

Abstract
Duchenne muscular dystrophy (DMD) is an X-linked recessive genetic disorder caused by mutations in the dystrophin gene. Affecting approximately 1 in 3,600-9337 boys, DMD patients exhibit progressive muscle degeneration leading to fatality as a result of heart or respiratory failure. Despite the severity and prevalence of the disease, there is no cure available. While murine models have been successfully used in illustrating the mechanisms of DMD, their utility in DMD research is limited due to their mild disease phenotypes such as lack of severe skeletal muscle and cardiac symptoms. To address the discrepancy between the severity of disease displayed by murine models and human DMD patients, dystrophin-deficient dog models with a splice site mutation in intron 6 were established. Examples of these are Golden Retriever muscular dystrophy and beagle-based Canine X-linked muscular dystrophy. These large animal models are widely employed in therapeutic DMD research due to their close resemblance to the severity of human patient symptoms. Recently, genetically tailored porcine models of DMD with deleted exon 52 were developed by our group and others, and can potentially act as a new large animal model. While therapeutic outcomes derived from these large animal models can be more reliably extrapolated to DMD patients, a comprehensive understanding of these models is still needed. This paper will discuss recent progress and future directions of DMD studies with large animal models such as canine and porcine models.
Keywords: Duchenne muscular dystrophy (DMD), golden reliever muscular dystrophy (GRMD), canine X-linked muscular dystrophy (CXMD), porcine (pig) model, exon skipping, hypertrophic feline muscular dystrophy (HFMD)
Introduction
Duchenne Muscular Dystrophy (DMD) is the most common lethal genetic disorder characterized by progressive muscle degeneration [1,2]. The early onsets of symptoms of DMD start at 2-4 years of age, including poor posture and weakness of the limb muscles, followed by gait disturbances [3]. As DMD progresses, it becomes fatal due to cardiac or respiratory failure by the age of 20-30 years. While the allelic mutation can affect any of the 79 exons pertaining to the dystrophin gene, exons 2-20 and exons 45-55 are common hotspots of large deletion and duplication mutations (≥1 exon) [4]. The most common form of mutations causing DMD are large deletion mutations (68%). However, large duplication mutations (11%), point mutations (11%), and small insertion/deletion mutations (7%) are also frequent [4]. These gene mutations often lead to a frameshift mutation to produce a premature stop codon (out-of-frame) and result in a lack of dystrophin protein - a protein essential to stabilize muscle membrane [3]. Another form of muscular dystrophy called Becker Muscular Dystrophy (BMD) is the milder version of DMD and is caused by in-frame mutations in most cases. This leads to a shorter, but functional dystrophin protein. While corticosteroid treatment is currently available for DMD patients to reduce muscle strength deterioration, there is still no cure for DMD [5]. Currently, therapeutic strategies for DMD focus on dystrophin replacement, compensation for dystrophin, reduction of fibrosis, and muscle regeneration [6]. Although many promising therapeutics reach the clinical trials [7], they do not always yield expected results [8]. To date, various mammalian models of DMD including mice, rats, dogs, cats, monkeys, and pigs with mutations in the DMD gene have been developed to assist in our understanding of pathogenesis and translating basic research findings into clinically useful therapeutics [9-14]. Murine DMD models are the most widely used, but there are several important limitations such as gaping differences in physiology from humans and display of milder phenotypes compared to patients. To address this issue, canine DMD models emerged as an important alternative in testing in vivo efficacy and toxicity of therapeutic applications. As well, recent genetic engineering allows for generating of a new pig DMD model with a deletion mutation in the hot spot region of the human DMD gene [13]. While outcomes from these large animal models could be more reliable to be extrapolated to patients, comprehensive understanding of their characteristics is needed for promoting development of new drugs and therapeutic approaches. In this review, we focus on recent progress and future directions of DMD studies with the dog (canine) and newly developed pig (porcine) models of DMD.
Canine models
In DMD patients, one-third of cases are due to de novo mutations in the DMD gene which is composed of 79 exons made up of 2.2 million base pairs [15,16]. Similarly, DMD mutations within dystrophin-deficient dogs occur spontaneously. Dystrophin-deficient muscular dystrophy in dogs has only been reported in several breeds as summarized in Table 1: Alaskan Malamute [17], Bergamasco [18], Belgian Groenendaeler Shepherd [19], Cavalier King Charles Spaniel [20], Cocker Spaniel [21], German Short-haired Pointer [22], Golden Retriever [23], Grand Basset Griffon Vendéen [24], Irish Terriers [25], Japanese Spitz [26], Labrador Retriever [27-29], Lurcher Siblings [30], Norfolk Terrier [31], Old English Sheepdog [32], Pembroke Welsh Corgi [33], Rat Terrier [34], Rottweiler [21], Tibetan Terrier [21], Unknown Mix [18], and Weimaraner [35]. While correlation between incidence of the disease and the particular breed of dogs has yet to be determined, it seems that the occurrence of dystrophin deficiency in dogs is unrelated to its genetic background [36]. Additionally, the mutation spectrum is unknown in dystrophin-deficient dogs due to limited numbers of cases and unidentified mutations in most of the affected dogs. In 1992, Sharp et al. first identified a mutation of the canine DMD gene in the Golden Retriever muscular dystrophy (GRMD) and it has become the most extensively examined and characterized canine model in several institutes [23]. Since then at least nine patterns of spontaneous dystrophin mutations have been reported in different breeds: a large deletion mutation of whole exons in the dystrophin gene of German Short-haired Pointers [22], an acceptor splice site mutation of intron 6 in the Golden Retriever [23], a deletion mutation of exons 8-29 in Tibetan Terriers [21], long interspersed repetitive element-1 insertion in intron 13 of Pembroke Welsh Corgis [33], an inversion mutation with a break point in intron 19 of the Japanese Spitz [26], an insertion of repetitive element in intron 19 of Labrador Retrievers [27], a point mutation at donor splice site in intron 50 of Cavalier King Charles Spaniels [20], a nonsense mutation in exon 58 of the Rottweiler [21] and lastly a small deletion mutation of 4 nucleotides in exon 65 of Cocker Spaniels [21]. Over the years, several dystrophin-deficient canine models have been established. The GRMD dogs were backcrossed with Beagle breed by Shimatsu et al. at National Center of Neurology and Psychiatry (NCNP) Japan to produce the Canine X-linked muscular dystrophy in Japan (CXMDJ) [37]. Although a colony has not been established for DMD-like Cavalier King Charles Spaniel Muscular Dystrophy (CKCS-MD), another canine model candidate, it has been reported and tested for exon skipping therapy in vitro [20]. At Auburn University (USA) a colony of dystrophin-deficient Pembroke Welsh Corgi was established by outbreeding with Beagles [33]. Of the various canine models, GRMD and CXMDJ are currently maintained as active colonies for analysis of muscular dystrophy pathogenesis and new drug development. The biggest advantage of these canine models compared to mouse models is that they show phenotypes closer to human DMD in skeletal muscle and cardiac muscles at a young age. While cardiac symptoms are one of the main causes of death for human patients [38], mouse models do not show severe cardiac symptoms. On the other hand, GRMD and CXMDJ models show severe clinical symptoms such as body wide muscle weakness and cardiac symptoms (Table 2). They also show various human DMD-like phenotypes such as joint contracture and kyphosis. The early onset of disease phenotypes in GRMD and CXMDJ enables more detailed analysis such as clinical grading, magnetic resonance imaging, electrocardiogram (ECG) and echocardiography. The models also have larger body weights and longer lifespan compared to mouse models making them more reflective of human disease for use in toxicological studies. Overall the canine models display more accurate representations of human DMD symptoms compared to the widely used mouse models.
Table 1.
Dystrophin-deficient dogs and their mutations
| Breed & original ref. | Mutation | Note |
|---|---|---|
| Alaskan Malamute [17] | Not identified | |
| Beagle [37,42] | A point mutation in intron 6 accepter splice site | Naturally occurring affected dogs not reported. CXMD and CXMDJ models were generated by outbreeding with GRMD an dartificial insemination with GRMD semen, respectively. Employed to test exon skipping and gene transfer [55,63,65] |
| Bergamasco [18] | Not identified | Affected female with high CK level (12,336 IU/l) and truncated dystrophin protein probably lacking C-terminal |
| Belgian Groenendaeler Shepherd [19] | Not identified | |
| Cavalier King Charles Spaniel [20] | A point mutation of a spicing donor site in intron 50 | Exon skipping tested with Eteplirsen in cultured skeletal muscle cells |
| Cocker Spaniel [21] | Small deletion mutation of four nucleotides in exon 65 | |
| German Short-haired Pointer [22] | Deletion mutation of the entire dystrophin gene including promoter and 3’ untranslated regions | |
| Golden Retriever [23] | A point mutation in intron 6 accepter splice site | GRMD is the most widely characterized and employed in preclinical trials for various therapeutic applications [52] |
| Grand Basset Griffon Vendéen [24] | Not identified | |
| Irish Terriers [25] | Not identified | |
| Japanese Spitz [26] | An inversion with break points in intron 19 of the dystrophin gene and in the RPGR gene | |
| Labrador Retriever [27] | Repetitive element insertion in intron 19 | |
| Labrador Retriever [29] | Not identified | Reduced dystrophin expression like BMD |
| Lurcher Sibling [30] | Not identified | |
| Norfolk Terrier [31] | Not identified | |
| Old English Sheepdog [32] | Not identified | |
| Pembroke Welsh Corgi [33] | Long interspersed repetitive element-1 insertion in intron 13 | Outbred to Beagle breed to establish a colony |
| Rat Terrier [34] | Not identified | Decreased level of dystrophin protein |
| Rottweiler [21] | A nonsense point mutation in exon 58 | |
| Tibetan Terrier [21] | Deletion mutation of exons 8-29 | |
| Unknown mix [18] | Not identified | Affected female with high CK level (45,000 IU/l) and no dystrophin expression |
| Weimaraner [35] | Not identified |
Table 2.
Clinical and pathological features with age in GRMD and CXMDJ
| GRMD | CXMDJ | ||
|---|---|---|---|
| General | Breed | Golden Retrievers | Beagles at 3rd or more generation by AI with GRMD semen |
| Mortality rate in pups | 4.8-32.1% within 14 days after birth (n=74-106) *The rate increases with inbreeding% [47] | 32.3% (10/31) by 3 days after birth (n=31) (13.3% in normal pups from carrier female; n=45) [48] | |
| Body weight | Mean 18.9 kg ± 1.1 (± SEM) at <9-12 mo (n=14) (Normal, 28.1 kg ± 2.3 [± SEM], n=8) [58] 7.5 kg ± 1.2 (± SD, normal 10.7 kg ± 1.8), 11.4 kg ± 2.2 (16.6 kg ± 2.0), 12.9 kg ± 3.1 (20.2 kg ± 2.3), 18.2 kg ± 3.2 (23.8 kg ± 1.7) at 3, 4.5, 6 and 12 mo, respectively (n=9-17) [6] [105] | Mean 11.9 kg ± 2.0 (± SD) between 6 and 21 mo (n=8) (Normal, 13.3 kg ± 1.0 [SD] between 6 and 21 mo [n=4]) [49] | |
| Serum CK level | Mean 24.2 K IU/l at 2 day after birth (n=2) Temporal decrease to mean 3.9 K IU/l (n=4) at 1 wk and then increase in range of 6.5-162 K IU/l by 6 mo (n=2-4) (0.2 - 1.0 K IU/L in normal dogs by 6 mo [n=3-9]) [42] | Significant increase to around 100 K U/l within 1 h after birth (n=6) [51] Mean 120 K IU/l at 1 day after birth, temporal decrease to mean 11 K IU/l at 3 weeks, increase to mean 85 K IU/l at 2 mo, and then gradual decrease by 12 mo [48] 12.5-138 K IU/l between 6 and 21 mo (n=8) (60 - 515 IU/l in normal dogs at 6-21 mo [n=4]) [49] | |
| Skeletal muscle | Gait/mobility disturbance | Initial sign at 8 wks [103] Significant impairments in speed, stride length, and power/force from 2 mo (n=12) [104] Significant tarsal joint contracture from 3 mo (n=9-17) [105] A loss of ambulation in 85% (n=94) dogs by 6 mo [104] | Initial sign of sitting or bunny hops at 2 mo Languor or difficulty in jumping/moving in 100% (5/5) dogs from 4 mo Bunny hops with hind limbs, shuffling walk or inability to walk in 100% (5/5) dogs from 6 mo No complete loss of locomotion by 12 mo (n=5) [48] |
| Muscle atrophy | Marked atrophy in truncal and temporal muscles at 2-3 mo followed by firm limb muscles Progress of atrophy in body-wide muscles by 6 mo [42] | Initial sign in distal limb and temporal muscles at 2 mo and in proximal limb muscle at 4 mo Hardness or thin in the limb and temporal muscles of 100% (5/5) dogs at 12 mo [48] | |
| Abnormal posture | Small tarsal joint angle from 3 mo [105] Lumbar kyphosis at 6 mo to lordosis by 14 mo [42] | Kyphosis at 12 mo [48] | |
| Respiratory dysfunction | Significant increase in expiratory flows (n=10) and abnormal abdominal respiration in 70% (7/10) dogs within 2.5 mo [45] Reduced ventilatory capacity with diaphragmatic abnormality in morphology and function in dogs between 6 and 17 mo (n=6-7) [46] | ||
| Dysphagia | Difficulty in eating foods with progressive macroglossia and continuous drooling at ~14 mo [42] | Initial sign with macroglossia and drooling at 4 mo Difficulty in taking foods and severe enlarged and thickened tongue in 100% (5/5) dogs at 12 mo [48] | |
| Jaw mobility | Inability to open jaw at 2-3 mo [42] | Significant inability from 4 mo (n=5) [48] | |
| Skeletal muscle | Histology | Severe degeneration (hyalinization, calcium accumulation, necrosis and/or central nucleation) in proximal limb, respiratory (diaphragm and intercostal), truncal and cervical muscles and enlarged fibers in tongue at 1-8 days after birth Invasion of histiocytic cells in the severe muscles from 4-6 days after birth [40] Extensive fibrosis and fiber size variation invastus lateralis muscle from 15 days (n=3) [41] Slight fibrosis and marked variation of fiber size with the above degeneration types in most skeletal muscles from 2 mo [40] Progression of the above histological features at least by 8 mo [103] Occasional fat infiltration from more than 13 mo [106] | Degeneration of hyaline/opaque fibers and slight infiltration of neutrophils in diaphragm within 1 h after birth Diffuse necrosis with oedema in diaphragm of neonates at 3 days [51] Increase in necrosis, fibrosis, and invasion of inflammatory cells in TA muscle at 2 mo Marked fibrosis, fiber size variation, centrally nucleated fibers in diaphragm and TA muscles at 6 mo [48] |
| Cardiac muscle | Electrocardiogram | Significantly increased heart rate at 9-12 mo (n=14) [58] Deep Q wave and increased Q/R ratio in 3/6 dogs between 6 mo and >2 yrs [107] Significantly shortened PR interval and increased Q/R ratio in dogs between 12-18 mo (n=8) [108] | Normal heart rate by 12 mo and the increase rate from 15 mo Deep Q wave and increased Q/R ratio from 6 mo Shortened PQ intervals from 15 mo [49] |
| Echocardiogram | Hyper echogenic lesion of LV wall in 73% (8/11) dogs at 6-7 mo [107] Significant thickening of free wall and septum in LV diastolic and systolic at 9-12 mo (n=14) Significantly reduced fractional shortening in dogs between 9-12 mo (n=14) [58] DCM in 55% (6/11) dogs between 7 mo and 9 yrs. (the earliest age at 22 mo) [21] | Hyper echogenic lesion of LV posterior wall in 60% (3/5) dogs at 12 mo No change in thickness of by 21 mo No obvious abnormality in fractional shortening by 21 mo [49] | |
| Hemodynamics | Significant hypertension with high pressure of ventricular and artery in dogs between 12 and 18 mo (n=8) [108] | ||
| Histology | Mineralization in LV papillary and free wall and septum from 6.5 mo [44] Increase in abnormal mitochondria and attenuated myofibrils from ~6.5 mo Fibrosis in LV and RV myocardium from 12 mo [44] Occasional fat infiltration from 1-1.5 mo [44] | Vacuole degeneration of Purkinje fibers in 100% (4/4) dogs at 4 mo Fragmentation of mitochondria and disruption of myofibrils in ventricular myocardium and Purkinje fibers at 4 mo No fibrosis by 13 mo and mild fibrosis in LV myocardium from 15 mo. Intact in RV myocardium by 21 mo [50] |
AI, artificial insemination; CK, creatine kinase, LV, left ventricular; RV, right ventricular; DCM, dilated cardiomyopathy.
The Golden Retriever muscular dystrophy (GRMD) model
The most extensively studied and best-characterized canine model is the GRMD. Its clinical and pathological features are summarized in Table 2. CRMD is the result of a point mutation on the intron 6 splice acceptor site which leads to exon 7 skipping (Figure 1) [23]. The point mutation results in a premature stop codon in exon 8, leading to a lack of dystrophin protein. Splice site mutations are reported in 3.0-5.8% of human DMD patients [4,39]. The advantage of the GRMD model over mouse models is that it exhibits similar symptoms to human DMD patients. The GRMD experiences progressive muscle degeneration in both the skeletal muscles and cardiac muscles resulting in abnormal gait with joint contracture, muscle atrophy and respiratory dysfunction at an early age (Table 2). In particular, degenerative fibers are observed soon after birth in active skeletal muscles such as the tongue, respiratory, limb, truncal and cervical muscles in the GRMD model [40]. Extensive fibrosis and size variation of muscle fibers are also reported in the vastus lateralis muscle 15 days after birth [41]. As a result of muscle damages, neonates with the GRMD depict high levels of serum creatine kinase (CK) [40,42]. The GRMD heart exhibits abnormalities similar to DMD patients in electrocardiogram and echocardiogram at the age of 6 months (Table 2) [43]. Calcification and fibrosis in ventricular myocardium are also observed at an early age of 6.5 and 12 months, respectively [44]. Mouse models however usually do not show such histological change in the heart at such an early age [9]. In addition, the GRMD model starts to exhibit respiratory difficulties between 2.5 and 17 months of age due to reduced function of the diaphragm as a result of degeneration [45,46]. Since cardiac and respiratory failures are the two leading causes of death for human DMD patients, the GRMD is useful as an animal model to study disease state of DMD and to test the efficacy of novel therapeutics. However, some differences are reported in symptoms between humans and dogs such as high mortality rate during breeding as a result of severe respiratory failure and intestinal infection at the neonatal stage [47]. Nevertheless, the advantage of the GRMD model to exhibit disease states at 6 month of age comparable to that of human DMD patients is clear.
Figure 1.
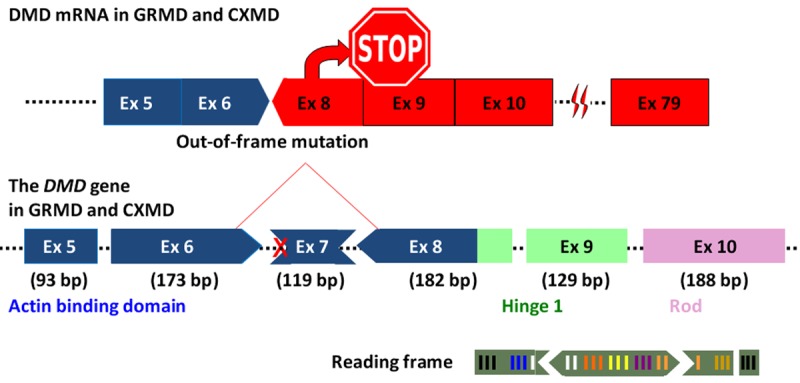
The mutation in GRMD and CXMD. The image illustrates the mutation pattern in dystrophic dogs GRMD and CXMD harboring a splice site mutation at the border of intron 6 and exon 7. Dotted lines indicate introns.
Canine X-linked muscular dystrophy (CXMD) model
The beagle-based CXMD models are generated by crossbreeding with the GRMD or artificial insemination with the GRMD spermatozoa [37,42]. Hence, it shares the same splice site mutation in intron 6 as the GRMD, leading to a lack of exon 7 in mRNA and subsequent absence of dystrophin protein (Figure 1). Since CXMD are smaller and easier to handle than the GRMD model, they are a useful animal model for DMD studies (Table 2). Currently, an active group of CXMD is maintained at NCNP in Japan to help with understanding and developing new therapeutic applications for DMD (called CXMDJ). Researchers at NCNP produced three generations of the CXMDJ model and characterized each generation physiologically and histologically [37,48-50]. By the third generation, the mortality rate of healthy beagles is 13.3% after 3 days whereas mortality in the dystrophic CXMDJ is significantly higher at 32.3% [37]. This high mortality rate is similar to 32.1% of neonatal mortality observed in GRMD dogs with high inbreeding rate (Table 2). Additionally, the serum CK level of the neonatal CXMDJ model significantly increased to approx. 100 K U/L within 1 hour after birth [51]. The CK level from venous blood of the newborn CXMDJ dogs is 7 times higher compared to the blood from umbilical cord. The observed high serum CK levels could be the result of diaphragm damage due to stress from initial pulmonary respiration. The change of serum CK levels with age is a common feature amongst CXMDJ and GRMD models (Table 2). The pulmonary stress triggers over-expression of inflammation and immune response stimulators such as IL-6, IL-8, COX-2, and selectin E as well as expression of immediate-early genes: c-fos and egr-1. Symptoms include gait disturbance, muscle atrophy, kyphosis, dysphagia, macroglossia, drooling, and jaw joint contracture. However, these clinical symptoms progress slower than that of the GRMD model (Table 2). Degenerative myofibers are observed in diaphragm of CXMDJ and selective muscles such as respiratory and limb muscles within a few days after birth [40,51]. Interstitial fibrosis in skeletal muscle is also observed in both GRMD and CXMDJ dog models at a young age [40,41,48]. In addition, studies of the conduction system in the heart called Purkinje fibres show vacuolar degeneration by 4 months of age as well as an overexpression of Dp71, a shorter isoform product of the DMD gene [50]. As a result of Purkinje fibre degeneration, abnormalities in cardiac signal conduction become clinically obvious in ECG and echocardiogram as early as 6 months of age in the CXMDJ model. Similar to DMD patients, both CXMDJ and GRMD models exhibit deep Q-waves and an increase in Q/R ratio in ECG and hyperechogenic lesion in the left ventricular wall at a young age (Table 2). Fibrosis in the left ventricular myocardium is also observed from the age of 12 and 15 months in GRMD and CXMDJ models, respectively. As summarized in Table 2, the CXMDJ model has milder symptoms and slower progression of disease in skeletal and cardiac muscles compared to the GRMD model.
Application of GRMD and CXMDJ models in preclinical trials
As described above, GRMD and CXMDJ models have the same acceptor splice site mutation in intron 6 and similar clinical and histological traits. Thus, basic strategies to treating dystrophic symptoms of these models are shared. Prior studies on these models include adeno-associated virus (AAV)-mediated and antisense oligo (AO)-mediated exon skipping, gene therapies, cell therapy, and drug therapy [52]. Amongst these potential therapies, exon skipping demonstrates successful therapeutic outcomes in GRMD and CXMDJ models. Specifically, to treat the splice site mutation in intron 6 within the dog models, exon skipping can be used to splice out exons 6 and 8 of dystrophin mRNA to correct the reading frame (Figure 2). This will leads to a truncated but still functional dystrophin protein [53-55]. In the GRMD model, exon skipping with AAV vectors showed restored dystrophin levels and improved muscle function. Specifically, Vulin et al. conducted skipping of exons 6 and 8 of mutated dystrophin mRNA using AAV-U7 [56]. The study reveals restoration of dystrophin-glycoprotein complex, down regulation of utrophin levels and doubling of tetanic strength of contralateral muscle four months after injection. Similarly, recombinant AAV6- and optimized AAV-U7-mediated exon skipping enable restoration of cardiac dystrophin levels in the GRMD [57]. Additional therapies explored using the GRMD model include restoration of left ventricular function using Bradykinin [58,59], increased α7 integrin to modify disease progression using prednisone, stabilization of basal lamina using laminin-α2 [60] and decreased inflammation and rescued dystrophin-glycoprotein complex using Bortezomib [61].
Figure 2.
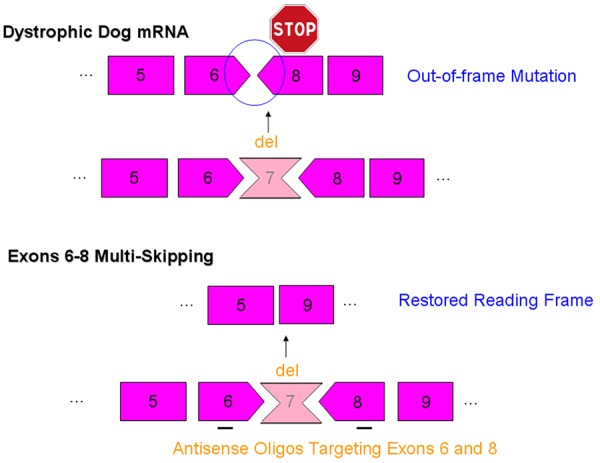
Exons 6-8 multi-skipping of dystrophic dog mRNA. The loss of function of dystrophin protein due to an out-of-frame mutation in dystrophin mRNA can be rescued using an antisense oligo cocktail targeting exons 6 and 8.
The CXMDJ model has been utilized to examine the therapeutic efficiency of exons 6 and 8 skipping with a cocktail of AOs with phosphorodiamidate morpholino oligomers (PMOs), 2’O-methyl AOs (phosphorothioate), or octa-guanidine dendrimer conjugated PMOs (Vivo-morpholinos or vPMOs) [55,62-64]. Intramuscular injections of cocktail PMOs targeting exons 6 and 8 on CXMDJ resulted in dystrophin expression up to 50% of normal levels [55,63]. As well, dystrophin-associated proteins such as nNOS and alpha-sarcoglycan, were restored and muscle inflammation was extensively diminished. After systemic injections of the PMO cocktail, physical examinations on treatment group revealed functional recovery. Specifically, treated dogs were able to run faster in 15 meter timed test compared to the control untreated CXMDJ group. Additionally, vPMO cocktail injection into limb muscles was able to induce extensive dystrophin protein expression [64]. As well, a recent study with the CXMDJ model illustrated that injection of an internally truncated but still functional dystrophin called micro-dystrophin via intra-amniotic injections with the recombinant AAV vector for CXMDJ fetuses led to significant improvement of skeletal muscle and cardiac functions [65,66].
Cavalier King Charles Spaniel model
Three cases of Cavalier King Charles Spaniel muscular dystrophy (CKCS-MD) discovered in the United Kingdom and the United States were characterized molecularly and histologically [20]. It was determined that the CKCS-MD has a point mutation at 5’ donor splice site in intron 50 of the canine DMD gene. The mutation leads to a deletion of exon 50 in mRNA, leading to a premature stop codon in exon 51 and finally results in a lack of dystrophin protein. Since exon 50 falls along the mutation hot spot of the human DMD gene, therapeutic methods examined on CKCS-MD can be more easily translated for application to human DMD patients. Clinical features in CKCS-MD include high CK level (approx. 33-65 KU/L), macroglossia, restriction of jaw movement, and progressive dysphagia. However, there was no observed histologicalchange in the heart of CKCS-MD. While the GRMD and CXMDJ models are commonly used in translational research, these models are large, weighing approximately 19 kg and 12 kg respectively. On the other hand, the CKCS-MD only weighs 5-8 kg so that they are more manageable and more economically feasible to use in preclinical tests. Importantly, the CKCS-MD is amenable to exon 51 skipping, which can potentially address the need of the largest number of DMD patients [4]. Indeed, Walmsley et al. tested a DMD drug candidate called eteplirsen to induce exon 51 skipping in skeletal muscle cells of the CKCS-MD. The results indicate presence of exon 51-skipped dystrophin transcript and induction of the dystrophin protein [20]. Intriguingly, although eteplirsen has a difference of 2 bps at the position of canine DMD exon 51, it could still successfully induce skipping of the exon and expression the protein. While the CKCS-MD model is useful for preclinical testing of antisense-mediated exon skipping therapy targeting mutational hot spot of exons 45-55 region in DMD, it could also be useful for other mutation specific therapies such as gene therapy with AAV vectors. Due to its shared mutation region with the human DMD gene, the CKCS-MD model can reasonably be expected to become an effective dog model for testing DMD therapies in preclinical trials.
Challenges in canine models
Canine models such as GRMD and CXMDJ possess many of the clinical manifestations seen in human DMD patients including progressive muscle weakness, fibrosis, joint contractures, and eventual death from cardiac and respiratory failure as summarized in Table 2. However, variability in disease severity ranging from mild to extremely severe phenotype is found among littermates [21,42,49,67,68]. The inconsistency in disease phenotype most likely occurs due to differences in the genetic background of the models as a result of modifier genes. As such, modifier genes can complicate our interpretation of the effectiveness of therapeutic interventions [69]. The increase in mortality rate during inbreeding of a GRMD colony may also be associated with modifier genes through genetic drift. A solution for the modification issue may be to mate with xenoplastic dogs or with different breeds in order to reduce coinheritance of modifier genes from one consanguinity or breed. Indeed, experiments have been conducted by outbreeding the GRMD with Yellow Labrador Retrievers, which have large body size close to the GRMD. However, the outbred-GRMD exhibits milder degeneration of dystrophic muscles than the inbred-GRMD [42,70]. The second hurdle with current canine models is that they are limited to a point mutation in intron 6 accepter splice site. While the use of genetic engineering to generate mutations more representative of human DMD are reported in mice, rats, monkeys, and pigs, it is not used to generate canine models. Genetic engineering is currently not a viable option for dogs because somatic cell nuclear transfer (SCNT) to generate new transgenic canine models is cumbersome and inefficient compared to other animals [71]. However, companion dogs do make frequent visitation to veterinary clinics, as reported in Table 1. Hence, by diligently identifying mutations and keeping carrier female dogs to produce offspring, we can potentially generate new dog models with mutant DMD genes of interest. Finally, since dogs are common companion animals, the lack of social acceptance to the use of dogs as experimental subjects and greater emotional attachment of researchers will become logistical and workplace challenges. Although there are some restrictions, generating canine DMD models with different mutations and/or severity will allow researchers to examine DMD pathogenesis more profoundly and efficacy of new therapeutic applications.
Porcine models
Currently, domestic pigs are widely accepted as an adequate model in biomedical research. Efficient technology now exists for gene targeting in pigs, whose advantages include uniform genetic backgrounds and the similarity in size, anatomy and physiology to humans [72,73]. Particularly, similarity in the cardiovascular system of pigs compared to those of humans is helpful for understanding human cardiovascular diseases and developing new drugs and therapies [74]. Wolf et al. has recently generated genetically modified porcine model of DMD using a combination of gene targeting with nuclear transfer techniques [13]. Our group in collaboration with Exemplar Genetics LLC has also developed a porcine DMD model (http://www.exemplargenetics.com/) (Unpublished). Both pig models have a deletion mutation of exon 52 in the DMD gene. The porcine DMD model established by Wolf et al. shows similar phenotypes to human DMD patients. With recent advancement towards establishing novel animal DMD models, preclinical trials with pig model may become an unavoidable step for developing new drugs and therapeutic applications to DMD. In the following sections, we will describe and discuss the recent developments and future applications of dystrophin-deficient pig models.
Dystrophin-deficient porcine models
In domesticated pigs, a spontaneous point mutation in the DMD gene has been identified by genome-wide association analysis. It is associated with a novel porcine stress syndrome characterized by muscle rigidity, muscle degeneration, and labored breathing [75]. In affected pigs, a missense mutation (C>T, R1958W) in exon 41 of the DMD gene was identified as the causative gene. Affected pigs exhibit reduction of dystrophin protein in both skeletal and cardiac muscles. A colony of this mutant pig was then established to examine their phenotypes [14]. Detailed analysis of the pig model shows 70% reduction of dystrophin protein in the diaphragm and psoas muscles, 80-85% reduction of dystrophin-associated alpha-sarcoglycan, fibrosis, and five times higher serum CK level compared to normal litters. The observed reduction in dystrophin protein makes this the first pig model of BMD. Although this porcine model is useful to study dystrophinopathy, missense mutations present in the porcine model do not lead to severe DMD symptoms [39]. In addition, missense mutations only account for 0.4% of all of the DMD mutations in humans [4].
In order to create a mutation model more relevant to DMD patients, Wolf et al. successfully generated dystrophin-deficient pigs with deletion in exon 52 of the DMD gene. This is accomplished by using a combination of gene targeting with SCNT [13]. SCNT is a technique involving the removal of the nucleus of oocyte and then implanting it with a donor cell nucleus from a somatic cell. The use of SCNT to genetically engineer donor cells such as fetal fibroblasts is currently the most widely used strategy to generate transgenic pigs [76]. The genetically modified pig model of DMD exhibits absence of dystrophin in skeletal muscles as well as reduced levels of dystrophin-associated proteins such as alpha-sarcoglycan and beta-dystroglycan. The serum CK level of two affected newborn piglets at 3-day-old is at 1,647 and 2,117 U/L, which is at a level almost ten times higher than normal piglets at the same age. The elevated serum CK level is similar to differences in CK level found in DMD newborn boys (2,003-2,791 U/L) and normal newborn boys (251.5 ± 113.8) [77]. At 4 weeks old, the serum CK level in affected pigs dramatically increased to 21,000 and 63,000 U/L. Additionally, histological examination of skeletal muscles revealed similarity to human pathology which includes increased muscle regeneration (centrally nucleated fibers), interstitial fibrosis, and mononuclear inflammatory cells. Physical examination of the pig DMD model demonstrates impaired mobility during walk, trout and gallop tests. In particular, these pigs exhibited prominent features of shortened strides and stiff movements. In addition, they have greater difficulty climbing platforms compared to wild-type pigs. Beyond muscle weakness, pig DMD models have a maximum life span of 3 months. The lifespan of affected pigs with low birth weight are even shorter. The correlation suggests that the severity of DMD phenotype at birth may determine their life expectancy. Overall, this transgenic pig model exhibits comparative phenotypes to human patients. However, one area of concerns in this study is that only the first generation was subjected to characterization of their phenotypes. In transgenic pigs generated by SCNT with genetically modified cells, the first generation has been reported to show phenotypic instability such as pulmonary hypertension, respiratory distress, malformed limbs, and contracted tendons, due to epigenetic dysregulation [78]. Abnormalities found in the first generation of transgenic pigs are then improved in the second or third generations [79]. To improve upon the study, Wolf et al. have generated female porcine cells with the same deletion mutation (DMD +/Δex52). The female pig model will allow us to sufficiently examine phenotype of affected pigs after the first generation, ones with normal epigenetic regulation. Although detailed characterization of the pig model still remains to be determined (e.g., cardiac symptoms), it should be noted that desired mutations could be more conveniently introduced in pigs compared to canines.
Future applications of the porcine DMD model
Currently several clinical trials for therapies targeting DMD are in progress or have been completed. This includes antisense-mediated exon skipping and rAAV-mediated mini-dystrophin gene transfer [7]. In exon skipping with antisense oligonucleotide, phase lll clinical trial of drisapersen (ID: NCT01480245 and NCT01803412 by Prosensa Therapeutics) have been terminated and eteplirsen (ID: NCT02255552 by Sarepta Therapeutics) for skipping of exon 51 are scheduled in the future. Clinical trials of exon 53 skipping are also being conducted by NCNP, Japan (ID: NCT02081625) and Prosensa Therapeutics (ID: NCT01957059). Therapies focusing on exon 51 and 53 skipping could be applicable to 20.5% (14.0%) and 14.7% (10.1%) of patients with large deletion mutations (all of patients), respectively [4]. However, one of the major challenges in single exon skipping is the unknown function of various forms of truncated dystrophin proteins. In the current clinical trials, the efficacy of exon 51 and 53 skipping therapies were tested with patient skeletal muscle cells and/or by injections into transgenic mice with the human DMD gene, or with an exon 52 deletion mutation (mdx52 mice) [80-83]. The current porcine DMD model with exon 52 deletion mutation treatable by either exon 51 and 53 skipping is an encouraging alternative to examine the function of truncated dystrophin protein instead of the mdx52 mouse which exhibits milder phenotypes and different physiology from that of human patients (Figure 3) [9]. Likewise, the current porcine model can assist in the development of multiple exon skipping therapy targeting the entire exons 45-55 region to produce a more functional type of truncated dystrophin proteins (Figure 4). Of patients with in-frame mutations, those expressing a truncated dystrophin protein encoded by exons 45-55 are reported to be associated with milder phenotypes or asymptomatic course in 95% of cases examined [84-86]. The potential of exons 45-55 skipping extends to treating 63% of DMD patients with deletion mutations [84]. The feasibility of this multiple exon skipping has been demonstrated in mdx52 mouse treated with 10-Vivo-morpholino cocktail [87,88]. Following exon 45-55 skipping, results indicate induction of truncated dystrophin protein and dystrophin-associated proteins in the body wide skeletal muscles. However, due to milder phenotypic manifestations of commonly used mdx52 mouse model, functional recovery has not been fully examined. Hence, the use of porcine DMD model can allow us to fully investigate the effectiveness of novel multiple exon skipping therapy. Multiple exon skipping using a cocktail of antisense oligonucleotides is technically challenging [89]. To achieve efficacious skipping, more toxic new generation morpholinos such as Vivo-morpholinos and peptide-conjugated morpholinos may need to be used [90]. As such, the availability of porcine DMD model with similar physiology to human DMD patients will become important in assessing the toxicity of novel therapies. All in all, exon 52-deleted pig model with similar severity to DMD patients will enable us to assess recovery of function and safety of new drugs/applications such as exons 45-55 skipping.
Figure 3.
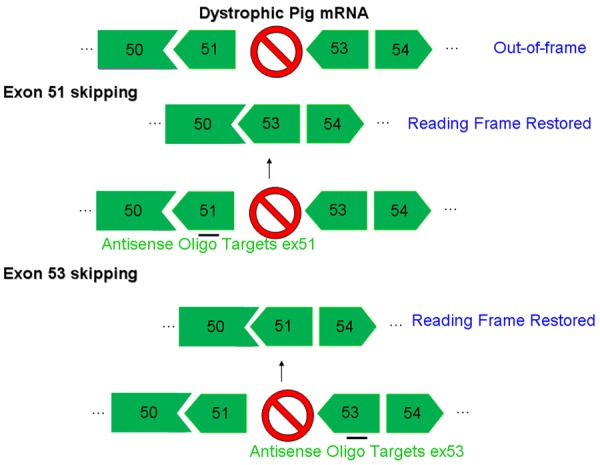
Exon 51/53 skipping of dystrophic pig mRNA. The loss of dystrophin protein due to the frame shift mutation in dystrophin mRNA can be rescued using an antisense oligo targeting either exon 51 or 53 to restore the reading frame.
Figure 4.
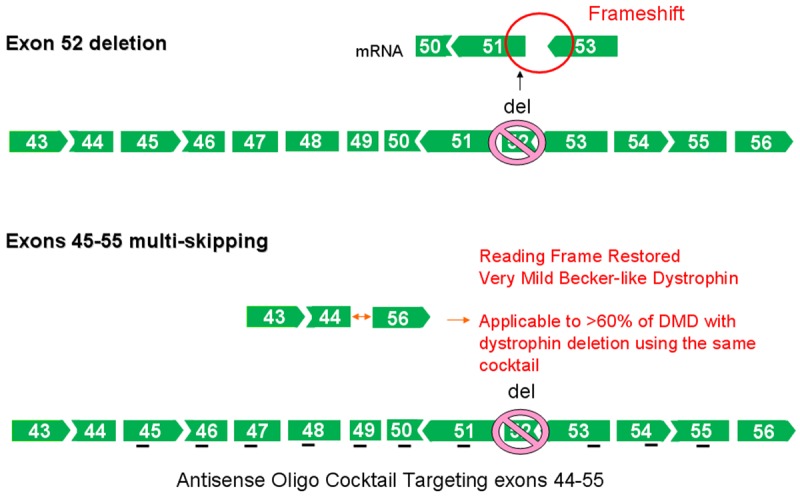
Exons 45-55 multi-skipping of exon 52 mRNA deletion. The loss of dystrophin protein as a result of the frame shift mutation in dystrophin mRNA can be rescued using a cocktail of antisense oligos targeting exons 44-55. This therapy is applicable for treating >60% of DMD patients with deletion mutations.
Feline models
Dystrophin-deficient feline muscular dystrophy, which is also called the hypertrophic feline muscular dystrophy (HFMD), was first reported in mixed shorthair male cats by Carpenter et al. [91]. Subsequently study by Winand et al. identified the feline DMD mutation as a deletion of promoter region and exon 1 in muscle and Purkinje isoforms [92]. Similar deletion mutation of promoter region and first exon in muscle and neuronal dystrophin isoforms was recently reported in a different pedigree [93]. While a colony of another HFMD was established at the University of Bern, Switzerland for detailed characterization, the mutation of the DMD gene has not been identified within the model [94-99]. Phenotypically, the HFMD is characterized by marked muscle hypertrophy, less fibrosis, and stiff gait. Marked histological change in HFMD heart is indicated by mineralization of myocardium [100]. Cardiac enlargement and hypertrophy were also found in affected cats with the HFMD at 9 and 12 months old, respectively. However, examination via electrocardiogram did not show the obvious abnormalities observed commonly in humans and canine models. A feature of the HFMD is lethal peracute rhabdomyolysis with severe hyperkalemia during anesthesia with an inhalation anesthetic called isoflurane [101]. Rhabdomyolysis is also reported in DMD and BMD patients as one of life-threatening perioperative complications [102]. While the response appears to be due to increased sensitivity of dystrophic muscles to anesthetic agents, the exact mechanism has not been revealed. Similar rhabdomyolysis during inhalation of anesthesia has not been reported in other dystrophin-deficient animal models. The unique symptom displayed by the feline model will be useful for examining preventative measures against perioperative complications in DMD and BMD patients. While the HFMD cats have unique advantages for use in DMD studies, their mutations and phenotypes still require greater understanding prior to common use in translational research.
Conclusion
The current GRMD and CXMDJ models possess clinical and histological phenotypes that mirror the severe phenotypes in skeletal and cardiac muscles of human DMD patients. However, in the current canine models, remarkable phenotypic variation and a limitation of one rare mutation still need to be considered for studies. Recently a dystrophin-deficient porcine model with a common mutation in the human DMD gene is developed using a combination of genetic engineering with cloning technology. While no therapeutic uses of the porcine model have been reported, this model could become a new large animal model of DMD because of its mutation type and phenotypic similarity to DMD patients. The potential of porcine models is highly anticipated and its success will be enhanced by our ability to stably produce genetically identical (clone) dystrophic pigs. This will allow us to examine pathogenesis of the disease and therapeutic effects of new drugs and applications more closely than the existing mammalian models. Together the dystrophin-deficient canine and porcine models will be important tools for future studies into therapies for DMD.
Acknowledgements
This work is supported by Muscular Dystrophy Canada, The Friends of Garret Cumming Research Fund, HM Toupin Neurological Research Fund, Canadian Institutes of Health Research (CIHR), Alberta Innovates: Health Solutions (AIHS), and Women and Children’s Health Research Institute (WCHRI). The project is supported financially through AIHS Summer Studentship Award, JSPS Postdoctoral Fellowships for Research Abroad.
References
- 1.Duchenne The Pathology of Paralysis with Muscular Degeneration (Paralysie Myosclerotique), or Paralysis with Apparent Hypertrophy. Br Med J. 1867;2:541–542. doi: 10.1136/bmj.2.363.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moser H. Duchenne muscular dystrophy: pathogenetic aspects and genetic prevention. Hum Genet. 1984;66:17–40. doi: 10.1007/BF00275183. [DOI] [PubMed] [Google Scholar]
- 3.Flanigan KM. Duchenne and Becker Muscular Dystrophies. Neurol Clin. 2014;32:671–688. doi: 10.1016/j.ncl.2014.05.002. [DOI] [PubMed] [Google Scholar]
- 4.Bladen CL, Salgado D, Monges S, Foncuberta ME, Kekou K, Kosma K, Dawkins H, Lamont L, Roy AJ, Chamova T, Guergueltcheva V, Chan S, Korngut L, Campbell C, Dai Y, Wang J, Barisic N, Brabec P, Lahdetie J, Walter MC, Schreiber-Katz O, Karcagi V, Garami M, Viswanathan V, Bayat F, Buccella F, Kimura E, Koeks Z, van den Bergen JC, Rodrigues M, Roxburgh R, Lusakowska A, Kostera-Pruszczyk A, Zimowski J, Santos R, Neagu E, Artemieva S, Rasic VM, Vojinovic D, Posada M, Bloetzer C, Jeannet PY, Joncourt F, Diaz-Manera J, Gallardo E, Karaduman AA, Topaloglu H, Sherif RE, Stringer A, Shatillo AV, Martin AS, Peay HL, Bellgard MI, Kirschner J, Flanigan KM, Straub V, Bushby K, Verschuuren J, Aartsma-Rus A, Beroud C, Lochmuller H. The TREAT-NMD DMD Global database: Analysis of More Than 7000 Duchenne Muscular Dystrophy Mutations. Hum Mutat. 2015;36:395–402. doi: 10.1002/humu.22758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goemans N, Buyse G. Current treatment and management of dystrophinopathies. Curr Treat Options Neurol. 2014;16:287. doi: 10.1007/s11940-014-0287-4. [DOI] [PubMed] [Google Scholar]
- 6.Kornegay JN, Spurney CF, Nghiem PP, Brinkmeyer-Langford CL, Hoffman EP, Nagaraju K. Pharmacologic management of Duchenne muscular dystrophy: target identification and preclinical trials. ILAR J. 2014;55:119–149. doi: 10.1093/ilar/ilu011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jarmin S, Kymalainen H, Popplewell L, Dickson G. New developments in the use of gene therapy to treat Duchenne muscular dystrophy. Expert Opin Biol Ther. 2014;14:209–230. doi: 10.1517/14712598.2014.866087. [DOI] [PubMed] [Google Scholar]
- 8.Lu QL, Cirak S, Partridge T. What Can We Learn From Clinical Trials of Exon Skipping for DMD? Mol Ther Nucleic Acids. 2014;3:e152. doi: 10.1038/mtna.2014.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Willmann R, Possekel S, Dubach-Powell J, Meier T, Ruegg MA. Mammalian animal models for Duchenne muscular dystrophy. Neuromuscul Disord. 2009;19:241–249. doi: 10.1016/j.nmd.2008.11.015. [DOI] [PubMed] [Google Scholar]
- 10.Nakamura A, Takeda S. Mammalian models of Duchenne Muscular Dystrophy: pathological characteristics and therapeutic applications. J Biomed Biotechnol. 2011;2011:184393. doi: 10.1155/2011/184393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nakamura K, Fujii W, Tsuboi M, Tanihata J, Teramoto N, Takeuchi S, Naito K, Yamanouchi K, Nishihara M. Generation of muscular dystrophy model rats with a CRISPR/Cas system. Sci Rep. 2014;4:5635. doi: 10.1038/srep05635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Larcher T, Lafoux A, Tesson L, Remy S, Thepenier V, Francois V, Le Guiner C, Goubin H, Dutilleul M, Guigand L, Toumaniantz G, De Cian A, Boix C, Renaud JB, Cherel Y, Giovannangeli C, Concordet JP, Anegon I, Huchet C. Characterization of Dystrophin Deficient Rats: A New Model for Duchenne Muscular Dystrophy. PLoS One. 2014;9:e110371. doi: 10.1371/journal.pone.0110371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klymiuk N, Blutke A, Graf A, Krause S, Burkhardt K, Wuensch A, Krebs S, Kessler B, Zakhartchenko V, Kurome M, Kemter E, Nagashima H, Schoser B, Herbach N, Blum H, Wanke R, Aartsma-Rus A, Thirion C, Lochmuller H, Walter MC, Wolf E. Dystrophin-deficient pigs provide new insights into the hierarchy of physiological derangements of dystrophic muscle. Hum Mol Genet. 2013;22:4368–4382. doi: 10.1093/hmg/ddt287. [DOI] [PubMed] [Google Scholar]
- 14.Hollinger K, Yang CX, Montz RE, Nonneman D, Ross JW, Selsby JT. Dystrophin insufficiency causes selective muscle histopathology and loss of dystrophin-glycoprotein complex assembly in pig skeletal muscle. FASEB J. 2014;28:1600–9. doi: 10.1096/fj.13-241141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caskey CT, Nussbaum RL, Cohan LC, Pollack L. Sporadic occurrence of Duchenne muscular dystrophy: evidence for new mutation. Clin Genet. 1980;18:329–341. doi: 10.1111/j.1399-0004.1980.tb02293.x. [DOI] [PubMed] [Google Scholar]
- 16.Barbujani G, Russo A, Danieli GA, Spiegler AW, Borkowska J, Petrusewicz IH. Segregation analysis of 1885 DMD families: significant departure from the expected proportion of sporadic cases. Hum Genet. 1990;84:522–526. doi: 10.1007/BF00210802. [DOI] [PubMed] [Google Scholar]
- 17.Ito D, Kitagawa M, Jeffery N, Okada M, Yoshida M, Kobayashi M, Nakamura A, Watari T. Dystrophin-deficient muscular dystrophy in an Alaskan malamute. Vet Rec. 2011;169:127. doi: 10.1136/vr.d2693. [DOI] [PubMed] [Google Scholar]
- 18.Shelton GD, Liu LA, Guo LT, Smith GK, Christiansen JS, Thomas WB, Smith MO, Kline KL, March PA, Flegel T, Engvall E. Muscular Dystrophy in Female Dogs. J Vet Intern Med. 2001;15:240–244. doi: 10.1892/0891-6640(2001)015<0240:mdifd>2.3.co;2. [DOI] [PubMed] [Google Scholar]
- 19.van Ham LML, Desmidt M, Tshamala M, Hoorens JK, Mattheeuws DRG. Canine X-linked muscular-dystrophy in belgian groenendaeler shepherds. J Am Anim Hosp Assoc. 1993;29:570–574. [Google Scholar]
- 20.Walmsley GL, Arechavala-Gomeza V, Fernandez-Fuente M, Burke MM, Nagel N, Holder A, Stanley R, Chandler K, Marks SL, Muntoni F, Shelton GD, Piercy RJ. A duchenne muscular dystrophy gene hot spot mutation in dystrophin-deficient cavalier king charles spaniels is amenable to exon 51 skipping. PLoS One. 2010;5:e8647. doi: 10.1371/journal.pone.0008647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kornegay JN, Bogan JR, Bogan DJ, Childers MK, Li J, Nghiem P, Detwiler DA, Larsen CA, Grange RW, Bhavaraju-Sanka RK, Tou S, Keene BP, Howard JF Jr, Wang J, Fan Z, Schatzberg SJ, Styner MA, Flanigan KM, Xiao X, Hoffman EP. Canine models of Duchenne muscular dystrophy and their use in therapeutic strategies. Mamm Genome. 2012;23:85–108. doi: 10.1007/s00335-011-9382-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schatzberg SJ, Olby NJ, Breen M, Anderson LV, Langford CF, Dickens HF, Wilton SD, Zeiss CJ, Binns MM, Kornegay JN, Morris GE, Sharp NJ. Molecular analysis of a spontaneous dystrophin ‘knockout’ dog. Neuromuscul Disord. 1999;9:289–295. doi: 10.1016/s0960-8966(99)00011-5. [DOI] [PubMed] [Google Scholar]
- 23.Sharp NJ, Kornegay JN, Van Camp SD, Herbstreith MH, Secore SL, Kettle S, Hung WY, Constantinou CD, Dykstra MJ, Roses AD, et al. An error in dystrophin mRNA processing in golden retriever muscular dystrophy, an animal homologue of Duchenne muscular dystrophy. Genomics. 1992;13:115–121. doi: 10.1016/0888-7543(92)90210-j. [DOI] [PubMed] [Google Scholar]
- 24.Klarenbeek S, Gerritzen-Bruning MJ, Rozemuller AJ, van der Lugt JJ. Canine X-linked muscular dystrophy in a family of Grand Basset Griffon Vendeen dogs. J Comp Pathol. 2007;137:249–252. doi: 10.1016/j.jcpa.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 25.Wentink GH, van der Linde-Sipman JS, Meijer AEFH, Kamphuisen HAC, van Vorstenbosch CJAHV, Hartman W, J HH. Myopathy with a possible recessive X-linked inheritance in a litter of Irish Terriers. Vet Pathol. 1972;9:328–349. doi: 10.1177/030098587200900504. [DOI] [PubMed] [Google Scholar]
- 26.Atencia-Fernandez S, Shiel RE, Mooney CT, Nolan CM. Muscular dystrophy in the Japanese Spitz: an inversion disrupts the DMD and RPGR genes. Anim Genet. 2015;46:175–84. doi: 10.1111/age.12266. [DOI] [PubMed] [Google Scholar]
- 27.Shin JH, Greer B, Hakim CH, Zhou Z, Chung YC, Duan Y, He Z, Duan D. Quantitative phenotyping of Duchenne muscular dystrophy dogs by comprehensive gait analysis and overnight activity monitoring. PLoS One. 2013;8:e59875. doi: 10.1371/journal.pone.0059875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bergman RL, Inzana KD, Monroe WE, Shell LG, Liu LA, Engvall E, Shelton GD. Dystrophin-deficient muscular dystrophy in a Labrador retriever. J Am Anim Hosp Assoc. 2002;38:255–261. doi: 10.5326/0380255. [DOI] [PubMed] [Google Scholar]
- 29.Baroncelli AB, Abellonio F, Pagano TB, Esposito I, Peirone B, Papparella S, Paciello O. Muscular dystrophy in a dog resembling human becker muscular dystrophy. J Comp Pathol. 2014;150:429–433. doi: 10.1016/j.jcpa.2013.12.006. [DOI] [PubMed] [Google Scholar]
- 30.Giannasi C, Tappin SW, Guo LT, Shelton GD, Palus V. Dystrophin-deficient muscular dystrophy in two lurcher siblings. J Small Anim Pract. 2015;56:577–580. doi: 10.1111/jsap.12331. [DOI] [PubMed] [Google Scholar]
- 31.Beltran E, Shelton GD, Guo LT, Dennis R, Sanchez-Masian D, Robinson D, De Risio L. Dystrophin-deficient muscular dystrophy in a Norfolk terrier. J Small Anim Pract. 2015;56:351–4. doi: 10.1111/jsap.12292. [DOI] [PubMed] [Google Scholar]
- 32.Wieczorek LA, Garosi LS, Shelton GD. Dystrophin-deficient muscular dystrophy in an old English sheepdog. Vet Rec. 2006;158:270–273. doi: 10.1136/vr.158.8.270. [DOI] [PubMed] [Google Scholar]
- 33.Smith BF, Yue Y, Woods PR, Kornegay JN, Shin JH, Williams RR, Duan D. An intronic LINE-1 element insertion in the dystrophin gene aborts dystrophin expression and results in Duchenne-like muscular dystrophy in the corgi breed. Lab Invest. 2011;91:216–231. doi: 10.1038/labinvest.2010.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wetterman CA, Harkin KR, Cash WC, Nietfield JC, Shelton GD. Hypertrophic muscular dystrophy in a young dog. J Am Vet Med Assoc. 2000;216:878–881. 864. doi: 10.2460/javma.2000.216.878. [DOI] [PubMed] [Google Scholar]
- 35.Baltzer WI, Calise DV, Levine JM, Shelton GD, Edwards JF, Steiner JM. Dystrophin-deficient muscular dystrophy in a Weimaraner. J Am Anim Hosp Assoc. 2007;43:227–232. doi: 10.5326/0430227. [DOI] [PubMed] [Google Scholar]
- 36.Mah JK, Korngut L, Dykeman J, Day L, Pringsheim T, Jette N. A systematic review and meta-analysis on the epidemiology of Duchenne and Becker muscular dystrophy. Neuromuscul Disord. 2014;24:482–491. doi: 10.1016/j.nmd.2014.03.008. [DOI] [PubMed] [Google Scholar]
- 37.Shimatsu Y, Katagiri K, Furuta T, Nakura M, Tanioka Y, Yuasa K, Tomohiro M, Kornegay JN, Nonaka I, Takeda S. Canine X-linked muscular dystrophy in Japan (CXMDJ) Exp Anim. 2003;52:93–97. doi: 10.1538/expanim.52.93. [DOI] [PubMed] [Google Scholar]
- 38.Passamano L, Taglia A, Palladino A, Viggiano E, D’Ambrosio P, Scutifero M, Rosaria Cecio M, Torre V, DE Luca F, Picillo E, Paciello O, Piluso G, Nigro G, Politano L. Improvement of survival in Duchenne Muscular Dystrophy: retrospective analysis of 835 patients. Acta Myol. 2012;31:121–125. [PMC free article] [PubMed] [Google Scholar]
- 39.Flanigan KM, Dunn DM, von Niederhausern A, Soltanzadeh P, Gappmaier E, Howard MT, Sampson JB, Mendell JR, Wall C, King WM, Pestronk A, Florence JM, Connolly AM, Mathews KD, Stephan CM, Laubenthal KS, Wong BL, Morehart PJ, Meyer A, Finkel RS, Bonnemann CG, Medne L, Day JW, Dalton JC, Margolis MK, Hinton VJ, Weiss RB. Mutational spectrum of DMD mutations in dystrophinopathy patients: application of modern diagnostic techniques to a large cohort. Hum Mutat. 2009;30:1657–1666. doi: 10.1002/humu.21114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nguyen F, Cherel Y, Guigand L, Goubault-Leroux I, Wyers M. Muscle lesions associated with dystrophin deficiency in neonatal golden retriever puppies. J Comp Pathol. 2002;126:100–108. doi: 10.1053/jcpa.2001.0526. [DOI] [PubMed] [Google Scholar]
- 41.Passerini L, Bernasconi P, Baggi F, Confalonieri P, Cozzi F, Cornelio F, Mantegazza R. Fibrogenic cytokines and extent of fibrosis in muscle of dogs with X-linked golden retriever muscular dystrophy. Neuromuscul Disord. 2002;12:828–835. doi: 10.1016/s0960-8966(02)00071-8. [DOI] [PubMed] [Google Scholar]
- 42.Valentine BA, Cooper BJ, de Lahunta A, O’Quinn R, Blue JT. Canine X-linked muscular dystrophy. An animal model of Duchenne muscular dystrophy: clinical studies. J Neurol Sci. 1988;88:69–81. doi: 10.1016/0022-510x(88)90206-7. [DOI] [PubMed] [Google Scholar]
- 43.Spurney CF. Cardiomyopathy of Duchenne muscular dystrophy: current understanding and future directions. Muscle Nerve. 2011;44:8–19. doi: 10.1002/mus.22097. [DOI] [PubMed] [Google Scholar]
- 44.Valentine BA, Cummings JF, Cooper BJ. Development of Duchenne-type cardiomyopathy. Morphologic studies in a canine model. Am J Pathol. 1989;135:671–678. [PMC free article] [PubMed] [Google Scholar]
- 45.DeVanna JC, Kornegay JN, Bogan DJ, Bogan JR, Dow JL, Hawkins EC. Respiratory dysfunction in unsedated dogs with golden retriever muscular dystrophy. Neuromuscul Disord. 2014;24:63–73. doi: 10.1016/j.nmd.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mead AF, Petrov M, Malik AS, Mitchell MA, Childers MK, Bogan JR, Seidner G, Kornegay JN, Stedman HH. Diaphragm remodeling and compensatory respiratory mechanics in a canine model of Duchenne muscular dystrophy. J Appl Physiol (1985) 2014;116:807–815. doi: 10.1152/japplphysiol.00833.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kornegay JN, Bogan JR, Bogan DJ, Childers MK, Grange RW. Golden retriever muscular dystrophy (GRMD): Developing and maintaining a colony and physiological functional measurements. Methods Mol Biol. 2011;709:105–123. doi: 10.1007/978-1-61737-982-6_7. [DOI] [PubMed] [Google Scholar]
- 48.Shimatsu Y, Yoshimura M, Yuasa K, Urasawa N, Tomohiro M, Nakura M, Tanigawa M, Nakamura A, Takeda S. Major clinical and histopathological characteristics of canine X-linked muscular dystrophy in Japan, CXMDJ. Acta Myol. 2005;24:145–154. [PubMed] [Google Scholar]
- 49.Yugeta N, Urasawa N, Fujii Y, Yoshimura M, Yuasa K, Wada MR, Nakura M, Shimatsu Y, Tomohiro M, Takahashi A, Machida N, Wakao Y, Nakamura A, Takeda S. Cardiac involvement in Beagle-based canine X-linked muscular dystrophy in Japan (CXMDJ): electrocardiographic, echocardiographic, and morphologic studies. BMC Cardiovasc Disord. 2006;6:47. doi: 10.1186/1471-2261-6-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Urasawa N, Wada MR, Machida N, Yuasa K, Shimatsu Y, Wakao Y, Yuasa S, Sano T, Nonaka I, Nakamura A, Takeda S. Selective vacuolar degeneration in dystrophin-deficient canine Purkinje fibers despite preservation of dystrophin-associated proteins with overexpression of Dp71. Circulation. 2008;117:2437–2448. doi: 10.1161/CIRCULATIONAHA.107.739326. [DOI] [PubMed] [Google Scholar]
- 51.Nakamura A, Kobayashi M, Kuraoka M, Yuasa K, Yugeta N, Okada T, Takeda S. Initial pulmonary respiration causes massive diaphragm damage and hyper-CKemia in Duchenne muscular dystrophy dog. Sci Rep. 2013;3:2183. doi: 10.1038/srep02183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Duan D. Duchenne muscular dystrophy gene therapy in the canine model. Hum Gene Ther Clin Dev. 2015;26:57–69. doi: 10.1089/humc.2015.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yokota T, Pistilli E, Duddy W, Nagaraju K. Potential of oligonucleotide-mediated exonskipping therapy for Duchenne muscular dystrophy. Expert Opin Biol Ther. 2007;7:831–842. doi: 10.1517/14712598.7.6.831. [DOI] [PubMed] [Google Scholar]
- 54.Pramono ZA, Takeshima Y, Alimsardjono H, Ishii A, Takeda S, Matsuo M. Induction of exon skipping of the dystrophin transcript in lymphoblastoid cells by transfecting an antisense oligodeoxynucleotide complementary to an exon recognition sequence. Biochem Biophys Res Commun. 1996;226:445–449. doi: 10.1006/bbrc.1996.1375. [DOI] [PubMed] [Google Scholar]
- 55.Yokota T, Hoffman E, Takeda S. Antisense oligo-mediated multiple exon skipping in a dog model of Duchenne muscular dystrophy. Methods Mol Biol. 2011;709:299–312. doi: 10.1007/978-1-61737-982-6_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vulin A, Barthelemy I, Goyenvalle A, Thibaud JL, Beley C, Griffith G, Benchaouir R, le Hir M, Unterfinger Y, Lorain S, Dreyfus P, Voit T, Carlier P, Blot S, Garcia L. Muscle function recovery in golden retriever muscular dystrophy after AAV1-U7 exon skipping. Mol Ther. 2012;20:2120–2133. doi: 10.1038/mt.2012.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bish LT, Sleeper MM, Forbes SC, Wang B, Reynolds C, Singletary GE, Trafny D, Morine KJ, Sanmiguel J, Cecchini S, Virag T, Vulin A, Beley C, Bogan J, Wilson JM, Vandenborne K, Kornegay JN, Walter GA, Kotin RM, Garcia L, Sweeney HL. Long-term restoration of cardiac dystrophin expression in golden retriever muscular dystrophy following rAAV6-mediated exon skipping. Mol Ther. 2012;20:580–589. doi: 10.1038/mt.2011.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Su JB, Cazorla O, Blot S, Blanchard-Gutton N, Ait Mou Y, Barthelemy I, Sambin L, Sampedrano CC, Gouni V, Unterfinger Y, Aguilar P, Thibaud JL, Bize A, Pouchelon JL, Dabire H, Ghaleh B, Berdeaux A, Chetboul V, Lacampagne A, Hittinger L. Bradykinin restores left ventricular function, sarcomeric protein phosphorylation, and e/nNOS levels in dogs with Duchenne muscular dystrophy cardiomyopathy. Cardiovasc Res. 2012;95:86–96. doi: 10.1093/cvr/cvs161. [DOI] [PubMed] [Google Scholar]
- 59.Barbash IM, Cecchini S, Faranesh AZ, Virag T, Li L, Yang Y, Hoyt RF, Kornegay JN, Bogan JR, Garcia L, Lederman RJ, Kotin RM. MRI roadmap-guided transendocardial delivery of exon-skipping recombinant adeno-associated virus restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Gene Ther. 2013;20:274–282. doi: 10.1038/gt.2012.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wuebbles RD, Sarathy A, Kornegay JN, Burkin DJ. Levels of alpha7 integrin and laminin-alpha2 are increased following prednisone treatment in the mdx mouse and GRMD dog models of Duchenne muscular dystrophy. Dis Model Mech. 2013;6:1175–1184. doi: 10.1242/dmm.012211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Araujo KP, Bonuccelli G, Duarte CN, Gaiad TP, Moreira DF, Feder D, Belizario JE, Miglino MA, Lisanti MP, Ambrosio CE. Bortezomib (PS-341) treatment decreases inflammation and partially rescues the expression of the dystrophin-glycoprotein complex in GRMD dogs. PLoS One. 2013;8:e61367. doi: 10.1371/journal.pone.0061367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Saito T, Nakamura A, Aoki Y, Yokota T, Okada T, Osawa M, Takeda S. Antisense PMO found in dystrophic dog model was effective in cells from exon 7-deleted DMD patient. PLoS One. 2010;5:e12239. doi: 10.1371/journal.pone.0012239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yokota T, Lu QL, Partridge T, Kobayashi M, Nakamura A, Takeda S, Hoffman E. Efficacy of systemic morpholino exon-skipping in Duchenne dystrophy dogs. Ann Neurol. 2009;65:667–676. doi: 10.1002/ana.21627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yokota T, Nakamura A, Nagata T, Saito T, Kobayashi M, Aoki Y, Echigoya Y, Partridge T, Hoffman EP, Takeda S. Extensive and prolonged restoration of dystrophin expression with vivo-morpholino-mediated multiple exon skipping in dystrophic dogs. Nucleic Acid Ther. 2012;22:306–315. doi: 10.1089/nat.2012.0368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hayashita-Kinoh H, Yugeta N, Okada H, Nitahara-Kasahara Y, Chiyo T, Okada T, Takeda S. Intra-amniotic rAAV-mediated microdystrophin gene transfer improves canine X-linked muscular dystrophy and may induce immune tolerance. Mol Ther. 2015;23:627–37. doi: 10.1038/mt.2015.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sakamoto M, Yuasa K, Yoshimura M, Yokota T, Ikemoto T, Suzuki M, Dickson G, Miyagoe-Suzuki Y, Takeda S. Micro-dystrophin cDNA ameliorates dystrophic phenotypes when introduced into mdx mice as a transgene. Biochem Biophys Res Commun. 2002;293:1265–1272. doi: 10.1016/S0006-291X(02)00362-5. [DOI] [PubMed] [Google Scholar]
- 67.Ambrosio CE, Valadares MC, Zucconi E, Cabral R, Pearson PL, Gaiad TP, Canovas M, Vainzof M, Miglino MA, Zatz M. Ringo, a Golden Retriever Muscular Dystrophy (GRMD) dog with absent dystrophin but normal strength. Neuromuscul Disord. 2008;18:892–893. doi: 10.1016/j.nmd.2008.06.385. [DOI] [PubMed] [Google Scholar]
- 68.Ambrosio CE, Fadel L, Gaiad TP, Martins DS, Araujo KP, Zucconi E, Brolio MP, Giglio RF, Morini AC, Jazedje T, Froes TR, Feitosa ML, Valadares MC, Beltrao-Braga PC, Meirelles FV, Miglino MA. Identification of three distinguishable phenotypes in golden retriever muscular dystrophy. Genet Mol Res. 2009;8:389–396. doi: 10.4238/vol8-2gmr581. [DOI] [PubMed] [Google Scholar]
- 69.Bello L, Kesari A, Gordish-Dressman H, Cnaan A, Morgenroth LP, Punetha J, Duong T, Henricson EK, Pegoraro E, McDonald CM, Hoffman EP. Genetic modifiers of ambulation in the CINRG duchenne natural history study. Ann Neurol. 2015;77:684–96. doi: 10.1002/ana.24370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Miyazato LG, Moraes JR, Beretta DC, Kornegay JN. Muscular dystrophy in dogs: does the crossing of breeds influence disease phenotype? Vet Pathol. 2011;48:655–662. doi: 10.1177/0300985810387070. [DOI] [PubMed] [Google Scholar]
- 71.Jang G, Kim MK, Lee BC. Current status and applications of somatic cell nuclear transfer in dogs. Theriogenology. 2010;74:1311–1320. doi: 10.1016/j.theriogenology.2010.05.036. [DOI] [PubMed] [Google Scholar]
- 72.Fan N, Lai L. Genetically modified pig models for human diseases. J Genet Genomics. 2013;40:67–73. doi: 10.1016/j.jgg.2012.07.014. [DOI] [PubMed] [Google Scholar]
- 73.Gun G, Kues WA. Current progress of genetically engineered pig models for biomedical research. Biores Open Access. 2014;3:255–264. doi: 10.1089/biores.2014.0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lelovas PP, Kostomitsopoulos NG, Xanthos TT. A comparative anatomic and physiologic overview of the porcine heart. J Am Assoc Lab Anim Sci. 2014;53:432–438. [PMC free article] [PubMed] [Google Scholar]
- 75.Nonneman DJ, Brown-Brandl T, Jones SA, Wiedmann RT, Rohrer GA. A defect in dystrophin causes a novel porcine stress syndrome. BMC Genomics. 2012;13:233. doi: 10.1186/1471-2164-13-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kurome M, Geistlinger L, Kessler B, Zakhartchenko V, Klymiuk N, Wuensch A, Richter A, Baehr A, Kraehe K, Burkhardt K, Flisikowski K, Flisikowska T, Merkl C, Landmann M, Durkovic M, Tschukes A, Kraner S, Schindelhauer D, Petri T, Kind A, Nagashima H, Schnieke A, Zimmer R, Wolf E. Factors influencing the efficiency of generating genetically engineered pigs by nuclear transfer: multi-factorial analysis of a large data set. BMC Biotechnol. 2013;13:43. doi: 10.1186/1472-6750-13-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mendell JR, Shilling C, Leslie ND, Flanigan KM, al-Dahhak R, Gastier-Foster J, Kneile K, Dunn DM, Duval B, Aoyagi A, Hamil C, Mahmoud M, Roush K, Bird L, Rankin C, Lilly H, Street N, Chandrasekar R, Weiss RB. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann Neurol. 2012;71:304–313. doi: 10.1002/ana.23528. [DOI] [PubMed] [Google Scholar]
- 78.Zhao J, Whyte J, Prather RS. Effect of epigenetic regulation during swine embryogenesis and on cloning by nuclear transfer. Cell Tissue Res. 2010;341:13–21. doi: 10.1007/s00441-010-1000-x. [DOI] [PubMed] [Google Scholar]
- 79.Cho SK, Kim JH, Park JY, Choi YJ, Bang JI, Hwang KC, Cho EJ, Sohn SH, Uhm SJ, Koo DB, Lee KK, Kim T, Kim JH. Serial cloning of pigs by somatic cell nuclear transfer: restoration of phenotypic normality during serial cloning. Dev Dyn. 2007;236:3369–3382. doi: 10.1002/dvdy.21308. [DOI] [PubMed] [Google Scholar]
- 80.Aartsma-Rus A, Bremmer-Bout M, Janson AA, den Dunnen JT, van Ommen GJ, van Deutekom JC. Targeted exon skipping as a potential gene correction therapy for Duchenne muscular dystrophy. Neuromuscul Disord. 2002;12(Suppl 1):S71–77. doi: 10.1016/s0960-8966(02)00086-x. [DOI] [PubMed] [Google Scholar]
- 81.Arechavala-Gomeza V, Graham IR, Popplewell LJ, Adams AM, Aartsma-Rus A, Kinali M, Morgan JE, van Deutekom JC, Wilton SD, Dickson G, Muntoni F. Comparative analysis of antisense oligonucleotide sequences for targeted skipping of exon 51 during dystrophin pre-mRNA splicing in human muscle. Hum Gene Ther. 2007;18:798–810. doi: 10.1089/hum.2006.061. [DOI] [PubMed] [Google Scholar]
- 82.Aoki Y, Nakamura A, Yokota T, Saito T, Okazawa H, Nagata T, Takeda S. In-frame dystrophin following exon 51-skipping improves muscle pathology and function in the exon 52-deficient mdx mouse. Mol Ther. 2010;18:1995–2005. doi: 10.1038/mt.2010.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Aoki Y, Nagata T, Yokota T, Nakamura A, Wood MJ, Partridge T, Takeda S. Highly efficient in vivo delivery of PMO into regenerating myotubes and rescue in laminin-alpha2 chain-null congenital muscular dystrophy mice. Hum Mol Genet. 2013;22:4914–4928. doi: 10.1093/hmg/ddt341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Beroud C, Tuffery-Giraud S, Matsuo M, Hamroun D, Humbertclaude V, Monnier N, Moizard MP, Voelckel MA, Calemard LM, Boisseau P, Blayau M, Philippe C, Cossee M, Pages M, Rivier F, Danos O, Garcia L, Claustres M. Multiexon skipping leading to an artificial DMD protein lacking amino acids from exons 45 through 55 could rescue up to 63% of patients with Duchenne muscular dystrophy. Hum Mutat. 2007;28:196–202. doi: 10.1002/humu.20428. [DOI] [PubMed] [Google Scholar]
- 85.Nakamura A, Yoshida K, Fukushima K, Ueda H, Urasawa N, Koyama J, Yazaki Y, Yazaki M, Sakai T, Haruta S, Takeda S, Ikeda S. Follow-up of three patients with a large in-frame deletion of exons 45-55 in the Duchenne muscular dystrophy (DMD) gene. J Clin Neurosci. 2008;15:757–763. doi: 10.1016/j.jocn.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 86.Yokota T, Takeda S, Lu QL, Partridge TA, Nakamura A, Hoffman EP. A renaissance for antisense oligonucleotide drugs in neurology: exon skipping breaks new ground. Arch Neurol. 2009;66:32–38. doi: 10.1001/archneurol.2008.540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Aoki Y, Yokota T, Nagata T, Nakamura A, Tanihata J, Saito T, Duguez SM, Nagaraju K, Hoffman EP, Partridge T, Takeda S. Bodywide skipping of exons 45-55 in dystrophic mdx52 mice by systemic antisense delivery. Proc Natl Acad Sci U S A. 2012;109:13763–13768. doi: 10.1073/pnas.1204638109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Echigoya Y, Aoki Y, Miskew B, Panesar D, Touznik A, Nagata T, Tanihata J, Nakamura A, Nagaraju K, Yokota T. Long-term efficacy of systemic multiexon skipping targeting dystrophin exons 45-55 with a cocktail of vivo-morpholinos in mdx52 mice. Mol Ther Nucleic Acids. 2015;4:e225. doi: 10.1038/mtna.2014.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Aoki Y, Yokota T, Wood MJ. Development of multiexon skipping antisense oligonucleotide therapy for Duchenne muscular dystrophy. Biomed Res Int. 2013;2013:402369. doi: 10.1155/2013/402369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Echigoya Y, Yokota T. Skipping Multiple Exons of Dystrophin Transcripts Using Cocktail Antisense Oligonucleotides. Nucleic Acid Ther. 2014;24:57–68. doi: 10.1089/nat.2013.0451. [DOI] [PubMed] [Google Scholar]
- 91.Carpenter JL, Hoffman EP, Romanul FC, Kunkel LM, Rosales RK, Ma NS, Dasbach JJ, Rae JF, Moore FM, McAfee MB, et al. Feline muscular dystrophy with dystrophin deficiency. Am J Pathol. 1989;135:909–919. [PMC free article] [PubMed] [Google Scholar]
- 92.Winand NJ, Edwards M, Pradhan D, Berian CA, Cooper BJ. Deletion of the dystrophin muscle promoter in feline muscular dystrophy. Neuromuscul Disord. 1994;4:433–445. doi: 10.1016/0960-8966(94)90082-5. [DOI] [PubMed] [Google Scholar]
- 93.Gambino AN, Mouser PJ, Shelton GD, Winand NJ. Emergent presentation of a cat with dystrophin-deficient muscular dystrophy. J Am Anim Hosp Assoc. 2014;50:130–135. doi: 10.5326/JAAHA-MS-5973. [DOI] [PubMed] [Google Scholar]
- 94.Gaschen FP, Hoffman EP, Gorospe JR, Uhl EW, Senior DF, Cardinet GH 3rd, Pearce LK. Dystrophin deficiency causes lethal muscle hypertrophy in cats. J Neurol Sci. 1992;110:149–159. doi: 10.1016/0022-510x(92)90022-d. [DOI] [PubMed] [Google Scholar]
- 95.Gaschen F, Burgunder JM. Changes of skeletal muscle in young dystrophin-deficient cats: a morphological and morphometric study. Acta Neuropathol. 2001;101:591–600. doi: 10.1007/s004010000299. [DOI] [PubMed] [Google Scholar]
- 96.Seiler G, Welle M, Busato A, Lin S, Gaschen FP. Mast cell proliferation and alterations in bFGF amount and localization are involved in the response of muscle to dystrophin deficiency in hypertrophic feline dystrophy. Neuromuscul Disord. 2001;11:56–71. doi: 10.1016/s0960-8966(00)00151-6. [DOI] [PubMed] [Google Scholar]
- 97.Radojevic V, Oppliger C, Gaschen F, Burgunder JM. Restoration of dystrophin expression in cultured hybrid myotubes. Neuropathol Appl Neurobiol. 2002;28:397–409. doi: 10.1046/j.1365-2990.2002.00409.x. [DOI] [PubMed] [Google Scholar]
- 98.Howard J, Jaggy A, Busato A, Gaschen F. Electrodiagnostic evaluation in feline hypertrophic muscular dystrophy. Vet J. 2004;168:87–92. doi: 10.1016/S1090-0233(03)00080-7. [DOI] [PubMed] [Google Scholar]
- 99.Chetboul V, Blot S, Sampedrano CC, Thibaud JL, Granger N, Tissier R, Bruneval P, Gaschen F, Gouni V, Nicolle AP, Pouchelon JL. Tissue Doppler imaging for detection of radial and longitudinal myocardial dysfunction in a family of cats affected by dystrophin-deficient hypertrophic muscular dystrophy. J Vet Intern Med. 2006;20:640–647. doi: 10.1892/0891-6640(2006)20[640:tdifdo]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 100.Gaschen L, Lang J, Lin S, Ade-Damilano M, Busato A, Lombard CW, Gaschen FP. Cardiomyopathy in dystrophin-deficient hypertrophic feline muscular dystrophy. J Vet Intern Med. 1999;13:346–356. doi: 10.1892/0891-6640(1999)013<0346:ciddhf>2.3.co;2. [DOI] [PubMed] [Google Scholar]
- 101.Gaschen F, Gaschen L, Seiler G, Welle M, Jaunin VB, Jmaa DG, Neiger-Aeschbacher G, Ade-Damilano M. Lethal peracute rhabdomyolysis associated with stress and general anesthesia in three dystrophin-deficient cats. Vet Pathol. 1998;35:117–123. doi: 10.1177/030098589803500205. [DOI] [PubMed] [Google Scholar]
- 102.Segura LG, Lorenz JD, Weingarten TN, Scavonetto F, Bojanic K, Selcen D, Sprung J. Anesthesia and Duchenne or Becker muscular dystrophy: review of 117 anesthetic exposures. Paediatr Anaesth. 2013;23:855–864. doi: 10.1111/pan.12248. [DOI] [PubMed] [Google Scholar]
- 103.Valentine BA, Cooper BJ, Cummings JF, deLahunta A. Progressive muscular dystrophy in a golden retriever dog: light microscope and ultrastructural features at 4 and 8 months. Acta Neuropathol. 1986;71:301–310. doi: 10.1007/BF00688053. [DOI] [PubMed] [Google Scholar]
- 104.Barthelemy I, Pinto-Mariz F, Yada E, Desquilbet L, Savino W, Silva-Barbosa SD, Faussat AM, Mouly V, Voit T, Blot S, Butler-Browne G. Predictive markers of clinical outcome in the GRMD dog model of Duchenne muscular dystrophy. Dis Model Mech. 2014;7:1253–1261. doi: 10.1242/dmm.016014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kornegay JN, Bogan DJ, Bogan JR, Childers MK, Cundiff DD, Petroski GF, Schueler RO. Contraction force generated by tarsal joint flexion and extension in dogs with golden retriever muscular dystrophy. J Neurol Sci. 1999;166:115–121. doi: 10.1016/s0022-510x(99)00118-5. [DOI] [PubMed] [Google Scholar]
- 106.Kornegay JN, Cundiff DD, Bogan DJ, Bogan JR, Okamura CS. The cranial sartorius muscle undergoes true hypertrophy in dogs with golden retriever muscular dystrophy. Neuromuscul Disord. 2003;13:493–500. doi: 10.1016/s0960-8966(03)00025-7. [DOI] [PubMed] [Google Scholar]
- 107.Moise NS, Valentine BA, Brown CA, Erb HN, Beck KA, Cooper BJ, Gilmour RF. Duchenne’s cardiomyopathy in a canine model: electrocardiographic and echocardiographic studies. J Am Coll Cardiol. 1991;17:812–820. doi: 10.1016/s0735-1097(10)80202-5. [DOI] [PubMed] [Google Scholar]
- 108.Townsend D, Turner I, Yasuda S, Martindale J, Davis J, Shillingford M, Kornegay JN, Metzger JM. Chronic administration of membrane sealant prevents severe cardiac injury and ventricular dilatation in dystrophic dogs. J Clin Invest. 2010;120:1140–1150. doi: 10.1172/JCI41329. [DOI] [PMC free article] [PubMed] [Google Scholar]