mM sodium fluoride. Cells were subsequently scraped, lysates homogenized using a Teflon homogenizer, and centrifuged at 15,000 rpm for 30 min at 4°C. Supernatant was collected for western blotting. Protein content of cell lysates in comparison to BSA protein standards provided with the kit was determined by BCA assay using plate reader Synergy HT at 562 nm (BioTek, Winooski, VT). Fifty-seventy μg of protein sample was then mixed with sample loading buffer and β-mercaptoethanol, boiled for 10 min at 80°C, loaded onto pre-cast appropriate gels and run in running buffer at 100 V for 90 min to fractionate proteins. A standard protein marker was run along with the samples. Gels were then transferred onto PVDF membrane at 16 V current for 90 min in the transfer buffer. After completion of transfer, blots were stained with Ponceau S to verify equivalent sample loading. The blots were blocked with 5% nonfat dry milk in TBST (10 mM Tris, pH 8.0/150 mM NaCl/0.05% Tween 20) for 1 h and washed with TBST. Blots were kept overnight at 4°C in 1:1000 working dilution of primary antibodies, pro-caspase-9, cleaved caspase-9, Bax, Bcl2, p-P38, P-38 and β-actin. Blots were washed and incubated with secondary anti-rabbit IgG or anti-mouse IgG conjugated to horseradish peroxidise at 1:2000 working dilution in PBS plus 0.1% Tween 20 (PBST). Samples were detected by chemiluminescence with super signal west femto max substrate. Relative quantitative expression of protein was measured by densitometric quantification of blots using Versa Doc Gel Imaging System (Bio-Rad, Hercules, CA).
Immunocytochemistry
Expression of cleaved caspase-9, GFAP, IL-1, IL-1R1 and p-P38 in the C6 cells were detected through immunocytochemistry following a previously described protocol [33]. Briefly, C6 cells were allowed to adhere on Poly-L-Lysine coated 4-well chamber slides and treated with As+Cd+Pb or control (vehicle, water). Cells were then fixed in 4% PFA for 20 minutes at 4°C. Cells were blocked with 5% BSA in 1X-PBS and washed with PBST. Cells were then incubated in cleaved caspase-9, GFAP, IL-1, IL-1R1 or P-P38 antibodies at 1:100 dilution at 4°C overnight. After rinsing in PBST, the cells were incubated with Alexa Fluor secondary antibodies at 1:200 for 60 min. Cells were re-rinsed and counterstained with nuclear Hoechst 33258 (0.2 mM) for 10 min. Following cell mounting with Vectashield medium, cells were visualized and image captured under a fluorescence microscope. Three-six fields were captured for each slide. For co-immunolabeling of GFAP with cleaved caspase-9, or the GFAP with IL-1 and IL-1R1, the respective primary antibodies/secondary antibodies were mixed and added to the cells; the rest was the same as single antibody labeling.
Statistical analysis
Data are presented as mean ± S.E. of the specified number of experiments. Statistical analysis was performed using SPSS 9.0 software (SPSS, Inc., Chicago, IL). Data were analyzed through one-way ANOVA, and then SNK post hoc test or Student’s t-test as required.
Results
Effect of As+Cd+Pb on viability and apoptosis of C6 glioma cells
We investigated whether As+Cd+Pb affects survival of C6 glioma cells. We determined the effect of individual As, Cd or Pb on C6 cell viability through MTT assay. Individual metals induced loss in cell viability, and the lethal concentrations, LC5, LC10 and LC20, are as depicted in Table 1. We then treated C6 with As+Cd+Pb at three different concentrations, LC5 (As: 5 μM + Cd: 2.5 μM + Pb: 15 μM), LC10 (As: 10 μM + Cd: 2.5 μM + Pb: 30 μM) or LC 20 (As: 20 μM + Cd: 10 μM + Pb: 60 μM), and measured cell viability. As+Cd+Pb induced a dose-dependent loss in cell viability (Table 2). To assess whether the combinatorial effect of the metals was additive, synergistic or anatagonistic, we calculated the CI values. CI-value < 1.0 for all three combinations indicated a synergistic effect (Table 3).
Table 1.
LC Values of As, Cd, and Pb in C6 glial cells
| LC | LC-5 (mM) | LC-10 (mM) | LC-20 (mM) |
|---|---|---|---|
| As | 5 | 10 | 20 |
| Cd | 2.5 | 5 | 10 |
| Pb | 15 | 30 | 60 |
Table 2.
As+Cd+Pb-induced loss in C6 cell viability
| As+Cd+Pb-conc | Loss in viability (%) |
|---|---|
| 5 mM + 2.5 mM + 15 mM | 20±2 |
| 10 mM + 5 mM + 30 mM | 45±4 |
| 20 mM + 10 mM + 60 mM | 78±7 |
Table 3.
CI values-Values of As+Cd+Pb in C6 glial cells
| As+Cd+Pb-conc | CI values |
|---|---|
| 5 mM + 2.5 mM + 15 mM | 0.87 |
| 10 mM + 5 mM + 30 mM | 0.84 |
| 20 mM + 10 mM + 60 mM | 0.89 |
To determine whether As+Cd+Pb-induced cytotoxicity is mediated through apoptosis, we performed TUNEL assay, a quantitative technique that detects DNA fragmentation. We found that As+Cd+Pb caused a dose-dependent increase in TUNEL staining of C6 cells (Figure 1A). To assess the number of TUNEL-positive nuclei, we counterstained C6 with nuclear Hoechst. Merged image of TUNEL and Hoechst demonstrated an increase in TUNEL-postive nuclei for As+Cd+Pb (Figure 1A), indicating enhanced nuclear fragmentation. We quantitated Apoptotic Index, i. e. number of TUNEL-positive cells per 100 nuclei. As+Cd+Pb caused a dose-dependent increase in Apoptotic Index (Figure 1B).
Figure 1.
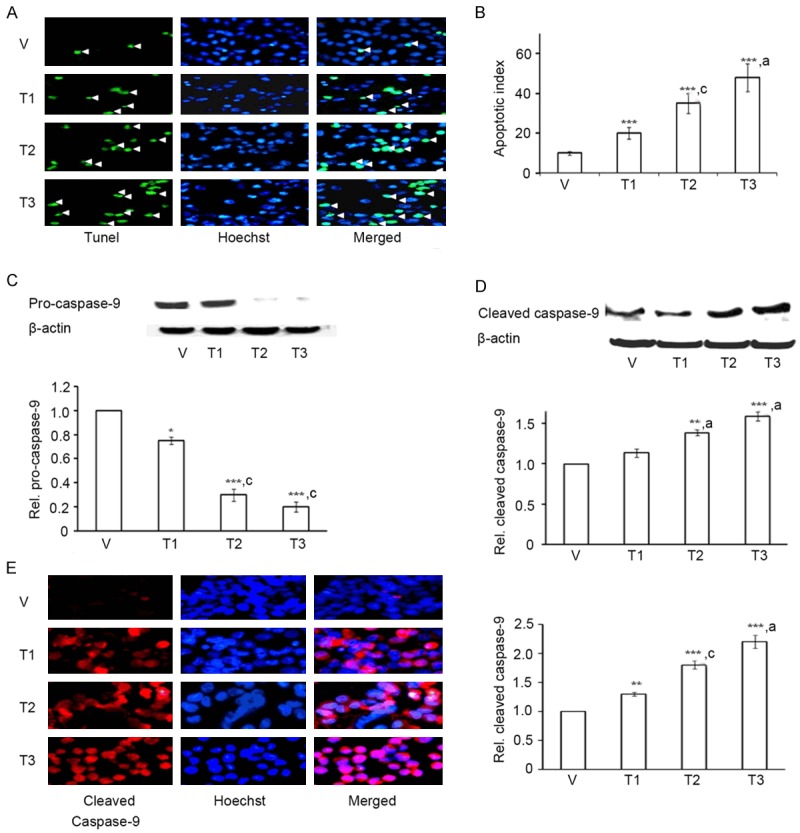
As+Cd+Pb induces dose-dependent apoptosis in C6 glial cells. Eighty percent confluent C6 cells were incubated with vehicle (V, water) or As+Cd+Pb at the three different concentrations (refer to Results) overnight, and apoptosis was analyzed. A. Representative TUNEL staining for apoptotic cells (green fluorescence, white arrowhead). Hoechst staining: blue fluorescence. B. Quantitation of dose-dependent increase of apoptotic cells. C. Dose-dependent decrease of pro-caspase-9 by Western blot. D. Dose-dependent increase in cleaved caspase-9 by Western blot. E. Immunofluorescence staining of cleaved caspase-9 (red fluorescence) and Hoechst (blue fluorescence).
To test whether As+Cd+Pb-induced apoptosis is caspase mediated we checked expression levels of initiator caspase-9 through western blotting. As+Cd+Pb stimulated a dose-dependent reduction in pro-caspase-9 (Figure 1C) while causing induction of cleaved capase-9 (Figure 1D), indicating a proteolytic cleavage of the pro-caspase-9. We verified caspase-9 cleavage through immunocytochemistry. An increased expression of cleaved caspase-9 in C6 by As+Cd+Pb corroborated western blot data (Figure 1E).
Effect of As+Cd+Pb on Bax and Bcl2 protein levels in C6 glioma cells
Bcl2 family of proteins control mitochondrial permeability and regulate caspase-induced apoptosis [37]. We thus tested whether Bcl2 family was involved in As+Cd+Pb-induced apoptosis. For this, we assessed the expression levels of pro-apoptotic Bax and anti-apoptotic Bcl2 through western blotting. As+Cd+Pb promoted Bax (Figure 2A) and suppressed Bcl2 (Figure 2B) expression levels in a dose-dependent manner, indicating induction of the mitochondrial apoptotic pathway. Ratio of Bax to Bcl2 being an important determinant of apoptotic cell death [38], we quantitated Bax/Bcl2 ratio from their western blot data. We observed that As+Cd+Pb caused a dose-dependent increase in Bax/Bcl2 ratio in C6 cells (Figure 2C), verifying apoptosis.
Figure 2.
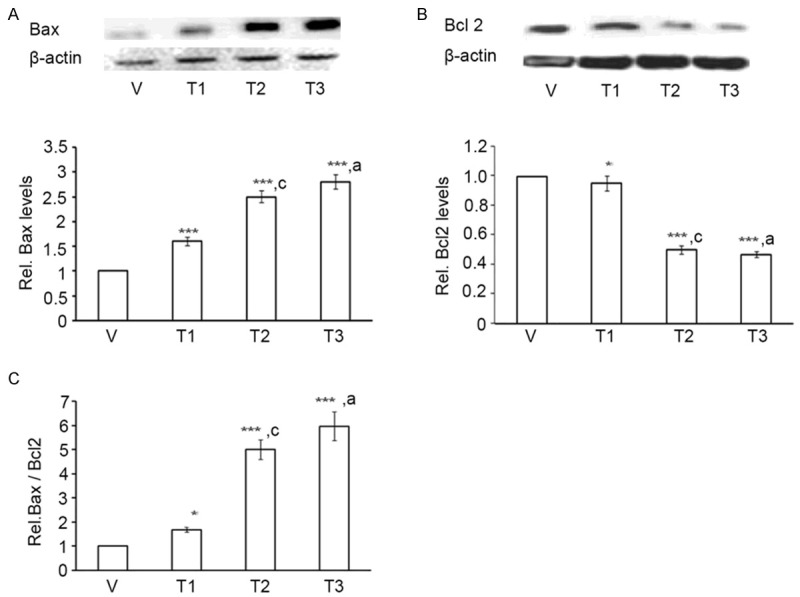
As+Cd+Pb induces Bax, reduces Bcl2 and increases Bax/Bcl2 ratio in C6 cells. Eighty percent confluent C6 cells were incubated with vehicle (V, water) or As+Cd+Pb at the three different concentrations overnight, and Bax and Bcl2 expression levels were analyzed. A. Dose-dependent increase of Bax by Western Blot. B. Dose-dependent decrease of Bcl2 by Western Blot. C. Dose-dependent increase of Bax/Bcl2 ratio.
Effect of As+Cd+Pb on astroglial activation and morphology of apoptotic C6 glioma cells
We next studied mechanism regulating C6 apoptosis. Astroglial activation characterized by enhanced GFAP expression and altered glial morphology participates in apoptosis [16,39]. We, therefore, tested whether As+Cd+Pb had any effect on astroglial activation and C6 cell morphology by performing immunocytochemistry with GFAP. As+Cd+Pb caused an increase in GFAP immunoreactivity in a dose-dependent manner (Figure 3A), suggesting enhanced astroglial activation. Decrease in C6 cell count (Figure 3A) validated the increase in cell death. In addition, alteration in C6 morphology characterized by increased globular and bulbous shape (represented by shape retention in the figure), and reduction in the number of projections and compactness was also evident (Figure 3B). We then tested whether astroglial activation correlates with C6 apoptosis by co-immunolabeling GFAP with cleaved caspase-9. As+Cd+Pb increased co-localization of GFAP with cleaved caspase-9, which was also dose-dependent (Figure 3C). This enhanced co-localization indicated that the astroglial activation and apoptosis were linked in the C6.
Figure 3.
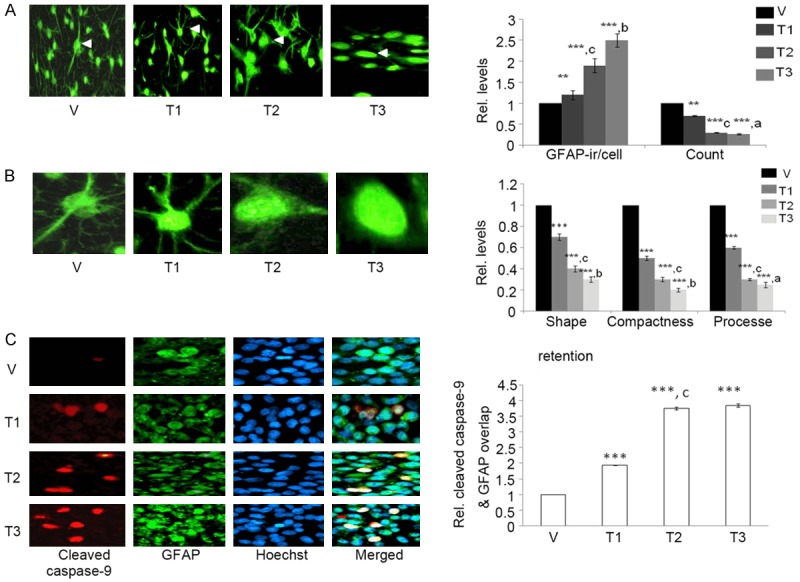
As+Cd+Pb induces dose-dependent GFAP immunoreactivity, decreases the cell count, alters cell morphology and increases cleaved caspase-9 expression in C6. Eighty percent confluent C6 cells were incubated with vehicle (V, water) or As+Cd+Pb at the three different concentrations overnight, and GFAP and cleaved caspase-9 expression were analyzed. A. Representative GFAP immunostaining (green fluorescence), and fold change in GFAP immunoreactivity (ir/cell) and cell count, shape, compactness and process. B. Magnified image of C6 cell (marked with white arrowhead in A). C. Representative cleaved caspase-9 (red fluorescence) and GFAP (green fluorescence) co-immunostaining. Hoechst staining: blue fluorescence.
Effect of As+Cd+Pb on inflammation in apoptotic C6 glioma cells
Astroglial activation coincides with up-regulation of pro-inflammatory cytokines, where IL-1 is a major player [40,41]. We thus examined whether As+Cd+Pb had any effect on IL-1 expression in C6 cells through immunocytochemistry. As+Cd+Pb induced dose-dependent increase in IL-1 expression (Figure 4A). IL-1R1 primarily mediates the inflammatory effects of IL-1 [42], and therefore, we examined the effect of As+Cd+Pb on IL-1R1 expression. Consistent with our observations on IL-1, As+Cd+Pb caused a dose-dependent increase in IL-1R1 expression (Figure 4B), indicating IL-1 mediated inflammation. We then assessed whether As+Cd+Pb-mediated astroglial activation coincides with IL-1 induction by co-immunolabeling GFAP with IL-1 or IL-1R1. We found that As+Cd+Pb stimulated co-localization of GFAP with IL-1 (Figure 4C) and IL-1R1 (Figure 4D), indicating that astroglial activation and inflammation were linked in the C6 cells.
Figure 4.
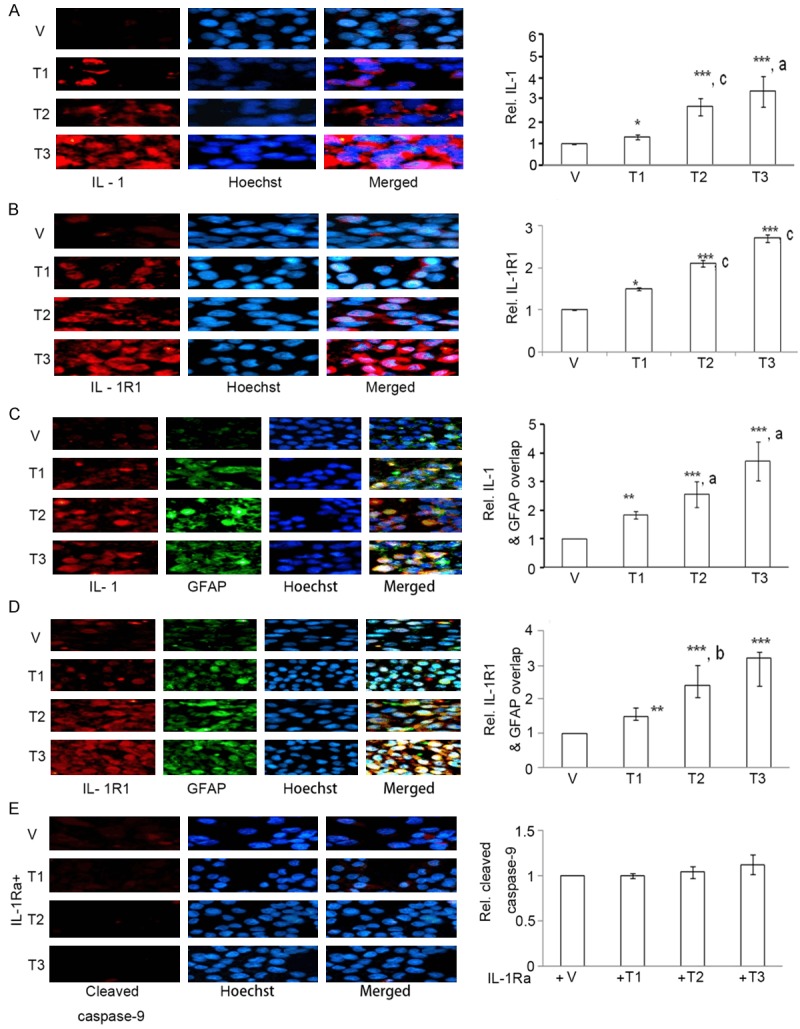
As+Cd+Pb up-regulates dose-dependent IL-1 and IL-1R1, inducing cleaved caspase-9 expression in C6. Eighty percent confluent C6 cells were incubated with vehicle (V, water) or As+Cd+Pb at the three different concentrations overnight, and IL-1, IL-1R1 and cleaved caspase-9 expressions were analyzed. A. Representative IL-1 immunostaining (red fluorescence). Hoechst staining: blue fluorescence. B. Representative IL-1R1 immunostaining (red fluorescence). Hoechst staining: blue fluorescence. C. Representative IL-1 (red fluorescence) and GFAP (green fluorescence) co-immunostaining. Hoechst staining: blue fluorescence. D. Representative IL-1R1 (red fluorescence) and GFAP (green fluorescence) co-immunostaining. Hoechst staining: blue fluorescence. E. Representative cleaved caspase-9 immunostaining (red fluorescence) upon co-treatment with IL-1Ra. Hoechst staining: blue fluorescence.
We examined whether As+Cd+Pb-induced inflammation could be responsible for C6 cell apoptosis. We co-treated As+Cd+Pb-treated cells with recombinant IL-1Ra, a member of IL-1 family that binds to IL-1 receptors to prevent inflammation [43], and checked for cleaved caspase-9 expression through immunocytochemistry. IL-1Ra prevented As+Cd+Pb-mediated induction of cleaved caspase-9 (Figure 4E), verifying the participation of IL-1 in C6 apoptosis.
Effect of As+Cd+Pb on MAPK activation in C6 glioma cells
Modulation in MAPK signaling induces glial cell apoptosis [20,21]. We thus assessed whether ERK, JNK and P-38 MAPKs are involved in As+Cd+Pb-induced C6 cell apoptosis. We co-treated As+Cd+Pb with PD98059, SP600125 or SB203580 that inhibits ERK, JNK and P38 signaling respectively, and performed immunocytochemistry of cleaved caspase-9. SB203580 prevented the increase in cleaved caspase-9 expression (Figure 5A), proving a P38-dependent induction of cleaved caspase-9. PD98059 (Figure 5B) and SP600125 had no effect on As+Cd+Pb-mediated induction of cleaved caspase-9 expression (Figure 5C), indicating the non-participation of ERK and JNK respectively in As+Cd+Pb-induced cleaved caspase-9. We proved activation of P38 signaling by measuring p-P38 expression through immunocytochemistry. As+Cd+Pb caused a dose-dependent increase in p-P38 expression (Figure 5D). We further verified dose-dependent increase in p-P38 expression through western blotting (Figure 5E).
Figure 5.
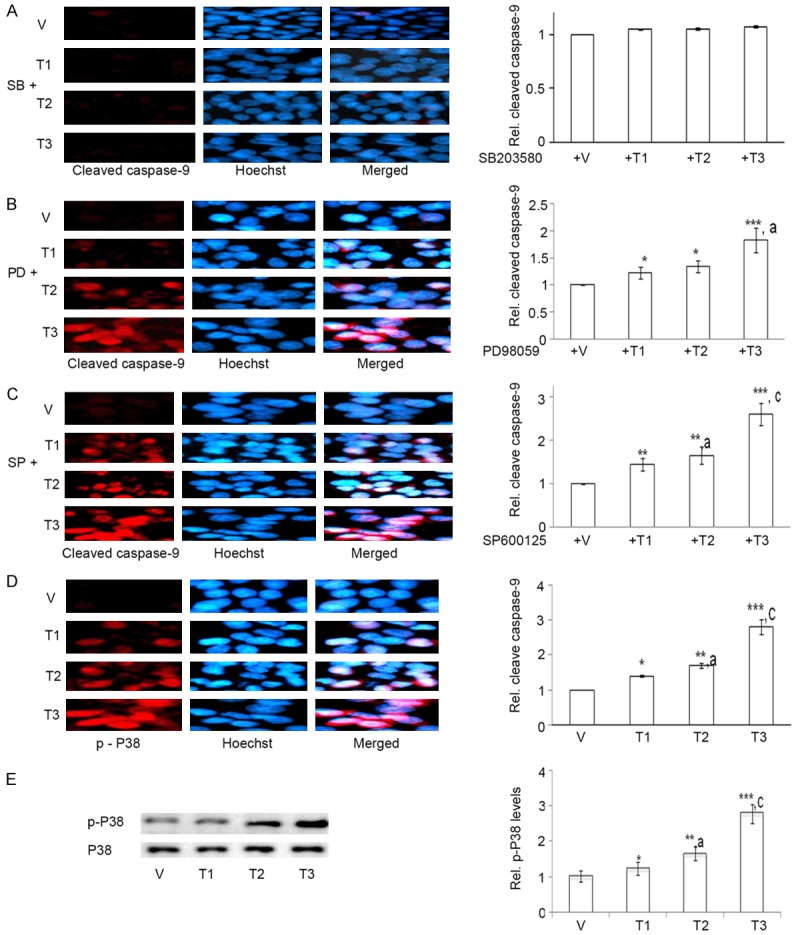
As+Cd+Pb activates dose-dependent P38 and not ERK and JNK-dependent cleaved caspase-9 expression in C6. Eighty percent confluent C6 cells were pre-incubated with SB203580, PD98059 or SP600125, and then co-incubated with vehicle (V, water) or As+Cd+Pb at the three different concentrations overnight, and cleaved caspase-9 expression was determined. A. Representative cleaved caspase-9 immunostaining (red fluorescence) for SB203580 (SB). Hoechst staining: blue fluorescence. B. Representative cleaved caspase-9 immunostaining (red fluorescence) for PD98059 (PD). Hoechst staining: blue fluorescence. C. Representative cleaved caspase-9 immunostaining (red fluorescence) for SP600125 (SP). Hoechst staining: blue fluorescence. Eighty percent confluent C6 cells were incubated with vehicle (V, water) or As+Cd+Pb at the three different concentrations overnight, and p-P38 expression was determined. D. Representative p-P38 immunostaining: red fluorescence. Hoechst: blue fluorescence. E. Dose-dependent increase of p-P38 by Western Blot.
Discussion
The current study reveals that a mixture of As, Cd and Pb at optimized concentration is toxic to malignant glial tumor cell line, C6. A dose-dependent up-regulation of apoptosis appears responsible for the reduction in C6 cell count. The apoptotic effect is mitochondrial with an increase in Bax/Bcl2 ratio that executes nuclear fragmentation through a caspase-dependent pathway. Factors regulating C6 apoptosis include astroglial activation, accompanied with altered glial morphology, inflammation and P38-MAPK signaling (Figure 6). Overall, our findings are important because they provide insight for proposing As+Cd+Pb as novel therapeutic against glioma.
Figure 6.
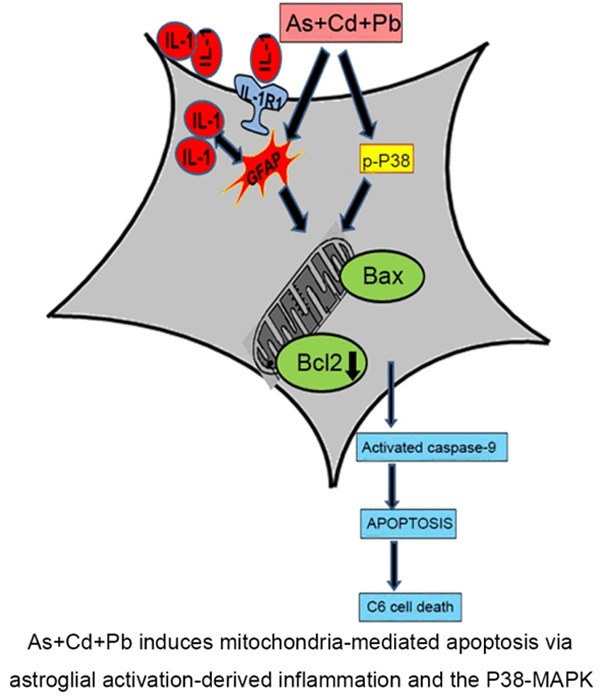
Schematic diagram of the mechanism that induces apoptosis in C6 glial cells. As+Cd+Pb activates GFAP, up-regulates IL-1 and IL-1R1 that stimulate inflammation. As+Cd+Pb promotes P38-MAPK signaling. Inflammation and P38-MAPK converge on the mitochondria and increase Bax and suppress Bcl2 expression that activates caspase-9 and triggers mitochondrial apoptosis.
Concurrent exposure to As, Cd and Pb reduce viability of primary astrocytes [27]. However, their effect on malignant glial cell lines remained unexplored. The current study evinces that As+Cd+Pb mixture is lethal to malignant glia, and brings forth the cytotoxic concentration as well. A concentration of 5-20 μM, 2.5-10 μM and 15-60 μM each of As, Cd and Pb respectively is found to induce 5-20% cell death in the C6 glia. At these concentrations, the metal mixture exhibited greater-than-additive effect, proving synergistic role of these metals in combination. This synergistic cytotoxicity justifies using the mixture rather than metals alone for our study.
BBB remains a major obstacle influencing therapeutic efficacies in malignant glioma [44]. Nevertheless, a notable aspect for As, C and Pb is their ability to cross BBB and reach the brain [27]. Metal deposition in the brain as high as 10-20% of the oral dose [27] is a major advantage of As+Cd+Pb exposure. Thus from this easy permeability viewpoint coupled with cytotoxicity, As+Cd+Pb at select doses may hold significant promise in targeting glioma tumors. Secondly, rather than oral exposure, stereotaxic insertion of the metal mixture may serve as a better option for efficient targeting at the malignant glial site. However, in vivo studies in tumor models for oral exposure and stereotaxic As+Cd+Pb-delivery are needed to prove the therapeutic potential of this metal mixture.
It is true, as commonly observed and accepted with cancer therapies, that the metal mixture may non-specifically affect neighbouring non-malignant brain cells. Nonetheless, metal doses used throughout our study being non-lethal, as reported in vivo [27], benefits of a novel suitable therapy against malignant glioma outweigh the associated side effects.
Our data reveal that As+Cd+Pb-mediated loss in C6 cell viability is due to apoptosis. While DNA fragmentation by As+Cd+Pb was earlier proved in primary astroglia [27], the present study proceeds a step ahead demonstrating involvement of the mitochondrial apoptotic pathway in glial cell lines. Proteolytic cleavage of caspase-9, responsible for activating executioner caspases and initiating the protease apoptotic cascade [45], was up-regulated in C6. In addition, levels of pro-apoptotic Bax and anti-apoptotic Bcl2, and the ratio of Bax/Bcl2 that induces mitochondrial damage through caspase activation in malignant cell lines [46,47] underwent a significant change. Hence it may be construed that disturbance in Bcl2 family rheostat [48] is the central event governing caspase-9 activation and mitochondrial apoptotic cascade by As+Cd+Pb.
Over-expression of Bcl2 and reduction in Bax has been reported in glial cell lines [49] and is linked with a reduced response to apoptotic stimuli [50,51]. Additionally, Bcl2 has been demonstrated to enhance the invasiveness of glioma (Wick et al. 1998). Thus, the ability to increase Bax and reduce Bcl2 draw attention towards As+Cd+Pb as a suitable curative to alleviate glial invasiveness.
The present study demonstrates that As+Cd+Pb stimulates dose-dependent astroglial activation, portrayed through GFAP up-regulation, that leads to C6 cell apoptosis. Consistent with an earlier study in C6 [16], our data depicting the transition to globular and bulbous cell shape complements increase in GFAP. Noticeably, unlike previous reports that solely claim association of GFAP with activated caspases in C6 [16,52,53], our data demonstrating a direct overlap of GFAP with cleaved caspase-9 highlights a strong link between astroglial activation and apoptosis in the cell line.
Likewise, IL-1 appeared as a major contributor to As+Cd+Pb-induced C6 apoptosis, as evident from a reduction in cleaved caspase-9 using IL-1Ra. Inflammation is known to promote GFAP immunoreactivity in astrocytes [54] and glial cell lines [54]. Correspondingly in the present study, immunohistochemical analysis demonstrating IL-1 juxtaposed to GFAP proves significant association of inflammation and glial activation. Additionally, akin to earlier reports claiming positive correlation between astroglial activation and IL-1 receptors [41], co-localization of IL-1R1 and GFAP substantiates IL-1-mediated immune activation by As+Cd+Pb. Thus, combined impact of closely linked inflammation and astroglial activation accounts for the significant apoptosis-mediated cytotoxicity in C6.
The current investigation demonstrates an augmentation of As+Cd+Pb-mediated P38 signaling, marked by increased p-P38 expression. It further depicts participation of P38 pathway in C6 apoptosis. Cadmium [55,56] and Pb [57] alone is proven to activate P38 pathway in the C6 glioma; however, impact of the three metals in combination on P38 signaling is unreported. In addition, to the best of knowledge, apoptotic effect of these metals via P38 is yet unidentified in any known glial cell lines. Thus, our study is the first to detect the significant contribution of P38 pathway in curbing glioma progression using these metals.
Our study reveals that As+Cd+Pb-induced apoptosis in C6 is independent of ERK and JNK-MAPKs. This observation on MAPKs in C6 is at variance with that in primary astrocytes, reported to exhibit an As+Cd+Pb-induced ERK and JNK rather than P38-MAPK-dependent apoptosis [27]. Thus, disparity in MAPK signaling may probably be one of the reasons causing As+Cd+Pb-mediated differential expression of GFAP; an increase for C6, as observed in the current study, in contrast to its’ reported reduction in primary astrocytes [27]. However, the specific role of MAPKs in diiferential GFAP expression calls for confirmation.
Taken as a whole, the significance of our investigation lies in the cytotoxic impact of As+Cd+Pb at selective doses on malignant glioma. The findings shed light on the mechanism linking astroglial activation, inflammation and P38-MAPK signaling to mitochondria-dependent caspase activation and apoptosis. Thus our study identifies the mechanistic targets and strongly claims the utility of As+Cd+Pb in managing malignant glioma.
Disclosure of conflict of interest
None.
References
- 1.Goodenberger ML, Jenkins RB. Genetics of adult glioma. Cancer Genet. 2012;205:613–621. doi: 10.1016/j.cancergen.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 2.Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 3.Jacobs VL, Valdes PA, Hickey WF, De Leo JA. Current review of in vivo GBM rodent models: emphasis on the CNS-1 tumour model. ASN Neuro. 2011;3:e00063. doi: 10.1042/AN20110014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou J, Atsina KB, Himes BT, Strohbehn GW, Saltzman WM. Novel delivery strategies for glioblastoma. Cancer J. 2012;18:89–99. doi: 10.1097/PPO.0b013e318244d8ae. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 6.Newton HB. Molecular neuro-oncology and the development of targeted therapeutic strategies for brain tumors. Part 3: brain tumor invasiveness. Expert Rev Anticancer Ther. 2004;4:803–821. doi: 10.1586/14737140.4.5.803. [DOI] [PubMed] [Google Scholar]
- 7.Steinbach JP, Weller M. Apoptosis in Gliomas: Molecular Mechanisms and Therapeutic Implications. J Neurooncol. 2004;70:247–256. doi: 10.1007/s11060-004-2753-4. [DOI] [PubMed] [Google Scholar]
- 8.Kroemer G, Petit P, Zamzami N, Vayssiere JL, Mignotte B. The biochemistry of programmed cell death. FASEB J. 1995;9:1277–1287. doi: 10.1096/fasebj.9.13.7557017. [DOI] [PubMed] [Google Scholar]
- 9.Bossy-Wetzel E, Green DR. Apoptosis: checkpoint at the mitochondrial frontier. Mutat Res. 1999;434:243–251. doi: 10.1016/s0921-8777(99)00032-4. [DOI] [PubMed] [Google Scholar]
- 10.Parrish AB, Freel CD, Kornbluth S. Cellular mechanisms controlling caspase activation and function. Cold Spring Harb Perspect Biol. 2013;5:a008672. doi: 10.1101/cshperspect.a008672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chiang MC, Ashraf QM, Mishra OP, Delivoria-Papadopoulos M. Mechanism of DNA fragmentation during hypoxia in the cerebral cortex of newborn piglets. Neurochem Res. 2008;33:1232–1237. doi: 10.1007/s11064-007-9574-8. [DOI] [PubMed] [Google Scholar]
- 12.Siddiqui WA, Ahad A, Ahsan H. The mystery of BCL2 family: Bcl-2 proteins and apoptosis: an update. Arch Toxicol. 2015;89:289–317. doi: 10.1007/s00204-014-1448-7. [DOI] [PubMed] [Google Scholar]
- 13.Brunelle JK, Letai A. Control of mitochondrial apoptosis by the Bcl-2 family. J Cell Sci. 2009;122:437–441. doi: 10.1242/jcs.031682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.D’Amico AG, Scuderi S, Saccone S, Castorina A, Drago F, D’Agata V. Antiproliferative effects of PACAP and VIP in serum-starved glioma cells. J Mol Neurosci. 2013;51:503–513. doi: 10.1007/s12031-013-0076-7. [DOI] [PubMed] [Google Scholar]
- 15.Kouri FM, Jensen SA, Stegh AH. The role of Bcl-2 family proteins in therapy responses of malignant astrocytic gliomas: Bcl2L12 and beyond. ScientificWorldJournal. 2012;2012:838916. doi: 10.1100/2012/838916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Swarnkar S, Singh S, Goswami P, Mathur R, Patro IK, Nath C. Astrocyte activation: a key step in rotenone induced cytotoxicity and DNA damage. Neurochem Res. 2012;37:2178–2189. doi: 10.1007/s11064-012-0841-y. [DOI] [PubMed] [Google Scholar]
- 17.Castigli E, Arcuri C, Giovagnoli L, Luciani R, Giovagnoli L, Secca T, Gianfranceschi GL, Bocchini V. Interleukin-1beta induces apoptosis in GL15 glioblastoma-derived human cell line. Am J Physiol Cell Physiol. 2000;279:C2043–2049. doi: 10.1152/ajpcell.2000.279.6.C2043. [DOI] [PubMed] [Google Scholar]
- 18.Sun W, Depping R, Jelkmann W. Interleukin-1beta promotes hypoxia-induced apoptosis of glioblastoma cells by inhibiting hypoxia-inducible factor-1 mediated adrenomedullin production. Cell Death Dis. 2014;5:e1020. doi: 10.1038/cddis.2013.562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kondo S, Barna BP, Morimura T, Takeuchi J, Yuan J, Akbasak A, Barnett GH. Interleukin-1 beta-converting enzyme mediates cisplatin-induced apoptosis in malignant glioma cells. Cancer Res. 1995;55:6166–6171. [PubMed] [Google Scholar]
- 20.Ding D, Wei S, Song Y, Li L, Du G, Zhan H, Cao Y. Osthole exhibits anti-cancer property in rat glioma cells through inhibiting PI3K/Akt and MAPK signaling pathways. Cell Physiol Biochem. 2013;32:1751–1760. doi: 10.1159/000356609. [DOI] [PubMed] [Google Scholar]
- 21.Tseng TH, Shen CH, Huang WS, Chen CN, Liang WH, Lin TH, Kuo HC. Activation of neutralsphingomyelinase, MAPKs, and p75 NTR-mediating caffeic acid phenethyl ester-induced apoptosis in C6 glioma cells. J Biomed Sci. 2014;21:61. doi: 10.1186/1423-0127-21-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feng Y, Huang J, Ding Y, Xie F, Shen X. Tamoxifen-induced apoptosis of rat C6 glioma cells via PI3K/Akt, JNK and ERK activation. Oncol Rep. 2010;24:1561–1567. doi: 10.3892/or_00001018. [DOI] [PubMed] [Google Scholar]
- 23.Levkovitz Y, Gil-Ad I, Zeldich E, Dayag M, Weizman A. Differential induction of apoptosis by antidepressants in glioma and neuroblastoma cell lines: evidence for p-c-Jun, cytochrome c, and caspase-3 involvement. J Mol Neurosci. 2005;27:29–42. doi: 10.1385/JMN:27:1:029. [DOI] [PubMed] [Google Scholar]
- 24.Wang DC, Zhang Y, Chen HY, Li XL, Qin LJ, Li YJ, Zhang HY, Wang S. Hyperthermia promotes apoptosis and suppresses invasion in C6 rat glioma cells. Asian Pac J Cancer Prev. 2012;13:3239–3245. doi: 10.7314/apjcp.2012.13.7.3239. [DOI] [PubMed] [Google Scholar]
- 25.Lee YJ, Kuo HC, Chu CY, Wang CJ, Lin WC, Tseng TH. Involvement of tumor suppressor protein p53 and p38 MAPK in caffeic acid phenethyl ester-induced apoptosis of C6 glioma cells. Biochem Pharmacol. 2003;66:2281–2289. doi: 10.1016/j.bcp.2003.07.014. [DOI] [PubMed] [Google Scholar]
- 26.Kugler W, Erdlenbruch B, Otten K, Jendrossek V, Eibl H, Lakomek M. MAP kinase pathways involved in glioblastoma response to erucylphosphocholine. Int J Oncol. 2004;25:1721–1727. [PubMed] [Google Scholar]
- 27.Rai A, Maurya SK, Khare P, Srivastava A, Bandyopadhyay S. Characterization of developmental neurotoxicity of As, Cd, and Pb mixture: synergistic action of metal mixture in glial and neuronal functions. Toxicol Sci. 2010;118:586–601. doi: 10.1093/toxsci/kfq266. [DOI] [PubMed] [Google Scholar]
- 28.Pettersson HM, Karlsson J, Pietras A, Ora I, Pahlman S. Arsenic trioxide and neuroblastoma cytotoxicity. J Bioenerg Biomembr. 2007;39:35–41. doi: 10.1007/s10863-006-9058-6. [DOI] [PubMed] [Google Scholar]
- 29.Waalkes MP, Diwan BA. Cadmium-induced inhibition of the growth and metastasis of human lung carcinoma xenografts: role of apoptosis. Carcinogenesis. 1999;20:65–70. doi: 10.1093/carcin/20.1.65. [DOI] [PubMed] [Google Scholar]
- 30.Corbit R, Ebbs S, King ML, Murphy LL. The influence of lead and arsenite on the inhibition of human breast cancer MCF-7 cell proliferation by American ginseng root (Panax quinquefolius L. ) Life Sci. 2006;78:1336–1340. doi: 10.1016/j.lfs.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 31.Esposito E, Iacono A, Muia C, Crisafulli C, Mattace Raso G, Bramanti P, Meli R, Cuzzocrea S. Signal transduction pathways involved in protective effects of melatonin in C6 glioma cells. J Pineal Res. 2008;44:78–87. doi: 10.1111/j.1600-079X.2007.00492.x. [DOI] [PubMed] [Google Scholar]
- 32.Bissell MG, Rubinstein LJ, Bignami A, Herman MM. Characteristics of the rat C-6 glioma maintained in organ culture systems. Production of glial fibrillary acidic protein in the absence of gliofibrillogenesis. Brain Res. 1974;82:77–89. doi: 10.1016/0006-8993(74)90894-4. [DOI] [PubMed] [Google Scholar]
- 33.Gupta S, Goswami P, Biswas J, Joshi N, Sharma S, Nath C, Singh S. 6-Hydroxydopamine and lipopolysaccharides induced DNA damage in astrocytes: involvement of nitric oxide and mitochondria. Mutat Res Genet Toxicol Environ Mutagen. 2015;778:22–36. doi: 10.1016/j.mrgentox.2014.12.007. [DOI] [PubMed] [Google Scholar]
- 34.Green AL, Ramkissoon SH, McCauley D, Jones K, Perry JA, Hsu JH, Ramkissoon LA, Maire CL, Hubbell-Engler B, Knoff DS, Shacham S, Ligon KL, Kung AL. Preclinical antitumor efficacy of selective exportin 1 inhibitors in glioblastoma. Neuro Oncol. 2015;17:697–707. doi: 10.1093/neuonc/nou303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sinha RA, Khare P, Rai A, Maurya SK, Pathak A, Mohan V, Nagar GK, Mudiam MK, Godbole MM, Bandyopadhyay S. Anti-apoptotic role of omega-3-fatty acids in developing brain: perinatal hypothyroid rat cerebellum as apoptotic model. Int J Dev Neurosci. 2009;27:377–383. doi: 10.1016/j.ijdevneu.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 36.Zhao L, Wientjes MG, Au JL. Evaluation of combination chemotherapy: integration of nonlinear regression, curve shift, isobologram, and combination index analyses. Clin Cancer Res. 2004;10:7994–8004. doi: 10.1158/1078-0432.CCR-04-1087. [DOI] [PubMed] [Google Scholar]
- 37.Lindsay J, Esposti MD, Gilmore AP. Bcl-2 proteins and mitochondria--specificity in membrane targeting for death. Biochim Biophys Acta. 2011;1813:532–539. doi: 10.1016/j.bbamcr.2010.10.017. [DOI] [PubMed] [Google Scholar]
- 38.Gross A, McDonnell JM, Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13:1899–1911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- 39.Das A, Banik NL, Ray SK. N-(4-Hydroxyphenyl) retinamide induced both differentiation and apoptosis in human glioblastoma T98G and U87MG cells. Brain Res. 2008;1227:207–215. doi: 10.1016/j.brainres.2008.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baudry M, Yao Y, Simmons D, Liu J, Bi X. Postnatal development of inflammation in a murine model of Niemann-Pick type C disease: immunohistochemical observations of microglia and astroglia. Exp Neurol. 2003;184:887–903. doi: 10.1016/S0014-4886(03)00345-5. [DOI] [PubMed] [Google Scholar]
- 41.Lin HW, Basu A, Druckman C, Cicchese M, Krady JK, Levison SW. Astrogliosis is delayed in type 1 interleukin-1 receptor-null mice following a penetrating brain injury. J Neuroinflammation. 2006;3:15. doi: 10.1186/1742-2094-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ericsson A, Liu C, Hart RP, Sawchenko PE. Type 1 interleukin-1 receptor in the rat brain: distribution, regulation, and relationship to sites of IL-1-induced cellular activation. J Comp Neurol. 1995;361:681–698. doi: 10.1002/cne.903610410. [DOI] [PubMed] [Google Scholar]
- 43.Arend WP, Malyak M, Guthridge CJ, Gabay C. Interleukin-1 receptor antagonist: role in biology. Annu Rev Immunol. 1998;16:27–55. doi: 10.1146/annurev.immunol.16.1.27. [DOI] [PubMed] [Google Scholar]
- 44.Zhan C, Lu W. The blood-brain/tumor barriers: challenges and chances for malignant gliomas targeted drug delivery. Curr Pharm Biotechnol. 2012;13:2380–2387. doi: 10.2174/138920112803341798. [DOI] [PubMed] [Google Scholar]
- 45.Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sawada M, Nakashima S, Banno Y, Yamakawa H, Hayashi K, Takenaka K, Nishimura Y, Sakai N, Nozawa Y. Ordering of ceramide formation, caspase activation, and Bax/Bcl-2 expression during etoposide-induced apoptosis in C6 glioma cells. Cell Death Differ. 2000;7:761–772. doi: 10.1038/sj.cdd.4400711. [DOI] [PubMed] [Google Scholar]
- 47.Cheng AC, Tsai ML, Liu CM, Lee MF, Nagabhushanam K, Ho CT, Pan MH. Garcinol inhibits cell growth in hepatocellular carcinoma Hep3B cells through induction of ROS-dependent apoptosis. Food Funct. 2010;1:301–307. doi: 10.1039/c0fo00134a. [DOI] [PubMed] [Google Scholar]
- 48.Korsmeyer SJ, Shutter JR, Veis DJ, Merry DE, Oltvai ZN. Bcl-2/Bax: a rheostat that regulates an anti-oxidant pathway and cell death. Semin Cancer Biol. 1993;4:327–332. [PubMed] [Google Scholar]
- 49.Wybranska I, Polus A, Mikolajczyk M, Knapp A, Sliwa A, Zapala B, Staszel T, Dembinska-Kiec A. Apoptosis-related gene expression in glioblastoma (LN-18) and medulloblastoma (Daoy) cell lines. Hum Cell. 2013;26:137–148. doi: 10.1007/s13577-011-0029-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Papi A, Tatenhorst L, Terwel D, Hermes M, Kummer MP, Orlandi M, Heneka MT. PPARgamma and RXRgamma ligands act synergistically as potent antineoplastic agents in vitro and in vivo glioma models. J Neurochem. 2009;109:1779–1790. doi: 10.1111/j.1471-4159.2009.06111.x. [DOI] [PubMed] [Google Scholar]
- 51.Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2:647–656. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- 52.Mares V, Lisa V, Malik R, Kozakova H, Sedo A. Cisplatin induced gamma-glutamyltransferase up-regulation, hypertrophy and differentiation in astrocytic glioma cells in culture. Histol Histopathol. 2003;18:687–693. doi: 10.14670/HH-18.687. [DOI] [PubMed] [Google Scholar]
- 53.Bernardi A, Frozza RL, Hoppe JB, Salbego C, Pohlmann AR, Battastini AM, Guterres SS. The antiproliferative effect of indomethacinloaded lipid-core nanocapsules in glioma cells is mediated by cell cycle regulation, differentiation, and the inhibition of survival pathways. Int J Nanomedicine. 2013;8:711–728. doi: 10.2147/IJN.S40284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kam AY, Tse TT, Kwan DH, Wong YH. Formyl peptide receptor like 1 differentially requires mitogen-activated protein kinases for the induction of glial fibrillary acidic protein and interleukin-1alpha in human U87 astrocytoma cells. Cell Signal. 2007;19:2106–2117. doi: 10.1016/j.cellsig.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 55.Park YK, Hong H, Jang BC. Transcriptional and translational regulation of COX-2 expression by cadmium in C6 glioma cells. Int J Mol Med. 2012;30:960–966. doi: 10.3892/ijmm.2012.1052. [DOI] [PubMed] [Google Scholar]
- 56.Kim SM, Park JG, Baek WK, Suh MH, Lee H, Yoo SK, Jung KH, Suh SI, Jang BC. Cadmium specifically induces MKP-1 expression via the glutathione depletion-mediated p38 MAPK activation in C6 glioma cells. Neurosci Lett. 2008;440:289–293. doi: 10.1016/j.neulet.2008.05.064. [DOI] [PubMed] [Google Scholar]
- 57.Posser T, de Aguiar CB, Garcez RC, Rossi FM, Oliveira CS, Trentin AG, Neto VM, Leal RB. Exposure of C6 glioma cells to Pb(II) increases the phosphorylation of p38(MAPK) and JNK1/2 but not of ERK1/2. Arch Toxicol. 2007;81:407–414. doi: 10.1007/s00204-007-0177-6. [DOI] [PubMed] [Google Scholar]